

Abstract
Plitidepsin is a host-targeted compound known for inducing a strong anti-SARS-CoV-2 activity, as well as for having the capacity of reducing lung inflammation. Since IL-6 is one of the main cytokines involved in acute respiratory distress syndrome, the effect of plitidepsin in IL-6 secretion in different in vitro and in vivo experimental models was studied. A strong plitidepsin-mediated reduction of IL-6 was found in human monocyte- derived macrophages exposed to non-productive SARS-CoV-2. In resiquimod (a ligand of TLR7/8) stimulated THP1 human monocytes, plitidepsin-mediated reductions of IL-6 mRNA and IL-6 levels were also noticed. Additionally, although resiquimod-induced binding to DNA of NF-kB family members was unaffected by plitidepsin, a decrease in the regulated transcription by NF-kB (a key transcription factor involved in the inflammatory cascade) was observed. Furthermore, the phosphorylation of p65 that is required for full transcriptional NF-kB activity, was significantly reduced by plitidepsin. Moreover, decreases of IL-6 levels and other pro-inflammatory cytokines were also seen in either SARS-CoV-2 or H1N1 influenza virus infected mice, which were treated at low enough plitidepsin doses to not induce antiviral effects. In summary, plitidepsin is a promising therapeutic agent for the treatment of viral infections, not only because of its host-targeted antiviral effect, but also for its immunomodulatory effect, which were evidenced in vitro and in vivo by the decrease of pro-inflammatory cytokines.
INTRODUCTION
Very shortly after the report of the first outbreak of the ongoing Coronavirus disease 2019 (COVID-19) pandemic caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a set of host proteins that could be potential targets for COVID-19 treatment was recognized based on their ability to interact with one or more viral proteins (1). Among others, elongation factor-1A (eEF1A), a protein initially implicated in the mechanism through which plitidepsin exerts its antitumor activity (2), was identified. Plitidepsin consists of a natural cyclic depsipeptide currently obtained by total synthesis and originally isolated from the marine tunicate Aplidium albicans (2). Plitidepsin-induced antiviral effect against SARS-CoV-2 was fully confirmed in different in vitro and in vivo experiments (3–5) in which plitidepsin demonstrated a strong impact on SARS-CoV-2 replication regardless of the experimental setting used. Notably, the strong antiviral activity of plitidepsin against SARS-CoV-2 was proven to be mediated by eEF1A inhibition (4).
Based on these results, a randomized, parallel, open-label, proof-of-concept clinical trial (APLICOV; ClinicalTrials.gov Identifier: NCT04382066) was conducted (6). Preliminary efficacy data in COVID-19 patients gathered from this trial were consistent with the previously described preclinical anti-SARS-CoV-2 activity of plitidepsin. An international Phase II trial exploring the efficacy and safety of plitidepsin in immunosuppressed hospitalized patients with symptomatic COVID-19 is currently ongoing (NEREIDA; ClinicalTrials.gov Identifier: NCT05705167).
Highly pathogenic respiratory viruses, such as Middle East respiratory coronavirus (MERS-CoV), SARS-CoV, influenza viruses and SARS-CoV-2, cause inflammatory airway disease, that, if becoming excessive, can eventually lead to acute respiratory distress syndrome (ARDS) in patients (7, 8) and non-human primates (9, 10). In this regard, a direct comparison of gene expression profiles in the lungs of cynomolgus macaques that were experimentally infected with SARS-CoV, demonstrated the key role of NF-kB in acute lung inflammation and ARDS, with phosphorylated-NF-kB being highly detected in the nuclei of lung cells, regardless of the age of the infected cynomolgus (10). Interestingly, in vitro experiments indicate that treatment of tumor cells with plitidepsin inhibits NF-kB transactivation in response to TNF-α at concentrations of this cytokine as high as 50 ng/mL for 6 hours (11), suggesting that in addition to its antiviral activity, plitidepsin might also have anti-inflammatory properties.
In experimental models and patients, plitidepsin evidenced a direct antiviral effect that was associated with a reduction of lung inflammation (4, 6). While reduction of viral load is likely responsible for the reduction in inflammation, plitidepsin treatment might have also a direct anti-inflammatory effect. This might result in a therapeutic impact of plitidepsin at late stages of severe disease when viral replication is no longer the driver of pathogenesis, but disease is mainly due to prolonged and exacerbated inflammation. We then aimed at characterizing more in depth the anti-inflammatory properties of plitidepsin using in vitro and in vivo models of airway diseases.
This article summarizes the results obtained from a series of in vitro experiments aimed at describing the effect of physiologically relevant concentrations of plitidepsin on the activation of NF-kB, as well as its effect in the secretion of IL-6, a key cytokine specially over-secreted in COVID-19 patients (12) and implicated in ARDS (13). Results gathered from in vivo models, in which mice were infected with either SARS-CoV-2 or H1N1 influenza virus and treated with plitidepsin at dose levels low enough to not induce a direct antiviral effect, are also presented.
MATERIALS AND METHODS
Materials
Plitidepsin (C57H87N7O15; MW: 1110.34) was synthetized and provided by PharmaMar, S.A. (Colmenar Viejo, Madrid, Spain). For in vitro experiments, stock solutions (at 1 mg/mL in dimethyl sulfoxide) were stored at −20°C until use. For in vivo experiments, lyophilized vials were reconstituted with a solution of polyoxyl 35 hydrogenated castor oil:ethanol:water (15:15:70; v/v/v) and further diluted with sterile water for injection.
Human tumor necrosis factor-α (TNF-α) was obtained from Cell Signaling Technologies (Danvers, MA, USA). Resiquimod (RQ), lipopolysaccharide from Escherichia coli 055:B5 (LPS), interferon α (IFN-α), QUANTI-Blue™ and QUANTI-Luc™ were from InvivoGen (San Diego, CA, USA). Cell Proliferation Kit I (MTT) was from Roche Diagnostic (Basel, Switzerland). Anti-NF-κB p65 and anti-Phospho-NF-κB p65 (Ser536) antibodies were from Cell Signaling Technologies (Danvers, MA, USA). Anti-α-Tubulin antibody was from Merck (Darmstadt, Germany). HRP-conjugated goat anti-rabbit and goat anti-mouse antibodies were purchased from SouthernBiotech (Birmingham, AL, USA).
Biosafety statements
SARS-CoV-2 experiments were performed at the biosafety level 3 (BSL-3) facilities of the IRTA-CReSA Biocontainment Unit or CMCiB (Barcelona, Spain), in accordance with the Institutional Biosafety requirements and Committee (registration No. CSB 40/2020 and CSB-20–015-M3, respectively). Additional experiments in SARS-CoV-2 infected mice were conducted in a pathogen-free animal BSL-3 facility at Icahn School of Medicine at Mount Sinai, adhering to the guidelines from Institutional Animal Care and Use Committee (IACUC) of the Icahn School of Medicine at Mount Sinai (registration No. IPROTO202100000021 and SPROTO202100000032, respectively). Influenza virus infection in mice were carried out at the biosafety level 2 (BSL-2) facilities of the Institute for Research in Biomedicine (Bellinzona, Switzerland). Experiments were performed in accordance with the Swiss Federal Veterinary Office guidelines and animal protocols were approved by the local veterinary authorities (authorization No. TI 11/19).
Virus isolates
SARS-CoV-2 virus isolate GISAID ID EPI_ISL_510689 (coded as Cat01) was used in the in vitro experiments carried out with human monocyte-derived macrophages (MDM). Cat01 was isolated from a nasopharyngeal swab in March 2020 (3). The virus isolate with GISAID ID EPI_ISL_47147 (coded as Cat02) was used in animal experiments that involved SARS-CoV-2 infection. Cat02 was isolated from a patient (nasopharyngeal swab) from Spain in April 2020 (14).
Production of virus stocks, isolation, and titration were performed in Vero E6 cell line (ATCC® CRL-1586™). Virus titres were determined using a standard TCID50 assay based on the Reed and Muench method and expressed as TCID50/mL.
Influenza virus infection in mice was performed with the A/PR8/34 (H1N1) isolate, which was grown and titrated as described previously (15).
Cells
THP1-Dual™ cells (InvivoGen, San Diego, CA, USA; reference thpd-nfis) were derived from the human THP-1 monocyte cell line by stable integration of two inducible reporter constructs. THP1-Dual™ cells feature the Lucia gene, a secreted luciferase reporter gene (under control of an ISG54 minimal promoter in conjunction with IFN-stimulated response elements), and a secreted embryonic alkaline phosphatase (SEAP) reporter gene (driven by an IFN-β minimal promoter fused to five copies of the NF-κB consensus transcriptional response element and three copies of the c-Rel binding site).
Peripheral blood mononuclear cells (PBMC) were obtained with a Ficoll-Hypaque gradient (Alere Technologies AS, Oslo, Norway) from human blood donors (Blood and Tissue Bank of Catalonia) and monocyte populations (>90% CD14+) were isolated with CD14-negative selection magnetic beads (Miltenyi Biotec, Cologne, Germany). A total of 3×105 monocytes per well were seeded in 48 well plates. Human monocyte-derived macrophages (MDM) were obtained by culturing these cells in the presence of 100 μg/mL of macrophage colony-stimulating factor (PeproTech EC, Ltd., London, UK) for seven days and replacing media and cytokines every two days. On day 7, both non-exposed and SARS-CoV-2 exposed MDM (Cat 01; MOI=2) were incubated with increasing concentrations of plitidepsin (1.1, 3.3 or 10 nM).
In vitro assays
In THP1-Dual™ cell experiments, cellular viability was determined at either 18 or 24 hours after plitidepsin exposure (2.5, 5 or 10 nM) using the Cell Proliferation Kit I (Roche Diagnostic, Basel, Switzerland) and, cell survival was expressed as percentage of control cell growth. To determine whether these increasing concentrations of plitidepsin may affect the transactivation of either NF-κB, or IFN, or both, THP1-Dual™ cells were treated with RQ (10 μg/mL), TNF-α (50 ng/mL) or IFN-α (20 ng/mL) for 18 hours. Then, transactivation levels were determined by monitoring the activity of SEAP (for NF-κB) and secreted luciferase (for IFN) according to manufactureŕs instructions of QUANTI- Blue™ and QUANTI-Luc™, respectively. IL-6 levels were quantified by ELISA in the supernatants (400g, 5°C, 10 minutes) of THP1-Dual™ cells, which were pre-incubated for 8 hours with increasing concentrations of plitidepsin (1, 10 or 50 nM) and then, exposed to RQ (10 μg/mL) for an additional 16-hour period.
All measurements (luminescence or absorbance for SEAP and ELISA) were made in a Perkin-Elmer® EnVision® reader (Waltham, MA, USA).
In human MDM experiments, plitidepsin-induced (at 1.1, 3.3 or 10 nM) effects (both cytotoxic and cytokine-modulating) were measured at 24 hours after viral addition. Cytotoxicity was measured using the CellTiter-Glo luminescent cell viability assay (Promega Co, WI, USA). Luminescence was measured in a Fluoroskan Ascent FL luminometer (ThermoFisher Scientific, MA, USA). At the same time, cell supernatants were centrifuged and stored at −20°C until the cytokines were quantified according to the detailed method below.
Animal experiments
All animal protocols were reviewed and approved according to regional Institutional Animal Care and Use Committees and conducted by certified staff.
In all experiments, animals were housed in ventilated cages on a 12-hour light-dark cycle at 21 to 23°C and 40 to 60% humidity. Mice were allowed free access to an irradiated diet and sterilized water.
SARS-CoV-2 virus infection model in mice.
The key pathological features induced by SARS-CoV-2 in infected mice have been extensively described in the experimental model used here (16). We performed two independent mouse infection experiments in two different laboratories (Spain and USA). Briefly, 5- to 6-week-old B6.Cg-Tg (K18-ACE2)2Prlmn/J (k18-hACE2) mice purchased from Charles River Lab (both sexes, Spain) or from JAX labs (female, USA) were used, as well as 129S1 mice from JAX labs (female, USA). Then, isoflurane-anesthetized animals were inoculated intranasally with 103 TCID50/mouse of SARS-CoV-2 (strain Cat02, Spain) or with 104 plaque forming units (PFU) of USA-WA1/2020 (K18hACE2, USA). On day 0, mice were randomly assigned to treatment groups (plitidepsin or placebo) and then administered subcutaneously with plitidepsin at 0.15 mg/kg on days 0, 1 and 2 (Spain) or at 0.15 mg/kg or 0.3 mg/kg on days 0, 1 and 2 (USA). Body weight and clinical respiratory and neurological signs were monitored daily to the end of the experimental period (day 7 for Spain, day 5 for USA) when animals were euthanized, their lungs dissected out and, divided into three equivalent portions (Spain) or whole lungs, nasal turbinates and brain were collected and processed for virus titration and cytokine analysis (USA).
The first lung portion was fixed by immersion in 10% buffered formalin and paraffin-embedded for histological and immunohistochemistry (IHC) studies. Summarizing, the formalin-fixed, paraffin-embedded lung portion was hematoxylin and eosin (H&E) stained, examined by optical microscopy and, SARS-CoV-2 related inflammation findings semi-quantified (none, mild, moderate, or severe) as previously described (16, 17). Immunohistochemistry methodology for staining the SARS-CoV-2 N protein required the application (at 1:15000 dilution) of rabbit monoclonal antibody (40143-R019, Sino Biological, Inc., Beijing, China) on the lungs and, then the viral antigen was semi-quantitatively scored (none, low, moderate, high) as already published (16, 17).
The second lung portion was processed for virus molecular detection (Spain) or titration by plaque assay (USA). For molecular detection, SARS-CoV-2 RNA was extracted from the tissue using the IndiMag Pathogen Kit (Indical Bioscience, Leipzig, Germany) on a Biosprint 96 workstation (QIAGEN, Hilden, Germany) according to Manufacturer`s instructions. Viral genomic RNA (gRNA) was detected by a RT-PCR previously published (18), which was slightly modified to be adapted to the AgPath-ID™ One-Step RT-PCR (ThermoFisher Scientific, Waltham, MA, USA) kit. RT–qPCR targets a portion of the envelope protein gene (position 26,141–26,253 of GenBank NC_004718). The primers and probes used, and their final concentration are the follow: forward, 5′-ACAGGTACGTTAATAGTTAATAGCGT-3′ (400 nM); reverse, 5′-ATATTGCAGCAGTACGCACACA-3′ (400 nM); and, probe, 5′-FAM-ACACTAGCCATCCTTA CTGCGCTTCG-TAMRA-3′ (200 nM). Thermal cycling was performed at 55°C for 10 minutes for reverse transcription, followed by 95°C for 3 minutes and then, 45 cycles of 94°C for 15 seconds and 58°C for 30 seconds.
For virus titration by plaque assay, 10-fold serial dilutions were prepared from cleared lung homogenates in 0.2% bovine serum albumin (BSA)/phosphate buffer saline (PBS) and plated onto VeroE6 cells followed by incubation for 1 hour while shaking. Inoculum was removed and plates were overlaid with Minimal Essential Media (MEM) containing 2% FBS/0.05% oxoid agar and incubated for 72 hours at 37°C. Plates were fixed with 4% formaldehyde overnight, stained with a SARS-CoV-2 nucleoprotein monoclonal antibody (1C7) followed by anti-Mouse IgG-HRP (Abcam) and developed using KPL TrueBlue peroxidase substrate (Seracare).
Finally, the third snap-frozen lung portion was assessed for cytokine determinations with Luminex, as previously described (19). Briefly, lung samples were thawed, weighed (100 mg) and homogenized on ice with lysis buffer (Spain 0.05% sodium azide, 0.5% Triton X-100, 1:500 Protease inhibitor cocktail (all from Sigma-Aldrich)) or without lysis buffer (USA) in sterile PBS (final volume, 1 mL). Homogenates were incubated for 1 hour at 4°C and centrifuged at 3000g for 10 minutes. Supernatants were collected and processed for cytokines quantitation as described below.
Influenza virus infection model in mice.
On day 0, 4- to 8-weeks old C57BL/6 female mice (Charles River Lab) were randomly assigned to receive the first dose of placebo or plitidepsin (at 0.15 mg/kg), subcutaneously. One hour later, ketamine/xylazine-anesthetized mice were intranasally inoculated (total volume, 40 μL; 20 μL/nare) with PBS or with 4×105 PFU of A/PR8/34 (H1N1) virus and subsequently treated once daily for two additional consecutive days (treatment period: days 0, 1 and 2). Body weight and clinical respiratory were monitored daily to the end of the experimental period (day 3), when all animals were euthanized, lungs were dissected free and bronchoalveolar lavage fluid (BALF) were obtained as described elsewhere (20, 21). In short, after euthanizing the mice by intraperitoneal administration of a short-acting barbiturate, the muscle surrounding the neck was gently removed, the trachea was exposed, and a catheter (Introcan Safety® 20–22G; B. Braun, Frankfurt, Germany) was inserted and securely tied to the trachea with a silk suture. Then, 0.5 to 0.8 mL of cold PBS (Sigma-Aldrich) was slowly injected and aspirated two to four times and, the recovered fluid was kept on ice. Then, BALF were centrifuged (1500 rpm for 5 minutes) and cytokine levels were quantified in 25 μL of the supernatants as detailed below.
Once obtained, lungs were processed for the molecular detection of the RNA encoding the viral nucleoprotein NP (PR8) as outlined below: lungs were disrupted in lysing matrix D 1.4 mm ceramic sphere tubes using FastPrep®−24 tissue disruption (MP Biomedicals, Illkirch-Graffenstaden, France) and RNA was isolated using an RNAeasy Mini kit (Qiagen, Hilden, Germany). One microgram of cDNA was synthesized using a cDNA synthesis kit (Applied Biosystems, Foster City, CA, USA) following the Manufacturer’s recommendations. For the RQ-PCR reaction, a SYBR® Master Mix (Applied Biosystems) was used and samples were run on a QuantStudio™ 3 Real-Time PCR system (Applied Biosystems). The expression levels of NP (PR8) were measured with the following sets of designed primers: forward, 5´-TGCCTGCCTGTGTGTATGG-3´ (20μM); reverse: 5´-AGGCTGTACACTTGGCTGTTT-3´ (20μM).
ELISA and Multiplex for cytokine quantitation
Regardless of the matrix (e.g., culture supernatants, lungs or BALF) in which cytokines were quantified, determinations were always performed according to the manufactureŕs instructions.
In the supernatants generated in the THP1-Dual™ experiments, human IL-6 was quantified by using the corresponding OptEIA™ ELISA sets (Becton Dickinson, Franklin Lakes, NJ, USA). In the supernatants generated in MDM incubations, cytokine IL-6 was analyzed with HCYTOMAG-60 kit (Merck KGaA, Darmstadt, Germany), measured by Luminex xMAP technology and analyzed with xPONENT 3.1 software (Luminex Corporation, Austin, TX, USA).
Cytokines in the lung supernatants of SARS-CoV-2 infected and treated mice were determined by using HCYTOMAG-70K kit (Merck KGaA) or Mouse ProcartaPlex (Thermofisher Scientific) measured by Luminex xMAP technology and analyzed with xPONENT 3.1 software (Luminex Corporation).
In BALF obtained from A/PR8/34 (H1N1) virus infected and treated mice, cytokines were quantified by using LEGENDplex™ Mouse Inflammation Panel (BioLegen, San Diego, CA, USA).
IL-6 mRNA expression in THP-1 cells
For a 6-hour period, THP-1 cells were treated either with plitidepsin (100 nM), with RQ (10 μg/mL) or with a combination of both compounds (plitidepsin, 100 nM; RQ, 10 μg/mL); a negative control (DMSO at 0.1%) was also included. IL-6 mRNA expression was then analyzed by qPCR. In short, total RNA was isolated from the cells with the RNeasy Mini Kit (Qiagen). For each sample, 2 μg of total RNA was reverse-transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Milan, Italy), according to the Manufacturer’s instructions. mRNA levels were analyzed by quantitative real-time PCR using the TaqMan Gene Expression Master Mix (Applied Biosystems) according to the Manufacturer’s protocol. TaqMan gene expression assay (Applied Biosystems) was used to quantify gene expression levels of human IL-6 (Hs00174131_m1), and the human HPRT (hypoxanthine phosphoribosyltransferase 1) (Hs02800695_m1) was used as a housekeeping gene. The analysis was carried out on a 7500 Real-Time PCR System (Applied Biosystems). The relative expression levels of each transcript were determined from three independent experiments, each performed in quadruplicate. Amplification efficiency was determined for each qPCR assay, and subsequently used to calculate relative expression values using the SDS System 7500 software (Applied Biosystems). Relative expression levels were calculated using the ΔΔCt method. In addition, the levels of IL-6 secreted were quantified by ELISA in the supernatants of these cultures.
NF-kβ transcription factor binding assay
A previous published protocol (22) using the NF-κβ Transcription Colorimetric Assay Kit (ab207216, Abcam) was performed following manufactureŕs instructions (abcam plc, Cambridge, UK). Briefly, the ab113474 nuclear isolation kit (abcam plc) was used to first obtain 5 μg of nuclear extracts which were incubated with oligonucleotides containing NF-κβ consensus binding sites for 1 h at room temperature. After washing, they were incubated with primary antibodies for 1 h at room temperature. Then, they were washed, and samples were incubated with HRP-conjugated secondary antibody for 1 h at room temperature and washed again. After adding the developing solution, the formation of the antigen-antibody complex was quantified on a Perkin-Elmer® EnVision® (Perkin Elmer) microplate reader at a wavelength of 450 nm.
Western blotting
Stimulated THP-1 cells with RQ (at 10 μg/mL), TNF-α (100 ng/mL) or LPS (at 10 μg/mL) were incubated without and with plitidepsin (at 100 nM) for 1 hour. Then, cells were collected, washed with cold PBS and treated with lysis buffer (20 mM Tris-HCl, pH 7.5; 150 mM NaCl; 1% (v/v) Nonidet P-40, 2 mM EDTA; 1 mM PMSF; 10 μg/mL aprotinin; and 10 μg/mL leupeptin) for 15 minutes in an ice bath. In the supernatants obtained after centrifugation (14000g for 30 minutes at 4 °C), the protein content was determined by a modified Bradford method. Twenty micrograms of total protein were resolved in 10% SDS-PAGE and transferred onto activated Immobilon-P PVDF membranes following standard techniques (23). Then, membranes were incubated with the appropriate primary antibody namely, anti-NF-κB p65, anti-Phospho-NF-κB p65 (Ser536) and Anti-α-Tubulin antibodies for 1 hour, washed and incubated with the secondary antibody. Proteins were visualized using the ECL System (GE Healthcare, Fairfield, CT, USA).
Statistical analysis
Statistical analyses and graph plotting were done using GraphPad Prism, version 9.2.0 (GraphPad Software Inc., La Jolla, CA, USA). Statistical differences between groups were evaluated using the non-parametric Mann–Whitney U test or Kruskall Wallis followed by Dunn’s post-test for multiple comparisons as mentioned in Figure 4 legend.
Figure 4. Anti-inflammatory cytokine milieu in the lung induced by plitidepsin.
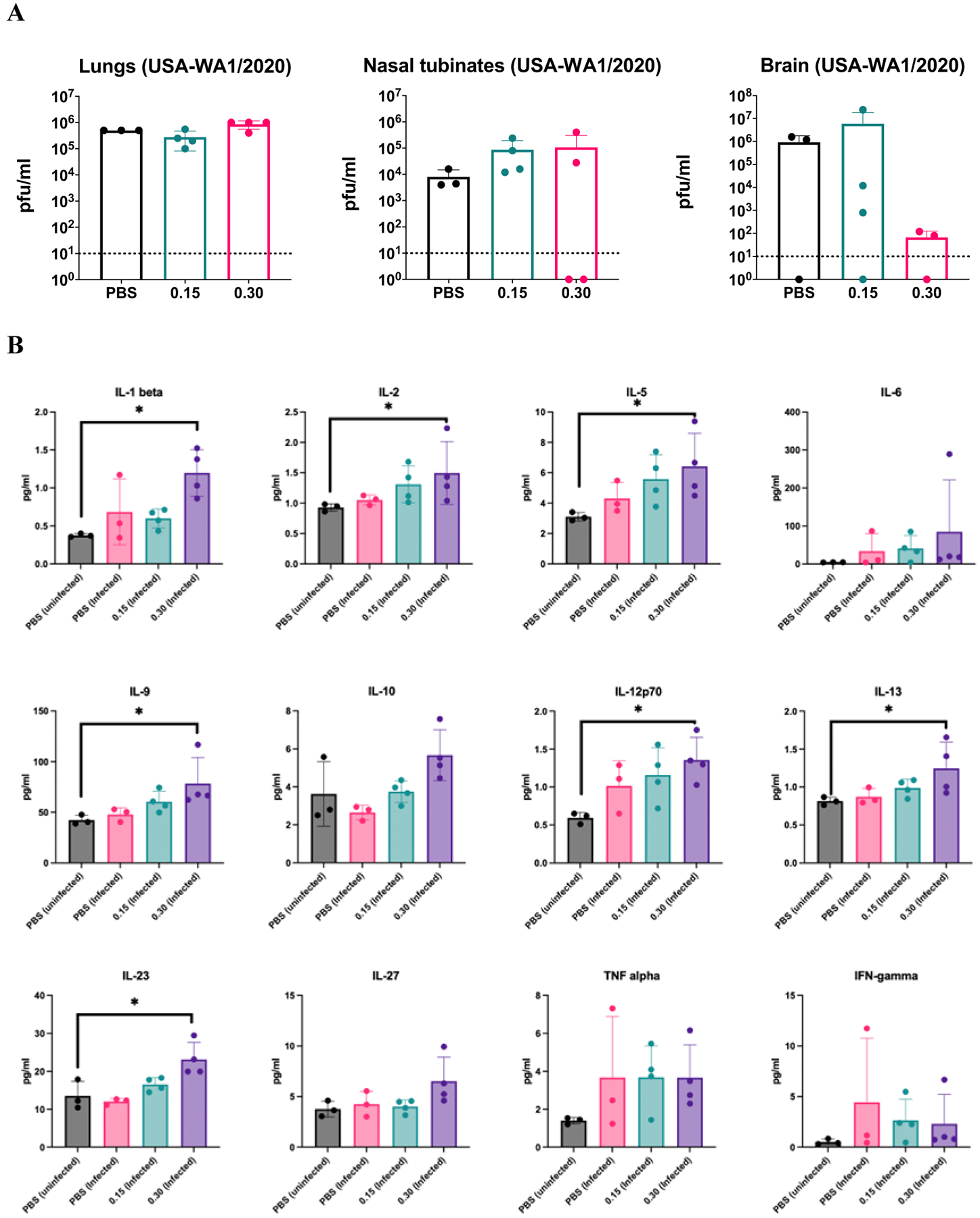
(A) Virus titers determined by plaque assay at 5 days post infection in lung, nasal turbinates and brain of hACE2 transgenic mice infected with 104 PFU of USA-WA1/2020 SARS-CoV-2 and treated with 0.15 or 0.30 mg/kg of plitidepsin for three consecutive days or placebo. Every dot represents an individual mouse. (B) Cytokine profile in cleared lung homogenates from SARS-CoV-2 infected lungs at day 5 post-infection and treated with 0.15 or 0.30 mg/kg of plitidepsin for three consecutive days or placebo. Every dot represents an individual mouse. Statistical differences between groups were tested using non-parametric Kruskall-Wallis test followed by Dunn’s multiple comparisons post-test. * P<0.05. Bars represent means and error bars represent standard deviations.
Experimental values were fitted according to an exponential decay equation model.
RESULTS
Plitidepsin inhibits NF-κB mediated transcription in response to TLR7 stimulation in vitro.
Plitidepsin-related effects on pro-inflammatory NF-κB and IFN pathways were assessed in vitro using THP1-Dual™ cells, stably transfected with two different reporter plasmids, namely NF-κB SEAP and IFN-stimulated response luciferase. In cell cultures exposed to plitidepsin, a very modest decrease in cell viability (compared to control, plitidepsin untreated cells) was obtained (3%, 9% and 16% at 2.5, 5.0 and 10.0 nM, respectively). Figure 1A (top panel) shows that RQ (ligand of TLR 7/8; mimicking ssRNA viral genomes) induced NF-κB transactivation in THP1-Dual™ cells (as determined by induction of an alkaline phosphatase reporter gene), which was reduced by plitidepsin in a dose-dependent manner (Figure 1B) without affecting IFN-α-mediated transcription of an IFN-inducible luciferase reporter gene (Figure 1A; bottom panel). In addition, RQ induced an increase in IFN-regulated transcription, likely due to the induction of an activated paracrine loop, mediated by an NF-κB-induced increase in IFN secretion. This induction was also inhibited by plitidepsin. Although statistically significant, a minimal effect on IFN-regulated transcription was observed only at 10 nM of plitidepsin (median fold-increases: 29.7 and 24.3 without and with plitidepsin, respectively), probably owing to the marginal plitidepsin-induced cytotoxicity. These results strongly suggest that NF-κB-regulated transcription is specifically inhibited by plitidepsin in RQ-stimulated THP1- Dual™ monocytes.
Figure 1. Plitidepsin modulates the NF-κB-regulated transcription.
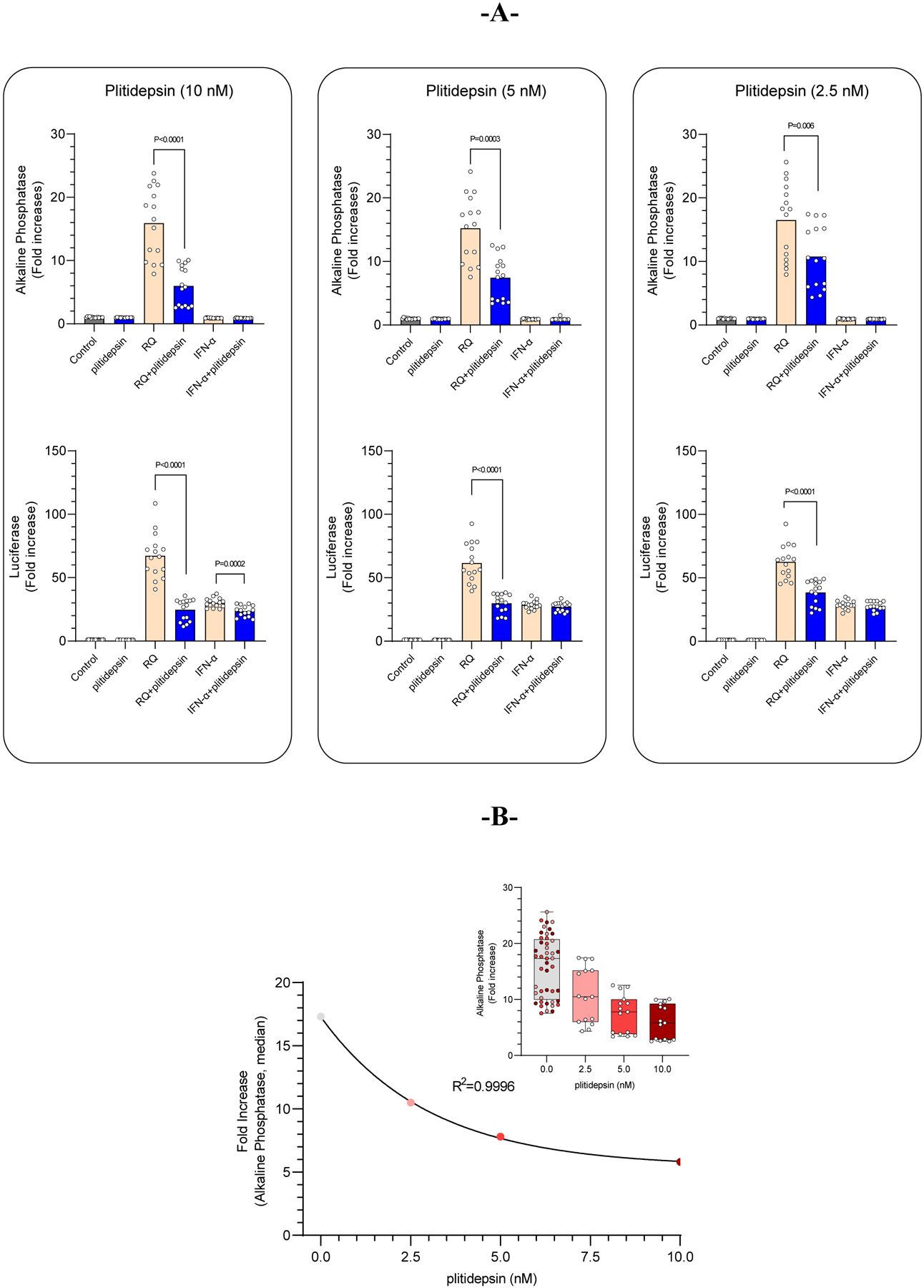
Resiquimod (10 μg/mL; NF-κB signaling) or Interferon-α (20 ng/mL; Interferon signaling) -challenged THP1-Dual™ cells were incubated with increasing concentrations of plitidepsin for 18 hours. (A) Only a marginal plitidepsin-mediated effect was observed in luciferase levels (a marker of the interferon-regulated transcription) in the interferon-challenged THP1- Dual™ cell supernatants. However, a statistically significant reduction of alkaline phosphatase (a marker of NF-κB-regulated transcription) levels were quantified in the supernatants of resiquimod-challenged THP1-Dual™ cells, with plitidepsin-mediated inhibition of NF-κB-regulated transcription being specific and concentration-dependent (B).
In a separate set of in vitro experiments, we monitored secretion of the NF-κB-inducible cytokine IL-6. A concentration-dependent decrease of IL-6 concentration (Figure 2A) was found in the supernatants of THP1-Dual™ monocytes stimulated with RQ and incubated with increasing concentrations of plitidepsin (1, 10 and 50 nM). Results demonstrated that even at a plitidepsin concentration as low as 1 nM, a significant reduction of IL-6 concentration was observed as compared to plitidepsin untreated RQ-stimulated cells (1.2-fold reduction; p=0.0317, n=12). This effect was more pronounced at the two highest plitidepsin concentrations used, which induced an IL-6 reduction of 1.7- and 4.3-fold (at 10 nM and 50 nM, respectively; both, p≤0.0001) as compared to plitidepsin untreated RQ-stimulated cells. Of note, this plitidepsin-induced effect on IL-6 secretion was produced without affecting THP1-Dual™ monocyte viability. A very similar behavior was noticed following the incubation of human MDM exposed to non-productive SARS-CoV-2 infection and that were incubated with increasing concentrations of plitidepsin, leading to a clear concentration-related reduction in IL-6 levels (Figure 2B), also without plitidepsin-related cytotoxicity.
Figure 2. Plitidepsin-mediated reduction of IL-6 secretion in THP-1 human monocytes and primary macrophages.
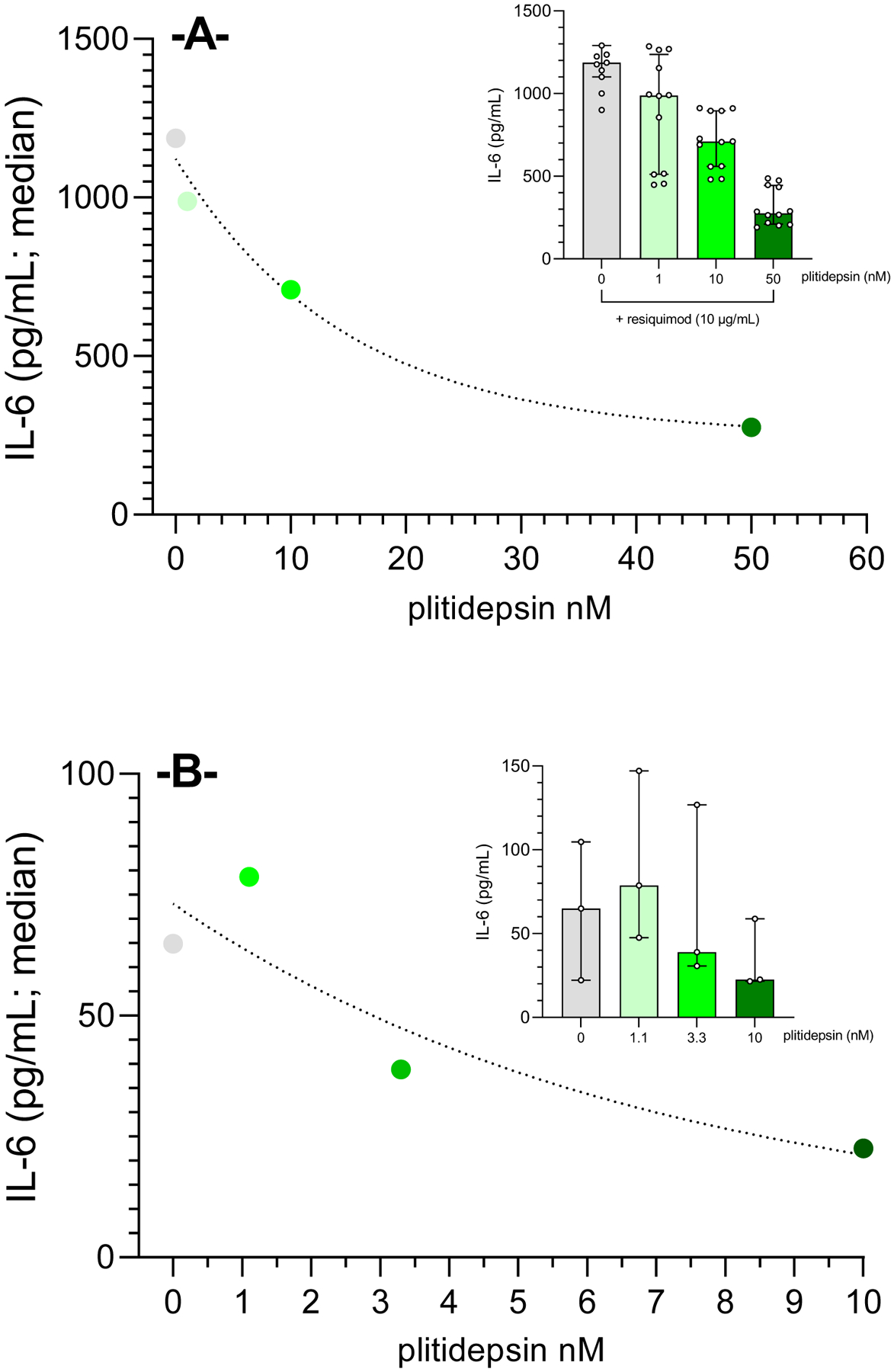
IL-6 levels quantified in the supernatants of resiquimod-challenged THP1-Dual™monocytes (A) and human monocyte-derived macrophages (MDM) exposed to non-productive SARS-CoV-2 (B) show a very strong concentration-dependent reduction when exposed to increasing concentrations of plitidepsin (THP-1 cells: 1, 10 and 50 nM for 16 h; MDM: 1.1, 3.3 and 10 nM for 24 h). Results show median values of n=12 and n=3 for THP-1 and MDM, respectively.
Inlets display scatter dot plot for individual samples obtained in each experiment.
Resiquimod, RQ (10 μg/mL) induced an increase of IL-6 mRNA expression (>200-fold) in THP-1 cells, which was statistically significantly reverted by plitidepsin (at 100 nM) to values that only reached a 30-fold increase (Figure 3A; left panel). This effect was translated into a significant decrease on IL-6 cytokine concentrations in the supernatants of the RQ-stimulated THP-1 cells in the presence of plitidepsin. IL-6 cytokine levels in the presence of plitidepsin was reduced to basal levels in RQ-treated cells (Figure 3A; right panel).
Figure 3. Plitidepsin regulates the NF-κB signaling pathway.
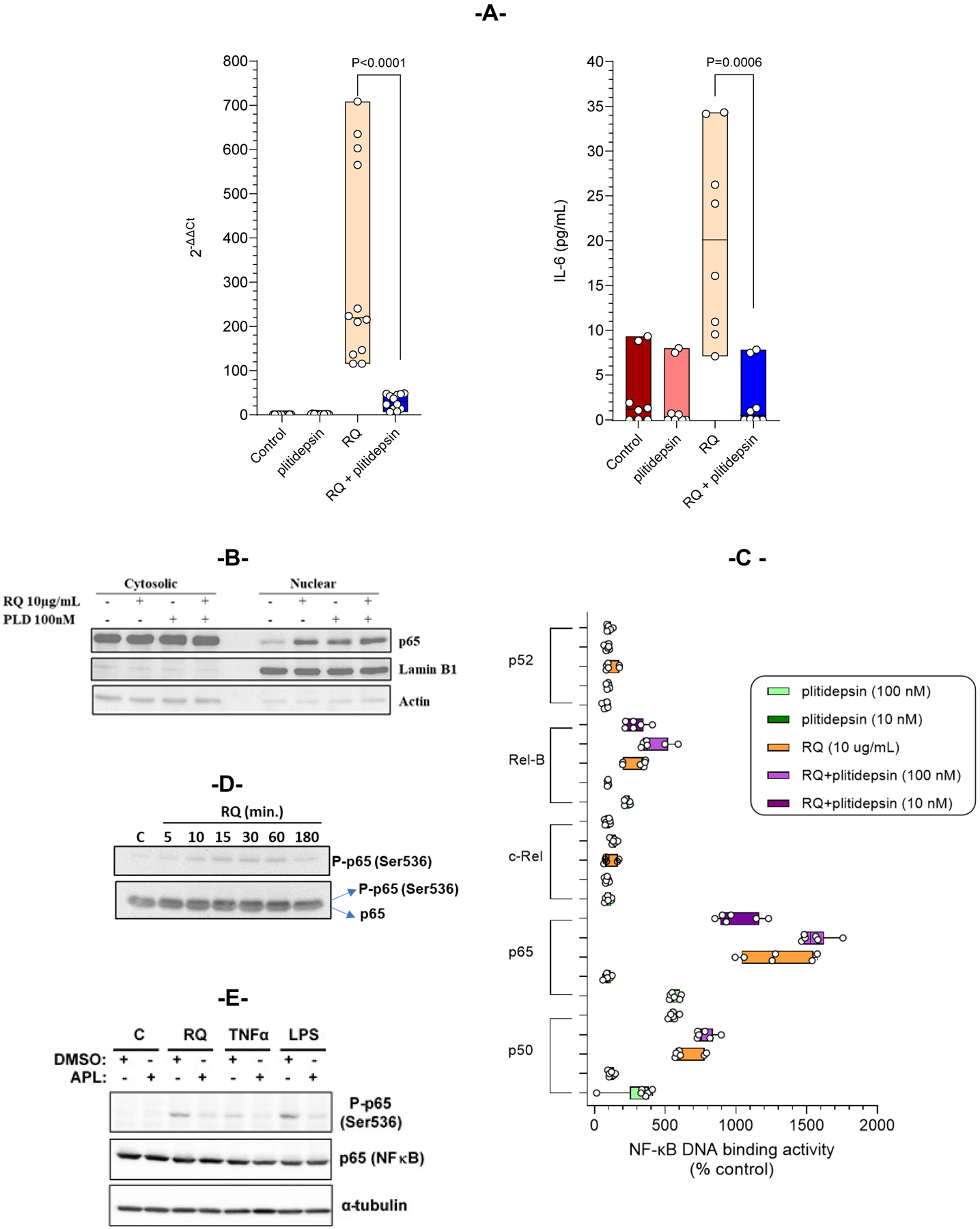
(A) THP-1 cells were co-treated with 100 nM of plitidepsin and 10 μg/mL RQ for 6 hours or only with plitidepsin or RQ. IL-6 RNA expression (left panel) was analyzed by qPCR and secreted IL-6 (right panel) were analyzed by ELISA. Data presented are the median of three independent experiments performed in quadruplicate. (B) THP-1 cells were stimulated with RQ (10 μg/mL) alone or in combination with plitidepsin (100 nM) for 1 hour, and nuclear and cytoplasmic fractions were obtained and analyzed by immunoblotting. Western blots revealed p65 nuclear import. The purity of the fractions was controlled with an Ab recognizing the nuclear Lamin B1 protein. (C) Binding of NF-kB family members (p50, p65, c-Rel, RelB and p52) to a specific double stranded DNA sequence containing the NF-kβ consensus binding site was quantified. THP1 cells were treated with RQ (10 μg/mL) alone or in combination with plitidepsin (10nM and 100 nM) for 2 hours. Nuclear extracts were obtained and tested for NF-κB binding. The data are the median of three independent experiments performed in duplicate. (D) THP-1 cells were stimulated for the indicated periods with RQ. Phosphorylation of p65 (Ser536) and total p65 were analyzed by Western blot. (E) Immunoblot assay showing p65 and phosphorylation of p65 (Ser536) in THP1 cells after stimulation with RQ (10 μg/mL), TNF-α (100 ng/mL) and LPS (10 μg/mL) with or without plitidepsin (100 nM) for 1 hour. Tubulin was analyzed as loading control.
Plitidepsin inhibits in vitro NF-κB phosphorylation in response to multiple stimuli.
As previously shown, even in the presence of 10 μg/mL RQ, plitidepsin inhibited the production of SEAP, indicating that transactivation from NF-κB was inhibited in the presence of the drug. Then, cellular localization of NF-κB in THP-1 cells treated with RQ 10 μg/mL or with plitidepsin 100 nM (Figure 3B) was analyzed by subcellular fractionation into cytosolic and nuclear extracts, for 1 hour. The activation of the pathway was demonstrated through the shuttling of the NF-κB to the nucleus in THP-1 cells after treatment with either RQ or plitidepsin. Combination of both treatments induced the same nuclear translocation of NF-κB. The ability of plitidepsin to modify the NF-κB DNA binding was examined after observing NF-kB localized to the nuclei in RQ, in plitidepsin-treated cells or in co-treated cells. Therefore, a specific double stranded DNA sequence containing the NF-κB consensus binding site (5´-GGGACTTTCC-3´) immobilized onto a 96-well plate was used. Results showed that NF-κB present in nuclear extracts specifically binds to the oligonucleotide and it is detected by primary antibodies, which recognize p50, p52, p65, c-Rel or RelB, which are only accessible when NF-κB is bound to its target DNA. THP-1 cells were treated and nuclear extracts assayed for NF-κB DNA binding. Intriguingly, plitidepsin (at 10 and 100 nM) was able to induce binding of the NF-κB members p50, p65 (RelA) and RelB to DNA, although to a lesser extent than RQ (at 10 μg/mL). Moreover, the RQ-induced binding of p50, p65 (RelA) and RelB to DNA resulted unaffected by the presence of any plitidepsin concentration (Figure 3C). Results strongly suggest plitidepsin downregulates the NF-κB regulated transcription not by preventing the binding of NF-κB to DNA, but rather by preventing the post-translational modifications of NF-κB responsible for the further recruitment of RNA polymerase to the NF-κB-responsive promoters. Specifically, optimal induction of NF-κB target genes also requires phosphorylation of NF-κB proteins, such as p65, within their transactivation domain by a variety of kinases (24). This said, the phosphorylation of p65 induced by RQ was analyzed at multiple time points. THP-1 cells were stimulated with RQ for indicated time periods and p-p65 (Ser536) levels were examined by Western blot. RQ induced p65 phosphorylation, occurring very rapidly within 10 to 60 min of RQ treatment (Figure 3D). We thus asked whether p65 phosphorylation at serine 536 (S536) could be modulated by plitidepsin. THP-1 cells were stimulated with RQ, TNFα or LPS and incubated with plitidepsin, and the levels of p-p65 (Ser536) were then checked by Western blot. It was observed that plitidepsin decreased the p65 phosphorylation of Ser536 induced by the different stimulating compounds (Figure 3E).
Plitidepsin inhibits inflammation in vivo in response to SARS-CoV-2 and influenza virus infection.
In order to see whether plitidepsin also has anti-inflammatory properties in vivo, we next treated SARS-CoV-2 infected K18-hACE2 mice with a dose of plitidepsin below the concentration previously known to have antiviral activity in vivo (4). For this purpose, we infected K18-hACE2 mice at the animal BSL3 facilities at ISMMS (USA) and treated them with two doses of plitidepsin at 0.15 or 0.30 mg/kg/day for three consecutive days or placebo. The experiment was stopped at day 5 post infection and lungs, nasal turbinates and brain were harvested for virus titration by plaque assay (Figure 4A). We and others have shown before that brain infection occurs during experimental SARS-CoV-2 infection in the K18-hACE2 mouse model (25). Our results confirmed that the low doses of plitidepsin used here did not have a direct antiviral effect in the lungs and only for half of the animals in the nasal turbinates; however, viral load was reduced, albeit not with statistical significance, in brain of mice treated with 0.30 mg/kg/day. Despite no impact in lung viral titers, this dose of plitidepsin was able to create an anti-inflammatory cytokine milieu in the lungs (Figure 4B) as indicated by enhanced levels in total lung homogenates of anti-inflammatory cytokines IL9, IL10 (Kruskall-Wallis test showed significant differences between groups at the 5% level but no statistical difference between groups was reached when compared with the Dunn’s post-test), and IL13. There was also increases in the IL12 cytokine family members IL12, IL23, IL27, known to stimulate T cell differentiation. However, at this time point post infection (5 DPI) no differences in IL6, TNF and IFNγ levels in lung homogenates were observed yet in infected mice with placebo or plitidepsin treatment, suggesting that this time point was too early in infection to see a clear plitidepsin effect on these cytokines. We also observed enhanced levels of IL1β, IL2 and IL5, which was significant for mice treated with 0.30 mg/kg/day of plitidepsin and suggests plitidepsin might contribute to lymphocyte activation in the lung through a balanced pro- and anti-inflammatory cytokine environment upon infection, instead of the more skewed pro-inflammatory environment in SARS-CoV-2-infected, not treated, animals. Since we have measured the cytokine levels in whole lung homogenates for this experiment, it is however not possible to exclude that the source of these cytokines is in the blood, interstitial lung or alveolar space.
In an independent experiment, we observed that SARS-CoV-2 infection at day 7 in mice caused a variable degree of broncho-interstitial pneumonia that is an inflammation of lung characterized by mononuclear inflammatory infiltration in the parenchyma, especially around the bronchi and bronchioles, as well as type 2 pneumocyte hyperplasia and hypertrophy (Figure 5A; bottom panel). This pneumonia was multifocally distributed when mild lesions were observed and was multifocal to diffuse in moderate to severe lesions. To determine whether plitidepsin treatment can reduce lung inflammation in response to SARS-CoV-2 infection, SARS-CoV-2 infected mice were treated with a low dose of plitidepsin unable to reduce viral replication. On day 7, regardless of the administered treatment (plitidepsin at 0.15 mg/kg/day for three consecutive days or placebo), a clear antiviral effect was not seen, as expressed by viral RNA to detect SARS-CoV-2 nucleoprotein in the lung of mice (Figure 5B; right panel). However, a statistically significantly reduced lung inflammatory score was recorded in plitidepsin compared to placebo-treated mice (0.25 versus 1.75; p<0.028) (Figure 5A; top). In the Fig 4 experiment, we measured cytokines at day 5, and we found a more balanced anti- and pro-inflammatory cytokine environment in the lungs of plitidepsin treated animals. To see whether the in vivo impact in cytokines of plitidepsin treatment could be better captured at later time points at the peak of inflammation, we measured IFN-γ, MCP-1 and IL-6 levels in lungs at day 7 post-infection. We observed a statistically significantly reduction of IFN-γ (14-fold), MCP-1 (4.2-fold) and IL-6 (3.6-fold) in the lungs of plitidepsin-treated SARS-CoV-2 mice at day 7 in 75 % of the mice treated at 0.15 mg/kg/day (Figure 5B, left panel) and a similar reduction in 100% of the mice treated at 0.30 mg/kg/day (Supplemental Figure 1).
Figure 5. Reduction of lung-inflammatory damage as well as cytokine levels by a non-antiviral dose of plitidepsin.
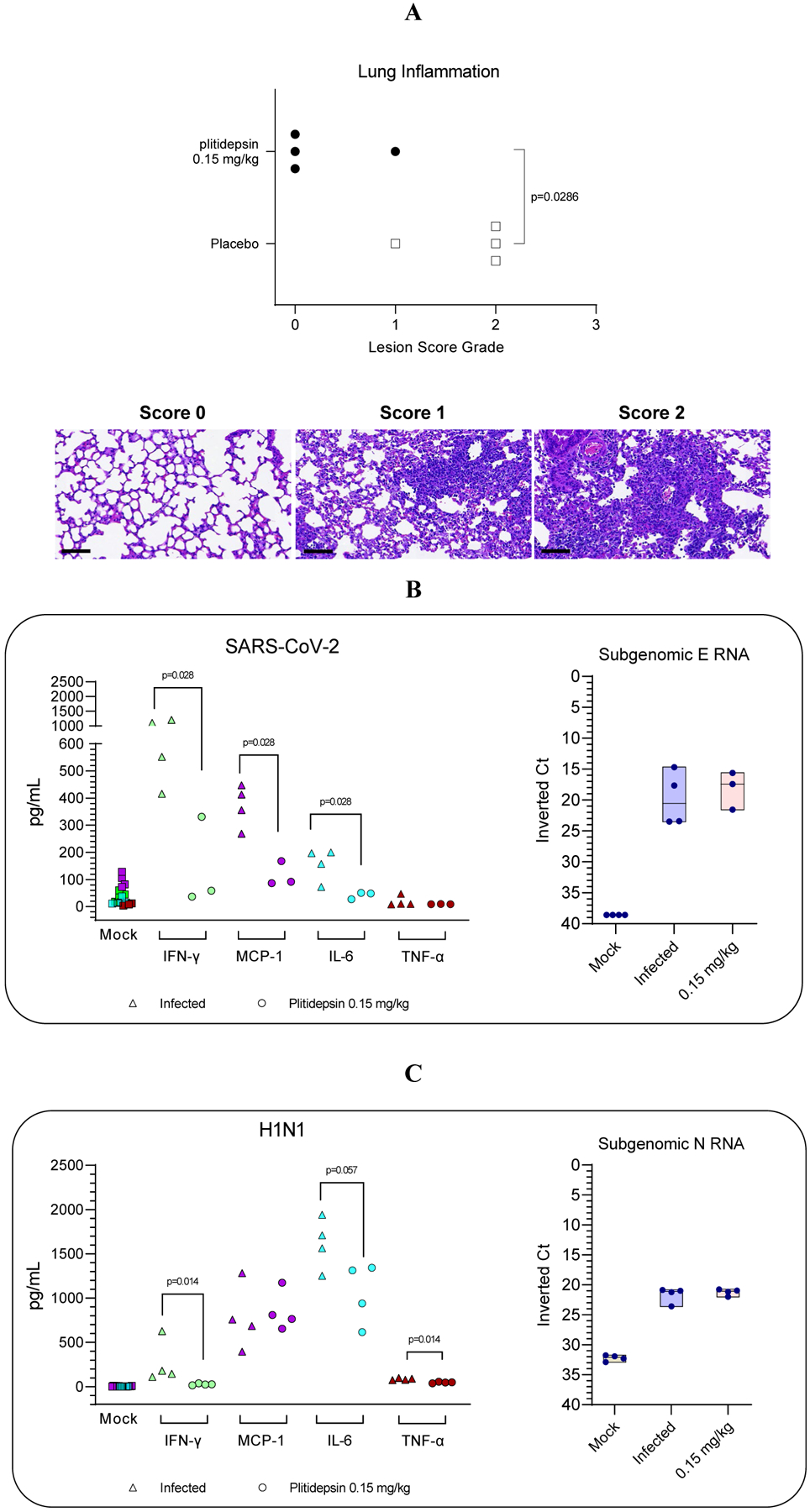
(A) Lung inflammation score (as by H&E) determined in plitidepsin and placebo-treated experimental groups at day 7 postinfection. Bottom: representative H&E stains (bar = 80 μm) of non-damaged lungs (score 0) or lungs affected by different severity score (1 or 2) of multifocal broncho-interstitial pneumonia characterized by mononuclear inflammatory cells within alveoli and alveolar walls and hyperplasia and hypertrophy of type II pneumocytes. (B, C left) Down regulation of cytokines in lungs and BALF of SARS- CoV-2 and H1N1 virus infected-mice, respectively treated either with non- antiviral doses (B, C right) of plitidepsin or placebo, at day 7 post-infection for SARS-CoV-2 and day 3 for H1N1. Inverted Ct were determined by qPCR of subgenomic RNA for E and N protein of SARS-CoV-2 and influenza virus, respectively.
It is worth to highlight that the samples from both SARS-CoV-2 experiments were taken in different DPI which may explain the apparent different results.
Similar results were observed when cytokines were determined in BALF obtained from H1N1-infected mice at day 3 DPI in which treatment with plitidepsin induced reduction of IFN-γ, IL-6 and TNF-α (6.4-fold, 1.5-fold, and 1.8-fold, respectively) levels compared to animals treated with placebo (Figure 5C).
Taken together, all these results strongly support the idea that plitidepsin can balance pro- and anti-inflammatory cytokine levels under different experimental conditions, leading to the induction of an in vivo anti-inflammatory effect in two different experimental models of pathogenic respiratory virus infections.
DISCUSSION
Pathogenic respiratory viruses, such as influenza virus, MERS-CoV, SARS-CoV and SARS-CoV-2, share a similar pathological pattern: a primary viral infection that replicates in the respiratory epithelium and gives rise to an immune response critical to control the spread of infection (26–28). In some patients, however, the immune response can evolve into a fulminant and fatal hyperinflammatory syndrome, characterized by severe increase in systemic cytokines (e.g., IL-6, IL-1β and TNFα) and chemokines (e.g., CCL2, CCL3, CCL20, CXCL1, CXCL3, CXCL10, IL-8) that leads to a cytokine storm followed by multiple-organ failure (29–31). Recently, this hyperinflammatory state of the lungs in severe COVID-19 has been linked to the activity of lung-resident human macrophages, where SARS-CoV-2 recognition activates the inflammasome response and the release of cytokines, which leads to pyroptosis and cell death in the absence of active viral replication (32).
Here we found that the induction of NF-κB-regulated transactivation mediated by RQ, a TRL7 mimicking agent, is inhibited by plitidepsin in the monocytic THP-1 cells in vitro, with a marginal effect on IFN-regulated transcription. This inhibition of NF-κB-regulated transcription could be explained by a plitidepsin-mediated decrease in RQ-induced phosphorylation of p65 at Ser536, with no effect on either the translocation to the nucleus nor the binding of p50, p65 (RelA) and RelB to DNA. A similar effect has been observed through the use of p65-S536A mutant to abolish phosphorylation in serine 536. It has been described that p65- S536A mutant protein showed similar DNA binding capacity when comparing to wild type (33). It was also observed that by using p65- S536A mutant cells, NF-kB transcriptional activity is severely inhibited (34, 35), but nuclear import is unmodified after 30 min of stimulation (36). Reduction in Ser536 phosphorylation has a negative impact on p65-dependent post- translational modifications of NF-κB required to achieve its full transcriptional activity (37), and may therefore decrease the subsequent production of cytokines. These aspects may explain why IL-6 levels quantified in the supernatants of RQ-challenged cells incubated with plitidepsin were identical to those obtained in unchallenged cells. Taken together, these results strongly support the regulatory role of plitidepsin in an inflammatory scenario. Moreover, a similar effect was observed in MDM exposed to SARS-CoV-2, in which plitidepsin could decrease the release of IL-6 induced by viral exposure in the absence of active viral replication. Hence, the immunomodulatory effect of plitidepsin is not linked to its antiviral activity, as human macrophages are resistant to SARS-CoV-2 infection (38, 39).
Increasing evidence indicates the role of the NF-κB signal transduction pathway in SARS-CoV-2-induced pro-inflammatory cytokines and chemokines production (40, 41). In agreement, a statistically significant reduction of IL-6, IFN-γ, TNF-α and MCP-1 was detected in the lungs or BALF obtained from influenza- or SARS-CoV-2-infected mice that were treated with plitidepsin at doses with marginal antiviral effect.
An alternative mechanism could implicate EF1A inhibition, since it has been implicated in IL-6 production. It has been previously described in different studies (42, 43) that eEF1A1-dependent STAT3 phosphorylation at serine 727 is crucial for NF-κB/STAT3 complex formation and subsequent IL-6 expression. Although currently there are no experimental evidences, the contribution of plitidepsin to this alternative mechanism cannot be completely ruled out.
An ideal therapeutic strategy for the successful treatment of respiratory viral infections should have two complementary components. First, a direct antiviral effect may decrease the viral load and reduce the likelihood of an immune over-response. Second, an immune modulating effect to control hyperinflammatory responses induced by viral replication (8). The clinical utility to reduce mortality in severe COVID-19 patients has already been demonstrated for similar host-directed therapies, with a reported dual antiviral and immunomodulatory activity (44). Here, we further support the widely reported potent antiviral activity of plitidepsin against SARS-CoV-2 both in vitro and in vivo (3–6) and expand its indication as an immunomodulatory agent as well. The host-directed effect of plitidepsin, which is safe to treat COVID-19 patients (6), reduces the probability of the emergence of resistance to treatment, and has a strain-independent antiviral profile in vitro (5, 6).
Results presented herein showed a decrease in pro-inflammatory cytokine release or induction of an anti-inflammatory cytokine environment after treatment with plitidepsin, which was observed in non-productive SARS-CoV-2 infections in vitro, as well as in respiratory virus-infected mice that were treated with different doses of plitidepsin.
Supplementary Material
KEY POINTS.
Plitidepsin downregulates NF-kB transcriptional activity
Plitidepsin decreases proinflammatory cytokine levels
Plitidepsin alleviates respiratory virus disease independent of its antiviral activity
ACKNOWLEDGMENTS
We acknowledge J. Díaz from the CMCiB for his constant help at the BSL3 facility. We thank R. Albrecht for support with the BSL3 facility and procedures at the Icahn School of Medicine at Mount Sinai, New York.
FUNDING SOURCES
This work was partly supported by a research agreement with PharmaMar S.A., by CRIPT (Center for Research on Influenza Pathogenesis and Transmission), a NIAID funded Center of Excellence for Influenza Research and Response (CEIRR, contract # 75N93021C00014), by NIAID grants U19AI142733, U19AI135972, U19AI168631, by a supplement to DoD grant W81XWH-20-1-0270, by the JPB and OPP foundations and an anonymous philanthropic donor to AG-S. SARS-CoV-2 research in the M.S. laboratory is supported by NIH grants R01AI160706 and R01DK130425. NI-U is supported by the Spanish Ministry of Science and Innovation (grant PID2020-117145RB-I00, Spain), EU HORIZON-HLTH-2021CORONA-01 (grant 101046118, European Union) and by institutional funding of PharmaMar, HIPRA, Amassence, Grifols, and Palobiofarma (Spain). The authors also acknowledge the crowdfunding initiative #Yomecorono (https://www.yomecorono.com) and Foundation Dormeur for financial support for the acquisition of the QuantStudio-5 real-time PCR system and an Eclipse Ts2RFL inverted research microscope.
REFERENCES
- 1.Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O’Meara MJ, Rezelj VV, Guo JZ, Swaney DL, Tummino TA, Huttenhain R, Kaake RM, Richards AL, Tutuncuoglu B, Foussard H, Batra J, Haas K, Modak M, Kim M, Haas P, Polacco BJ, Braberg H, Fabius JM, Eckhardt M, Soucheray M, Bennett MJ, Cakir M, McGregor MJ, Li Q, Meyer B, Roesch F, Vallet T, Mac Kain A, Miorin L, Moreno E, Naing ZZC, Zhou Y, Peng S, Shi Y, Zhang Z, Shen W, Kirby IT, Melnyk JE, Chorba JS, Lou K, Dai SA, Barrio-Hernandez I, Memon D, Hernandez-Armenta C, Lyu J, Mathy CJP, Perica T, Pilla KB, Ganesan SJ, Saltzberg DJ, Rakesh R, Liu X, Rosenthal SB, Calviello L, Venkataramanan S, Liboy-Lugo J, Lin Y, Huang XP, Liu Y, Wankowicz SA, Bohn M, Safari M, Ugur FS, Koh C, Savar NS, Tran QD, Shengjuler D, Fletcher SJ, O’Neal MC, Cai Y, Chang JCJ, Broadhurst DJ, Klippsten S, Sharp PP, Wenzell NA, Kuzuoglu-Ozturk D, Wang HY, Trenker R, Young JM, Cavero DA, Hiatt J, Roth TL, Rathore U, Subramanian A, Noack J, Hubert M, Stroud RM, Frankel AD, Rosenberg OS, Verba KA, Agard DA, Ott M, Emerman M, Jura N, von Zastrow M, Verdin E, Ashworth A, Schwartz O, d’Enfert C, Mukherjee S, Jacobson M, Malik HS, Fujimori DG, Ideker T, Craik CS, Floor SN, Fraser JS, Gross JD, Sali A, Roth BL, Ruggero D, Taunton J, Kortemme T, Beltrao P, Vignuzzi M, Garcia-Sastre A, Shokat KM, Shoichet BK, and Krogan NJ. 2020. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583: 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Losada A, Munoz-Alonso MJ, Garcia C, Sanchez-Murcia PA, Martinez-Leal JF, Dominguez JM, Lillo MP, Gago F, and Galmarini CM. 2016. Translation Elongation Factor eEF1A2 is a Novel Anticancer Target for the Marine Natural Product Plitidepsin. Sci Rep 6: 35100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rodon J, Muñoz-Basagoiti J, Perez-Zsolt D, Noguera-Julian M, Paredes R, Mateu L, Quiñones C, Perez C, Erkizia I, Blanco I, Valencia A, Guallar V, Carrillo J, Blanco J, Segalés J, Clotet B, Vergara-Alert J, and Izquierdo-Useros N. 2021. Identification of Plitidepsin as Potent Inhibitor of SARS-CoV-2-Induced Cytopathic Effect After a Drug Repurposing Screen. Frontiers in Pharmacology 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White KM, Rosales R, Yildiz S, Kehrer T, Miorin L, Moreno E, Jangra S, Uccellini MB, Rathnasinghe R, Coughlan L, Martinez-Romero C, Batra J, Rojc A, Bouhaddou M, Fabius JM, Obernier K, Dejosez M, Guillen MJ, Losada A, Aviles P, Schotsaert M, Zwaka T, Vignuzzi M, Shokat KM, Krogan NJ, and Garcia-Sastre A. 2021. Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science 371: 926–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sachse M, Tenorio R, Fernandez de Castro I, Munoz-Basagoiti J, Perez-Zsolt D, Raich-Regue D, Rodon J, Losada A, Aviles P, Cuevas C, Paredes R, Segales J, Clotet B, Vergara-Alert J, Izquierdo-Useros N, and Risco C. 2022. Unraveling the antiviral activity of plitidepsin against SARS-CoV-2 by subcellular and morphological analysis. Antiviral Res 200: 105270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Varona JF, Landete P, Lopez-Martin JA, Estrada V, Paredes R, Guisado-Vasco P, Fernandez de Orueta L, Torralba M, Fortun J, Vates R, Barberan J, Clotet B, Ancochea J, Carnevali D, Cabello N, Porras L, Gijon P, Monereo A, Abad D, Zuniga S, Sola I, Rodon J, Vergara-Alert J, Izquierdo-Useros N, Fudio S, Pontes MJ, de Rivas B, Giron de Velasco P, Nieto A, Gomez J, Aviles P, Lubomirov R, Belgrano A, Sopesen B, White KM, Rosales R, Yildiz S, Reuschl AK, Thorne LG, Jolly C, Towers GJ, Zuliani-Alvarez L, Bouhaddou M, Obernier K, McGovern BL, Rodriguez ML, Enjuanes L, Fernandez-Sousa JM, Krogan NJ, Jimeno JM, and Garcia-Sastre A. 2022. Preclinical and randomized phase I studies of plitidepsin in adults hospitalized with COVID-19. Life Sci Alliance 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, Wang B, Xiang H, Cheng Z, Xiong Y, Zhao Y, Li Y, Wang X, and Peng Z. 2020. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. Jama 323: 1061–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang Y, Wang ML, Chien CS, Yarmishyn AA, Yang YP, Lai WY, Luo YH, Lin YT, Chen YJ, Chang PC, and Chiou SH. 2020. Highlight of Immune Pathogenic Response and Hematopathologic Effect in SARS-CoV, MERS-CoV, and SARS-Cov-2 Infection. Frontiers in immunology 11: 1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rockx B, Kuiken T, Herfst S, Bestebroer T, Lamers MM, Oude Munnink BB, de Meulder D, van Amerongen G, van den Brand J, Okba NMA, Schipper D, van Run P, Leijten L, Sikkema R, Verschoor E, Verstrepen B, Bogers W, Langermans J, Drosten C, Fentener van Vlissingen M, Fouchier R, de Swart R, Koopmans M, and Haagmans BL. 2020. Comparative pathogenesis of COVID-19, MERS, and SARS in a nonhuman primate model. Science 368: 1012–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smits SL, de Lang A, van den Brand JM, Leijten LM, van IWF, Eijkemans MJ, van Amerongen G, Kuiken T, Andeweg AC, Osterhaus AD, and Haagmans BL. 2010. Exacerbated innate host response to SARS-CoV in aged non-human primates. PLoS Pathog 6: e1000756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Losada A, Munoz-Alonso MJ, Martinez-Diez M, Gago F, Dominguez JM, Martinez-Leal JF, and Galmarini CM. 2018. Binding of eEF1A2 to the RNA-dependent protein kinase PKR modulates its activity and promotes tumour cell survival. Br J Cancer 119: 1410–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, Jordan TX, Oishi K, Panis M, Sachs D, Wang TT, Schwartz RE, Lim JK, Albrecht RA, and tenOever BR. 2020. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 181: 1036–1045 e1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Del Valle DM, Kim-Schulze S, Huang HH, Beckmann ND, Nirenberg S, Wang B, Lavin Y, Swartz TH, Madduri D, Stock A, Marron TU, Xie H, Patel M, Tuballes K, Van Oekelen O, Rahman A, Kovatch P, Aberg JA, Schadt E, Jagannath S, Mazumdar M, Charney AW, Firpo-Betancourt A, Mendu DR, Jhang J, Reich D, Sigel K, Cordon-Cardo C, Feldmann M, Parekh S, Merad M, and Gnjatic S. 2020. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med 26: 1636–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katsande PM, Fernandez-Bastit L, Ferreira WT, Vergara-Alert J, Hess M, Lloyd-Jones K, Hong HA, Segales J, and Cutting SM. 2022. Heterologous Systemic Prime-Intranasal Boosting Using a Spore SARS-CoV-2 Vaccine Confers Mucosal Immunity and Cross-Reactive Antibodies in Mice as well as Protection in Hamsters. Vaccines (Basel) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez SF, Lukacs-Kornek V, Kuligowski MP, Pitcher LA, Degn SE, Kim YA, Cloninger MJ, Martinez-Pomares L, Gordon S, Turley SJ, and Carroll MC. 2010. Capture of influenza by medullary dendritic cells via SIGN-R1 is essential for humoral immunity in draining lymph nodes. Nat Immunol 11: 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vidal E, Lopez-Figueroa C, Rodon J, Perez M, Brustolin M, Cantero G, Guallar V, Izquierdo-Useros N, Carrillo J, Blanco J, Clotet B, Vergara-Alert J, and Segales J. 2022. Chronological brain lesions after SARS-CoV-2 infection in hACE2-transgenic mice. Vet Pathol 59: 613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brustolin M, Rodon J, Rodriguez de la Concepcion ML, Avila-Nieto C, Cantero G, Perez M, Te N, Noguera-Julian M, Guallar V, Valencia A, Roca N, Izquierdo-Useros N, Blanco J, Clotet B, Bensaid A, Carrillo J, Vergara-Alert J, and Segales J. 2021. Protection against reinfection with D614- or G614-SARS-CoV-2 isolates in golden Syrian hamster. Emerg Microbes Infect 10: 797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corman VM, Landt O, Kaiser M, Molenkamp R, Meijer A, Chu DK, Bleicker T, Brunink S, Schneider J, Schmidt ML, Mulders DG, Haagmans BL, van der Veer B, van den Brink S, Wijsman L, Goderski G, Romette JL, Ellis J, Zambon M, Peiris M, Goossens H, Reusken C, Koopmans MP, and Drosten C. 2020. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benet S, Galvez C, Drobniewski F, Kontsevaya I, Arias L, Monguio-Tortajada M, Erkizia I, Urrea V, Ong RY, Luquin M, Dupont M, Chojnacki J, Dalmau J, Cardona P, Neyrolles O, Lugo-Villarino G, Verollet C, Julian E, Furrer H, Gunthard HF, Crocker PR, Tapia G, Borras FE, Fellay J, McLaren PJ, Telenti A, Cardona PJ, Clotet B, Vilaplana C, Martinez-Picado J, and Izquierdo-Useros N. 2021. Dissemination of Mycobacterium tuberculosis is associated to a SIGLEC1 null variant that limits antigen exchange via trafficking extracellular vesicles. J Extracell Vesicles 10: e12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Hoecke L, Job ER, Saelens X, and Roose K. 2017. Bronchoalveolar Lavage of Murine Lungs to Analyze Inflammatory Cell Infiltration. Journal of Visualized Experiments. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun F, Xiao G, and Qu Z. 2017. Murine Bronchoalveolar Lavage. Bio Protoc 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fafian-Labora JA, and O’Loghlen A. 2021. NF-kappaB/IKK activation by small extracellular vesicles within the SASP. Aging Cell 20: e13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sambrook J, Russell DW, and Sambrook J. 2006. The condensed protocols from Molecular cloning : a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. [Google Scholar]
- 24.Viatour P, Merville M-P, Bours V, and Chariot A. 2005. Phosphorylation of NF-κB and IκB proteins: implications in cancer and inflammation. Trends in Biochemical Sciences 30: 43–52. [DOI] [PubMed] [Google Scholar]
- 25.Rathnasinghe R, Strohmeier S, Amanat F, Gillespie VL, Krammer F, Garcia-Sastre A, Coughlan L, Schotsaert M, and Uccellini MB. 2020. Comparison of transgenic and adenovirus hACE2 mouse models for SARS-CoV-2 infection. Emerg Microbes Infect 9: 2433–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ng DL, Al Hosani F, Keating MK, Gerber SI, Jones TL, Metcalfe MG, Tong S, Tao Y, Alami NN, Haynes LM, Mutei MA, Abdel-Wareth L, Uyeki TM, Swerdlow DL, Barakat M, and Zaki SR. 2016. Clinicopathologic, Immunohistochemical, and Ultrastructural Findings of a Fatal Case of Middle East Respiratory Syndrome Coronavirus Infection in the United Arab Emirates, April 2014. The American journal of pathology 186: 652–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding Y, Wang H, Shen H, Li Z, Geng J, Han H, Cai J, Li X, Kang W, Weng D, Lu Y, Wu D, He L, and Yao K. 2003. The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. The Journal of pathology 200: 282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalil AC, and Thomas PG. 2019. Influenza virus-related critical illness: pathophysiology and epidemiology. Critical care (London, England) 23: 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, and Manson JJ. 2020. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet (London, England) 395: 1033–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryabkova VA, Churilov LP, and Shoenfeld Y. 2021. Influenza infection, SARS, MERS and COVID-19: Cytokine storm - The common denominator and the lessons to be learned. Clin Immunol 223: 108652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chua RL, Lukassen S, Trump S, Hennig BP, Wendisch D, Pott F, Debnath O, Thurmann L, Kurth F, Volker MT, Kazmierski J, Timmermann B, Twardziok S, Schneider S, Machleidt F, Muller-Redetzky H, Maier M, Krannich A, Schmidt S, Balzer F, Liebig J, Loske J, Suttorp N, Eils J, Ishaque N, Liebert UG, von Kalle C, Hocke A, Witzenrath M, Goffinet C, Drosten C, Laudi S, Lehmann I, Conrad C, Sander LE, and Eils R. 2020. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat Biotechnol 38: 970–979. [DOI] [PubMed] [Google Scholar]
- 32.Sefik E, Qu R, Junqueira C, Kaffe E, Mirza H, Zhao J, Brewer JR, Han A, Steach HR, Israelow B, Blackburn HN, Velazquez SE, Chen YG, Halene S, Iwasaki A, Meffre E, Nussenzweig M, Lieberman J, Wilen CB, Kluger Y, and Flavell RA. 2022. Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature 606: 585–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buss H, Dorrie A, Schmitz ML, Hoffmann E, Resch K, and Kracht M. 2004. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-kappaB at serine 536 is mediated by multiple protein kinases including IkappaB kinase (IKK)-alpha, IKKbeta, IKKepsilon, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J Biol Chem 279: 55633–55643. [DOI] [PubMed] [Google Scholar]
- 34.O’Mahony AM, Montano M, Van Beneden K, Chen LF, and Greene WC. 2004. Human T-cell lymphotropic virus type 1 tax induction of biologically Active NF-kappaB requires IkappaB kinase-1-mediated phosphorylation of RelA/p65. J Biol Chem 279: 18137–18145. [DOI] [PubMed] [Google Scholar]
- 35.Yang F, Tang E, Guan K, and Wang CY. 2003. IKK beta plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J Immunol 170: 5630–5635. [DOI] [PubMed] [Google Scholar]
- 36.Mattioli I, Sebald A, Bucher C, Charles RP, Nakano H, Doi T, Kracht M, and Schmitz ML. 2004. Transient and selective NF-kappa B p65 serine 536 phosphorylation induced by T cell costimulation is mediated by I kappa B kinase beta and controls the kinetics of p65 nuclear import. J Immunol 172: 6336–6344. [DOI] [PubMed] [Google Scholar]
- 37.Zhong H, May MJ, Jimi E, and Ghosh S. 2002. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Molecular cell 9: 625–636. [DOI] [PubMed] [Google Scholar]
- 38.Dalskov L, Møhlenberg M, Thyrsted J, Blay-Cadanet J, Poulsen ET, Folkersen BH, Skaarup SH, Olagnier D, Reinert L, Enghild JJ, Hoffmann HJ, Holm CK, and Hartmann R. 2020. SARS-CoV-2 evades immune detection in alveolar macrophages. EMBO reports 21: e51252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perez-Zsolt D, Munoz-Basagoiti J, Rodon J, Elosua-Bayes M, Raich-Regue D, Risco C, Sachse M, Pino M, Gumber S, Paiardini M, Chojnacki J, Erkizia I, Muniz-Trabudua X, Ballana E, Riveira-Munoz E, Noguera-Julian M, Paredes R, Trinite B, Tarres-Freixas F, Blanco I, Guallar V, Carrillo J, Blanco J, Telenti A, Heyn H, Segales J, Clotet B, Martinez-Picado J, Vergara-Alert J, and Izquierdo-Useros N. 2021. SARS-CoV-2 interaction with Siglec-1 mediates trans-infection by dendritic cells. Cell Mol Immunol 18: 2676–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirano T, and Murakami M. 2020. COVID-19: A New Virus, but a Familiar Receptor and Cytokine Release Syndrome. Immunity 52: 731–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neufeldt CJ, Cerikan B, Cortese M, Frankish J, Lee JY, Plociennikowska A, Heigwer F, Prasad V, Joecks S, Burkart SS, Zander DY, Subramanian B, Gimi R, Padmanabhan S, Iyer R, Gendarme M, El Debs B, Halama N, Merle U, Boutros M, Binder M, and Bartenschlager R. 2022. SARS-CoV-2 infection induces a pro-inflammatory cytokine response through cGAS-STING and NF-kappaB. Commun Biol 5: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schulz I, Engel C, Niestroj AJ, Kehlen A, Rahfeld JU, Kleinschmidt M, Lehmann K, Rossner S, and Demuth HU. 2014. A non-canonical function of eukaryotic elongation factor 1A1: regulation of interleukin-6 expression. Biochim Biophys Acta 1843: 965–975. [DOI] [PubMed] [Google Scholar]
- 43.Xu S, Wu X, Zhang X, Chen C, Chen H, and She F. 2020. CagA orchestrates eEF1A1 and PKCdelta to induce interleukin-6 expression in Helicobacter pylori-infected gastric epithelial cells. Gut Pathog 12: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barnette KG, Gordon MS, Rodriguez D, Bird TG, Skolnick A, Schnaus M, Skarda PK, Lobo S, Sprinz E, Arabadzhiev G, Kalaydzhiev P, and Steiner M. 2022. Oral Sabizabulin for High-Risk, Hospitalized Adults with Covid-19: Interim Analysis. NEJM Evidence 1: EVIDoa2200145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.