

Abstract
Impairing the BET-family co-activator BRD4 with small molecule inhibitors (BETi) showed encouraging pre-clinical activity in treating acute myeloid leukemia (AML). However, dose-limiting toxicities and limited clinical activity dampened the enthusiasm for BETi as a single agent. BETi resistance in AML myeloblasts was found to correlate with maintaining mitochondrial respiration, suggesting that identifying the metabolic pathway sustaining mitochondrial integrity could help develop approaches to improve BETi efficacy. Herein, we demonstrated that mitochondria-associated lactate dehydrogenase allows AML myeloblasts to utilize lactate as a metabolic bypass to fuel mitochondrial respiration and maintain cellular viability. Pharmacologically and genetically impairing lactate utilization rendered resistant myeloblasts susceptible to BET inhibition. Low-dose combinations of BETi and oxamate, a lactate dehydrogenase inhibitor, reduced in vivo expansion of BETi-resistant AML in cell line and patient-derived murine models. These results elucidate how AML myeloblasts metabolically adapt to BETi by consuming lactate and demonstrate that combining BETi with inhibitors of lactate utilization may be useful in AML treatment.
Introduction
Acute myeloid leukemia (AML) is a malignant disease of the bone marrow characterized by arrest of hematopoietic precursors in the early stage of development and uncontrolled proliferation of myeloblasts. The dysregulation of MYC has been identified across genomic subtypes of AML and associated with many pathogenic genetic abnormalities including: aberrations in NPM1 (1,2) and FLT3-ITD (3,4), MYC amplifications, as well as common translocations such as RUNX1-ETO, MLL, and PML-RARα (5,6). The transcription of MYC target genes is regulated by the BET family protein, BRD4, an epigenetic modifier that recognizes hyperacetylated chromatin regions and promotes the assembly of a large platform of transcription regulating proteins that facilitates transcriptional elongation (7). BRD4 is enriched at large enhancer elements, called super-enhancers, which are frequently co-opted by tumor cells that drive the expression of genes critical to cell fate, proliferation, and survival (8,9). While small molecule BET inhibitors (BETi) showed initial promise as a therapeutic strategy in AML (10,11), a small proportion of AML myeloblasts display rapid transcriptional plasticity to survive the initial BETi challenge and subsequently become the dominant population (12,13). As a result, early clinical trials with BETi were unsuccessful in both solid and hematologic malignancies (14–18), but the use of BETi in combination regimens continues to garner enthusiasm (19–22).
AML cells possess a unique metabolic signature with higher anabolic pathway precursors including, increased glycolysis, glutaminolysis, and intermediates of the tricarboxylic acid cycle (TCA) and pentose phosphate pathway (23,24). The oncogene MYC has been shown to drive cells toward elevated glycolysis (25–27) and glutaminolysis (28–30). As such, inhibition of BRD4 reduces MYC gene expression, cell proliferation (4,31–34), and glycolytic metabolism (35–37). While upregulation of MYC renders AML cells resistant to BET inhibition (13), we previously identified some BETi resistant AML cells where MYC reactivation does not occur (20). Rather, we demonstrated that BET inhibition disrupts mitochondrial respiration and ATP production in some but not all AML samples, which coincides with escape from BETi-mediated toxicity (20). These findings suggest alternative metabolic pathways that sustain mitochondrial respiration, ATP production, and proliferation are utilized in response to BETi.
Here, we show that BETi-resistant AML cells localize a higher abundance of lactate dehydrogenase (LDH) to their mitochondrial structures compared to BETi-susceptible AML cells and are predisposed to utilize lactate as a metabolic bypass to maintain mitochondrial respiration during BET inhibition. As such, we identified novel metabolic biomarkers related to lactate utilization in AML patients that predict BETi responsiveness. We also employed multiple genetic models and chemical inhibitors to verify that disrupting lactate utilization stymies BETi resistance. These findings were translated in vivo using the broad LDH inhibitor, oxamate (38,39), where combined treatment with BETi and oxamate blunts BETi-resistance AML chimerism in both cell line (CDX) and patient derived xenograft (PDX) transplantation models. To date, this is the first observation of AML myeloblasts utilizing lactate to maintain metabolic integrity and circumvent antileukemic therapy. These findings support the concept of testing lactate utilization inhibitors in clinical settings, especially in conjunction with BETi.
Materials and Methods
Reagents
Antibodies specific for hCD45-APC and anti-rabbit Alexa 488 were purchased from Biolegend, hCD33-Cy7 from BD Biosciences, LDHA/B from Abcam, SOD1 and VDAC from Cell Signaling Technologies, and MCT1 and MCT4 from Invitrogen. Anti-rabbit and anti-goat antibodies were purchased from LI-COR. Helix NP Blue was purchased from Biolegend, BioTracker 488 Green Nuclear Dye from Sigma-Millipore, CellTrace Violet, mitoTracker Deep Red, and CellMask Orange from Invitrogen and Hoechst 33342 from Thermo Scientific. Paraformaldehyde was from Electron Microscopy Sciences. INCB054329 was provided by Incyte Corporation and AZD3965, UK-5099, sodium oxamate, CPI-613, and sodium L-lactate were purchased from Selleckchem. Lactate and pyruvate assays kits were purchased from Sigma-Aldrich. RPMI and IMDM were purchased from Gibco. Seahorse XF RPMI, glucose, glutamine, sodium pyruvate, oligomycin, FCCP, rotenone, and antimycin A were purchased from Agilent.
Cell Lines
The MV-4–11 cell line was purchased from American Type Culture Collection (ATCC) and MOLM-13 cells purchased from Deutsche Sammelung von Mikroorganismen und Zellkulturen (DSMZ). ATCC and DSMZ cell lines are authenticated by short tandem repeat profiling and cytochrome C oxidase gene analysis, and per lab standard of practice, cell lines were tested for Mycoplasma using the Universal Mycoplasma Detection Kit (ATCC). Cell cultures were split every 3–4 days to maintain cells in exponential growth phase and experiments conducted within 10–30 passages from thawing. Unless otherwise described, MV-4–11 and MOLM-13 cells were cultured in IMDM and RPMI, respectively, which was supplemented with 10% FBS and 100 U/mL penicillin and 100 μg/mL streptomycin. For pyruvate supplementation experiments, the culture media was supplemented with sodium pyruvate so that the final pyruvate concentrations were 2x (220 mg/L) or 5x (550 mg/L) above standard culture media. For defined carbon source experiments, Seahorse XF RPMI with 10% FBS was supplemented with glucose (4.5 mg/dL)/glutamine (584 mg/L)/pyruvate (110 mg/L) and/or sodium lactate (4.5 mg/L). Cells were cultured in a 37°C incubator at 5% CO2.
Patient Samples
Primary patient samples were provided by the Vanderbilt-Ingram Cancer Center Hematopoietic Malignancies Repository and in accordance with the tenets of the Declaration of Helsinki. Written informed consent was obtained from all patients, and experimentation performed on these samples was approved by the Vanderbilt University Medical Center Institutional Review Board.
Global Metabolomics
Sample Processing for Mass Spectrometry:
Cells were collected and pelleted into 1.5 mL Eppendorf tubes with media aspirated. Cell pellets were immediately placed in dry ice, and 1 mL 80% HPLC-grade MeOH/H2O cooled to −80 °C was added to each cell pellet. Samples were incubated at −80C for 15 minutes for further enzymatic inactivation. Samples were removed from −80 °C, placed on water ice, and vortexed twice for 30 seconds to extract metabolites. Samples were centrifuged at. 20 krcf for 10 minutes at 4 °C for protein and oligonucleotide precipitation. Sample supernatant was then evaporated using a speed vacuum and reconstituted in 30 uL sample solvent (water:methanol:acetonitrile, 2:1:1, v/v). 5 uL was further analyzed by liquid chromatography-mass spectrometry (LC-MS).
LC-MS method:
Ultimate 3000 UHPLC (Dionex) is coupled to Q Exactive Plus-Mass spectrometer (QE-MS, Thermo Scientific) for metabolite profiling. A hydrophilic interaction chromatography method (HILIC) employing an Xbridge amide column (100 × 2.1 mm i.d., 3.5 μm; Waters) is used for polar metabolite separation. Detailed LC method was described previously (40) except that mobile phase A was replaced with water containing 5 mM ammonium acetate (pH 6.8). The QE-MS is equipped with a HESI probe with related parameters set as below: heater temperature, 120 °C; sheath gas, 30; auxiliary gas, 10; sweep gas, 3; spray voltage, 3.0 kV for the positive mode and 2.5 kV for the negative mode; capillary temperature, 320 °C; S-lens, 55; A scan range (m/z) of 70 to 900 was used in positive mode from 1.31 to 12.5 minutes. For negative mode, a scan range of 70 to 900 was used from 1.31 to 6.6 minutes and then 100 to 1,000 from 6.61 to 12.5 minutes; resolution: 70000; automated gain control (AGC), 3×106 ions. Customized mass calibration was performed before data acquisition.
Analysis of Data:
LC-MS peak extraction and integration were performed using commercially available software Sieve 2.2 (Thermo Scientific). The peak area was used to represent the relative abundance of each metabolite in different samples. The missing values were handled as described in previous study (40). Mass spectrometry data was analyzed by first normalizing to sample cell count. Fold changes were determined for MV-4–11 and MOLM-13 treated with DMSO or 300 nM BETi for 48 hr. This was achieved by dividing the average of the three replicate values for BETi 300 nM by the average of the three replicate values for DMSO.
Cellular Replication Assay
Cell lines were stained with CellTrace Violet (5 μM) for 20 min in PBS, washed, and plated in specific culture media or treatments. After 72 hr, cells were resuspended in FACS media (PBS, 2% FBS, 0.02% NaAz) and median fluorescent intensity quantified by flow cytometry.
Cellular Viability Assay
Cells were stained with a membrane permeable (BioTracker 488 Green Nuclear Dye) and impermeable (Sytox Blue; 0.5 μM) nuclear fluorescent dyes for 20 min is cell culture media. Cells were washed, resuspended in 100 μL PBS, and imaged using a fluorescent plate reader (ex/em: 435/480, 490/525 nm). Viability was quantified by dividing the Sytox and BioTracker relative fluorescent units and normalizing to mock treated samples.
Cell Fractionation and LDH Immunoblot
Approximately 20×106 cells were fractionated per the manufacturer’s instructions (Thermo Fisher Scientific) and mitochondria lysed in cell lysis buffer (PBS, 1% NP40, protease inhibitors). Lysates were resuspended in reducing SDS/PAGE sample buffer and fractionated by a 4–20% SDS/PAGE gel. Separated proteins were transferred to a nitrocellulose membrane. Membranes were blocked in LI-COR PBS Blocking Buffer, incubated with various immunoblotting antibodies followed by the appropriate fluorophore-conjugated secondary antibodies. Membranes were imaged in a GelDoc Go Imaging System (Bio-Rad) and densitometry quantified by ImageJ software.
Lactate Utilization Assays
Cells were treated in standard culture media for 24, 48, or 72 with/without BETi (0.15 μM). Cells were washed, resuspended in minimal media (Seahorse Agilent media (pH 7.4)) supplemented with BETi and/or oxamate (25 μM) or CPI-613 (200 μM), and cultured for 2 hr at 37 °C (5% CO2). After 2 hr, sodium lactate (20 μM) was added to the culture for 2 hr. Cells were lysed, deproteinated (Sigma Aldrich), and intracellular lactate or pyruvate levels were quantified using assay kits per manufacturer’s instructions (Sigma Aldrich). Concentrations were quantified relative to a standard provided by the kit using a fluorescent plate reader.
Seahorse Analysis
Seahorse plates were coated with poly-L lysine for 20 min, washed using sterile water, and aspirated prior to addition of cells. On the day of the experiment, cells were resuspended in mitochondria stress test media (Seahorse Agilent media (pH 7.4), 1 mM pyruvate, 10 mM glucose, 2 mM glutamine) and transferred to the Seahorse plate. Each plate included at least 4 wells with no cells as a background control. The cartridge was hydrated overnight prior to the day of the experiment at 37 °C (no CO2). On the day of the experiment the water in the cartridge plate was exchanged for Seahorse Calibrant media (Agilent Technologies). The cartridge was sequentially loaded with oligomycin (15 mM), carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP, 15 mM), and rotenone/antimycin A (5 mM) all diluted in mitochondria assay media for injection into the culture. To normalize across wells by cell density, the Seahorse plate was imaged using a BioTek Cytation 5. Oxygen consumption rates were collected using a Seahorse XFe96 Analyzer and analyzed using Wave Desktop software (Agilent Technologies).
CRISPR/Cas9 Experiments
For CRISPR knockout experiments, MV-4–11 and MOLM-13 cells were electroporated with Cas9/gRNA ribonucleic protein (RNP) using a Neon transfection system (Thermo Fischer Scientific). Briefly, Alt-R® S.p. Cas9 Nuclease V3 (IDT) was complexed with gRNA at a 1:2.5 ratio for 10 minutes at room temperature. RNP was added to achieve a final concentration of 250 μg/mL to 100 μL of cells (107 cells/mL) plus Alt-R® Cas9 Electroporation Enhancer (IDT) in Buffer R. MV-4–11 cells were electroporated at 1350 V, 35 ms pulse width, 1 pulse. MOLM-13 cells were electroporated at 1600 V, 10 ms pulse width, 3 pulses. The sgRNAs were synthesized through IDT and the protospacer sequences are as follows: BRD4 – GAGTGGTGCTCAAGACACTA; SLC16A1 – TATCCATGACACTTCGCTGG; LDHB – GGACTGTACTTGACGATCTG. The Alt-R® CRISPR-Cas9 Negative Control crRNA #1 (IDT) was used as a control in all experiments. Three independent experiments were conducted for each condition.
To validate CRISPR/Cas9 cutting, genomic DNA was isolated using DNA Blood Mini Kit (Qiagen). A PCR was performed using Q5® High-Fidelity 2X Master Mix (NEB). For BRD4, primers For: TCTGTGGGCCTTCCTTTCTC and Rev: CTTGTCATTCTGAGCGGTGC, with initial denaturation at 98°C, followed by 30 cycles of 98°C for 10 seconds, annealing at 67°C for 30 seconds, then extension at 72°C for 40 seconds. For SLC16A1, PCR was similarly performed with primers For: ACATTTCAAGAGTCCAGCAGA and Rev: AAAGAGTTTTATAGGTGTGCCTT, with an annealing temperature of 62°C. For LDHB, PCR was similarly performed with primers For: ATAACCAAGCCAGATGAAGC and Rev: TCGAAAAAGGACACACTTCC, with an annealing temperature of 63°C. PCR products were purified using QIAquick PCR Purification Kit (Qiagen). Sanger sequencing was completed via Genewiz/Azenta and CRISPR indel efficiency was analyzed using TIDE software v3.3.0.
MCT4 Knockdown
MCT-4 expression was knocked down in MV-4–11 and MOLM-13 cells using the RNAiMAX siRNA transfection per manufacturer instructions (Thermo Fisher Scientific). In brief, cells were plated at 100,000 cells/200 μL in a 96-well plate. Then the cells were treated with three siRNAs targeting MCT-4 at equal concentrations (Assay ID: S17416, S17417, S17418) or scrambled siRNA for a negative control at a final concentration of 1 pmol/well. After 24 hr, cells were treated with BETi or vehicle control and harvested 72 hr after plating. Two independent experiments were conducted, each with technical duplicates and biological duplicates. MCT-4 knockdown was verified using flow cytometry.
Transcript Analysis by qPCR
RNA extraction and cDNA synthesis:
For AML cell line gene expression analyses, cells were plated at an initial density of 200,000 cells/mL and cultured for 72 hr across three separate experiments. Cell lines, cryopreserved bone marrow mononuclear cells, and cryopreserved peripheral blood leukocytes were washed twice in cold PBS and centrifuged at 200 × g for 10 minutes at room temperature. Total RNA was extracted from the cell pellets using the RNeasy Mini Kit (Qiagen, Hilden, Germany), 3 μg of RNA treated with RNase-free DNase I (Thermo Fisher Scientific, Waltham, MA, USA), and reverse transcribed to cDNA using the SuperScript™ III First-Strand Synthesis System (Thermo Fisher Scientific) by oligo(dT)20 priming.
Quantitative PCR (qPCR):
qPCR was performed using TaqMan assays (Thermo Fisher Scientific) targeting LDHA (Hs01378790_g1, FAM), SLC16A1 (Hs01560299_m1, FAM), and SLC16A3 (Hs00358829_m1, FAM). Assays were prepared in triplicate wells with the TaqMan Gene Expression Master Mix (Thermo Fisher Scientific), and the amplified products were detected on the QuantStudio™ 3 Real-Time PCR System (Thermo Fisher Scientific) using the following cycling protocol: 2 minutes at 50°C, 10 minutes at 95°C, and 40 cycles of 15 seconds at 95°C and 60 seconds at 60°C. Relative transcript abundance was calculated using the 2−ΔΔCt method, normalizing each target’s cycle threshold (Ct) to the endogenous control gene, ACTB (Hs99999903_m1, VIC). Gene expression for each of the AML cell lines was analyzed across 3 separate experimental days, and the gene expression of each AML patient sample was analyzed in triplicate across three qPCR reactions.
In vivo murine modeling
NSGS [NOD-scid IL2Rgnull3Tg(hSCF/hGM-CSF/hIL3)] mice were maintained in an accredited animal facility. Mice were housed 2–5 animals to a cage in specific pathogen-free conditions. Food and water were provided ad libitum. All animal experiments were approved by the Vanderbilt Institutional Animal Care and Use Committee and IRB.
Female NSGS mice, 6–8 weeks old, were irradiated with 100 cGy microwave radiation. After 24 hr, mice were transplanted via tail vein injection with cells of interest (CDX = 1×106 MOLM-13 cells; PDX = 2×106 AML mononuclear cells). Mice were randomized post cell injection into cages of 5. Prior to treatment, peripheral microchimerism was documented at week 1 in CDX and at week 2 of PDX models. Mice showing no peripheral chimerism by 2 weeks in CDX or 3 weeks in PDX were considered engraftment failures and removed from the study. Upon establishing microchimerism, mice were treated with either BETi (INCB054329, Incyte), oxamate (Selleckchem), UK5099 (Selleckchem), and/or vehicle control. BETi was dissolved in N,N-dimethylacetamide (DMAC) and diluted in 5% methylcellulose, oxamate dissolved in PBS, and UK5099 dissolved in DMSO and diluted into PBS. BETi (CDX = 50 mg/kg, PDX = 75 mg/kg) was dosed at by oral gavage, b.i.d., 5 days a week. Oxamate (200 mg/kg) was dosed daily by intraperitoneal injection, 5 days a week. UK5099 (40 mg/kg) were dosed daily by intraperitoneal injection, 3 days a week. Peripheral blood was assessed weekly for human chimerism by flow cytometry. Spleen/body ratio was calculated as organ weight (grams) per gram of body weight.
Statistics
Specific statistical details for each experiment can be found in the corresponding figure legend. Error bars for all experiments represent variation of the mean across mice. A minimum of three experimental replicates were performed for each assay and the specific number of replicates is noted in the corresponding figure legend. All P values were calculated using a two-way ANOVA (Sidak’s multiple comparisons test or Tukey multiple comparisons test) or t-test when applicable. Statistical work was performed using Prism 6 software (GraphPad) and significance is indicated on the graphs as follows: *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001, ns=not significant.
Data Availability
All data needed to evaluate the conclusions in the paper are present in the article and/or Supplementary Materials. Additional data are available upon request from the corresponding author.
Results
AML cells use neither glutaminolysis nor fatty acid oxidation to resist BETi
This study leverages the novel BET inhibitor INCB054329 (41) to identify how AML myeloblasts metabolically bypass BET inhibition using two different human AML cell lines: the BETi-resistant MOLM-13 and BETi-susceptible MV-4–11 cells (Fig. S1A). We have previously demonstrated that the increased resistance to BETi corresponds with maintenance of mitochondrial respiration in MOLM-13 cells but not in MV-4–11 cells during BET inhibition (20). This suggests that MOLM-13 cells utilize carbon sources in alternative metabolic pathways to allow persistence during BETi treatment. To identify changes in metabolic pathways, AML cell lines were treated with 300 nM BETi for 48 hr and the resulting cell lysates subjected to global untargeted metabolic mass spectrometry (Fig. 1A, Table S1). Consistent with previous reports that BET inhibition disrupts glycolysis (35–37), many of the glycolytic metabolites were similarly decreased in MV-4–11 and MOLM-13 following BETi treatment even though intracellular glucose levels were increased (Fig. 1B). The metabolic intermediates of the tricarboxylic acid (TCA) cycle were better maintained in MOLM-13 cells compared to MV-4–11 during BET inhibition (Fig. 1C), which is similar to what we have previously observed during extracellular flux analysis (20). The capacity to sustain mitochondrial metabolic activity coincided with drastically higher levels of ATP observed in MOLM-13 than MV-4–11 cells (Fig. 1D). These results demonstrate that BETi-resistant MOLM-13 cells better sustain TCA cycle integrity and ATP homeostasis during BET inhibition.
Figure 1:
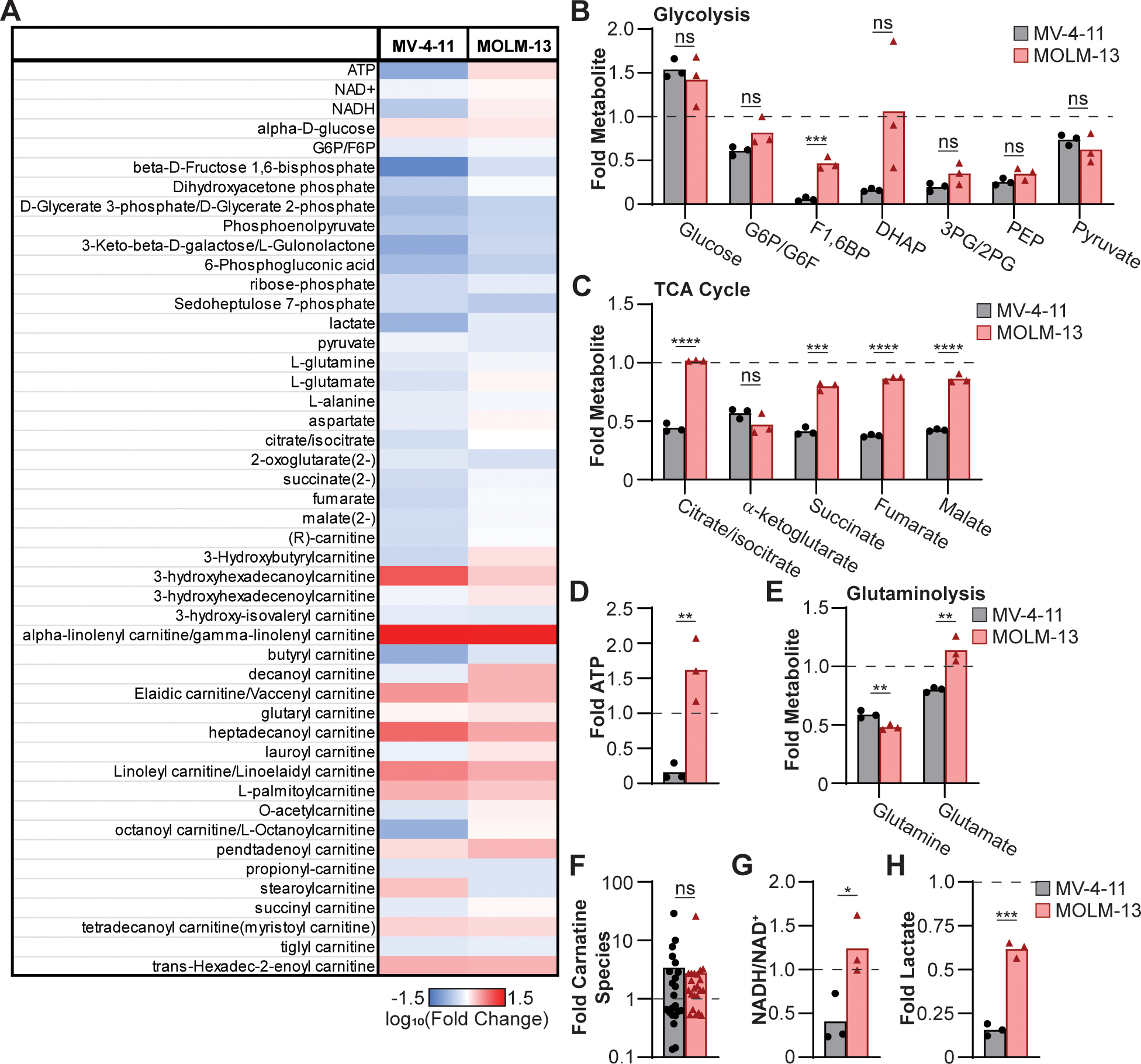
MOLM-13 cells better maintain TCA cycle metabolic activity following BET inhibition. MV-4–11 and MOLM-13 cells were treated with 300 nM of BETi for 48 hr and then subjected to mass spectrometry to assess global metabolite concentrations. (A) Data is presented as log10(Fold change) relative to mock-treated cells. Fold change in (B) glycolytic and (C) TCA cycle metabolites, (D) ATP, (E) glutaminolysis metabolites, (F) total carnitine species, (G) NADH/NAD+ ratio, and (H) lactate is presented. Each point represents (B-E, G, H) independent biological replicates or (F) mean result of individual carnitine species (biological triplicate) (n=3). (B-H) unpaired t test (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, ns = not significant).
One possible metabolic pathway sustaining TCA cycle activity in the absence of glycolysis is glutaminolysis. Glutamate levels were elevated in MOLM-13 cells compared to MV-4–11 (Fig. 1E), which could indicate heightened glutaminolysis. Glutamate dehydrogenase 1 (GLUD1) is the enzyme responsible for converting glutamate into α-ketoglutarate and is MYC regulated (42,43), and α-ketoglutarate levels were decreased in MV-4–11 and MOLM-13 cells (Fig. 1C). Thus, an accumulation of glutamate in BETi-treated MOLM-13 cells may reflect a blockade in glutaminolysis. This led us to query whether increased glutaminolysis was the cause of BETi resistance in MOLM-13 cells, by treating both cell lines with BETi and/or V-9302, a small-molecule inhibitor of glutamine transport (44). The addition of V-9302 alone disrupted cellular replication of MOLM-13 cells but had a minimal additive effect during co-treatment with BETi when quantifying MOLM-13 cellular replication and viability (Fig. S1B, C). These results demonstrate that glutaminolysis is essential for MOLM-13 cells to maintain cellular replication but is not leveraged to maintain viability during BETi treatment.
Given that glutaminolysis was not the cause of BETi resistance in MOLM-13 cells, fatty acid oxidation was considered as alternative metabolic pathway as fatty acids can be used to sustain mitochondrial respiration by being converted to acetyl-CoA. The addition of carnitine is necessary to transfer fatty acids into the mitochondria. However, following BETi treatment, MV-4–11 and MOLM-13 have similar abundances of carnitine species (Fig. 1F) indicating comparable fatty acid oxidation. We next blocked the transfer of carnitine species into the mitochondria using the transport inhibitor etomoxir (45) in combination with BETi, and comparably disrupted cellular replication without an effect on viability (Fig. S1D, E) in MOLM-13 and MV-4–11 cells. Taken together, these results demonstrate that the BETi-resistant MOLM-13 cells leverage neither glutaminolysis nor fatty acid oxidation to maintain mitochondrial respiration during BET inhibition.
BETi-resistant cells are better equipped to utilize lactate
The utilization of lactate by eukaryotic cells is a rare metabolic process that has been observed in skeletal muscle (46,47), cardiac tissue (46,48), and some cancers (49–51) in nutrient-deplete environments. Lactate utilization requires the association of lactate dehydrogenase (LDH) to the mitochondria where it preferentially facilitates the conversion of lactate into pyruvate resulting in the oxidation of NAD+ to form NADH and transferring electrons and protons into the electron transport chain thereby driving increased superoxide production (52–54). Consistent with heightened lactate utilization, MOLM-13 cells have an increased NADH/NAD+ ratio (Fig. 1G) as well as higher levels of intracellular lactate (Fig. 1H) compared to MV-4–11. These results imply that MOLM-13 cells are consuming lactate to maintain mitochondrial respiration and coincide with previous observations that resistance of MOLM-13 cells to BETi coincides with increased mitochondrial superoxide production (20).
To determine whether MOLM-13 cells had an increased capacity to utilize lactate compared to MV-4–11, we used quantitative PCR (qPCR) to measure expression of key lactate transporters. Monocarboxylate transporter 1 (MCT1) is the high affinity bi-directional lactate transporter that allows for lactate uptake (55), while MCT4 is an inducible low affinity lactate exporter (56,57). MOLM-13 cells express higher levels of MCT1 and lower levels of MCT4 indicating they are primed to import and retain lactate compared to MV-4–11 cells and maintained MCT1 and MCT4 transcript levels following BETi treatment (Fig. 2A, S1F). In the absence of BETi treatment, expression of LDHA and LDHB was decreased in MOLM-13 cells relative to MV-4–11 indicating decreased expression of LDH. While BETi treatment had minimal effect on LDHB levels, LDHA transcript was increased in MOLM-13 compared to MV-4–11. However, the regulation of lactate utilization is not dictated by LDHA/B expression but rather changes to the subcellular localization of LDH, where translocation of LDH protein into the mitochondria facilitates utilization of lactate to sustain mitochondrial respiration (52–54). Therefore, MV-4–11 and MOLM-13 cells were fractionated and the abundance of LDH protein quantified by immunoblot. The abundance of the cytosolic superoxide dismutase 1 (SOD1) and mitochondrial voltage-dependent anion-selective channel (VDAC) were quantified to ensure clean fractionation. While LDH protein levels were similar in the cytosolic fractions, LDH protein abundance was elevated in the mitochondrial fraction from MOLM-13 cells compared to MV-4–11 (Fig. 2B). Similarly, cells were treated with BETi for 48 hr and fluorescently imaged to spatially localize LDHA/B with mitochondria. MOLM-13 cells localized more LDH to mitochondria compared to MV-4–11 when mock treated or treated with BETi (Fig. 2C). In addition, treatment with BETi heightened localization of LDH to mitochondrial structures in MOLM-13 cells but had no effect on LDH localization in MV-4–11 cells. These results demonstrate that while LDHA/B transcript is decreased in MOLM-13 cells compared to MV-4–11, total LDH protein is elevated with a higher fraction of the LDH protein associated with mitochondrial structures, consistent with MOLM-13 cells being better equipped to utilize lactate during metabolic stress.
Figure 2:
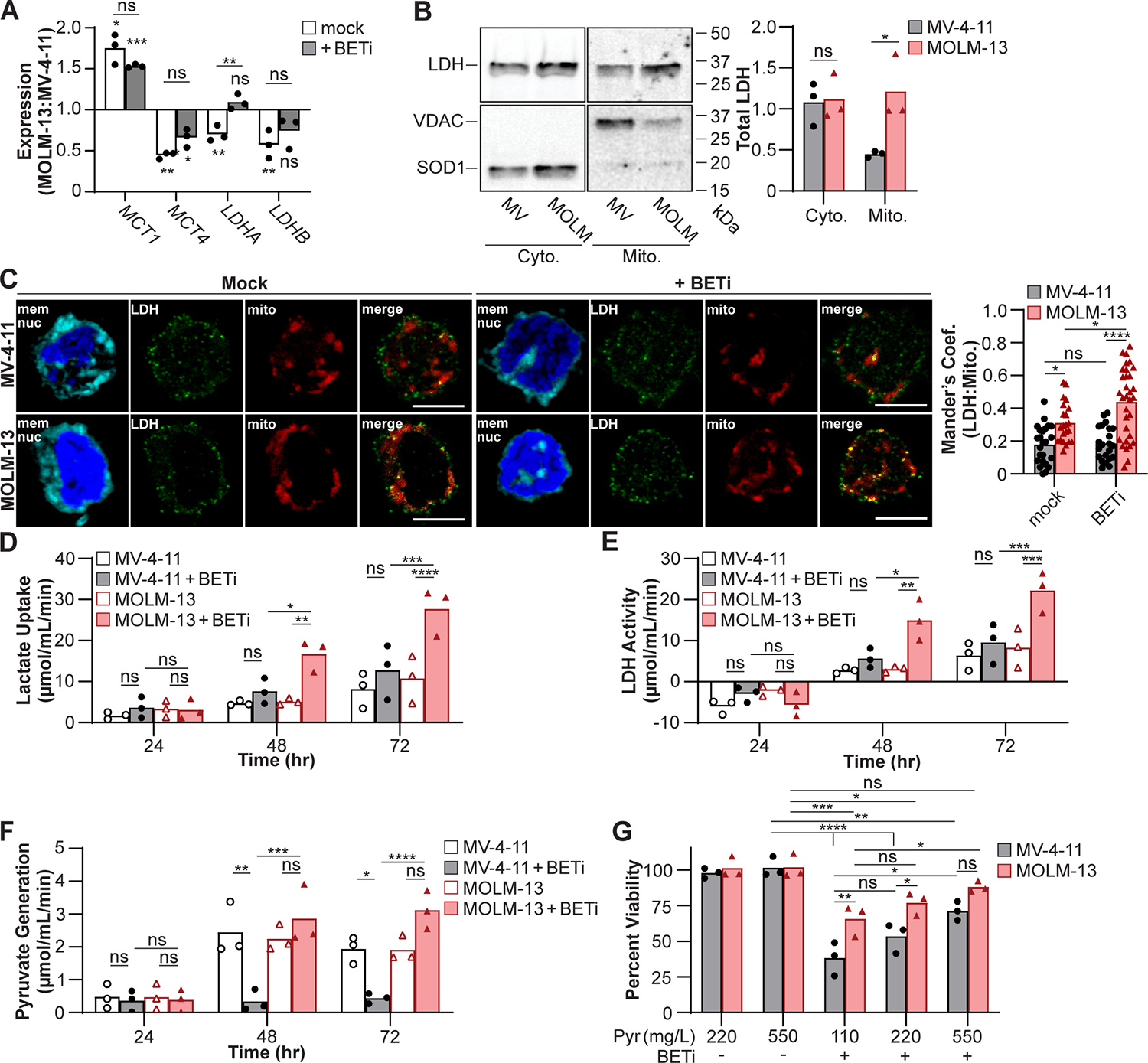
MOLM-13 cells are predisposed to utilize lactate upon BETi treatment. (A) Cells were mock treated or treated with BETi for 48 hr and gene expression associated with lactate homeostasis quantified in MOLM-13 cells relative to MV-4–11 by qPCR. (B) MV-4–11 (MV) and MOLM-13 (MOLM) cells were fractionated and LDH protein in the cytosolic (Cyto.) and mitochondrial (Mito.) fractions quantified by immunoblot. Superoxide dismutase 1 (SOD1) and voltage-dependent anion channel (VDAC) protein were immunoblotted to ensure clean fractionation. (C) Cells were treated with BETi for 48 hr and Mander’s coefficient of colocalization between LDH and mitochondria quantified by fluorescent imaging. (D-F) Cells were treated with BETi, and the media exchanged at indicated timepoints with minimal media with and without oxamate (25 μM) or CPI-613 (200 μM) or lysed to quantify baseline metabolite levels. Minimal media was subsequently supplemented with lactate (20 μM) for 2 hr, cells lysed, and intracellular lactate and pyruvate levels quantified using a fluorescent plate reader (C, Lactate Uptake = oxamate-treated – baseline; D, LDH Activity = oxamate-treated – mock-treated; E, Pyruvate Generation = CPI-613-treated – baseline). (F) Cells were cultured for 72 hr with BETi in media supplemented with sodium pyruvate (Pyr) and viability quantified using a fluorescent plate reader. Viability was quantified relative to vehicle control treated cells at 110 mg/L pyruvate. (A, D-G) Each point represents the mean (technical triplicate), (B) total obtained from an individual experiment (n=3), (C) or individual cell. (A-B) Paired t test or two-way ANOVA with (C, G) Sidak’s or (D-F) Tukey multiple comparisons test (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, ns = not significant).
We recognized that the heightened association of LDH to mitochondria may allow MOLM-13 cells to utilize lactate to maintain cellular pyruvate levels during the metabolic stress induced by BET inhibition. Therefore, MV-4–11 and MOLM-13 cells were treated with BETi (0.15 μM) to determine if and when lactate utilization is observed. Following BETi treatment, cells were provided lactate as the sole carbon source in the presence of the LDH inhibitor oxamate (38,39) or the pyruvate dehydrogenase inhibitor CPI-613 (58) and the abundance of lactate or pyruvate quantified using a fluorescence plate reader. After 24 hrs of BET inhibition, lactate uptake was similar across cells with and without BETi treatment (Fig. 2D). In addition, LDH activity was comparable and negative 24 hr post-BETi treatment (Fig. 2E), indicating that LDH was primarily acting at the backend of glycolysis rather than utilizing lactate in the mitochondria. The generation of pyruvate was also comparable 24 hr post-BETi treatment (Fig. 2F). However, at 48 and 72 hr, a significant increase in lactate uptake and LDH activity was observed in BETi-treated MOLM-13 cells compared to untreated MOLM-13 cells, a phenotype not observed in MV-4–11 cells (Fig. 2D–E). Consistent with the utilization of lactate, MOLM-13 cells treated with BETi for 48 and 72 hr generated higher levels of pyruvate following the addition of lactate compared to BETi-treated MV-4–11 cells (Fig. 2F). These results demonstrate that a 48–72 hr treatment with BETi drives MOLM-13 cells to utilize lactate to maintain their intracellular pyruvate levels.
Lactate utilization is a metabolic switch that AML may employ to resist BETi toxicity
Inhibition of BET may starve AML myeloblasts of pyruvate by disrupting glycolysis, which is necessary to sustain TCA cycle activity. The utilization of lactate bypasses this metabolic disruption by converting lactate to pyruvate and allowing BETi-resistant cells to maintain cellular viability. Supplementing excess pyruvate during BETi treatment should render BETi-susceptible AML myeloblasts metabolically resistant to BETi. Therefore, the cell lines were treated with BETi in culture media supplemented with 2- (220 mg/L) or 5-fold (550 mg/L) excess sodium pyruvate for 72 hr and viability quantified in a fluorescent plate reader. MOLM-13 cells maintained cellular viability better than MV-4–11 cells during BETi treatment; however, supplementing the culture media with 5-fold excess pyruvate was sufficient for MV-4–11 cells to maintain cellular viability at levels comparable to MOLM-13 cells (Fig. 2G). These findings demonstrate that BETi treatment starves myeloblasts of pyruvate, a necessary metabolic precursor to the TCA cycle, and that the utilization of lactate acting as a metabolic bypass provides pyruvate and may establish BETi resistance.
To determine whether the conversion of lactate into pyruvate allows MOLM-13 cells to better sustain mitochondrial respiration in response to BETi, cell lines were simultaneously treated with BETi and inhibitors of lactate utilization: AZD3965 to impair extracellular lactate uptake by the MCT1 (59), UK5099 to prevent the translocation of lactate into the mitochondria through the mitochondrial pyruvate carrier (MPC) (46,60), and oxamate to broadly impair LDH activity (54). Following treatment for 72 hr, oxygen consumption rates were quantified in the cell lines by extracellular flux analysis by Seahorse during a mitochondrial stress test (Fig. 3A). While MV-4–11 cells were sensitive to BETi treatment, MOLM-13 cells better maintained basal respiration and ATP synthase activity (Fig. 3B). Furthermore, treatment with inhibitors of lactate utilization sensitized ATP synthase activity in MOLM-13 cells to BET inhibition, significantly reducing oxygen consumption rates compared to BETi treatment alone and to levels comparable to BETi-treated MV-4–11 cells (Fig. 3B). Co-treatment with BETi and oxamate was sufficient to significantly decrease basal respiration compared BETi treatment alone, while the addition of AZD3965 and UK5099 in combination with BETi strongly trended toward decreasing basal respiration (P = 0.07 and 0.08 respectively). Not all parameters of mitochondrial respiration were disrupted as maximal respiration was not affected by the addition of lactate utilization inhibitors in MOLM-13 cells (Fig. S2) indicating that BETi and lactate utilization does not alter the capacity of mitochondria in MOLM-13 cells to shuttle electrons. Taken together, these results demonstrate that MOLM-13 cells leverage lactate utilization to maintain ATP-coupled respiration.
Figure 3:
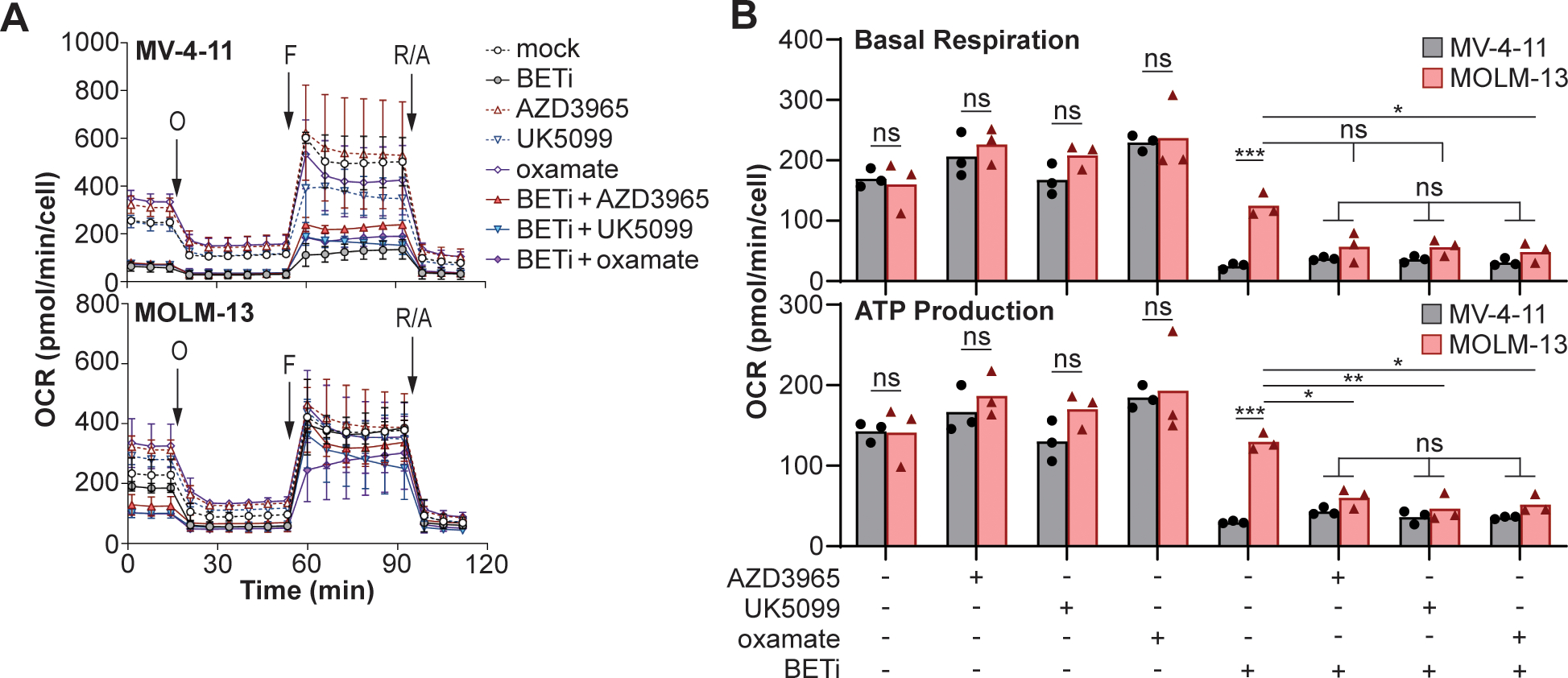
MOLM-13 cells utilize lactate to maintain mitochondrial respiration during BETi treatment. (A) MV-4–11 and MOLM-13 cells were cultured for 72 hr with indicated treatments (BETi = 0.15 μM; AZD3965, UK5099, oxamate = 0.1 μM), subjected to a mitochondrial stress test (O = oligomycin, F = FCCP, R/A = rotenone/antimycin A), and oxygen consumption rates (OCR) quantified by extracellular flux analysis. (B) Basal respiration and ATP production were quantified where each point represents individual experiments (n=3). (A) Error bars represent standard error across experiments and (B) two-way ANOVA with Sidak’s multiple comparisons test (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ns = not significant).
We have previously demonstrated that BET inhibition drives BETi sensitive cells to undergo apoptosis (20). To determine whether lactate consumption sustains cellular viability in MOLM-13 cells during BET inhibition, the cell lines were treated with BETi and the inhibitors of lactate utilization for 72 hr and viability quantified in a fluorescent plate reader. Coinciding with disruptions in mitochondrial respiration, treatment with inhibitors of lactate utilization significantly decreased viability of MOLM-13 cells during BETi treatment to levels comparable to BETi-treated MV-4–11 cells (Fig. 4A). To confirm the specificity of overcoming BET inhibition by utilizing lactate, we genetically disrupted BRD4 via CRISPR, and treated the cells with lactate utilization inhibitors. The rate of frame shift insertions/deletions (indels) in BRD4 in MV-4–11 cells was consistently lower than MOLM-13 after 72 hr (Fig. S3A), which could reflect differences in DNA damage repair efficiency or essentiality of BRD4. While MOLM-13 cells demonstrated increased viability relative to MV-4–11 following disruption of BRD4, inhibiting lactate utilization was sufficient to decrease cellular viability in MOLM-13 cells to levels comparable to MV-4–11 (Fig. 4B). Disrupting LDHB rather than LDHA via CRISPR had a similar effect on MOLM-13 cells, rendering them more susceptible to BETi-mediated toxicity (Fig. S3B–C), which is consistent with LDHB playing the dominate role in lactate utilization (61). Furthermore, disrupting the expression of the primary lactate importer, MCT1 (Fig. S3C), rendered MOLM-13 cells more susceptible to BETi-mediated toxicity (Fig. 4C). In contrast, siRNA knockdown of MCT4 had no effect on viability during BETi treatment (Fig. S3D). These results demonstrate that MOLM-13 cells utilize lactate as a metabolic bypass to resist BET inhibition.
Figure 4:
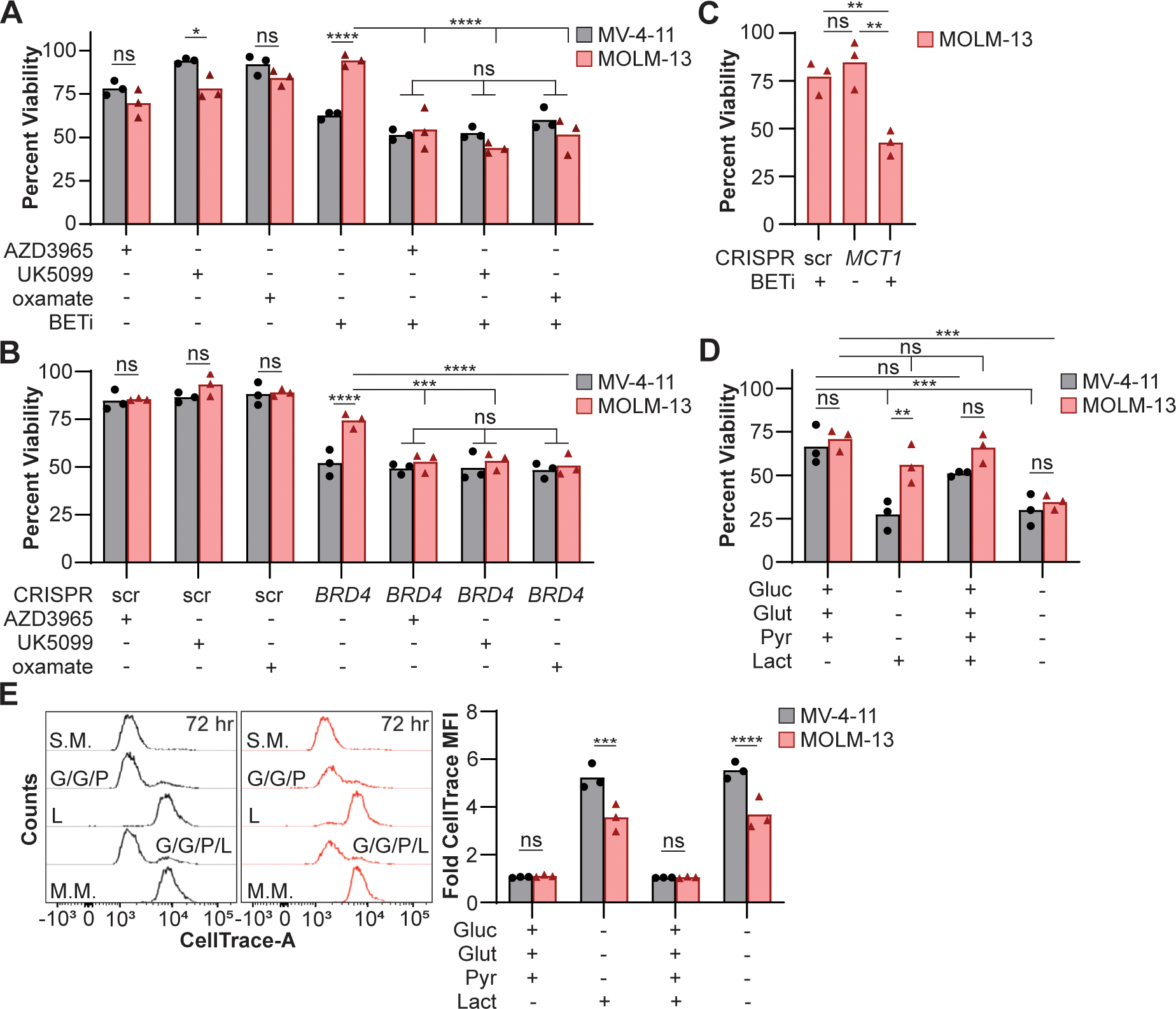
Lactate maintains cellular viability of MOLM-13 cells following metabolic inhibition. (A) MV-4–11 and MOLM-13 cells were cultured for 72 hr with BETi (0.15 μM) and/or lactate utilization inhibitors (AZD3965, UK5099, oxamate = 0.1 μM), and viability quantified using a fluorescent plate reader. Viability was quantified relative to cells treated with vehicle control. (B) BRD4 was disrupted, and cells treated with lactate utilization inhibitors (0.1 μM) or (C) MCT1 was disrupted, and cells treated with BETi (0.15 μM). After 48 hr, viability was quantified using a fluorescent plate reader relative to cells treated with vehicle and scramble (scr) control. (D-E) Cells were loaded with a CellTrace dye and cultured for 72 hr in minimal media supplemented with glucose (Gluc; 25 mM), glutamine (Glut; 4 mM), pyruvate (Pyr; 1 mM), and/or lactate (40 μM). (D) Viability was quantified in a fluorescent plate reader and (E) CellTrace quantified by flow cytometry relative to cells cultured in standard media. Each point represents the mean (technical (A-C) duplicate or (D) triplicate) value or (E) the median fluorescent intensity (MFI) from cells for an individual experiment (n=3). (A-B, D-E) Two-way ANOVA with Sidak’s multiple comparisons test or (C) paired t test (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, ns = not significant).
These findings led us to the conclusion that MOLM-13 cells utilize lactate when no other carbon sources are available or in high lactate environments. To further support this hypothesis, cell lines were cultured in minimal media supplemented with or without glucose, glutamine, and pyruvate at concentrations mimicking standard culture media and/or lactate (40 μM) for 72 hr. Cellular viability was comparable between MV-4–11 and MOLM-13 cells in minimal media containing glucose, glutamine, and pyruvate; however, MOLM-13 cells better maintained cellular viability when lactate was the sole carbon source (Fig. 4D). Furthermore, the addition of lactate to media containing glucose, glutamine, and pyruvate had no effect on cellular viability indicating that the presence of lactate in the culture was not toxic. While lactate supported cellular viability of MOLM-13 cells, lactate alone was not sufficient to maintain cellular replication to same degree as media containing glucose, glutamine, and pyruvate (Fig. 4E). These data demonstrate that lactate utilization allows BETi-resistant cells to maintain mitochondrial homeostasis and cellular viability during BET inhibition.
BETi-resistant cells do not survive in CDX model in presence of oxamate
Based on these findings in vitro, we tested the combined inhibition of BET and lactate utilization in a CDX transplantation model of the BETi-resistant MOLM-13 cells in NSGS mice. After establishing disseminated leukemia, mice were treated with 50 mg/kg of BETi twice a day and/or 200 mg/kg oxamate daily, 5 days a week for 2 weeks and then sacrificed (Fig. 5A). The percentage of MOLM-13 cells were quantified in the peripheral blood, spleen, and bone marrow using anti-human CD45 and CD33 monoclonal antibodies. Spleen weights were comparable across treatments (Fig. 5B). Monotherapy with BETi or oxamate treatment modestly decreased human leukemia burdens in the spleen and bone marrow; however, combining BETi with oxamate during treatment led to a greater than 5-fold reduction beyond monotherapy, and near resolution of the human leukemia (Fig 5C, D). Surprisingly, inhibiting lactate translocation into the mitochondria using UK5099 to block MPC was antagonistic with BETi (Fig. S4), which suggests that the specific targets within lactate utilization have differing efficacy in combination with BETi. Taken together, these results demonstrate the synergistic potential of BET inhibition and oxamate in treating BETi-resistant AML.
Figure 5:
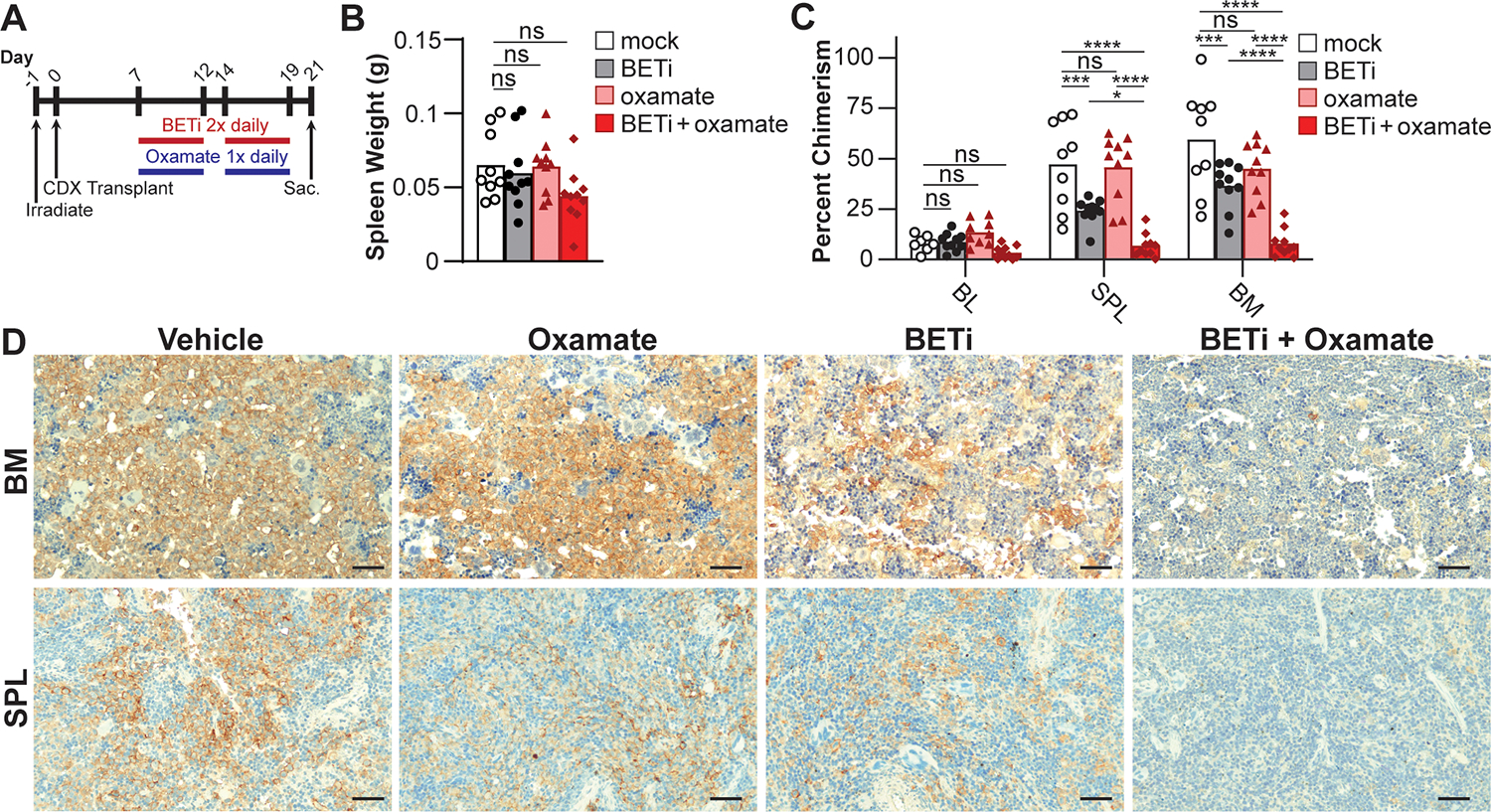
Oxamate enhances the efficacy of BETi in preventing MOLM-13 chimerism in CDX. (A) NSGS mice were engrafted with MOLM-13 cells and treated with vehicle control, BETi (50 mg/kg), and/or oxamate (200 mg/kg), and then sacrificed. (B) Spleens were weighed and (C) tumor burden assessed by chimerism analysis by quantifying hCD45+hCD33+ cells using flow cytometry and (D) immunohistochemistry (hCD45, scale bar = 50 μm). (B-C) Each point represents a single mouse (mock, n = 9; BETi or oxamate, n = 10; BETi+oxamate, n = 11). (B) One- or (C) two-way ANOVA with Tukey multiple comparisons test (*P ≤ 0.05, ***P ≤ 0.001, ****P ≤ 0.0001, ns = not significant).
BET resistance in patient samples correlates with an increased capacity to utilize lactate
Lactate utilization allows MOLM-13 cells to better resist BETi-mediated toxicity; however, MV-4–11 and MOLM-13 cells are immortalized cell lines and may not fully reflect primary AML myeloblasts. Therefore, we sought to determine if lactate utilization is a metabolic resistance mechanism to BET inhibition in AML patient samples (Table S2). Therefore, AML myeloblasts were fractionated to determine the abundance of LDH in the mitochondria relative to the GI50 concentration for BETi. A positive correlation between BETi resistance and the relative abundance of mitochondria-associated LDH (Fig. 6A) demonstrated that an increased resistance to BETi coincides with the potential to utilize lactate. To confirm increased lactate utilization in BETi resistant AML myeloblasts, cells were provided lactate as a sole carbon source in the presence/absence of oxamate and lactate uptake and LDH activity quantified using a fluorescence plate reader. Corresponding with increased mitochondrial LDH (Fig. 6A), BETi resistant patient samples had a higher rate of lactate internalization and LDH activity with lactate as a sole substrate (Fig. 6B). These results demonstrate that increased BETi resistance correlates with an increased capacity to consume and utilize lactate.
Figure 6:
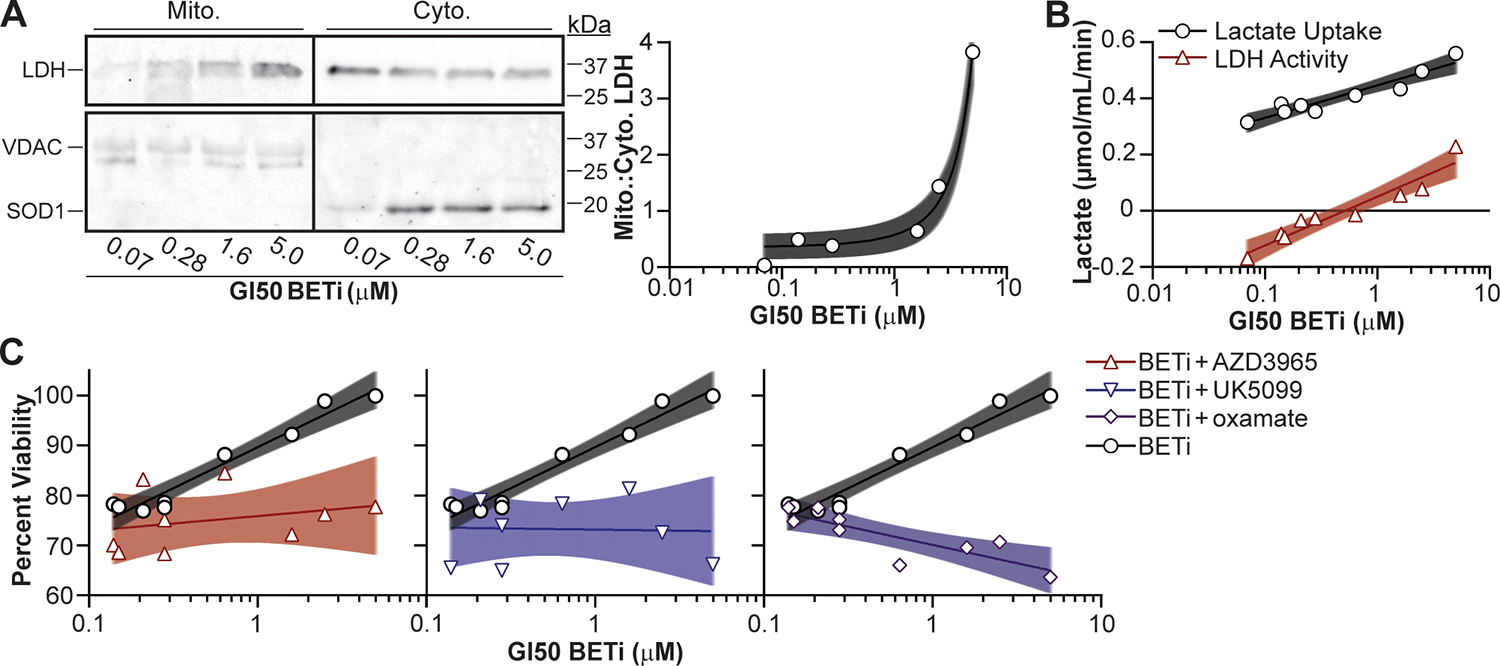
Lactate utilization facilitates BETi resistance in AML patient myeloblasts. (A) Patient myeloblasts with defined GI50 BETi concentrations were lysed, fractionated, and LDH protein in the cytosolic (Cyto.) and mitochondrial (Mito.) fractions quantified by immunoblot. SOD1 and VDAC protein were immunoblotted to ensure clean fractionation. (B) Cells were treated with BETi, and the media exchanged after 48 hr with minimal media with and without oxamate (25 μM) or lysed to quantify baseline metabolite levels. Minimal media was subsequently supplemented with lactate (20 μM) for 2 hr, cells lysed, and intracellular lactate levels quantified using a fluorescent plate reader (Lactate Uptake = oxamate-treated – baseline; LDH Activity = oxamate-treated – mock-treated). (C) Myeloblasts were cultured for 72 hr with BETi (0.15 μM) and/or lactate utilization inhibitors (AZD3965, UK5099, oxamate = 0.1 μM) and viability quantified using a fluorescent plate reader. Viability was quantified relative to vehicle control treated cells. Each point represents the mean (technical duplicate) value obtained from an individual patient. Shaded regions represent 95% confidence bands.
To verify whether AML myeloblasts with an increased GI50 concentration for BETi were utilizing lactate to overcome BET inhibition, cells were treated with BETi, and inhibitors of lactate utilization and cellular viability quantified. As predicted, a positive correlation was observed between viability and the GI50 concentration during BETi treatment alone (Fig. 6C). Treatment with AZD3965 alone was mildly toxic across AML myeloblasts but UK5099 and oxamate alone exhibited a slight positive correlation with BETi GI50 concentrations (Fig. S5). AZD3965 in combination with BETi showed a marginal decrease in viability compared to AZD3965 alone, while UK5099 in combination with BETi exhibited efficacy in decreasing the viability of BETi-resistant AML myeloblasts (Fig. 6C). Interestingly, oxamate was the only lactate utilization inhibitor in combination with BETi to display a negative correlation with the BETi GI50 concentration (Fig. 6C). These findings indicate that the addition of oxamate is not inherently toxic to AML myeloblasts but better enhances the susceptibility of BETi-resistant cells to BET inhibition.
Oxamate sensitizes BETi-resistant AML to BET inhibition in PDX model
Based on these findings in vitro, we tested the combined inhibition of BET and lactate utilization using a patient derived xenograft (PDX) transplantation model of BETi-resistant AML cells (GI50 BETi = 2.5 μM) in NSGS mice. After establishing disseminated leukemia, mice were treated with 50 mg/kg of BETi (4 weeks on, 1 week off, 4 weeks on) twice daily and/or 200 mg/kg oxamate daily 5 days a week for 9 weeks and then sacrificed (Fig. 7A). Spleens in the mice receiving combined BETi and oxamate were significantly smaller than vehicle control (Fig. 7B). The percentage of AML myeloblasts were quantified in the peripheral blood, spleen, and bone marrow using anti-human CD45 and CD33 monoclonal antibodies, and via hCD45 antibodies in immunohistochemistry. While monotherapy had minimal effect on tumor burden compared to the vehicle control, the combined treatment of BETi and oxamate substantially reduced AML burdens in the spleen and bone marrow compared to mock-treated mice with a strong trend in decreased AML burdens was observed relative to BETi-treated mice (Fig. 7C, D). Taken together, these results demonstrate combining BETi treatment with oxamate may be more efficacious for patients with AML than monotherapy alone.
Figure 7:
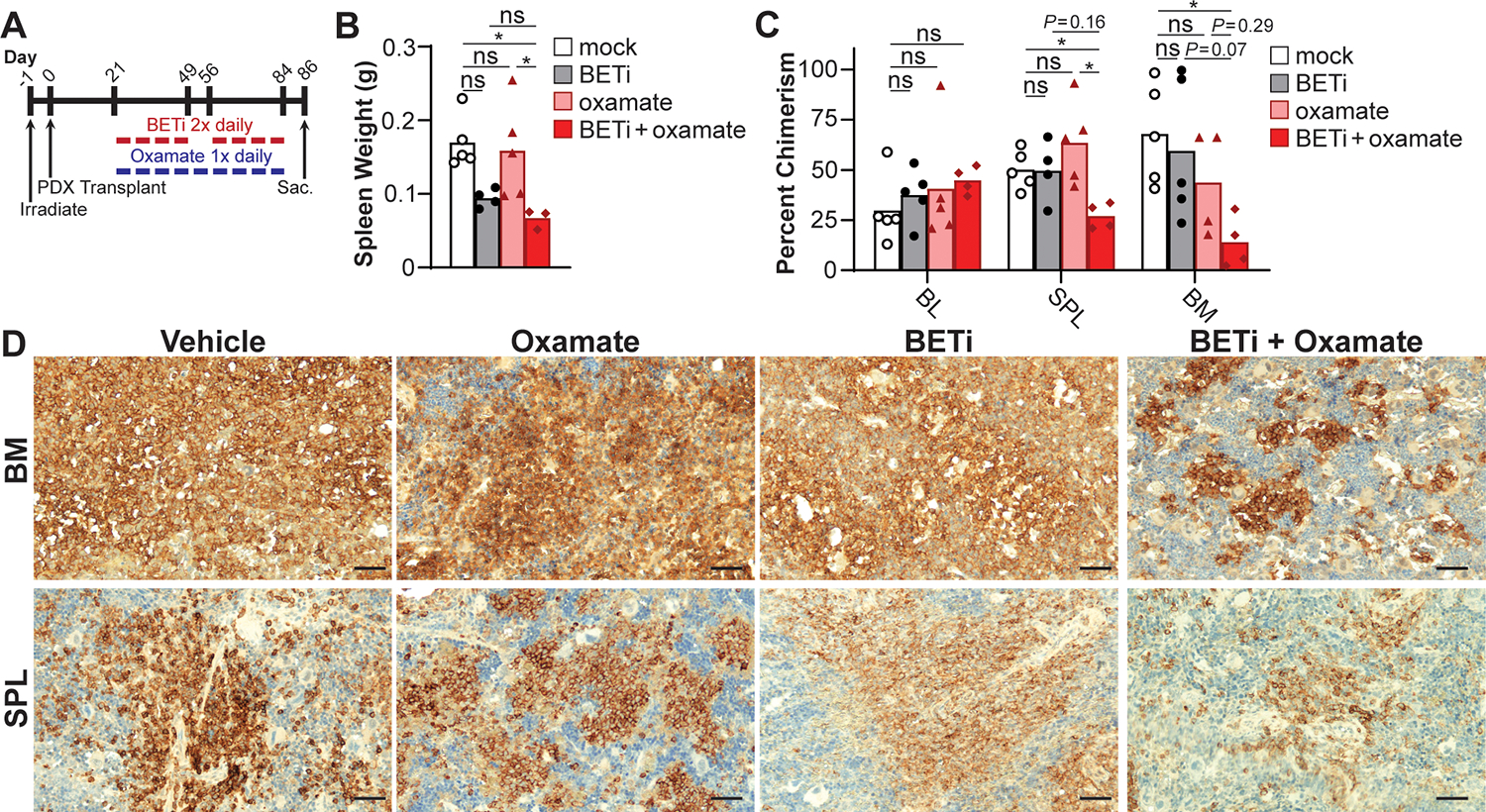
Lactate utilization facilitates BETi resistance in AML patient myeloblasts. (A) NSGS mice were engrafted with BETi-resistant AML myeloblasts (GI50 BETi = 2.5 μM). Mice were treated with vehicle control, BETi (50 mg/kg) and/or oxamate (200 mg/kg), and then sacrificed. (B) Spleens were weighed, and tumor burden assessed by chimerism analysis by quantifying hCD45+hCD33+ cells using (C) flow cytometry and (D) immunohistochemistry (hCD45, scale bar = 50 μm). Each point represents a single mouse (mock, n = 5; BETi or oxamate, n = 4 or 5; BETi+oxamate, n = 3 or 4). (B) One- or (C) two-way ANOVA with Tukey multiple comparisons test (*P ≤ 0.05, ns = not significant).
Discussion
Here, we report AML myeloblasts utilizing lactate as a metabolic bypass during BETi treatment. While previous studies highlight the transcriptional plasticity of malignant cells to escape the therapeutic pressures of BETi (12,13), this is the first report of lactate utilization allowing AML myeloblasts to maintain metabolic integrity and circumvent antileukemic therapy. BET inhibition disrupts glycolysis leading to collapses in mitochondrial respiration and cellular ATP levels (Fig. 8A). However, BETi-resistant cells leverage lactate as an alternative metabolic means to maintain cellular pyruvate levels, mitochondrial respiration, and cellular viability (Fig. 8B). These phenotypes were not just observed in AML cell lines, but a strong correlation was observed between GI50 concentrations for BETi and the capacity to utilize lactate from primary AML patient samples. As proof of principle, we translated these findings in vivo to demonstrate that the combination of simultaneous BETi and LDH inhibition was effective in killing xenografted BETi-resistant leukemia (Fig. 8C). These results demonstrate that some AML myeloblasts metabolically adapt to overcome BET inhibition by consuming lactate, but that combinations of BETi and lactate utilization inhibitors may stymie this metabolic escape.
Figure 8:
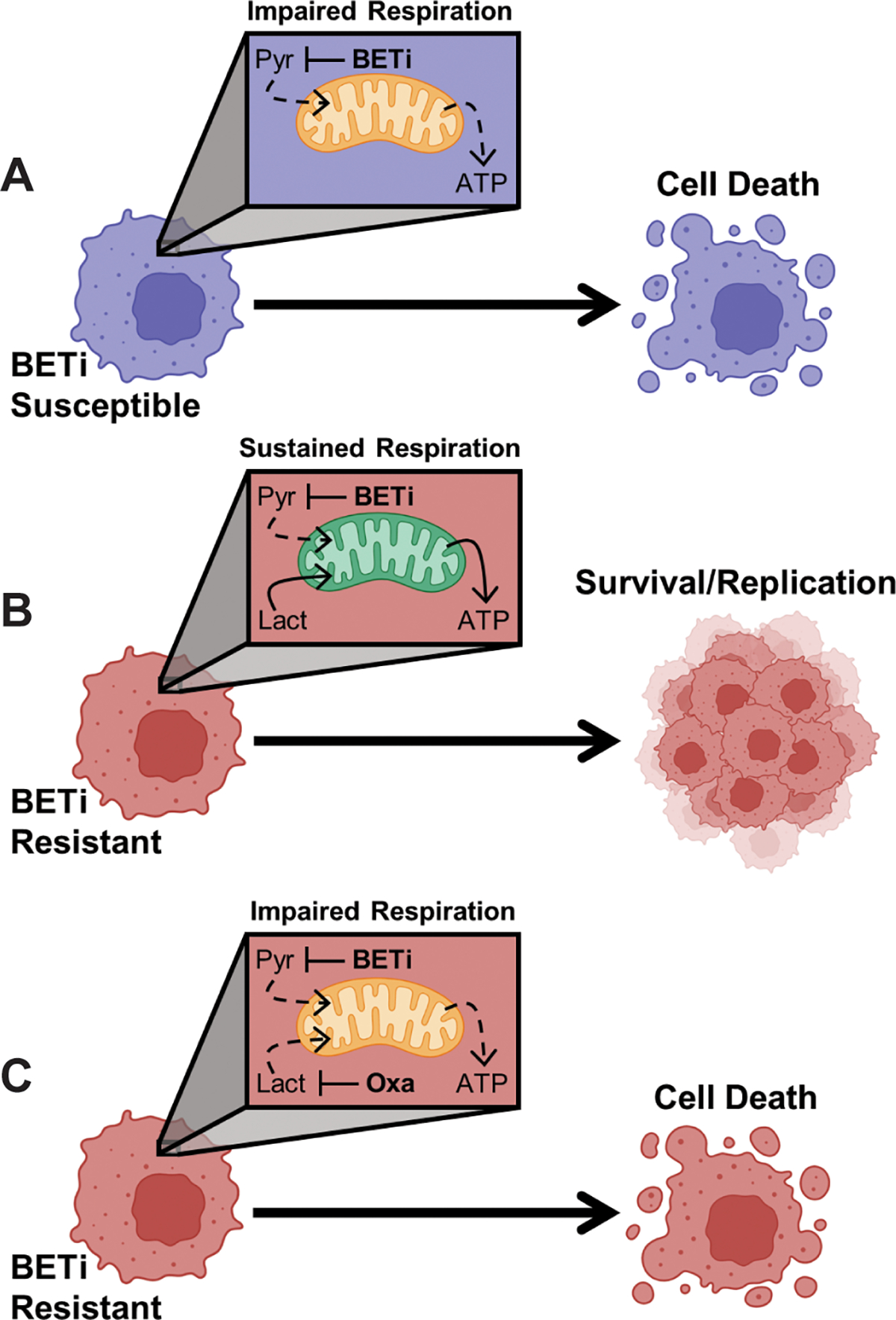
AML myeloblasts utilize lactate as a metabolic bypass to resist BET inhibition. (A) Treatment with BETi disrupts glycolysis resulting in a collapse in intracellular pyruvate (Pyr) and an inability to maintain mitochondrial respiration resulting in cell death. (B) BETi resistance coincides with an increased capacity for mitochondria to utilize lactate (Lact) as an alternative carbon source to sustain mitochondrial respiration. (C) However, co-treatment with BETi and oxamate (Oxa) to prevent utilization of lactate impairs mitochondrial respiration in BETi-resistant cells leading to cell death. Created with BioRender.com.
Both the MOLM-13 and MV-4–11 cells contain the FLT3-ITD+ mutation, which is a high risk feature that occurs in 20–30% of AML patients, and remains a challenge to treat in the clinic despite (62) improved outcomes with new FLT3 inhibitors (63,64). A recent paper described that disrupting the electron transport chain caused FLT3-ITD+ AML myeloblasts to import lactate from the extracellular environment as a metabolic response to inhibiting mitochondrial respiration (49). We extensively studied the FLT3-ITD+ cell lines MOLM-13 and MV-4–11 and identified a similar capacity to utilize lactate to sustain mitochondrial respiration. However, MOLM-13 cells were significantly better equipped to utilize lactate than MV-4–11 following BETi treatment. These findings suggest that FLT3 mutational status does not affect lactate utilization and that significant metabolic heterogeneity exists within the FLT3-ITD+ subset of AML.
While we focused on metabolic escapes to resist BETi treatment in gross myeloblasts, the metabolic plasticity of leukemic stem cells differs, as leukemic stem cells (LSCs) utilize amino acids such as glutamine to sustain mitochondrial respiration (65,66). Treatment with venetoclax and azacitidine (ven/aza) disrupts glutamine utilization, the preferred carbon source in LSCs, which collapses oxidative phosphorylation and cellular viability (65). However, resistance to ven/aza coincides with an upregulation of fatty acid oxidation as a metabolic bypass to maintain mitochondrial respiration (65,67). By contrast, we identified that in resistance to blocking glycolysis with BETi, the preferred metabolic pathway for sustaining mitochondrial respiration in non LSCs, lactate is readily used as an alternative carbon source. Taken together, these findings highlight metabolic complexity of AML as myeloblasts and LSCs preferentially employ different metabolic escape mechanisms to cope with inhibition of their preferred metabolic pathways for sustaining mitochondrial respiration.
We previously established that the intermittent dosing of BETi spared animals cardiac toxicity (20), which has been observed in rodents treated with BETi (68). While oxamate treatment together with BETi was effective in the AML xenograft models (Fig. 5), the addition of UK5099 was largely ineffective, and potentially antagonistic in facilitating BETi-mediated toxicity in vivo (Fig. S4). This was initially surprising as both UK5099 and oxamate synergized with BETi in vitro, suggesting that not all targets of lactate utilization are equally efficacious in vivo. One possible explanation is that oxygen tension and nutrient availability impart metabolic constraints within the blood, peripheral tissues, and bone marrow that are significantly different to tissue culture conditions. Both oxamate and UK5099 disrupt metabolic pathways other than lactate utilization including inhibiting LDH activity at the end of anaerobic glycolysis (oxamate) and blocking translocation of pyruvate into mitochondria (UK5099). These functions may result in unintended consequences in energy homeostasis and redox balancing that are not captured in in vitro assays. Previous studies used AZD3965 (69–72), UK5099 (55), and oxamate (73–75) as monotherapies to treat cancer in murine models, but these findings have not translated well to the clinic. We propose the alternative strategy to inhibit or modulate specific metabolic pathways to enhance the efficacy or limit the toxicity of antileukemic drugs. Recognition of the metabolic switching to utilize lactate by AML cells could lead to new targeted therapy and studying available lactate utilization inhibitors with BETi therapy in clinical settings should be considered.
Supplementary Material
Significance Statement.
Lactate utilization allows AML myeloblasts to maintain metabolic integrity and circumvent antileukemic therapy, which supports testing of lactate utilization inhibitors in clinical settings to overcome BET inhibitor resistance in AML.
Acknowledgements
The Vanderbilt-Ingram Cancer Center (VICC) Hematopoietic Malignancies Tissue Repository, the Vanderbilt University Medical Center Translational Pathology Shared Resource, and other VICC shared Core Services were critical in completion of this work.
This work was supported in part by the Incyte Corporation (M.R. Savona), Leukemia and Lymphoma Society (M.R. Savona), Adventure Alle Fund (M.R. Savona), Beverly and George Rawlings Directorship (M.R. Savona), and NIH grants 1R01CA262287 (M.R. Savona), P30 CA068485 (M.R. Savona), UTK Start-up Funds (A.J. Monteith), F30DK127699 (A.J. Silver), T32GM007347 (A.J. Silver), R01CA217987 (J.C. Rathmell), R01DK105550 (J.C. Rathmell), and R01CA193256 (J.W. Locasale). Flow cytometry was conducted in the VUMC Flow Cytometry Shared Resource, which is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404). Confocal microscopy was performed at the Advanced Microscopy and Imaging Center at the University of Tennessee. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Author Disclosures
M.C. Stubbs is an employee of Incyte Corporation/Incyte Research Institute. J.W. Locasale is an advisor for Nanocare Technologies and Cornerstone Pharmaceuticals. J.C. Rathmell is a founder, scientific advisory board member, and stockholder of Sitryx Therapeutics, a scientific advisory board member and stockholder of Caribou Biosciences, a member of the scientific advisory board of Nirogy Therapeutics, has consulted for Merck, Pfizer, and Mitobridge within the past three years, and has received research support from Incyte Corp., Calithera Biosciences, and Tempest Therapeutics. M.R. Savona is member on the board/advisory committee for Bristol Myers Squibb, CTI, Forma, Geron, GSK, Karyopharm, Ryvu, Taiho, Takeda, and TG Therapeutics, consultancy and equity ownership with Karyopharm and Ryvu, patents and royalties from Boehringer Ingelheim, and research funding from ALX Oncology, Astex, Incyte, Takeda, TG Therapeutics. All other authors declare no potential conflicts of interest.
References
- 1.Bonetti P, Davoli T, Sironi C, Amati B, Pelicci PG, Colombo E. Nucleophosmin and its AML-associated mutant regulate c-Myc turnover through Fbw7 gamma. J Cell Biol 2008;182:19–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bullinger L, Dohner K, Dohner H. Genomics of Acute Myeloid Leukemia Diagnosis and Pathways. J Clin Oncol 2017;35:934–46 [DOI] [PubMed] [Google Scholar]
- 3.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011;478:529–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fiskus W, Sharma S, Qi J, Shah B, Devaraj SG, Leveque C, et al. BET protein antagonist JQ1 is synergistically lethal with FLT3 tyrosine kinase inhibitor (TKI) and overcomes resistance to FLT3-TKI in AML cells expressing FLT-ITD. Mol Cancer Ther 2014;13:2315–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mathew S, Lorsbach RB, Shearer P, Sandlund JT, Raimondi SC. Double minute chromosomes and c-MYC amplification in a child with secondary myelodysplastic syndrome after treatment for acute lymphoblastic leukemia. Leukemia 2000;14:1314–5 [DOI] [PubMed] [Google Scholar]
- 6.Muller-Tidow C, Steffen B, Cauvet T, Tickenbrock L, Ji P, Diederichs S, et al. Translocation products in acute myeloid leukemia activate the Wnt signaling pathway in hematopoietic cells. Mol Cell Biol 2004;24:2890–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, et al. c-Myc regulates transcriptional pause release. Cell 2010;141:432–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell 2013;155:934–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013;153:320–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature 2010;468:1067–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011;478:524–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fong CY, Gilan O, Lam EY, Rubin AF, Ftouni S, Tyler D, et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015;525:538–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rathert P, Roth M, Neumann T, Muerdter F, Roe JS, Muhar M, et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 2015;525:543–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amorim S, Stathis A, Gleeson M, Iyengar S, Magarotto V, Leleu X, et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol 2016;3:e196–204 [DOI] [PubMed] [Google Scholar]
- 15.Berthon C, Raffoux E, Thomas X, Vey N, Gomez-Roca C, Yee K, et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol 2016;3:e186–95 [DOI] [PubMed] [Google Scholar]
- 16.Lewin J, Soria JC, Stathis A, Delord JP, Peters S, Awada A, et al. Phase Ib Trial With Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients With Selected Advanced Solid Tumors. J Clin Oncol 2018;36:3007–14 [DOI] [PubMed] [Google Scholar]
- 17.Piha-Paul SA, Hann CL, French CA, Cousin S, Brana I, Cassier PA, et al. Phase 1 Study of Molibresib (GSK525762), a Bromodomain and Extra-Terminal Domain Protein Inhibitor, in NUT Carcinoma and Other Solid Tumors. JNCI Cancer Spectr 2020;4:pkz093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Postel-Vinay S, Herbschleb K, Massard C, Woodcock V, Soria JC, Walter AO, et al. First-in-human phase I study of the bromodomain and extraterminal motif inhibitor BAY 1238097: emerging pharmacokinetic/pharmacodynamic relationship and early termination due to unexpected toxicity. Eur J Cancer 2019;109:103–10 [DOI] [PubMed] [Google Scholar]
- 19.Pericole FV, Lazarini M, de Paiva LB, Duarte A, Vieira Ferro KP, Niemann FS, et al. BRD4 Inhibition Enhances Azacitidine Efficacy in Acute Myeloid Leukemia and Myelodysplastic Syndromes. Front Oncol 2019;9:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramsey HE, Greenwood D, Zhang S, Childress M, Arrate MP, Gorska AE, et al. BET Inhibition Enhances the Antileukemic Activity of Low-dose Venetoclax in Acute Myeloid Leukemia. Clin Cancer Res 2021;27:598–607 [DOI] [PubMed] [Google Scholar]
- 21.Saenz DT, Fiskus W, Mill CP, Perera D, Manshouri T, Lara BH, et al. Mechanistic basis and efficacy of targeting the beta-catenin-TCF7L2-JMJD6-c-Myc axis to overcome resistance to BET inhibitors. Blood 2020;135:1255–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson AJ, Stubbs M, Liu P, Ruggeri B, Khabele D. The BET inhibitor INCB054329 reduces homologous recombination efficiency and augments PARP inhibitor activity in ovarian cancer. Gynecol Oncol 2018;149:575–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castro I, Sampaio-Marques B, Ludovico P. Targeting Metabolic Reprogramming in Acute Myeloid Leukemia. Cells 2019;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen WL, Wang JH, Zhao AH, Xu X, Wang YH, Chen TL, et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood 2014;124:1645–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 2012;15:110–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem 2000;275:21797–800 [DOI] [PubMed] [Google Scholar]
- 27.Bioenergetics Racker E. and the problem of tumor growth. Am Sci 1972;60:56–63 [PubMed] [Google Scholar]
- 28.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009;458:762–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem 1979;254:2669–76 [PubMed] [Google Scholar]
- 30.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 2008;105:18782–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang G, Deng W, Liu Y, Wang C. General mechanism of JQ1 in inhibiting various types of cancer. Mol Med Rep 2020;21:1021–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otto C, Schmidt S, Kastner C, Denk S, Kettler J, Muller N, et al. Targeting bromodomain-containing protein 4 (BRD4) inhibits MYC expression in colorectal cancer cells. Neoplasia 2019;21:1110–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park IH, Yang HN, Jeon SY, Hwang JA, Kim MK, Kong SY, et al. Anti-tumor activity of BET inhibitors in androgen-receptor-expressing triple-negative breast cancer. Sci Rep 2019;9:13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010;463:899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang MY, Liu SL, Huang WL, Tang DB, Zheng WW, Zhou N, et al. Bromodomains and Extra-Terminal (BET) Inhibitor JQ1 Suppresses Proliferation of Acute Lymphocytic Leukemia by Inhibiting c-Myc-Mediated Glycolysis. Med Sci Monit 2020;26:e923411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fan P, Wang B, Meng Z, Zhao J, Jin X. PES1 is transcriptionally regulated by BRD4 and promotes cell proliferation and glycolysis in hepatocellular carcinoma. Int J Biochem Cell Biol 2018;104:1–8 [DOI] [PubMed] [Google Scholar]
- 37.Song X, Zhang T, Ding H, Feng Y, Yang W, Yin X, et al. Non-genetic stratification reveals epigenetic heterogeneity and identifies vulnerabilities of glycolysis addiction in lung adenocarcinoma subtype. Oncogenesis 2022;11:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papaconstantinou J, Colowick SP. The role of glycolysis in the growth of tumor cells. II. The effect of oxamic acid on the growth of HeLa cells in tissue culture. J Biol Chem 1961;236:285–8 [PubMed] [Google Scholar]
- 39.Papaconstantinou J, Colowick SP. The role of glycolysis in the growth of tumor cells. I. Effects of oxamic acid on the metabolism of Ehrlich ascites tumor cells in vitro. J Biol Chem 1961;236:278–84 [PubMed] [Google Scholar]
- 40.Liu X, Ser Z, Locasale JW. Development and quantitative evaluation of a high-resolution metabolomics technology. Anal Chem 2014;86:2175–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stubbs MC, Burn TC, Sparks R, Maduskuie T, Diamond S, Rupar M, et al. The Novel Bromodomain and Extraterminal Domain Inhibitor INCB054329 Induces Vulnerabilities in Myeloma Cells That Inform Rational Combination Strategies. Clin Cancer Res 2019;25:300–11 [DOI] [PubMed] [Google Scholar]
- 42.Shroff EH, Eberlin LS, Dang VM, Gouw AM, Gabay M, Adam SJ, et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc Natl Acad Sci U S A 2015;112:6539–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011;35:871–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schulte ML, Fu A, Zhao P, Li J, Geng L, Smith ST, et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med 2018;24:194–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wolf HP, Engel DW. Decrease of fatty acid oxidation, ketogenesis and gluconeogenesis in isolated perfused rat liver by phenylalkyl oxirane carboxylate (B 807–27) due to inhibition of CPT I (EC 2.3.1.21). Eur J Biochem 1985;146:359–63 [DOI] [PubMed] [Google Scholar]
- 46.Brooks GA, Dubouchaud H, Brown M, Sicurello JP, Butz CE. Role of mitochondrial lactate dehydrogenase and lactate oxidation in the intracellular lactate shuttle. Proc Natl Acad Sci U S A 1999;96:1129–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dubouchaud H, Butterfield GE, Wolfel EE, Bergman BC, Brooks GA. Endurance training, expression, and physiology of LDH, MCT1, and MCT4 in human skeletal muscle. Am J Physiol Endocrinol Metab 2000;278:E571–9 [DOI] [PubMed] [Google Scholar]
- 48.Valenti D, de Bari L, Atlante A, Passarella S. L-Lactate transport into rat heart mitochondria and reconstruction of the L-lactate/pyruvate shuttle. Biochem J 2002;364:101–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Erdem A, Marin S, Pereira-Martins DA, Geugien M, Cunningham A, Pruis MG, et al. Inhibition of the succinyl dehydrogenase complex in acute myeloid leukemia leads to a lactate-fuelled respiratory metabolic vulnerability. Nat Commun 2022;13:2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Bari L, Chieppa G, Marra E, Passarella S. L-lactate metabolism can occur in normal and cancer prostate cells via the novel mitochondrial L-lactate dehydrogenase. Int J Oncol 2010;37:1607–20 [DOI] [PubMed] [Google Scholar]
- 51.Pizzuto R, Paventi G, Porcile C, Sarnataro D, Daniele A, Passarella S. l-Lactate metabolism in HEP G2 cell mitochondria due to the l-lactate dehydrogenase determines the occurrence of the lactate/pyruvate shuttle and the appearance of oxaloacetate, malate and citrate outside mitochondria. Biochim Biophys Acta 2012;1817:1679–90 [DOI] [PubMed] [Google Scholar]
- 52.Niu X, Chen YJ, Crawford PA, Patti GJ. Transport-exclusion pharmacology to localize lactate dehydrogenase activity within cells. Cancer Metab 2018;6:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu H, Wang Y, Ying M, Jin C, Li J, Hu X. Lactate dehydrogenases amplify reactive oxygen species in cancer cells in response to oxidative stimuli. Signal Transduct Target Ther 2021;6:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Young A, Oldford C, Mailloux RJ. Lactate dehydrogenase supports lactate oxidation in mitochondria isolated from different mouse tissues. Redox Biol 2020;28:101339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Corbet C, Bastien E, Draoui N, Doix B, Mignion L, Jordan BF, et al. Interruption of lactate uptake by inhibiting mitochondrial pyruvate transport unravels direct antitumor and radiosensitizing effects. Nat Commun 2018;9:1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Contreras-Baeza Y, Sandoval PY, Alarcon R, Galaz A, Cortes-Molina F, Alegria K, et al. Monocarboxylate transporter 4 (MCT4) is a high affinity transporter capable of exporting lactate in high-lactate microenvironments. J Biol Chem 2019;294:20135–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ullah MS, Davies AJ, Halestrap AP. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J Biol Chem 2006;281:9030–7 [DOI] [PubMed] [Google Scholar]
- 58.Zachar Z, Marecek J, Maturo C, Gupta S, Stuart SD, Howell K, et al. Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J Mol Med (Berl) 2011;89:1137–48 [DOI] [PubMed] [Google Scholar]
- 59.Murray CM, Hutchinson R, Bantick JR, Belfield GP, Benjamin AD, Brazma D, et al. Monocarboxylate transporter MCT1 is a target for immunosuppression. Nat Chem Biol 2005;1:371–6 [DOI] [PubMed] [Google Scholar]
- 60.Halestrap AP. The mitochondrial pyruvate carrier. Kinetics and specificity for substrates and inhibitors. Biochem J 1975;148:85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen YJ, Mahieu NG, Huang X, Singh M, Crawford PA, Johnson SL, et al. Lactate metabolism is associated with mammalian mitochondria. Nat Chem Biol 2016;12:937–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thiede C, Steudel C, Mohr B, Schaich M, Schakel U, Platzbecker U, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002;99:4326–35 [DOI] [PubMed] [Google Scholar]
- 63.Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N Engl J Med 2019;381:1728–40 [DOI] [PubMed] [Google Scholar]
- 64.Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med 2017;377:454–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018;34:724–40 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013;12:329–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stevens BM, Jones CL, Pollyea DA, Culp-Hill R, D’Alessandro A, Winters A, et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat Cancer 2020;1:1176–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Piquereau J, Boet A, Pechoux C, Antigny F, Lambert M, Gressette M, et al. The BET Bromodomain Inhibitor I-BET-151 Induces Structural and Functional Alterations of the Heart Mitochondria in Healthy Male Mice and Rats. Int J Mol Sci 2019;20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beloueche-Babari M, Casals Galobart T, Delgado-Goni T, Wantuch S, Parkes HG, Tandy D, et al. Monocarboxylate transporter 1 blockade with AZD3965 inhibits lipid biosynthesis and increases tumour immune cell infiltration. Br J Cancer 2020;122:895–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hong CS, Graham NA, Gu W, Espindola Camacho C, Mah V, Maresh EL, et al. MCT1 Modulates Cancer Cell Pyruvate Export and Growth of Tumors that Co-express MCT1 and MCT4. Cell Rep 2016;14:1590–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Polanski R, Hodgkinson CL, Fusi A, Nonaka D, Priest L, Kelly P, et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin Cancer Res 2014;20:926–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Quanz M, Bender E, Kopitz C, Grunewald S, Schlicker A, Schwede W, et al. Preclinical Efficacy of the Novel Monocarboxylate Transporter 1 Inhibitor BAY-8002 and Associated Markers of Resistance. Mol Cancer Ther 2018;17:2285–96 [DOI] [PubMed] [Google Scholar]
- 73.El-Sisi AE, Sokar SS, Abu-Risha SE, El-Mahrouk SR. Oxamate potentiates taxol chemotherapeutic efficacy in experimentally-induced solid ehrlich carcinoma (SEC) in mice. Biomed Pharmacother 2017;95:1565–73 [DOI] [PubMed] [Google Scholar]
- 74.Li X, Lu W, Hu Y, Wen S, Qian C, Wu W, et al. Effective inhibition of nasopharyngeal carcinoma in vitro and in vivo by targeting glycolysis with oxamate. Int J Oncol 2013;43:1710–8 [DOI] [PubMed] [Google Scholar]
- 75.Qiao T, Xiong Y, Feng Y, Guo W, Zhou Y, Zhao J, et al. Inhibition of LDH-A by Oxamate Enhances the Efficacy of Anti-PD-1 Treatment in an NSCLC Humanized Mouse Model. Front Oncol 2021;11:632364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the article and/or Supplementary Materials. Additional data are available upon request from the corresponding author.