

Abstract
Objective:
To provide a historical perspective and narrative review on research into the molecular pathogenesis of osteoarthritis pain.
Design:
PubMed databases were searched for combinations of “osteoarthritis”, “pain” and “animal models” for papers that represented key phases in the history of osteoarthritis pain discovery research including epidemiology, pathology, imaging, preclinical modelling and clinical trials.
Results:
The possible anatomical sources of osteoarthritis pain were identified over 50 years ago, but relatively slow progress has been made in understanding the apparent disconnect between structural changes captured by radiography and symptom severity. Translationally relevant animal models of osteoarthritis have aided in our understanding of the structural and molecular drivers of osteoarthritis pain, including molecules such as nerve growth factor (NGF) and C-C motif chemokine ligand 2 (CCL2). Events leading to persistent osteoarthritis pain appear to involve a two-step process involving changes in joint innervation, including neo-innervation of the articular cartilage, as well as sensitization at the level of the joint, dorsal root ganglion and central nervous system.
Conclusions:
There remains a great need for the development of treatments to reduce osteoarthritis pain in patients. Harnessing all that we have learned over the past several decades is helping us to appreciate the important interaction between structural disease and pain, and this is likely to facilitate development of new disease modifying therapies in the future.
Keywords: Osteoarthritis, pain, sensitization, animal models, nerve growth factor
Introduction.
Significant progress has been made in unravelling molecular pathogenic mechanisms in osteoarthritis (OA) over the past 30 years and beyond. These have been aided in recent decades by large scale genomic studies, clinically relevant preclinical models, refinement of clinical outcome measures and novel molecular targeting. Whilst we are still some way from having full disease modifying drugs that will modulate structural disease as well as symptoms, we have seen the emergence of a number of pharmacological approaches that modulate aspects of disease such as structure modification by sprifermin, a truncated FGF18 analogue, and strategies that neutralize nerve growth factor (NGF) in the treatment of OA pain. Accompanying these studies is a greater understanding of their mechanism of action, their site of action and other molecules that contribute to the pathway and which could themselves also be potential targets. These studies build on several earlier decades of careful observation, pathological investigation and early molecular discovery to end up where we are today. Harnessing this collective knowledge and working across research silos is bringing us closer to effect real change to benefit patients.
Pathological correlates with Clinical OA pain.
The study of OA pain has a long history. It was well established over 50 years ago that OA pain originated in the joint; indeed, both neurectomy and intra-articular injection of local anaesthetic were able to relieve pain 1. It was also appreciated that pain was mechanically induced; osteotomy, by altering the loaded regions of the joint, was able to alleviate symptoms 2. Pain thresholds varied between and within individuals and this was attributed to the influence of social and other patient factors that affected the pain experience. In 1989, Brandt speculated on the pathologies within the OA joint that could give rise to pain 3. These included synovitis (possibly induced by crystals or by ‘damage particles’ from abrided cartilage), stretching of the periosteum over osteophytes, microfractures within the subchondral bone, venous hypertension due to distortion of medullary blood flow in bone (synonymous with bone marrow oedema on magnetic resonance imaging), distension of the joint capsule and muscle spasm. Altman later added ligament insertions and damaged menisci to this list 4. These interpretations were partly based on the sensitivity of these tissues in a healthy joint to sharp pressure. Indeed, in their seminal paper of 1950, Kellgren and Samuel used healthy human volunteers and of patients undergoing arthrotomy under local anesthesia to document the degree to which capsular tissues and ligaments within the knee were sensitive to sharp pressure 5. They concluded that fibrous ligaments appeared to be a major source of joint pain. A later study performed by orthopedic surgeon brothers without intra-articular anesthesia confirmed the sensitivity of the cruciate ligament insertions and also noted the insensitivity of articular cartilage, consistent with it being aneural and avascular 6. As a result, cartilage was largely excluded as being relevant to the origin of pain in OA.
The role of inflammation, specifically synovitis, in OA pain has remained controversial. Whilst Kellgren and Samuel documented the relative insensitivity of most regions of the healthy synovium, the Dye study reported that palpation of the suprapatellar pouch, capsule, and the medial and lateral retinacula produced moderate to severe localized pain 6. Likewise, innervation of the synovium was demonstrated in 1990 7 and in OA tissue these fibres were strongly stained for substance P (also known as tachykinin), a neuropeptide that, along with CGRP, increased the sensitivity of pain fibres and also contributed to neurogenic inflammation through inciting cytokine release from mast cells and stromal cells 8–10. Such neurotransmitters and other inflammatory molecules such as prostaglandins were proposed to drive pain in arthritis 11. Part of this might also have been due to the oedematous response in the tissue, leading to increased mechano-sensitivity. Importantly, in the 1980s the concept of ‘silent nociceptors’ took shape - nociceptors that would not normally respond to mechanical stimuli became sensitized to do so after an inflammatory insult 12. Electrophysiology techniques have also been used to show that injection of a variety of inflammatory agents into the joint produced peripheral sensitization and increased activity of the nerve afferents innervating the joint (reviewed in 13). Whether these findings were relevant to osteoarthritis with lower levels of inflammation was unclear at the time.
Anti-inflammatory agents in OA pain:
The late 1970s and 1980s was the age of the non-steroidal anti-inflammatory drug (NSAID) revolution with many head-to-head studies comparing analgesic efficacy of different proprietary treatments. NSAIDs can promote analgesia through inhibition of the production of prostaglandin E2, reducing sensitization of nerves, as well as by reducing local inflammation. However, as a class, for the treatment of OA pain these drugs were analgesic but without evidence of a true anti-inflammatory effect (i.e. they didn’t reduce inflammatory cell infiltration). Indeed, it was noted that the synovium was little changed after treatment raising some authors to suggest that the mechanism of action was not synovium based. A similar phenomenon was also described following treatment with intraarticular and oral glucocorticoid use in OA; a number of studies have demonstrated a temporary analgesic response with i.a. use of long-acting glucocorticoids, but a direct correlation with synovial volume or vascularity, a surrogate marker for inflammation, was missing. Low dose oral prednisolone is not analgesic 14, but 10mg oral prednisolone daily was able to reduce symptoms 15. In the latter study there was a reduction in synovial volume but little effect on power doppler signal on ultrasound or vascularity of the synovium. The pain target for NSAIDs and corticosteroids therefore remains uncertain.
Of note, for both of these classes of anti-inflammatories, a concern over accelerating structural joint disease has been raised. In the case of NSAIDs, these were shown to accelerate structural disease in canine OA models and could suppress the normal anabolic response of damaged cartilage in vitro 16, 17 18, 19. This might be one of the mechanisms behind “indomethacin knee” or “indomethacin hip” a phenomenon where disease was seemingly accelerated by prolonged use of indomethacin 20, 21. Similarly, i.a. corticosteroid may accelerate cartilage loss in OA after prolonged use. In one study where individuals were given 3 monthly i.a. steroid injections over a two-year period, no analgesic effect over placebo was noted (time points earlier than 3 months were not measured) but a worsening of cartilage loss was observed by MRI in the steroid group compared with placebo 22.
Discordance between pain and pathology:
In recent decades much investment has been made in developing quantitative measures of OA damage within the joint using magnetic resonance imaging (MRI), and to a lesser extent computerised tomography (CT). MRI has the advantage of being able to reveal the soft tissues, e.g. synovium and bone marrow, and to correlate these pathologies with patient reported pain over time. Whilst these clinical studies have significantly increased our knowledge of pathology within the joint 23, how this changes with disease course 24, how age-related OA resembles post traumatic OA 25, and how individual pathologies relate to one another geographically within the joint 26–29, the studies have not helped to elucidate the primary pathology that drives pain in the OA joint. This is largely because all pathologies correlate moderately well with pain and they also correlate with one another 30–32. Building on previous earlier descriptions, it has become clear that much of the discordance of reported pain and pathological change in the joint is due to extra-articular factors that have a large and unpredictable impact on patient perceived pain. Indeed, controlling for these person level effects improves the correlation of pain score with radiographic score 33, 34.
Another area where clinical research has contributed to our understanding of OA pain is through the use of quantitative sensory testing to assess sensitization. From the body of work carried out in the past 15 years, a few important concepts have emerged: measures of peripheral and central sensitization are associated with OA-related pain 35, presence of both peripheral and central sensitization predicts development of persistent knee pain 36 and abnormalities of somatosensory perception normalize following successful surgical intervention 37 38, 39, as do functional MRI images of the brain 40, 41 indicating that ongoing pathology within the joint is driving these changes. Further understanding of the relationship between sensitization and persistent OA pain may provide opportunities for prevention, delay or reversal of chronic pain 42.
Pre-clinical models in OA pain discovery research.
In the early 2000s, a push to develop models of osteoarthritis suitable for pharmaceutical testing was made (with criteria listed at the time including: reproducibility, ability to predict efficacy in humans, similar pathology as human disease). The monoiodoacetate (MIA) rat model emerged which induced pathology by targeting chondrocyte metabolism and exhibited development of weight-bearing asymmetry 43, 44. Surgical rat 45 and mouse models of OA 46, 47 also began to be tested for behavioral changes associated with pain. Reversal of these behaviors were often tested using NSAIDs and behaviors were largely found to be sensitive in a dose-dependent manner. However, even at this time discrepancies in reports across laboratories in the responses of models to these standard analgesics were noted 43, 48. This highlights the challenges experienced when performing these types of tests, the dependence on the expertise within specific laboratories, and almost certainly, differences in bias mitigation strategies used.
In a 2018 review 49, we had noted that the number of studies examining pain in animal models of osteoarthritis was limited, and an updated search since that time indicates that the landscape in terms of numbers of studies has not increased much (Table 1). A PubMed search from 2018-August 2023 for “osteoarthritis mouse” yielded 2,306 papers, however adding the word ‘pain’ decreased this number to 381. Of these, only 145 papers used a model of OA (rather than an acute inflammatory joint pain model) and performed at least one pain behavior assessment. However, the types of studies being performed has changed over this time. Since 2018, of the studies that have measured any pain-related behavioral assessment, the inclusion of both evoked and non-evoked types of pain-related behavior has become more routine. Currently, the majority of mouse testing involves surgical induction of OA. For historical reasons this is generally performed in male mice where structural disease is significantly greater. Where comparisons have been made, female mice appear to develop pain at an earlier stage of joint (cartilage) damage and this can be seen in slower as well as more aggressive surgical models, such as that induced by DMM, partial medial meniscectomy (PMX) 50 and intact ACL rupture models 51. Continued use of models that facilitate development of joint damage in both sexes should help to improve this area of research moving forward. When it comes to reporting, most papers include specification regarding blinding when describing histological scoring, but less than 50% of papers performing behavioral testing stated whether these tests were performed blinded or not. In addition, the details included in these sections could be improved in order to increase replicability. Finally, in contrast to how the study of joint inflammatory pain started with a major focus on electrophysiological measurements, assessment of functional changes in nervous system tissues remains relatively uncommon in the setting of long-term osteoarthritis models (32%) – as the majority of studies include behavioral testing as a secondary outcome to structural joint changes. Through the use of both electrophysiology and calcium imaging, we have learned that osteoarthritis is associated with increased responsiveness of nociceptors to mechanical stimuli as well as with a lower action potential firing threshold 52–54. Moving forward, continued adoption of neuroscience technologies combined with the use of these well-established pre-clinical models of osteoarthritis is expected to yield novel analgesic targets.
Table 1.
PubMed search ‘osteoarthritis pain mouse’: 2018-Aug 13, 2023
| % Papers since 2018 (out of 145 papers total) | |
|---|---|
| Monosodium iodoacetate (MIA) model | 24 |
| Surgical joint destabilization model | 61 |
| Collagenase model | 6 |
| Systemic OA models (aging or obesity) | 6 |
| Non-surgical joint injury model | 6 |
| Two model comparison included | 7 |
| Included analysis (molecular or functional) of neural tissue | 32 |
| Assessed measure of sensitization (allodynia/hyperalgesia) | 71 |
| Assessed non-evoked behaviors | 61 |
| Pain assessed at more than one time point | 77 |
| Assessed pain and joint damage | 88 |
| Blinding of pain assessments included | 45 |
| Testing performed in both sexes | 12 |
The discovery of NGF as a principal driver of OA pain:
An important recent breakthrough in molecular pain pathogenesis, that has been facilitated by the use of murine models of disease, was the discovery that the neurotrophin, nerve growth factor (NGF) was an important driver of OA pain. NGF had a strong established role in neuronal development; causing sprouting and elongation along gradients of NGF 55, 56. The notion that NGF also had sensitizing activities at the level of nociceptive fibres was important and paved the way for considering it as an analgesic target 57, 58. In murine OA, NGF mRNA was transiently regulated in whole joint extracts immediately in the post operative phase and then again at late stages after destabilization of the joint (from 8-10 weeks post operation according to type of surgery), correlating with the time of spontaneous pain behaviour in mice 59, 60. Delivery of the soluble high affinity receptor (TrkA) to mice with established pain led to rapid analgesia 59. Analgesia through neutralization of NGF was also demonstrated using anti-NGF antibodies in rat surgically induced OA 61, and by NGF vaccination in mice 60.
These studies have been substantially validated by the success of clinical trials using neutralizing antibodies to NGF. First published in 2010, anti-NGF delivered subcutaneously (two doses over 12 months) demonstrated strong, dose-dependent analgesia, quite unlike anything seen previously 62. Anecdotal reports from patients recruited in this study from the lead author indicated that this treatment appeared to lead to a staggering improvement in symptoms such that patients were able to “go out dancing for the first time in years!”. However, the clinical trials that followed experienced a number of setbacks. The Federal Drug Association (FDA), noted early on that some trial participants (around 6%) were developing rapidly progressive joint damage. This was deemed to be due to rapidly progressive OA (RPOA), rather than a Charcot joint (in which the joint has lost its proprioception), or avascular necrosis 63. It was also observed that this risk appeared to increase at the higher doses of anti-NGF and especially when combined with NSAIDs. The risk was not only in index joints (the joint with the primary diagnosis of OA), but was also appearing in joints which were not known to have disease. Although phase II and phase III clinical studies were restarted (with mitigation strategies in place), RPOA was still seen in around 8% of participants 64, and in 2021 the FDA failed to grant a licence for anti-NGF treatment for OA pain. However, feline and canine versions have gone on to receive approval for treating joint pain in these species, which could help inform the longer-term effects of such agents 65.
The source of NGF in the OA joint has been subject to some debate over the years. The protein has been immunolocalized to human OA synovium 66, 67, while mRNA analyses of synovium from normal, painful and painless human OA joints failed to show NGF mRNA regulation by this tissue 68. Immunostaining was also evident in osteochondral channels, and associated with pain, in late human OA 69, 70 and more recently in surgically induced rat OA 71. This appears to accord with the discovery that NGF is strongly induced by injury to articular cartilage, both in vitro and in vivo after joint destabilization and correlates with the timing of pain behaviour 72. There was little evidence of an increase in inflammatory genes at the time NGF became elevated in late murine OA further implicating a non-inflammatory drive of NGF regulation when mice develop pain behaviour. While OA subchondral bone marrow lesions also failed to show regulation of NGF mRNA 73, production of NGF by other cells in the subchondral bone such as macrophages and osteoclasts 70 has been demonstrated. Together this supports the hypothesis whereby the injured basal cartilage and/or subchondral bone makes NGF and this is both necessary for the neoinnervation seen in advanced disease, as well as changing the firing threshold for neurons in the vicinity (reviewed in 74 75) (Figure 1). Evidence to support this is suggested by the appearance of CGRP-positive and NaV1.8-positive neuronal fibres that develop in the subchondral bone below damaged regions of the articular cartilage 67, 76, 77. This process may be facilitated by secretion of netrin-1 by subchondral bone osteoclasts. Netrin-1 promotes neuronal growth and its deletion protects mice from development of mechanical allodynia after surgically induced OA 78.
Figure 1. Molecular model of OA pain.
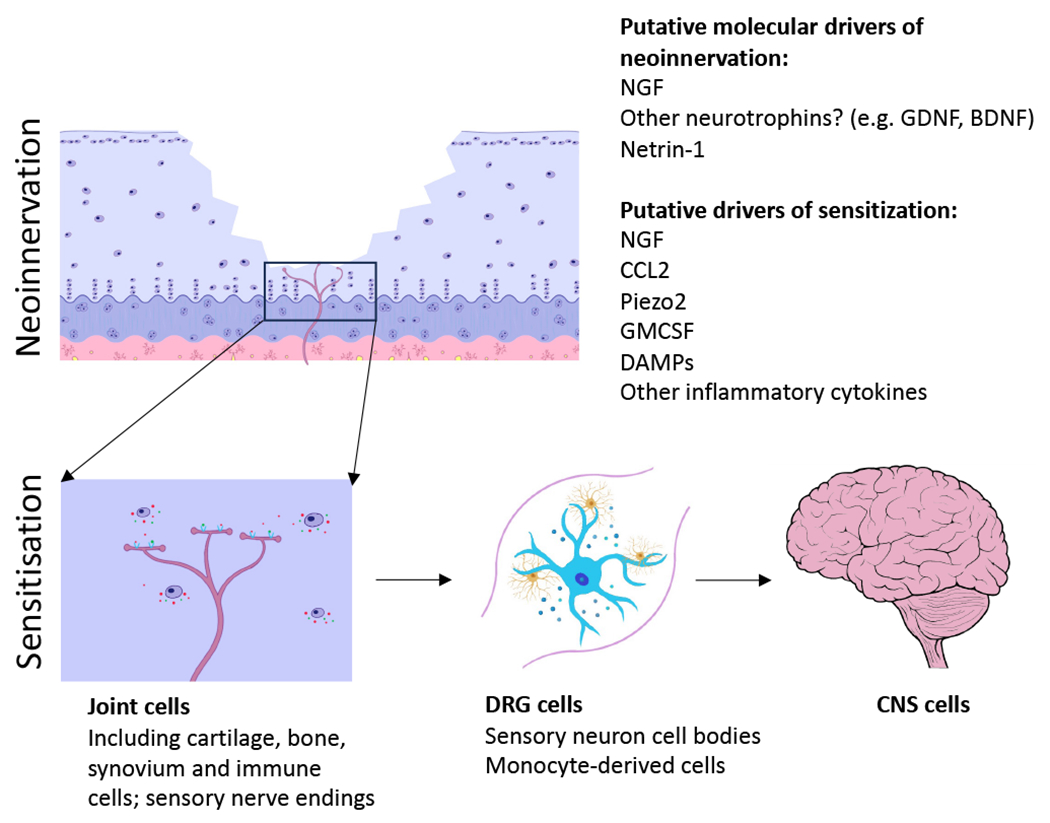
Model requires a two-step process: neoinnervation of joint tissues, including the osteochondral junction, and sensitization of the nociceptor response at the level of the joint and dorsal root ganglion (DRG) which may lead to central sensitization. Key molecular players include nerve growth factor (NGF), C-C chemokine ligand 2 (CCL2) and Piezo2. Netrin-1 has been implicated in guiding axons through the subchondral bone. A number of other molecules have been implicated in nociceptor sensitization in OA. Little is known about molecules that influence central sensitization. GDNF – glial derived neurotrophic factor; BDNF – brain derived neurotrophic factor; GMCSF – granulocyte-macrophage colony stimulating factor.
Other candidate molecular pathways:
Whilst the current evidence suggests that NGF is a primary driver of pain in OA, other robust pathways have also emerged that influence pain sensitivity in experimental OA. One of these is the C-C chemokine, CCL2 (also known as monocyte chemotactic protein-1, MCP-1) through its receptor, CCR2. CCL2 was transiently increased in the dorsal root ganglia (DRG) of mice at 8 weeks post joint destabilization (by DMM) and deletion of CCR2 was associated with loss of macrophage infiltration in the DRG and a failure to develop late spontaneous pain behaviour 79. In a separate study, both CCR2 and CCL2 null mice had delayed onset of late spontaneous OA pain behaviour despite structural damage in the joint being similar to wild type mice over the same period 80. Long-term systemic therapeutic targeting of CCR2 via a receptor antagonist also displayed an analgesic benefit 81. Further work has demonstrated that intra-articular targeting of CCR2 may also be sufficient to reduce symptoms in a mouse model of OA 82.
Strong evidence for Piezo2 has also emerged. This mechanosensitive ion channel expressed by nociceptors modulates sensitivity of murine OA pain and has a direct effect on NGF-mediated responses. Indeed, deletion of Piezo2 in NaV1.8-specific pain fibres reduced spontaneous pain behaviour in mice 12 weeks after surgical joint destabilization (without affecting structural joint disease) and abrogated pain sensitization (and joint swelling) induced by injecting NGF directly into the joint. As Piezo2 is a mechanosensor, it potentially provides another link to explain the highly mechanosensitive nature of OA pain 83.
Finally, multiple other candidate pathways have been identified that can influence sensitization potentially through neuroimmune interactions such as Toll-like receptor signaling, GM-CSF, and other inflammatory cytokines (reviewed in 84).
Influence of biological sex on pain.
It has been known for a long time from epidemiological studies that females experience increased symptomatic OA associated with higher pain scores and pain persistence. This is evidence largely obtained from post-menopausal women in whom most OA cases are described, raising the possibility that sex hormones may influence the experience of pain. Sexual pain dimorphism has also been described in murine OA (also noted above). It is striking and consistent that female mice develop pain at a seemingly earlier stage of structural (cartilage damage) disease 50, 85, 86. The molecular profiles within the whole joint as well as the synovium also differ between sexes 50, 51 and joint shape may also contribute to risk 87. Spontaneous OA associated with aging also demonstrates differences between the molecular and cellular profiles within the DRGs between sexes 88. Interestingly, while antibodies against CGRP failed to provide analgesic relief for human OA knee pain89, data in non-arthritis rodent models of pain have demonstrated more robust effects in female compared with male animals90. Functional MRI imaging has been used in rat OA, albeit in the monosodium iodoacetate model, where differences in the periaqueductal grey matter connectivity was seen in females. Such findings would be consistent with increasing the predisposition to developing chronic pain 91.
Future directions
It is fair to say that good progress has been made in the past few decades at unravelling molecular mechanisms that underly pain in OA, aided by careful mechanistic molecular studies and clinically relevant in vivo models. However, we still do not have treatments that improve the lives of patients and there is still a nudging concern that targeting pain without targeting structural disease may have detrimental outcomes regarding structural disease, at least in a subset of patients. It is intuitive that powerful analgesia per se is going to encourage increased use of a joint and thereby drive the very mechano-inflammatory pathways that drove disease in the first place 92. But there are other possible ways in which neutralizing targets, such as NGF, could worsen joint damage. In the case of NSAIDs, these were shown to suppress the normal anabolic response of damaged cartilage in vitro. In the case of NGF, its receptors (TrkA and p75) are expressed on a variety of non-neuronal cell types and may therefore have additional effects on mesenchymal tissues of the joint. For instance, p75 is known to be expressed on mesenchymal stem cells within the synovial fluid and osteoblasts. Chondrocytes also express both TrkA and p75 but the role of these has not been elucidated 93. On the other hand, while CCR2 is also expressed by multiple cell types within the joint, including nerves, its deletion does not appear to lead to worsening structural disease. Indeed in some studies mild structural protection has been demonstrated. Finally, there is some emerging evidence that local ablation of knee innervating nerve afferents, for example through the use of resiniferatoxin or cryoneurolysis, may provide analgesic relief, but whether this strategy will have adverse structural effects is not yet clear94. Therefore, considering how targeting may affect not only the target cell type but all cells within the joint would likely help to refine how we can harness the analgesic powers of a given drug whilst minimising effects on other joint tissues.
Acknowledgements:
1. We acknowledge Odeta Bareckiene for her graphical support. 2. TLV directs the Centre for OA Pathogenesis Versus Arthritis (grant nos. 20205 and 21612). REM receives support from the National Institutes of Health (R01AR077019). 3. This funding has contributed to some of the studies cited in the review.
Conflicts of interest declaration:
TLV has received research funding for the STEpUP OA Consortium from Pfizer, Novartis, Fidia, Biosplice and UCB. TLV is on the editorial board of OAC and is an associate editor for OAC Open. REM is on the editorial board of OAC.
References
- 1.Obletz BE. Relief of pain in osteo-arthritis of the hip by partial denervation of the hip joint. Ann Rheum Dis 1948; 7: 255. [PubMed] [Google Scholar]
- 2.Helal B. The pain in primary osteoarthritis of the knee. Its causes and treatment by osteotomy. Postgrad Med J 1965; 41: 172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brandt KD. Pain, synovitis, and articular cartilage changes in osteoarthritis. Semin Arthritis Rheum 1989; 18: 77–80. [DOI] [PubMed] [Google Scholar]
- 4.Altman RD, Dean D. Pain in osteoarthritis. Introduction and overview. Semin Arthritis Rheum 1989; 18: 1–3. [DOI] [PubMed] [Google Scholar]
- 5.Kellgren JH, Samuel EP. The sensitivity and innervation of the articular capsule. J Bone Joint Surg [Br] 1950; 32: : 84–92. [Google Scholar]
- 6.Dye SF, Vaupel GL, Dye CC. Conscious neurosensory mapping of the internal structures of the human knee without intraarticular anesthesia. Am J Sports Med 1998; 26: 773–777. [DOI] [PubMed] [Google Scholar]
- 7.Kidd BL, Mapp PI, Blake DR, Gibson SJ, Polak JM. Neurogenic influences in arthritis. Ann Rheum Dis 1990; 49: 649–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hukkanen M, Gronblad M, Rees R, Kottinen YT, Gibson SJ, Hietanen J, et al. Regional distribution of mast cells and peptide containing nerves in normal and adjuvant arthritic rat synovium. J Rheumatol 1991; 18: 177–183. [PubMed] [Google Scholar]
- 9.Saito T, Koshino T. Distribution of neuropeptides in synovium of the knee with osteoarthritis. Clin Orthop Relat Res 2000: 172–182. [DOI] [PubMed] [Google Scholar]
- 10.Saxler G, Loer F, Skumavc M, Pfortner J, Hanesch U. Localization of SP- and CGRP-immunopositive nerve fibers in the hip joint of patients with painful osteoarthritis and of patients with painless failed total hip arthroplasties. Eur J Pain 2007; 11: 67–74. [DOI] [PubMed] [Google Scholar]
- 11.Samad TA, Sapirstein A, Woolf CJ. Prostanoids and pain: unraveling mechanisms and revealing therapeutic targets. Trends Mol Med 2002; 8: 390–396. [DOI] [PubMed] [Google Scholar]
- 12.Schaible HG, Schmidt RF. Effects of an experimental arthritis on the sensory properties of fine articular afferent units. J Neurophysiol 1985; 54: 1109–1122. [DOI] [PubMed] [Google Scholar]
- 13.McDougall JJ. Arthritis and pain. Neurogenic origin of joint pain. Arthritis Res Ther 2006; 8: 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wenham CY, Hensor EM, Grainger AJ, Hodgson R, Balamoody S, Dore CJ, et al. A randomized, double-blind, placebo-controlled trial of low-dose oral prednisolone for treating painful hand osteoarthritis. Rheumatology (Oxford) 2012; 51: 2286–2294. [DOI] [PubMed] [Google Scholar]
- 15.Kroon FPB, Kortekaas MC, Boonen A, Bohringer S, Reijnierse M, Rosendaal FR, et al. Results of a 6-week treatment with 10 mg prednisolone in patients with hand osteoarthritis (HOPE): a double-blind, randomised, placebo-controlled trial. Lancet 2019; 394: 1993–2001. [DOI] [PubMed] [Google Scholar]
- 16.Watson M. The suppressing effect of indomethacin on articular cartilage. Rheumatol Rehabil 1976; 15: 26–30. [DOI] [PubMed] [Google Scholar]
- 17.Slowman-Kovacs SD, Albrecht ME, Brandt KD. Effects of salicylate on chondrocytes from osteoarthritic and contralateral knees of dogs with unilateral anterior cruciate ligament transection. Arthritis Rheum 1989; 32: 486–490. [DOI] [PubMed] [Google Scholar]
- 18.Palmoski MJ, Colyer RA, Brandt KD. Marked suppression by salicylate of the augmented proteoglycan synthesis in osteoarthritic cartilage. Arthritis Rheum 1980; 23: 83–91. [DOI] [PubMed] [Google Scholar]
- 19.Palmoski MJ, Brandt KD. In vivo effect of aspirin on canine osteoarthritic cartilage. Arthritis Rheum 1983; 26: 994–1001. [DOI] [PubMed] [Google Scholar]
- 20.Doherty M, Jones A. Indomethacin hastens large joint osteoarthritis in humans--how strong is the evidence? J Rheumatol 1995; 22: 2013–2016. [PubMed] [Google Scholar]
- 21.Rashad S, Revell P, Hemingway A, Low F, Rainsford K, Walker F. Effect of non-steroidal anti-inflammatory drugs on the course of osteoarthritis. Lancet 1989; 2: 519–522. [DOI] [PubMed] [Google Scholar]
- 22.McAlindon TE, LaValley MP, Harvey WF, Price LL, Driban JB, Zhang M, et al. Effect of Intra-articular Triamcinolone vs Saline on Knee Cartilage Volume and Pain in Patients With Knee Osteoarthritis: A Randomized Clinical Trial. JAMA : the journal of the American Medical Association 2017; 317: 1967–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tiderius CJ, Olsson LE, Leander P, Ekberg O, Dahlberg L. Delayed gadolinium-enhanced MRI of cartilage (dGEMRIC) in early knee osteoarthritis. Magnetic resonance in medicine 2003; 49: 488–492. [DOI] [PubMed] [Google Scholar]
- 24.Massardo L, Watt I, Cushnaghan J, Dieppe P. Osteoarthritis of the knee joint: an eight year prospective study. Ann Rheum Dis 1989; 48: 893–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bowes MA, Lohmander LS, Wolstenholme CBH, Vincent GR, Conaghan PG, Frobell RB. Marked and rapid change of bone shape in acutely ACL injured knees - an exploratory analysis of the Kanon trial. Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society 2019; 27: 638–645. [DOI] [PubMed] [Google Scholar]
- 26.Yusup A, Kaneko H, Liu L, Ning L, Sadatsuki R, Hada S, et al. Bone marrow lesions, subchondral bone cysts and subchondral bone attrition are associated with histological synovitis in patients with end-stage knee osteoarthritis: a cross-sectional study. Osteoarthritis Cartilage 2015; 23: 1858–1864. [DOI] [PubMed] [Google Scholar]
- 27.Carrino JA, Blum J, Parellada JA, Schweitzer ME, Morrison WB. MRI of bone marrow edema-like signal in the pathogenesis of subchondral cysts. Osteoarthritis Cartilage 2006; 14: 1081–1085. [DOI] [PubMed] [Google Scholar]
- 28.Guermazi A, Hayashi D, Roemer FW, Zhu Y, Niu J, Crema MD, et al. Synovitis in knee osteoarthritis assessed by contrast-enhanced magnetic resonance imaging (MRI) is associated with radiographic tibiofemoral osteoarthritis and MRI-detected widespread cartilage damage: the MOST study. J Rheumatol 2014; 41: 501–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neogi T, Felson D, Niu J, Lynch J, Nevitt M, Guermazi A, et al. Cartilage loss occurs in the same subregions as subchondral bone attrition: a within-knee subregion-matched approach from the Multicenter Osteoarthritis Study. Arthritis Rheum 2009; 61: 1539–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Englund M, Niu J, Guermazi A, Roemer FW, Hunter DJ, Lynch JA, et al. Effect of meniscal damage on the development of frequent knee pain, aching, or stiffness. Arthritis Rheum 2007; 56: 4048–4054. [DOI] [PubMed] [Google Scholar]
- 31.Felson DT, Niu J, Guermazi A, Roemer F, Aliabadi P, Clancy M, et al. Correlation of the development of knee pain with enlarging bone marrow lesions on magnetic resonance imaging. Arthritis Rheum 2007; 56: 2986–2992. [DOI] [PubMed] [Google Scholar]
- 32.Roemer FW, Guermazi A, Javaid MK, Lynch JA, Niu J, Zhang Y, et al. Change in MRI-detected subchondral bone marrow lesions is associated with cartilage loss: the MOST Study. A longitudinal multicentre study of knee osteoarthritis. Ann Rheum Dis 2009; 68: 1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neogi T, Felson D, Niu J, Nevitt M, Lewis CE, Aliabadi P, et al. Association between radiographic features of knee osteoarthritis and pain: results from two cohort studies. BMJ 2009; 339: b2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soni A, Kiran A, Hart DJ, Leyland KM, Goulston L, Cooper C, et al. Prevalence of reported knee pain over twelve years in a community-based cohort. Arthritis and rheumatism 2012; 64: 1145–1152. [DOI] [PubMed] [Google Scholar]
- 35.Neogi T, Frey-Law L, Scholz J, Niu J, Arendt-Nielsen L, Woolf C, et al. Sensitivity and sensitisation in relation to pain severity in knee osteoarthritis: trait or state? Ann Rheum Dis 2015; 74: 682–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carlesso LC, Segal NA, Frey-Law L, Zhang Y, Na L, Nevitt M, et al. Pain Susceptibility Phenotypes in Those Free of Knee Pain With or at Risk of Knee Osteoarthritis: The Multicenter Osteoarthritis Study. Arthritis Rheumatol 2019; 71: 542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kosek E, Ordeberg G. Abnormalities of somatosensory perception in patients with painful osteoarthritis normalize following successful treatment. Eur J Pain 2000; 4: 229–238. [DOI] [PubMed] [Google Scholar]
- 38.Soni A, Wanigasekera V, Mezue M, Cooper C, Javaid MK, Price AJ, et al. Central Sensitization in Knee Osteoarthritis: Relating Presurgical Brainstem Neuroimaging and PainDETECT-Based Patient Stratification to Arthroplasty Outcome. Arthritis & rheumatology (Hoboken, N.J.) 2019; 71: 550–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Graven-Nielsen T, Wodehouse T, Langford RM, Arendt-Nielsen L, Kidd BL. Normalization of widespread hyperesthesia and facilitated spatial summation of deep-tissue pain in knee osteoarthritis patients after knee replacement. Arthritis Rheum 2012; 64: 2907–2916. [DOI] [PubMed] [Google Scholar]
- 40.Gwilym SE, Keltner JR, Warnaby CE, Carr AJ, Chizh B, Chessell I, et al. Psychophysical and functional imaging evidence supporting the presence of central sensitization in a cohort of osteoarthritis patients. Arthritis and rheumatism 2009; 61: 1226–1234. [DOI] [PubMed] [Google Scholar]
- 41.Gwilym SE, Filippini N, Douaud G, Carr AJ, Tracey I. Thalamic atrophy associated with painful osteoarthritis of the hip is reversible after arthroplasty: a longitudinal voxel-based morphometric study. Arthritis and rheumatism 2010; 62: 2930–2940. [DOI] [PubMed] [Google Scholar]
- 42.Miller RE, Malfait AM. Can we prevent chronic osteoarthritis pain? A view from the bench. Osteoarthritis Cartilage 2021; 29: 1635–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bove SE, Calcaterra SL, Brooker RM, Huber CM, Guzman RE, Juneau PL, et al. Weight bearing as a measure of disease progression and efficacy of anti-inflammatory compounds in a model of monosodium iodoacetate-induced osteoarthritis. Osteoarthritis Cartilage 2003; 11: 821–830. [DOI] [PubMed] [Google Scholar]
- 44.Kobayashi K, Imaizumi R, Sumichika H, Tanaka H, Goda M, Fukunari A, et al. Sodium iodoacetate-induced experimental osteoarthritis and associated pain model in rats. J Vet Med Sci 2003; 65: 1195–1199. [DOI] [PubMed] [Google Scholar]
- 45.Bove SE, Laemont KD, Brooker RM, Osborn MN, Sanchez BM, Guzman RE, et al. Surgically induced osteoarthritis in the rat results in the development of both osteoarthritis-like joint pain and secondary hyperalgesia. Osteoarthritis Cartilage 2006; 14: 1041–1048. [DOI] [PubMed] [Google Scholar]
- 46.Inglis JJ, McNamee KE, Chia S-L, Essex D, Feldmann M, Williams RO, et al. Regulation of pain sensitivity in experimental osteoarthritis by the endogenous peripheral opioid system. Arthritis and rheumatology 2008; 58: 3110–3119. [DOI] [PubMed] [Google Scholar]
- 47.Malfait AM, Ritchie J, Gil AS, Austin JS, Hartke J, Qin W, et al. ADAMTS-5 deficient mice do not develop mechanical allodynia associated with osteoarthritis following medial meniscal destabilization. Osteoarthritis Cartilage 2010; 18: 572–580. [DOI] [PubMed] [Google Scholar]
- 48.Fernihough J, Gentry C, Malcangio M, Fox A, Rediske J, Pellas T, et al. Pain related behaviour in two models of osteoarthritis in the rat knee. Pain 2004; 112: 83–93. [DOI] [PubMed] [Google Scholar]
- 49.Miller RE, Malfait AM. Osteoarthritis pain: What are we learning from animal models? Best Pract Res Clin Rheumatol 2017; 31: 676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.von Loga IS, Batchelor V, Driscoll C, Burleigh A, Chia S-L, Stott B, et al. Does pain at an earlier stage of chondropathy protect female mice from structural progression after surgically induced osteoarthritis. Arthritis & Rheumatology 2020; Dec;72. : 2083–2093. [DOI] [PubMed] [Google Scholar]
- 51.Bergman RF, Lammlin L, Junginger L, Farrell E, Goldman S, Darcy R, et al. Sexual dimorphism of the synovial transcriptome underpins greater PTOA disease severity in male mice following joint injury. Osteoarthritis Cartilage 2023. [DOI] [PubMed] [Google Scholar]
- 52.Miller RE, Kim YS, Tran PB, Ishihara S, Dong X, Miller RJ, et al. Visualization of Peripheral Neuron Sensitization in a Surgical Mouse Model of Osteoarthritis by In Vivo Calcium Imaging. Arthritis Rheumatol 2018; 70: 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schuelert N, Johnson MP, Oskins JL, Jassal K, Chambers MG, McDougall JJ. Local application of the endocannabinoid hydrolysis inhibitor URB597 reduces nociception in spontaneous and chemically induced models of osteoarthritis. Pain 2011; 152: 975–981. [DOI] [PubMed] [Google Scholar]
- 54.Ai M, Hotham WE, Pattison LA, Ma Q, Henson FMD, Smith ESJ. Role of Human Mesenchymal Stem Cells and Derived Extracellular Vesicles in Reducing Sensory Neuron Hyperexcitability and Pain Behaviors in Murine Osteoarthritis. Arthritis Rheumatol 2023; 75: 352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ritter AM, Lewin GR, Kremer NE, Mendell LM. Requirement for nerve growth factor in the development of myelinated nociceptors in vivo. Nature 1991; 350: 500–502. [DOI] [PubMed] [Google Scholar]
- 56.Levi-Montalcini R Growth Control of Nerve Cells by a Protein Factor and Its Antiserum: Discovery of This Factor May Provide New Leads to Understanding of Some Neurogenetic Processes. Science 1964; 143: 105–110. [DOI] [PubMed] [Google Scholar]
- 57.Woolf CJ, Safieh-Garabedian B, Ma QP, Crilly P, Winter J. Nerve growth factor contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience 1994; 62: 327–331. [DOI] [PubMed] [Google Scholar]
- 58.McMahon SB, Bennett DL, Priestley JV, Shelton DL. The biological effects of endogenous nerve growth factor on adult sensory neurons revealed by a trkA-IgG fusion molecule. Nat Med 1995; 1: 774–780. [DOI] [PubMed] [Google Scholar]
- 59.McNamee KE, Burleigh A, Gompels LL, Feldmann M, Allen SJ, Williams RO, et al. Treatment of murine osteoarthritis with TrkAd5 reveals a pivotal role for nerve growth factor in non-inflammatory joint pain. Pain 2010; 149: 386–392. [DOI] [PubMed] [Google Scholar]
- 60.von Loga IS, El-Turabi A, Jostins L, Miotla-Zarebska J, Mackay-Alderson J, Zeltins A, et al. Active immunisation targeting nerve growth factor attenuates chronic pain behaviour in murine osteoarthritis. Annals of the rheumatic diseases 2019; 2019;78: 672–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.LaBranche TP, Bendele AM, Omura BC, Gropp KE, Hurst SI, Bagi CM, et al. Nerve growth factor inhibition with tanezumab influences weight-bearing and subsequent cartilage damage in the rat medial meniscal tear model. Annals of the rheumatic diseases 2017; 76: 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lane NE, Schnitzer TJ, Birbara CA, Mokhtarani M, Shelton DL, Smith MD, et al. Tanezumab for the treatment of pain from osteoarthritis of the knee. N Engl J Med 2010; 363: 1521–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hochberg MC, Tive LA, Abramson SB, Vignon E, Verburg KM, West CR, et al. When Is Osteonecrosis Not Osteonecrosis?: Adjudication of Reported Serious Adverse Joint Events in the Tanezumab Clinical Development Program. Arthritis Rheumatol 2016; 68: 382–391. [DOI] [PubMed] [Google Scholar]
- 64.Tive L, Bello AE, Radin D, Schnitzer TJ, Nguyen H, Brown MT, et al. Pooled analysis of tanezumab efficacy and safety with subgroup analyses of phase III clinical trials in patients with osteoarthritis pain of the knee or hip. Journal of pain research 2019; 12: 975–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Enomoto M, Mantyh PW, Murrell J, Innes JF, Lascelles BDX. Anti-nerve growth factor monoclonal antibodies for the control of pain in dogs and cats. Vet Rec 2019; 184: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stoppiello LA, Mapp PI, Wilson D, Hill R, Scammell BE, Walsh DA. Structural associations of symptomatic knee osteoarthritis. Arthritis & rheumatology (Hoboken, N.J.) 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aso K, Shahtaheri SM, Hill R, Wilson D, McWilliams DF, Nwosu LN, et al. Contribution of nerves within osteochondral channels to osteoarthritis knee pain in humans and rats. Osteoarthritis Cartilage 2020; 28: 1245–1254. [DOI] [PubMed] [Google Scholar]
- 68.Wyatt LA, Nwosu LN, Wilson D, Hill R, Spendlove I, Bennett AJ, et al. Molecular expression patterns in the synovium and their association with advanced symptomatic knee osteoarthritis. Osteoarthritis and cartilage 2019; 27: 667–675. [DOI] [PubMed] [Google Scholar]
- 69.Walsh DA, McWilliams DF, Turley MJ, Dixon MR, Franses RE, Mapp PI, et al. Angiogenesis and nerve growth factor at the osteochondral junction in rheumatoid arthritis and osteoarthritis. Rheumatology (Oxford) 2010; 49: 1852–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aso K, Shahtaheri SM, Hill R, Wilson D, McWilliams DF, Walsh DA. Associations of Symptomatic Knee Osteoarthritis With Histopathologic Features in Subchondral Bone. Arthritis Rheumatol 2019; 71: 916–924. [DOI] [PubMed] [Google Scholar]
- 71.Aso K, Walsh DA, Wada H, Izumi M, Tomitori H, Fujii K, et al. Time course and localization of nerve growth factor expression and sensory nerve growth during progression of knee osteoarthritis in rats. Osteoarthritis Cartilage 2022; 30: 1344–1355. [DOI] [PubMed] [Google Scholar]
- 72.Driscoll C, Chanalaris A, Knights C, Ismail H, Sacitharan PK, Gentry C, et al. Nociceptive Sensitizers Are Regulated in Damaged Joint Tissues, Including Articular Cartilage, When Osteoarthritic Mice Display Pain Behavior. Arthritis & rheumatology (Hoboken, N.J.) 2016; 68: 857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kuttapitiya A, Assi L, Laing K, Hing C, Mitchell P, Whitley G, et al. Microarray analysis of bone marrow lesions in osteoarthritis demonstrates upregulation of genes implicated in osteochondral turnover, neurogenesis and inflammation. Annals of the rheumatic diseases 2017; 76: 1764–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vincent TL. Peripheral Pain Mechanisms in Osteoarthritis. Pain 2020; 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Malfait AM, Miller RE, Block JA. Targeting neurotrophic factors: Novel approaches to musculoskeletal pain. Pharmacol Ther 2020; 211: 107553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Obeidat AM, Miller RE, Miller RJ, Malfait AM. The nociceptive innervation of the normal and osteoarthritic mouse knee. Osteoarthritis and cartilage 2019; 27: 1669–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Obeidat AM, Ishihara S, Li J, Lammlin L, Junginger L, Maerz T, et al. Intra-Articular Sprouting Of Nociceptors Accompanies Progressive Osteoarthritis: Comparative Evidence In Four Murine Models. BioRxiv preprint 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhu S, Zhu J, Zhen G, Hu Y, An S, Li Y, et al. Subchondral bone osteoclasts induce sensory innervation and osteoarthritis pain. J Clin Invest 2019; 129: 1076–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Miller RE, Tran PB, Das R, Ghoreishi-Haack N, Ren D, Miller RJ, et al. CCR2 chemokine receptor signaling mediates pain in experimental osteoarthritis. Proc Natl Acad Sci U S A 2012; 109: 20602–20607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Miotla Zarebska J, Chanalaris A, Driscoll C, Burleigh A, Miller RE, Malfait AM, et al. CCL2 and CCR2 regulate pain-related behaviour and early gene expression in post-traumatic murine osteoarthritis but contribute little to chondropathy. Osteoarthritis and cartilage 2017; 25: 406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Longobardi L, Temple JD, Tagliafierro L, Willcockson H, Esposito A, D’Onofrio N, et al. Role of the C-C chemokine receptor-2 in a murine model of injury-induced osteoarthritis. Osteoarthritis Cartilage 2017; 25: 914–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ishihara S, Obeidat AM, Wokosin DL, Ren D, Miller RJ, Malfait AM, et al. The role of intra-articular neuronal CCR2 receptors in knee joint pain associated with experimental osteoarthritis in mice. Arthritis Res Ther 2021; 23: 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Obeidat AM, Wood MJ, Adamczyk NS, Ishihara S, Li J, Wang L, et al. Piezo2 expressing nociceptors mediate mechanical sensitization in experimental osteoarthritis. Nat Commun 2023; 14: 2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Conaghan PG, Cook AD, Hamilton JA, Tak PP. Therapeutic options for targeting inflammatory osteoarthritis pain. Nat Rev Rheumatol 2019; 15: 355–363. [DOI] [PubMed] [Google Scholar]
- 85.Hwang HS, Park IY, Hong JI, Kim JR, Kim HA. Comparison of joint degeneration and pain in male and female mice in DMM model of osteoarthritis. Osteoarthritis Cartilage 2021; 29: 728–738. [DOI] [PubMed] [Google Scholar]
- 86.Temp J, Labuz D, Negrete R, Sunkara V, Machelska H. Pain and knee damage in male and female mice in the medial meniscal transection-induced osteoarthritis. Osteoarthritis Cartilage 2020; 28: 475–485. [DOI] [PubMed] [Google Scholar]
- 87.Javaheri B, Razi H, Piles M, de Souza R, Chang YM, Maric-Mur I, et al. Sexually dimorphic tibia shape is linked to natural osteoarthritis in STR/Ort mice. Osteoarthritis Cartilage 2018; 26: 807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Geraghty T, Obeidat AM, Ishihara S, Wood MJ, Li J, Lopes EBP, et al. Age-Associated Changes in Knee Osteoarthritis, Pain-Related Behaviors, and Dorsal Root Ganglia Immunophenotyping of Male and Female Mice. Arthritis Rheumatol 2023; 75: 1770–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jin Y, Smith C, Monteith D, Brown R, Camporeale A, McNearney TA, et al. CGRP blockade by galcanezumab was not associated with reductions in signs and symptoms of knee osteoarthritis in a randomized clinical trial. Osteoarthritis Cartilage 2018; 26: 1609–1618. [DOI] [PubMed] [Google Scholar]
- 90.Paige C, Plasencia-Fernandez I, Kume M, Papalampropoulou-Tsiridou M, Lorenzo LE, David ET, et al. A Female-Specific Role for Calcitonin Gene-Related Peptide (CGRP) in Rodent Pain Models. J Neurosci 2022; 42: 1930–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Da Silva JT, Tricou C, Zhang Y, Tofighbakhsh A, Seminowicz DA, Ro JY. Pain modulatory network is influenced by sex and age in a healthy state and during osteoarthritis progression in rats. Aging Cell 2021; 20: e13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vincent TL. Mechanoflammation in osteoarthritis pathogenesis | Elsevier Enhanced Reader. Seminars in arthritis and rheumatism 2019; 49: S36–38. [DOI] [PubMed] [Google Scholar]
- 93.Iannone F, De Bari C, Dell’accio F, Covelli M, Patella V, Lo Bianco G, et al. Increased expression of nerve growth factor (NGF) and high affinity NGF receptor (p140 TrkA) in human osteoarthritic chondrocytes. Rheumatology (Oxford, England) 2002; 41: 1413–1418. [DOI] [PubMed] [Google Scholar]
- 94.Walsh DA. Osteoarthritis: Nerve ablation - a new treatment for OA pain? Nat Rev Rheumatol 2017; 13: 393–394. [DOI] [PubMed] [Google Scholar]