

Summary
Here, we present Anchored-fusion, a highly sensitive fusion gene detection tool. It anchors a gene of interest, which often involves driver fusion events, and recovers non-unique matches of short-read sequences that are typically filtered out by conventional algorithms. In addition, Anchored-fusion contains a module based on a deep learning hierarchical structure that incorporates self-distillation learning (hierarchical view learning and distillation [HVLD]), which effectively filters out false positive chimeric fragments generated during sequencing while maintaining true fusion genes. Anchored-fusion enables highly sensitive detection of fusion genes, thus allowing for application in cases with low sequencing depths. We benchmark Anchored-fusion under various conditions and found it outperformed other tools in detecting fusion events in simulated data, bulk RNA sequencing (bRNA-seq) data, and single-cell RNA sequencing (scRNA-seq) data. Our results demonstrate that Anchored-fusion can be a useful tool for fusion detection tasks in clinically relevant RNA-seq data and can be applied to investigate intratumor heterogeneity in scRNA-seq data.
Graphical abstract
Highlights
-
•
Anchored-fusion detects fusion genes with high sensitivity in paired-end RNA-seq
-
•
Anchoring a gene of interest avoids over-filtering based on homology alignment
-
•
A deep learning module filters false positive chimeric reads
-
•
Anchored-fusion shows high sensitivity in single-cell applications
Motivation
Gene fusion is one of the key events driving cancer development. Identifying critical fusion genes using RNA sequencing (RNA-seq) data has been applied in clinical samples for diagnosis, subtyping, and targeted therapeutic purposes. However, current gene fusion detection algorithms of RNA-seq are limited by their lack of sensitivity, making it difficult to apply them to low-read-depth data, for example in single-cell and/or clinical contexts.
Yuan et al. present Anchored-fusion, a method for detecting fusion genes with high sensitivity from paired-end RNA-seq. Anchoring a gene of interest avoids over-filtering, and a deep learning model removes false positives. Anchored-fusion demonstrates superior sensitivity in various scenarios, particularly in detecting fusion genes from single-cell RNA-seq.
Introduction
Chromosomal translocations, deletions, and other structural variations can result in the fusion of partial sequences from two genes, which creates a novel chimeric gene. This occurrence is known as gene fusion, which can lead to the creation of abnormal transcripts or proteins during subsequent biological processes.1 Fusion genes play a crucial role in the occurrence and development of cancer and are often utilized for cancer diagnosis and classification. For example, the BCR-ABL1 fusion gene encodes a chimeric protein that induces the development of chronic myeloid leukemia (CML).2 Tyrosine kinase inhibitors that specifically target BCR-ABL1 fusion,3 such as imatinib,4 are the standard therapy for CML. The TMPRSS2-ERG gene fusion is a predictive biomarker for patients with prostate cancer and plays a critical role in evaluating the characteristics and prognosis of the disease.5 Neurotrophic tropomyosin kinase receptor (NTRK).
(NTRK) gene fusions have been reported in at least 34 cancer types and are considered biomarkers for predicting drug resistance and survival.6 The small-molecule inhibitor larotrectinib7 provides a targeted treatment option for NTRK gene fusions present in various cancer types. Targeted therapy drugs for fusion genes have been approved and widely used in clinical practice. Consequently, gene fusion events have been extensively employed as biomarkers in tumor classification, grading, prognosis evaluation, and therapeutic guidance for patients with cancer.
Several bioinformatics tools have been developed to detect gene fusion events de novo in transcriptome sequencing (RNA sequencing [RNA-seq]) data.8,9,10,11,12 However, compared to traditional methods such as fluorescence13 in situ hybridization (FISH) and quantitative real-time PCR (real-time qPCR),14 such fusion detection is limited in sensitivity, which hinders its clinical applications.15 Furthermore, these bioinformatics methods face challenges when applied to single-cell RNA-seq (scRNA-seq) data with low sequencing depth, the primary method for unraveling cellular heterogeneity.
Sequence homology is an important reason for the low detection sensitivity of existing fusion detection tools. Sequence homologies are widespread in the human genome, resulting in RNA read pairs potentially mapping to multiple locations. Most fusion gene detection tools exclude data from these multi-mapping read pairs, which poses a long-standing challenge in detecting fusion events involving sequence homology. Another widely accepted challenge is to effectively eliminate false positives. During library preparation and second-generation sequencing, accidental ligation of amplicons may happen, leading to the creation of artificial chimeric reads. These reads might be mistakenly identified as fusion genes by the analysis tools, resulting in false positive fusion evidences.16 However, implementing stricter hard filtering to avoid false positives may further reduce the sensitivity of detecting true fusions.17
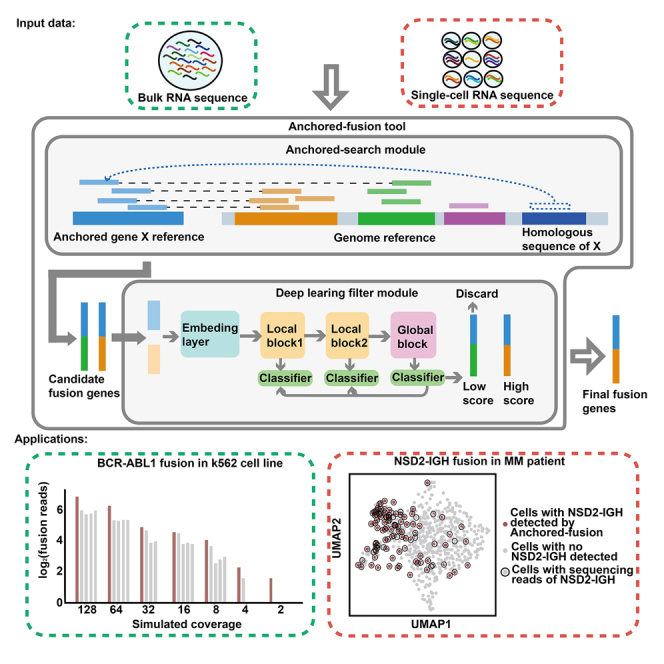
To address the challenges mentioned above, we present a fusion gene detection algorithm named Anchored-fusion (Figure 1). Anchored-fusion enables sensitive detection by anchoring a user-specified gene of interest and controls false positives through a deep learning framework using the hierarchical view learning and distillation (HVLD) architecture. We benchmarked Anchored-fusion against other methods in several conditions, including simulated fusion gene transcriptomic data, real bulk RNA-seq (bRNA-seq) data, and scRNA-seq data. The benchmark results demonstrate that Anchored-fusion is highly sensitive in detecting fusion genes. As a result, Anchored-fusion can serve as a valuable tool with clinical application to fusion detection in shallow sequenced bulk and scRNA-seq data. Anchored-fusion software is available at https://github.com/ShenLab-Genomics/Anchored-Fusion.
Figure 1.

Workflow of Anchored-fusion
(A) Anchored-fusion workflow to identify fusion genes and their fusion breakpoints, including identifying potential fusion gene candidates, determining fusion gene breakpoints, training the discrimination model, filtering out artificial chimeric reads, and, finally, confirming the final fusion gene and its breakpoints.
(B) The framework of the hierarchical view learning and distillation (HVLD) model used by Anchored-fusion consists of embedding layers, local blocks, and a global block. It also incorporates self-distillation learning, which involves using the output of the global block to guide the output of local blocks.
(C) Chromosome 22 and chromosome 9 fuse to generate the BCR-ABL1 fusion gene. The sequence near the fusion site of the BCR gene has a homologous sequence on chromosome 5. Some supporting evidences have been excluded because they are aligned to the homologous sequence.
(D) The DUX4 gene is composed of D4Z4 repeat sequences and is also present on chromosomes 10, 22, Y, etc., with hundreds of copies.
Results
Anchored-fusion overview
Anchored-fusion (Figure 1A) detects fusion events involving a user-specified gene of interest with high sensitivity. Firstly, the paired-end reads from the test sample are aligned to the “anchored” gene X. The process of anchoring maximally preserves all reads that may have been transcribed from anchored gene X, thus ensuring high sensitivity in detecting fusion genes. In contrast, existing tools mostly align against the whole genome or transcriptome, which often leads to compromised sensitivity due to sequence homology. Subsequently, Anchored-fusion selects the paired-end reads, which were partially matched to gene X, and aligns the unmatched part against the genome to search for the candidate partner gene set Y. Next, a refined alignment is performed on the anchored gene X and the potential fusion gene set Y to identify fusion breakpoints. Finally, instead of using a supporting read threshold to filter as most methods do, Anchored-fusion incorporates a model with a deep neural network called the HVLD model to filter out false positive fusions that may be caused by artificial factors during the experimental process (Figures 1B and S1). The anchoring approach enables highly sensitive detection of supporting reads for fusion events, while the HVLD deep learning framework effectively filters out false positive fusion events customized to the input data, which allows the tool to maintain highly sensitive detection of fusion genes with a small number of supporting evidences.. Notably, the HVLD model distinguishes true fusion reads from noise and artifacts based solely on fusion sequence features, without requiring other features such as read counts.
We tested fusion genes with existing detection challenges caused by sequence homology. For example, the classical BCR-ABL1 fusion gene contains a 47 bp short homologous sequence with chromosome 5 near its fusion breakpoint, which causes some supporting evidences at that location to be incorrectly matched or filtered out due to multiple alignments (Figures 1C and S2). In another example, fusion events involving the DUX4 gene with different fusion partners have been reported to drive the occurrence of cancer.18,19,20 The DUX4 gene is located within a region of macrosatellite repeats with each 3.3 kb repeat unit called D4Z4 in the subtelomere region of chromosome 4. This region contains dozens of D4Z4 copies and contains a significant number of highly homologous sequence matches throughout the genome. As a result, the supporting sequencing reads of the DUX4 fusion gene are matched to hundreds of loci, leading to few supporting evidences or even the erroneous removal of the fusion event (Figures 1D and S3). These problems have long been a challenge in the design of fusion gene detection algorithms.11 To overcome these challenges, Anchored-fusion utilizes an anchoring step that retains reads with sequence homology. Additionally, the HVLD module is used to dynamically filter out false positives based solely on sequence features, independent of the number of supporting evidences. Thus, Anchored-fusion achieves the highest sensitivity among all competing methods.
Anchored-fusion incorporates a hierarchical self-distilling deep learning framework
To eliminate false positives while maintaining true fusion genes with few supporting reads, Anchored-fusion incorporates a module with an HVLD network to filter artificial fusion reads based on sequence features alone. HVLD has a hierarchical structure that integrates both local and global information of the sequence, with a self-distillation learning framework to capture useful features. Previous studies have suggested that short sequences on chimeric sequences exhibit distinguishing features between artificially and naturally occurring fusion sequences. For example, natural fusion sites often exhibit splicing site characteristics, while some specific sequences are more likely to be present in fusion sequences generated by PCR misannealing and erroneous extension.9,12,16 Thus, we designed a local information extraction module based on 1D convolution to extract local sequence features. We also incorporated a global block with multi-head self-attention21 to capture the sequence features that are correlated but spatially distant. To further enhance the predictive performance, we integrated self-distillation learning.22 The self-distillation learning is designed to reduce the interference of noise on the shallow neural network and improve the model’s generalization capability. In summary, we developed a hierarchical deep learning model called HVLD that can dynamically capture both local and global information for the classification of true or false fusion genes.
Before we evaluated the full model, we first evaluated the ability of the HVLD model to discover artificially chimeric reads. We selected six bRNA-seq datasets from cancer cell lines of K562, TE441T, NALM6, NCI-H660, NCIH3122, and KM12. Additionally, we included two scRNA-seq datasets: one from the K562 cell line and the other from a clinical cohort of patients with multiple myeloma (MM). For each dataset, we selected chimeric reads from the input dataset to generate negative samples, assuming these chimeric reads depict the characteristics of artificially false fusions in the dataset. We believe this is a reasonable assumption since real fusion events produce very few chimeric reads, if any, and the vast majority of chimeric reads in the dataset are generated for artificial reasons. For all test cases, we took the real fusions downloaded from Pan-Cancer Analysis of Whole Genomes as positive samples (Figure S4).
We trained and evaluated the ability of HVLD and other competing models to distinguish between false and true fusion on the eight datasets downloaded. The positive and negative samples were generated through the above methods. We performed a random split of the total set into the training-validation set and the test set, with a 7:3 ratio. To mitigate model bias, we ensured that both sets contained an equal number of positive and negative samples for training and testing. For performance evaluation, accuracy, area under the curve (AUC), precision-recall AUC (PRAUC), precision, and recall were all applied as evaluation metrics. Furthermore, we compared the HVLD model with four existing methods: the bidirectional long short-term memory (bi-LSTM) model used in scFusion,12 the Transformer model,21 the multi-layer perceptron (MLP) model, the support vector machine (SVM) model, and our model framework without distillation learning (HVL).
Our HVLD model outperformed multiple machine learning and deep learning models across all metrics. As shown in Figure 2, in the bRNA-seq dataset, the average accuracy of our self-distillation model was 82.1% (maximum: 83.4%, median: 82.1%, minimum: 80.8%). Compared to the suboptimal Transformer model, our model HVLD achieved an average improvement of 8.85% in accuracy (with a maximum of 9.95%, a median of 9.05%, and a minimum of 7.40%). Furthermore, compared to HVL, HVLD achieved an average improvement of 0.75% (with a maximum of 1.27%, a median of 0.68%, and a minimum of 0.38%) on the bRNA-seq dataset. Similarly, in scRNA-seq datasets of patients with MM and the K562 cell line, HVLD also achieved the best performance with AUC values of 94.5% and 91.0%, representing improvements of 4.80% and 5.45% compared to the Transformer model, respectively. This phenomenon can be attributed to the following reasons: (1) HVLD, which combines 1D convolution and Transformer, is capable of simultaneously discovering short feature sequences of both natural and artificial fusion events, as well as capturing the relationships between these feature sequences. (2) The distillation learning part allows the shallow model to better capture discriminative features, thereby further enhancing the deep model’s ability to distinguish between these two types of fusion events.
Figure 2.

Performance of different machine learning and deep learning algorithms in distinguishing false fusion genes from true fusion genes across 6 test bRNA-seq datasets
(A) Indicator values of different algorithms on various bRNA-seq datasets.
(B) AUC plots of different algorithms on various scRNA-seq datasets.
Anchored-fusion detects fusion genes from simulated RNA-seq data with high sensitivity
To comprehensively evaluate the performance of Anchored-fusion, we benchmarked Anchored-fusion against other widely used tools including STAR-Fusion,9 FusionInspector,10 FusionCatcher,8 and Arriba11 using simulated reads for six fusion genes. The six fusion genes, namely BCR-ABL1, CIC-DUX4, DUX4-IGH, TMPRSS2-ERG, EML4-ALK, and TPM3-NTRK1, which are known as driver mutations of the previously mentioned six cell lines, respectively, were supported by ample research evidences. Due to different fusion breakpoints between fusion partners, each pair of fusion partners may form several different fusion genes. We chose the fusion gene that is supported by experimental evidence and exhibits the highest expression level in its corresponding cell line for simulation. We used the wgsim23 to generate fusion transcripts with different expression levels. The simulated fusion transcripts were then added to the RNA-seq data of the GM12878 cell line. We simulated different levels of expression for the fusion transcripts by varying the coverage, resulting in a total of seven different expression levels ranging from a base coverage of 128 to 2.
Anchored-fusion was able to find the most supporting evidences in both spanning and split reads in almost every fusion gene test case (Figure 3). Additionally, compared to other tools, Anchored-fusion had a lower coverage rate limit for identifying fusion genes. These results indicate that Anchored-fusion is better at capturing supporting evidences compared to other tools, due to its remarkably high sensitivity. Thus, Anchored-fusion can detect fusion genes with low expression and can be applied to RNA-seq samples with shallow sequencing. Furthermore, Anchored-fusion provided a balanced combination of spanning and split supporting evidences simultaneously, which contributed to the robustness of its results. In contrast, only FusionCatcher among the other tools guaranteed the provision of both types of supporting evidences. However, it showed a much higher requirement for coverage rate.
Figure 3.

Benchmark for fusion gene detection using simulated fusion transcripts
We simulated fusion gene bRNA-seq data with varying coverage and applied multiple fusion detection tools to identify supporting evidences for fusion genes. The x axis represents the ratio of simulated transcript base pairs to fusion gene base pairs.
(A)–(F) represent specific fusion genes, and the fusion breakpoints are annotated with GRCh38. The left section of the subgraph depicts all support evidences, and the y axis represents the logarithm base 2 of the quantity of evidences. The middle section corresponds to spanning evidences, while the right section represents split evidences. The y axis indicates the ratio of detected supporting evidences for each tool compared to the maximum supporting evidences among them.
Among all methods, Anchored-fusion demonstrated the best detection capability in the case of BCR-ABL1 and CIC-DUX4. In the test for another fusion gene, DUX4-IGH, only Anchored-fusion and FusionCatcher detected this fusion gene, where Anchored-fusion achieved only one-fourth of the detection limit of FusionCatcher. Taken together, Anchored-fusion outperformed other tools in the detection of fusion genes with homologous sequences. This advantage can be attributed to the fact that other tools may discard or misalign sequencing reads with multiple mapping against the genome whereas Anchored-fusion retains them completely through the anchoring approach.
Anchored-fusion demonstrates high sensitivity in detecting fusion genes in cancer cell line RNA-seq data
Next, we compared Anchored-fusion against other tools in bRNA-seq data of previously mentioned cell lines with varying sequencing depths. By progressively randomly downsampling half of the original cancer cell line bRNA-seq data, we simulated the gradient dilution to evaluate the detection sensitivity of different methods. In this experiment, we performed a total of 7 subsampling iterations, resulting in eight different datasets with varying sequencing depths, including the original data. Some pairs of fusion partners form multiple fusion genes with different fusion breakpoints, including DUX4-IGH in the NALM6 cell line, TMPRSS2-ERG in the NCI-H660 cell line, and TPM3-NTRK1 in the KM12 cell line. For fusion genes with multiple fusion transcripts identified, we selected the fusion transcript with the highest expression level to display in the results. The fusion breakpoints identified by each tool among the fusion partners are listed in Table S1.
Figure 4 showed that Anchored-fusion detected fusion genes with high sensitivity in cancer bRNA-seq datasets. Except for the TMPRSS2-ERG gene with a relatively low expression, Anchored-fusion successfully detected the corresponding fusion genes even when the datasets were subsampled 7 times with less than 1 M sequence depth. In contrast, none of the other tools were able to achieve this. The remarkable performance of Anchored-fusion in real cell line data demonstrates its sensitivity in the detection of fusion genes in reality. Because of the high sensitivity, Anchored-fusion is applicable in situations with lower sequencing depth, allowing for cost savings and enabling the detection of fusion genes with low expression levels.
Figure 4.

Benchmark results of fusion gene detection using bRNA-seq of cancer cell lines for 5 different tools
Each tool detects the supporting evidences of fusion genes using real cell line transcriptome data. The x axis represents the number of transcripts (in millions), and the labels correspond to subsampling ranges of , respectively.
(A)–(F) correspond to the detection results of a specific fusion gene in the cell line, and the fusion breakpoints are annotated with GRCh38. The left section of the subgraph depicts all support evidences, and the y axis represents the logarithm base 2 of the quantity of evidences. The middle section corresponds to spanning evidences, while the right section represents split evidences. The y axis indicates the ratio of detected supporting evidences for each tool compared to the maximum supporting evidences among them.
As an example, Anchored-fusion, FusionCatcher, and STAR-Fusion have detected the DUX4-IGH fusion gene. However, both Anchored-fusion and FusionCatcher reported that the breakpoint of DUX4 is located at nucleotide position 1224 within its transcript sequence (NM_001306068.3), while STAR-Fusion identified the DUX4 breakpoint at position 1511. The breakpoint identified by Anchored-fusion and FusionCatcher aligns with the DUX4-IGH fusion reported by Tian et al.,24 where the last 16 amino acids of the DUX4 protein were replaced by IGH. Thus, the accurate detection of fusion genes involving DUX4 by Anchored-fusion proves its advantage in identifying fusion events involving sequence homology.
Anchored-fusion detected BCR-ABL1 fusion in the K562 cell line scRNA-seq
scRNA-seq has revolutionized our understanding of cellular heterogeneity and has been widely used for exploring disease mechanisms and finding personalized therapeutic approaches. However, scRNA-seq often suffers from low sequencing depth, and the sensitive detection of fusion genes under such low-sequencing conditions remains a challenge for existing fusion gene detection tools. Among the current gene detection tools, only scFusion12 has been shown to have the ability to detect fusion genes in full-length-transcript-based scRNA-seq datasets.
To evaluate the fusion gene detection capability of Anchored-fusion in scRNA-seq data, we tested a K562 cell line Smart-seq2 dataset consisting of 350 cells, with an average of 1.3 M reads per cell. The K562 cell line harbors the BCR-ABL1 fusion gene. In this dataset, Anchored-fusion successfully detected 35 cells containing the BCR-ABL1 fusion gene (Figure 5A). In comparison, other methods reported lower numbers of cells with BCR-ABL fusion (Figure 5B).
Figure 5.

Benchmark of BCR-ABL1 fusion detection in K562 scRNA-seq dataset
(A) Uniform manifold approximation and projection (UMAP) visualization of all K562 scRNA-seq data, with cells containing the BCR-ABL1 fusion gene detected by Anchored-fusion highlighted.
(B) Bar chart showing the number of cells containing the BCR-ABL1 fusion gene detected by Anchored-fusion and five other methods in the K562 single-cell dataset.
(C) UMAP visualization plot of all cells in the K562 scRNA-seq data. All cells containing the BCR-ABL1 fusion gene are circled, and the highlighted ones indicate those detected by Anchored-fusion tool. We identified the cells containing the BCR-ABL1 fusion gene with its reference sequence.
(D) The number of single cells with the BCR-ABL1 fusion gene in different cell types detected by various tools. The single cells are categorized based on the amount of supporting evidences, and the bubble size represents the number of detected cells.
To further elucidate why Anchored-fusion detects more single cells with BCR-ABL1 fusion genes compared to other tools, we examined the supporting evidences detectable in this dataset. Specifically, we mapped the sequences of all cells in this dataset to the reference sequence of the BCR-ABL1 fusion gene and counted two types of supporting evidences. In 42 cells, we identified both spanning and split reads for BCR-ABL1, of which 83.3% were discovered by Anchored-fusion (Figure 5C). Subsequently, we classified these cells into categories of very low (1–2 supporting evidences), low (3–4 supporting evidences), moderate (5–6 supporting evidences), high (7–8 supporting evidences), and very high (9 or more supporting evidences) based on their total supporting evidences count. As depicted in Figure 5C, Anchored-fusion detected almost all cells apart from the very low category, of which it identified two-thirds of the cells (10 out of 15). In comparison, the suboptimal tools Arriba and scFusion only detected 2 and 3 cells of the very low category, respectively, whereas STAR-Fusion, FusionInspector, and FusionCatcher failed to detect cells in this category. Taken together, Anchored-fusion demonstrates ultra-high sensitivity compared to other tools in detecting fusion genes, especially for cells with low levels of supporting evidence. The high sensitivity of Anchored-fusion helps facilitate downstream analysis such as analyzing intratumor heterogeneity.
Anchored-fusion accurately detected the NSD2-IGH fusion gene in patients with MM
To further demonstrate the application of Anchored-fusion to patient stratification in clinical scRNA-seq data, we analyzed an MM scRNA-seq dataset from Jang et al.,25 which comprises 597 individual cells from 15 patients with MM. Our analysis focused on the NSD2-IGH fusion gene, which is caused by chromosome 4 and 14 translocations, written as t(4; 14). Previous studies have indicated that this fusion event can lead to NSD2 overexpression in 15%–20% of patients with MM, and these patients were reported to have lower survival rates and to exhibit poor response to cytotoxic chemotherapy.26 Using the FISH method, Jang et al. have revealed that three out of fifteen patients (including two patients with refractory MM, RRMM1 and RRMM2, and one patient with smoldering MM, SMM0) have t(4; 14) translocation. Their further gene expression analysis revealed that the cells of patients with t(4; 14) had high-risk characteristics.25
We successfully identified the NSD2-IGH fusion gene with Anchored-fusion in all of the three patients as reported in the original study. In total, we identified 99 single cells with the NSD2-IGH fusion gene in the MM dataset, which were all derived from the aforementioned three patients (Figure 6A). Specifically, we detected two distinct breakpoints in NSD2, located at the 464th and 1,091st nucleotides of the NSD2 transcript sequence (NM_133330.3), specifically at positions 1,902,353 and 1,905,943 on chromosome 4, respectively. The fusion gene with the breakpoint at 1,905,943 is derived from RRMM1 and RRMM2, and the one with the breakpoint at nucleotide 1,902,353 is derived from SMM0 (Figures 6B and S5).
Figure 6.

Fusion detection using different tools for scRNA-seq dataset from a clinical cohort of patients with multiple myeloma
(A) UMAP visualization of MM scRNA-seq data. The single cells identified by Anchored-fusion as having the NSD2-IGH fusion gene in each patient are highlighted.
(B) UMAP visualization of MM scRNA-seq data. The single cells identified by Anchored-fusion with different breakpoints of NSD2 are highlighted.
(C) UMAP visualization of MM scRNA-seq data. All cells containing NSD2-IGH fusion gene are circled. The highlighted cells indicate those detected by Anchored-fusion tool from three NSD2-IGH-positive patients. We identified the cells containing the NSD2-IGH fusion gene with its reference sequence.
(D) The percentage of cells positive for NSD2-IGH detected by Anchored-fusion among all NSD2-IGH-positive cells. In the lower part of the image are the indices of the number of the support evidences per 1 million reads in NSD2-IGH-positive cells detected by Anchored-fusion.
(E) Bar charts showing the total number of cells containing the NSD2-IGH fusion gene detected by Anchored-fusion and five other methods in the MM single-cell dataset, along with the number of cells detected in each patient.
Next, we plotted all single cells that were detected by Anchored-fusion as well as those that contained NSD2-IGH reads but were not detected by Anchored-fusion (Figures 6C and 6D). The results indicated that Anchored-fusion was able to detect 92.5% of the total NSD2-IGH-positive cells. Among all single cells from the three NSD2-IGH-positive patients, Anchored-fusion identified 60.3% of them with NSD2-IGH, accounting for 32.6%, 59.5%, and 77.6% in the three patients, respectively. In contrast, other methods have identified fewer cells with the fusion gene compared to Anchored-fusion (Figure 6E). Of note, Anchored-fusion identified many more fusion-containing cells in patient RRMM1 than other methods. This is consistent with Jang et al.'s analysis of its single-cell gene expression, which suggests a high risk.25 In contrast, scFusion and Arriba did not detect NSD2-IGH in this patient. Anchored-fusion identified the highest number of cells with this fusion gene in two other patients as well. In summary, these findings suggest that Anchored-fusion can serve as a valuable tool for single-cell fusion gene detection because of its high sensitivity.
Discussion
In this study, we present Anchored-fusion for sensitive detection of fusion genes. Anchored-fusion achieves highly sensitive fusion detection by anchoring a fusion gene of interest. Within the Anchored-fusion framework, we developed a deep learning module that effectively filters out artificially generated fusion reads using only sequence information. The evaluation of fusion gene discovery across multiple RNA-seq datasets demonstrates that Anchored-fusion effectively addresses the challenge of detecting fusion genes from genes with sequence homology. Additionally, the high sensitivity allows Anchored-fusion to detect fusion genes at lower expression levels or sequencing depths. In particular, we demonstrate the advantage of applying Anchored-fusion to analyze full-length-based scRNA-seq data.
Previous studies suggest that the number of fusion partners for each gene follows a power-law distribution, meaning that the majority of genes involved in fusion have only one or two fusion partners while a small number of genes have a large number of fusion partners. These genes that participate in a large number of fusion events are called central genes.27 For example, in non-small cell lung cancer, it has been discovered that the ALK gene has over 90 fusion partners.28 The prevalence of fusion events involving NTRK genes in solid tumors is as high as 1% and has been discovered in at least 34 types of cancer.6 Drugs targeting central genes have been proven effective against cancer driven by their fusion genes. As an example, TRK inhibitors such as entrectinib and larotrectinib have been shown to be effective in various NTRK-fusion-positive solid tumor diseases.7 Therefore, it is crucial to accurately and sensitively identify fusion events involving central fusion genes to guide clinical therapeutic strategies.
There is widespread sequence homology in the human genome, which inevitably affects the alignment of RNA-seq reads. Notably, dozens of known driver fusion genes contain homologous sequences in the genome. The supporting evidence for such events, which contain short homologous sequences, will be discarded by existing algorithms, resulting in a decrease in the number of detected supporting evidences. Additionally, existing algorithms often struggle to detect fusion genes when the sequencing depth or expression level is low. In contrast, Anchored-fusion retains all the sequencing reads that map or partially map to the anchored gene, thus gathering all relevant supporting evidence. This feature supports Anchored-fusion in discovering key fusion genes with high sensitivity.
The commonly used approach to filter artificial fusion fragments is to set a hard filtering cutoff of supporting reads. However, this approach may decrease the sensitivity of the tool. To address this issue, Anchored-fusion employed a deep learning framework to exclude false fusion genes using only sequence information. Artificial fusion sequences often result from accidental connections between PCR products and templates, followed by erroneous extensions. Unlike naturally occurring fusion genes, the fusion sites of artificial fusion products do not display characteristics of splice sites. Furthermore, certain sequences, such as polyA and polyT sequences, are more prone to mismatch errors. Therefore, considering both global and local information of the sequence helps distinguish between artificial and real fusion events.
Single-cell transcriptomics plays an important role in identifying cell types, analyzing cellular heterogeneity, and thereby revealing the dynamic gene expression and cell fate.29 In practice, cell- or cluster-type annotation is often performed based on known marker genes that are specifically expressed in those cells or clusters.30 Some fusion genes that function as driver genes in cancer serve as marker genes to distinguish between normal cells and cancer cells. For example, the BCR-ABL1 gene is the sole definitive marker to differentiate between normal hematopoietic stem cells and CML stem cells.31 However, previous methods have often struggled to detect fusion genes due to the low sequencing coverage in scRNA-seq.32 In contrast, Anchored-fusion detects fusion genes with high sensitivity in scRNA-seq data and adapts flexibly to situations where fusion breakpoints may vary.
Anchored-fusion needs users to provide the anchored gene of interest for fusion detection. Fusions not involving the anchored gene cannot be detected. This approach improves detection sensitivity by compromising the comprehensiveness of de novo fusion discovery. Compared to other methods that can discover multiple fusion genes at the same time, Anchored-fusion seems to perform worse in discovering new fusion genes. However, fusion genes involving the central gene are more likely to act as driver events in cancer development, and the high sensitivity of Anchored-fusion for such genes makes it easier to find these more important fusion genes. Additionally, in the clinical setting, it is more cost effective to use PCR to specifically detect target fusion genes when the fusion genes and fusion sites are known, as opposed to conducting RNA-seq. Therefore, we recommend using Anchored-fusion for cases where the other fusion partner is unknown or the fusion site is unknown. Alternatively, it can be used to detect fusion genes in RNA-seq data obtained before.
We anticipate that Anchored-fusion can be a useful tool to analyze bulk RNA-seq and scRNA-seq data from clinical samples from patients with cancer for cost-effective and efficient detection. Given the high sensitivity of Anchored-fusion, it can help to evaluate whether the fusion genes identified by different methods are real or not. Haas et al. pursued an approach called “wisdom of crowds,” which took the fusion genes found by over different methods as true fusions.9 We believe that Anchored-fusion can be regarded as one of the methods and that its high sensitivity could potentially aid in the verification of fusion genes that are challenging to detect. Additionally, the HVLD model may help distinguish true fusion genes from false positives. Moreover, we may reevaluate the frequency of different cancer driver fusion genes in public databases and accurately assess the correlation between fusion gene expression levels and clinical phenotypes with extensive application of Anchored-fusion to diverse tumor types and various cases of cancer.
Limitations of study
Anchored-fusion as implemented requires users to provide the reference sequence of their target gene. As a result, fusion genes that do not include the target gene will not be detected. Additionally, Anchored-fusion can only detect fusion genes based on paired-end RNA-seq data and cannot be used for single-end RNA-seq data or whole-genome or whole-exome sequencing data.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Cancer cell lines bRNA-seq for K562, TE441T, NALM6, NCI-H660, NCIH3122 and KM12 | Ghandi et al.33 | SRA:SRP186687 |
| K562 cell lines scRNA-seq | Gupta et al.34 | SRA:SRP291312 |
| Multiple myeloma patients scRNA-seq | Jang et al.25 | SRA:SRP158590 |
| True fusion genes | Aaltonen et al.35 | Synapse:syn10003873 |
| Software and algorithms | ||
| Anchored-fusion | This paper | https://doi.org/10.5281/zenodo.10677267 or https://github.com/ShenLab-Genomics/Anchored-Fusion |
| scFusion | Jin et al.12 | https://github.com/XiDsLab/scFusion |
| STAR-Fusion v1.12.0 | Haas et al.9 | https://github.com/STAR-Fusion |
| FusionInspector v2.8.0 | Haas et al.10 | https://github.com/FusionInspector/FusionInspector |
| FusionCatcher v1.30 | Nicorici et al.8 | https://github.com/ndaniel/fusioncatcher |
| Arriba v2.4.0 | Uhrig et al.11 | https://github.com/suhrig/arriba |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Ning Shen (shenningzju@zju.edu.cn).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
All original code has been deposited at GitHub and is publicly available as of the date of publication. URLs and DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Method details
The framework of Anchored-fusion
To detect fusion events involving the anchored gene X with high sensitivity, Anchored-fusion first maps all paired-end reads to the reference sequence of the anchored gene X. All reads aligned to gene X are collected, including those aligned to other copies of X or sequences have microhomology with X. This approach maximizes the retention of reads that are potentially transcribed from gene X. Next, it selects paired-end reads in which only one read can be mapped to the anchored gene X and aligns the remaining unmapped read to the entire genome. The set of genes they align to is referred to as a potential fusion partner gene set, called Y. Anchored-fusion removes genes with homologous regions to gene X from the gene set Y, as those genes may be erroneous template matched during annealing or incorrectly aligned during mapping. Due to its superior performance in terms of RNA-seq mapping accuracy, transcriptome coverage, and efficiency, we have chosen BWA-MEM as the alignment tool.33
Next, Anchored-fusion utilizes the BLAT tool36 to perform local alignment on candidate chimeric reads to accurately predict the precise location of gene fusion. Candidate fusion genes with uncertain fusion positions are filtered out. The process of searching for fusion genes considers paired-end reads spanning across fusion sites (referred to as spanning reads) and single reads containing fusion sites (referred to as split reads) as supporting evidences for the fusion gene. Candidates with a significantly imbalanced number of spanning and split reads will be filtered out. Finally, a deep neural network called HVLD is used to predict the probability that each candidate fusion gene is naturally occurring, and genes with a low probability (less than 0.1) will be filtered out. We train a decision model to learn the distribution of the artificial fusion genes of the current dataset for each dataset inputted by users. Given almost all of the chimeric reads obtained from the inputted dataset are artificial, we regard all of them as false fusion and process them into negative samples for training and testing the HVLD model. By comparing these artificial samples and real fusion samples, which are downloaded from PCAWG, our HVLD model can learn the distribution difference between the real fusions and false fusion of the current dataset. If the dataset provided by the user is too small, we recommend using the default model parameter provided on our GitHub.
HVLD model inference
We have designed a deep neural network called HVLD with a hierarchical structure from local to global, based on 1D convolution and multi-head self-attention,21 to learn to distinguish between artificial and natural fusion sequences. Additionally, we employed the technique of self-distillation learning22 to enhance the feature extraction capability of the shallow network. First, the fusion sequences containing fusion breakpoints are encoded into sequence matrices using the one-hot encoding method. Then, these matrices pass through the embedding module, local information extraction module, and global information extraction module sequentially. Except for the embedding module, each module outputs the sequence-splitting result through a classifier following it. Empirically, we choose the output of the last module serving as the final classification probability for the sequence.
-
(1)
Embedding block
The encoding layers encode the sequence represented by ‘A', ‘T', ‘G', and ‘C', as well as the splitting breakpoints ‘H', into a sequence matrix using one-hot encoding. Then, it is put into a fully connected layer (FC layer). The FC layers expand the feature dimension of each unit in the sequence from 5 to 256 dimensions.
-
(2)
Local sequence information extraction blocks
The local sequence information extraction block consists of two 1D convolutional blocks, each has a kernel of size 3. These convolutional operations are performed to extract local features from the sequence. Then, we use a max pooling layer to reduce redundancy and highlight significant features. Finally, the extracted local information of the sequence is passed to the next block. At the same time, the block’s classifier outputs the sequence classification probability based on the current block’s output.
-
(3)
Global information extraction block
The global information of the sequence is integrated with the Transformer encoder,21 which utilizes a multi-head attention mechanism to capture the global dependencies within the sequence. Based on the relationships between the current unit and other units including itself, the multi-head attention layer integrates information from all units to generate new features for the current unit. This block not only receives the local information extracted from the previous module but also integrates the local information based on their relevance. By doing so, it effectively captures the global information of the sequence, enabling more accurate sequence classification. The global information obtained from this block is also used to generate sequence classification probability through the classifier. However, unlike previous modules, this global information serves as the final output of the whole model.
-
(4)
Self-distill learning
Traditional distillation learning involves training a complex model as a teacher model and transferring knowledge from it to a relatively simpler model, known as the student model. The goal is to make the student model’s output as close as possible to that of the teacher model. Through distillation learning, the complexity of the student model is reduced while maintaining its performance comparable to the complex model.37 In self-distillation learning, both the deep and shallow networks are part of the same model. The deep network acts as the teacher model, while the shallow network serves as the student model. By making the output of the shallow network as close as possible to the output of the deep network, the knowledge learned by the deep network is transferred to the shallow network. Self-distillation learning can reduce the interference of noise on the shallow neural network, enhance the deep network’s ability to extract discriminative features and improve the model’s generalization capability. As a result, it leads to better predictive performance.22
Self-distillation learning employs two types of labels to train the shallow neural network: the ground-truth labels of the sequence itself (hard labels) and the output distributions of the classifier in the deepest layer of the neural network (soft labels), which in our work specifically refers to the output distributions of the Global information extraction block. The loss between the hard labels and all classifiers is calculated with cross-entropy loss (CE loss), and the loss between the soft labels and the shallow classifier is computed with Kullback-Leibler divergence loss (KL loss). Therefore, the loss function of the HVLD model can be represented as follows:
| (Equation 1) |
Where represents the output of the -th classifier, is the number of classifiers, and is the sequence label. And is the hyperparameter used to balance the two types of losses.
Data for HVLD generate process
We utilized six Cancer Cell Line Encyclopedia (CCLE) cancer cell line bRNA-seq datasets, namely K562, TE441T, NALM6, NCI-H660, NCIH3122, and KM12.38 These cell lines have been confirmed by CCLE to harbor the following fusion genes: BCR-ABL1,2 CIC-DUX4,39 DUX4-IGH,40 TMPRSS2-ERG,41 EML4-ALK,34 TPM3-NTRK1.35 We also collected two scRNA-seq datasets, including one K562 cell line with 350 cells42 and a clinical multiple myeloma (MM) dataset, including 15 patients and 597 cells.25
We used the subsequences containing fusion breakpoints from real fusion genes and technical artifacts as positive and negative training samples, respectively. The specific process of obtaining training and testing data is shown in Figure S4. For positive samples: We downloaded the 3540 fusion genes from Pan-Cancer Analysis of Whole Genomes (PCAWG) for positive samples, which contain positions of partner gene breakpoints. These fusion genes can be taken as the true golden standard because artificial fusion genes have been strictly excluded from this dataset.43 For negative samples: We regarded all of the chimeric reads found in the RNA-seq dataset provided by the user as negative samples. Because in sequencing data, there are ten thousand times more chimeric reads caused by artificial than ones generated from fusion genes.12 To be specific, first, we used BWA to map all of the RNA-seq reads in the user’s dataset to the whole genome, and those aligned to two different genes were considered candidate chimeric reads. Secondly, we aligned the candidates to the whole genome again with BLAT to remove the chimeric reads that were aligned incorrectly in the first step. Specifically, the reads generated from one gene but mistakenly mapped to two genes with BWA. Thirdly, reads that were aligned to homologous genes were also filtered out. Finally, we calculated the fusion breakpoint of these chimeric reads from their alignment position. Based on the breakpoint positions and the genome annotation, we inferred the input sequences of positive and negative samples for training and testing. The input sequences were combined by two subsequences of each partner gene. The subsequence was 100 base pairs long, starting from or ending at the fusion breakpoint, depending on whether the gene it derived from was located at the 5′ end or the 3′ end of the fusion gene. To explicitly include information about the fusion breakpoints, we inserted a fusion point marker 'H' between two subsequences of partner genes, resulting in a total length of 201 bp for the input sequence.
To avoid potential harmful impacts, all negative training samples containing anchored gene X were removed. While building the positive training and testing sets, we included all positive samples from the PCAWG fusion gene dataset. To maintain a balanced number of positive and negative samples, we randomly selected 3,540 chimeric reads in all from the negative samples for training and testing. Randomly selecting negative samples and using the entire set for training showed no significant differences in performance. (Figure S6). We randomly divided the total samples into a training-validation set, which comprised 70% of the data, and a testing set, which comprised 30% of the data. The training-validation set was used for model building and hyperparameter optimization, while the testing set was used for model evaluation. Then, we divided the training-validation set samples randomly into five equal parts and used each part as the validation set while using the remaining parts as training sets alternately. We saved the best parameters of the model when it achieved the best AUC score among the five validation sets. We repeated this process ten times, taking the mean of the results as the final outcome.
Simulation setup
We used six fusion genes BCR-ABL1,2 CIC-DUX4,39 DUX4-IGH,40 TMPRSS2-ERG41, EML4-ALK,34 TPM3-NTRK135 for simulation. We downloaded the transcription sequence reference files of these genes from NCBI (https://www.ncbi.nlm.nih.gov/) and aligned the RNA-seq reads from cancer cell lines K562, TE441T, NALM6, NCI-H660, NCIH3122 and KM1238 to these references. These cell lines have been confirmed by CCLE to harbor the above fusion genes. Through this approach, we identified the fusion breakpoints on the transcript sequences for these fusion genes. Next, we concatenated half of the reference sequences involved in the fusion as fusion partners to construct the transcriptional reference sequence for the fusion gene. We used wgsim23 to simulate the RNA-seq of these fusion genes from their transcription reference and set the length of short reads as 101bp. For each gene, we set seven levels of the simulated reads count, which range from 2 to 128. The number of simulated reads for a transcript gene can be obtained by multiplying the reference length by the simulated level and dividing it by the length of the short reads. It can be represented as follows:
| (Equation 2) |
The simulated level can be regarded as the base coverage of the fusion genes. Finally, we combined these simulated reads with the RNA-seq samples from GM12878 as the test data.
Brief description of competing methods
We used STAR-Fusion (v1.12.0), FusionInspector (v2.8.0), FusionCatcher (v1.30), and Arriba (2.4.0) as bRNA-seq competing methods. Together with scFusion, these methods formed the scRNA competition method.
STAR-Fusion bases on the STAR aligner to map the pair-end RNA-seq reads to the whole genome. STAR44 is a fast method that can align each part of the chimeric sequences to the appropriate position in the whole genome using sequential maximum mappable seed search. Those spanning reads and split reads discovered by STAR are inputted into STAR-Fusion as support evidences. Subsequently, STAR-Fusion9 utilizes a series of complex filters to discard fusion genes with homologous partners or with low supporting evidences. FusionInspector10 is used to evaluate a specified set of candidate fusions. The STAR-Fusion method uses the FusionInspector tool to further assess the candidate fusion genes it outputs. To evaluate the quality of the input fusion genes, FusionInspector assigns them to previously known clusters according to their expression levels and sequence features. These clusters are composed of known true fusion genes and false ones, which are clustered based on their expression levels and sequence features. FusionInspector describes the characteristics of the input fusion genes with the mutual features of the assigned clusters, such as quality, oncogenicity, and other relevant information. Arriba11 also utilizes the alignment results from STAR and applies expression filtering and homology filtering. Additionally, Arriba provides a blacklist of recurrent false fusion genes and a whitelist of known true fusions, which are used to filter out false fusions or rescue true fusions respectively. FusionCatcher8 uses four aligners, Bowtie, BLAT, STAR, and Bowtie2, to confirm fusion genes simultaneously. FusionCatcher allows reads to be aligned to intronic regions to discover fusion genes containing intronic sequences, but this approach also leads to higher false positive rates. scFusion is a fusion gene detection algorithm specifically designed for single-cell data. It aims to improve the precision of fusion gene detection. scFusion utilizes zero-inflated negative binomial (ZINB) distributions to calculate the expression distribution range of false fusions in single-cell sequencing. True fusion genes always exhibit higher expression levels compared the false ones, which allows them to be identified based on this distribution. In addition, scFusion12 also employs deep learning-based methods to filter out false fusions. We have provided the versions and download links for these methods in the STAR Methods: key resources table.
BRNA-seq and scRNA-seq analysis
We used STAR-Fusion (v1.12.0), FusionInspector (v2.8.0), FusionCatcher (v1.30), and Arriba (2.4.0) for bRNA-seq. For scRNA-seq, we added scFusion in the test, which can only be used to detect scRNA-seq data and cannot be applied to bRNA-seq data. All of them were run on the default parameters. Except for scFusion, all tools, including Anchored-fusion, searched for fusion genes in each cell’s RNA-seq data in the same way as in bRNA-seq data. The results from all cells were then aggregated to obtain the final result.
The parameter details of the bi-LSTM
We used the same model and parameters as scFusion.12 Specifically, the sequences were represented by five different 5-dimensional feature vectors. Next, they were passed on to the three sequence-to-sequence bi-LSTM layers (with 32,64 and 128 bi-LSTM units, respectively) and further to a sequence-to-one bi-LSTM layer with 256 bi-LSTM units. Finally, the outputs of the deepest bi-LSTM layer were fed to two fully connected layers followed by a softmax layer to produce the softmax probabilities of the read being classified to chimeric artifacts. The learning rate of it was 0.0001.
The parameter details of encoding the SVM
We referred to the method proposed by Bari et al.45 to convert the input DNA sequence into a feature tensor. Specifically, we represented the four nucleotides, ‘A', ‘T', ‘G', and ‘C', as vectors [1,0,0,0], [0,1,0,0], [0,0,1,0], and [0,0,0,1], respectively. The fusion site was represented as [1,1,1,1]. Additionally, we calculated the nucleotide density feature based on the number and distance of matching nucleotides to the fusion site as follows:
| (Equation 3) |
Where is the basic group of the sequence and represents the nucleotide density of basic group . is the length between and breakpoint. ,
Quantification and statistical analysis
In the process of evaluating the performance of HDVL and other models in distinguishing between naturally occurring fusion fragments and artificially created fusion fragments, we selected accuracy (ACC), area under the curve (AUC), precision-recall area under the curve (PRAUC), precision, and recall as metrics. We employed 5-fold cross-validation to obtain the optimal parameters and evaluated the model’s performance on the test set using these parameters. Specifically, we randomly divided the training dataset into five subsets. Each subset was used as a validation set in turns, while the remaining four subsets were used for training the model. The five subsets were rotated as validation sets, and the model parameters achieving the best AUC value on the validation set throughout the entire process were retained. This process was repeated 10 times, and the average value was taken as the final performance metric, with results showing a variance of less than 0.01.
Acknowledgments
We thank Tianyun Zhang, Dandan Hu, and the members of Shen Lab and Wang Lab for helpful discussions on the project. We are also thankful for the technical support by the Core Facilities of Liangzhu Laboratory. This work is supported by the National Key R&D Program of China (2022ZD0160703), the National Natural Science Foundation of China (grant no. 62202422), the Zhejiang Province Science and Technology Plan Project (2022C03134), the Liangzhu Laboratory, and the Starting Fund from Zhejiang University.
Author contributions
N.S. conceived the project. N.S. and S.C.C. carried out the proof-of-concept study. X.Y. and C.Z. developed the deep learning model and conducted the computational experiments. Z.S. provided additional biological insights into the experimental results. N.S. and H.W. supervised the project. X.Y., N.S., H.W., C.Z., S.C.C., and J.B. drafted, revised, and edited the manuscript. All authors read and approved the final manuscript.
Declaration of interests
S.C.C. has stock options from ROME Therapeutics. None of this work has been supported by that company. The authors have submitted a patent application for the method.
Published: March 18, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2024.100733.
Contributor Information
Haishuai Wang, Email: haishuai.wang@zju.edu.cn.
Ning Shen, Email: shenningzju@zju.edu.cn.
Supplemental information
References
- 1.Rowley J.D. A New Consistent Chromosomal Abnormality in Chronic Myelogenous Leukaemia identified by Quinacrine Fluorescence and Giemsa Staining. Nature. 1973;243:290–293. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- 2.Daley G.Q., Van Etten R.A., Baltimore D. Induction of Chronic Myelogenous Leukemia in Mice by the P210bcr/abl Gene of the Philadelphia Chromosome. Science. 1990;247:824–830. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- 3.Jabbour E., Kantarjian H. Chronic myeloid leukemia: 2020 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2020;95:691–709. doi: 10.1002/ajh.25792. [DOI] [PubMed] [Google Scholar]
- 4.Druker B.J. Advances in Cancer Research. Elsevier; 2004. Imatinib as a Paradigm of Targeted Therapies; pp. 1–30. [DOI] [PubMed] [Google Scholar]
- 5.Song C., Chen H. Predictive significance of TMRPSS2-ERG fusion in prostate cancer: a meta-analysis. Cancer Cell Int. 2018;18:177. doi: 10.1186/s12935-018-0672-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manea C.A., Badiu D.C., Ploscaru I.C., Zgura A., Bacinschi X., Smarandache C.G., Serban D., Popescu C.G., Grigorean V.T., Botnarciuc V. A review of NTRK fusions in cancer. Ann. Med. Surg. 2022;79 doi: 10.1016/j.amsu.2022.103893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drilon A., Laetsch T.W., Kummar S., DuBois S.G., Lassen U.N., Demetri G.D., Nathenson M., Doebele R.C., Farago A.F., Pappo A.S., et al. Efficacy of Larotrectinib in TRK Fusion–Positive Cancers in Adults and Children. N. Engl. J. Med. 2018;378:731–739. doi: 10.1056/NEJMoa1714448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicorici D., Satalan M., Edgren H., Kangaspeska S., Murumagi A., Kallioniemi O., Virtanen S., Kilkku O. Bioinformatics; 2014. FusionCatcher - a Tool for Finding Somatic Fusion Genes in Paired-End RNA-Sequencing Data. [DOI] [Google Scholar]
- 9.Haas B.J., Dobin A., Li B., Stransky N., Pochet N., Regev A. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019;20:213. doi: 10.1186/s13059-019-1842-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haas B.J., Dobin A., Ghandi M., Van Arsdale A., Tickle T., Robinson J.T., Gillani R., Kasif S., Regev A. Targeted in silico characterization of fusion transcripts in tumor and normal tissues via FusionInspector. Cell Rep. Methods. 2023;3 doi: 10.1016/j.crmeth.2023.100467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uhrig S., Ellermann J., Walther T., Burkhardt P., Fröhlich M., Hutter B., Toprak U.H., Neumann O., Stenzinger A., Scholl C., et al. Accurate and efficient detection of gene fusions from RNA sequencing data. Genome Res. 2021;31:448–460. doi: 10.1101/gr.257246.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin Z., Huang W., Shen N., Li J., Wang X., Dong J., Park P.J., Xi R. Single-cell gene fusion detection by scFusion. Nat. Commun. 2022;13:1084. doi: 10.1038/s41467-022-28661-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui C., Shu W., Li P. Fluorescence In situ Hybridization: Cell-Based Genetic Diagnostic and Research Applications. Front. Cell Dev. Biol. 2016;4 doi: 10.3389/fcell.2016.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guseva N.V., Jaber O., Tanas M.R., Stence A.A., Sompallae R., Schade J., Fillman A.N., Miller B.J., Bossler A.D., Ma D. Anchored multiplex PCR for targeted next-generation sequencing reveals recurrent and novel USP6 fusions and upregulation of USP6 expression in aneurysmal bone cyst. Genes. Chromosomes Cancer. 2017;56:266–277. doi: 10.1002/gcc.22432. [DOI] [PubMed] [Google Scholar]
- 15.Kerbs P., Vosberg S., Krebs S., Graf A., Blum H., Swoboda A., Batcha A.M.N., Mansmann U., Metzler D., Heckman C.A., et al. Fusion gene detection by RNA-sequencing complements diagnostics of acute myeloid leukemia and identifies recurring NRIP1-MIR99AHG rearrangements. Haematologica. 2022;107:100–111. doi: 10.3324/haematol.2021.278436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng Z., Yuan C., Zellmer L., Liu S., Xu N., Liao D.J. Hypothesis: Artifacts, Including Spurious Chimeric RNAs with a Short Homologous Sequence, Caused by Consecutive Reverse Transcriptions and Endogenous Random Primers. J. Cancer. 2015;6:555–567. doi: 10.7150/jca.11997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torres-García W., Zheng S., Sivachenko A., Vegesna R., Wang Q., Yao R., Berger M.F., Weinstein J.N., Getz G., Verhaak R.G.W. PRADA: pipeline for RNA sequencing data analysis. Bioinformatics. 2014;30:2224–2226. doi: 10.1093/bioinformatics/btu169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okimoto R.A., Wu W., Nanjo S., Olivas V., Lin Y.K., Ponce R.K., Oyama R., Kondo T., Bivona T.G. CIC-DUX4 oncoprotein drives sarcoma metastasis and tumorigenesis via distinct regulatory programs. J. Clin. Invest. 2019;129:3401–3406. doi: 10.1172/JCI126366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Satomi K., Ohno M., Kubo T., Honda-Kitahara M., Matsushita Y., Ichimura K., Narita Y., Ichikawa H., Yoshida A. Central nervous system sarcoma with ATXN1::DUX4 fusion expands the concept of CIC-rearranged sarcoma. Genes Chromosomes Cancer. 2022;61:683–688. doi: 10.1002/gcc.23080. [DOI] [PubMed] [Google Scholar]
- 20.Siegele B.J., Stemmer-Rachamimov A.O., Lilljebjorn H., Fioretos T., Winters A.C., Dal Cin P., Treece A., Gaskell A., Nardi V. N-terminus DUX4-immunohistochemistry is a reliable methodology for the diagnosis of DUX4-fused B-lymphoblastic leukemia/lymphoma (N-terminus DUX4 IHC for DUX4-fused B-ALL) Genes Chromosomes Cancer. 2022;61:449–458. doi: 10.1002/gcc.23033. [DOI] [PubMed] [Google Scholar]
- 21.Vaswani A., Shazeer N., Parmar N., Uszkoreit J., Jones L., Gomez A.N., Kaiser Ł., Polosukhin I. Proceedings of the 31st International Conference on Neural Info rmation Processing Systems NIPS’17. Curran Associates Inc.); 2017. Attention is all you need; pp. 6000–6010. [Google Scholar]
- 22.Zhang L., Song J., Gao A., Chen J., Bao C., Ma K. 2019 IEEE/CVF International Conference on Computer Vision. ICCV); 2019. Be Your Own Teacher: Improve the Performance of Convolutional Neural Networks via Self Distillation; pp. 3712–3721. [DOI] [Google Scholar]
- 23.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian L., Shao Y., Nance S., Dang J., Xu B., Ma X., Li Y., Ju B., Dong L., Newman S., et al. Long-read sequencing unveils IGH-DUX4 translocation into the silenced IGH allele in B-cell acute lymphoblastic leukemia. Nat. Commun. 2019;10:2789. doi: 10.1038/s41467-019-10637-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jang J.S., Li Y., Mitra A.K., Bi L., Abyzov A., van Wijnen A.J., Baughn L.B., Van Ness B., Rajkumar V., Kumar S., Jen J. Molecular signatures of multiple myeloma progression through single cell RNA-Seq. Blood Cancer J. 2019;9:2–10. doi: 10.1038/s41408-018-0160-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lhoumaud P., Badri S., Rodriguez-Hernaez J., Sakellaropoulos T., Sethia G., Kloetgen A., Cornwell M., Bhattacharyya S., Ay F., Bonneau R., et al. NSD2 overexpression drives clustered chromatin and transcriptional changes in a subset of insulated domains. Nat. Commun. 2019;10:4843. doi: 10.1038/s41467-019-12811-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Latysheva N.S., Oates M.E., Maddox L., Flock T., Gough J., Buljan M., Weatheritt R.J., Babu M.M. Molecular Principles of Gene Fusion Mediated Rewiring of Protein Interaction Networks in Cancer. Mol. Cell. 2016;63:579–592. doi: 10.1016/j.molcel.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ou S.-H.I., Zhu V.W., Nagasaka M. Catalog of 5’ Fusion Partners in ALK-positive NSCLC Circa 2020. JTO Clin. Res. Rep. 2020;1 doi: 10.1016/j.jtocrr.2020.100015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adil A., Kumar V., Jan A.T., Asger M. Single-Cell Transcriptomics: Current Methods and Challenges in Data Acquisition and Analysis. Front. Neurosci. 2021;15 doi: 10.3389/fnins.2021.591122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clarke Z.A., Andrews T.S., Atif J., Pouyabahar D., Innes B.T., MacParland S.A., Bader G.D. Tutorial: guidelines for annotating single-cell transcriptomic maps using automated and manual methods. Nat. Protoc. 2021;16:2749–2764. doi: 10.1038/s41596-021-00534-0. [DOI] [PubMed] [Google Scholar]
- 31.Ma J., Pettit N., Talburt J., Wang S., Weissman S.M., Yang M.Q. Integrating Single-Cell Transcriptome and Network Analysis to Characterize the Therapeutic Response of Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2022;23 doi: 10.3390/ijms232214335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giustacchini A., Thongjuea S., Barkas N., Woll P.S., Povinelli B.J., Booth C.A.G., Sopp P., Norfo R., Rodriguez-Meira A., Ashley N., et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat. Med. 2017;23:692–702. doi: 10.1038/nm.4336. [DOI] [PubMed] [Google Scholar]
- 33.Ghandi M., Huang F.W., Jané-Valbuena J., Kryukov G.V., Lo C.C., McDonald E.R., Barretina J., Gelfand E.T., Bielski C.M., Li H., et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569:503–508. doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gupta K., Lalit M., Biswas A., Sanada C.D., Greene C., Hukari K., Maulik U., Bandyopadhyay S., Ramalingam N., Ahuja G., et al. Modeling expression ranks for noise-tolerant differential expression analysis of scRNA-seq data. Genome Res. 2021;31:689–697. doi: 10.1101/gr.267070.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aaltonen L.A., Abascal F., Abeshouse A., Aburatani H., Adams D.J., Agrawal N., Ahn K.S., Ahn S.-M., Aikata H., Akbani R., et al. Pan-cancer analysis of whole genomes. Nature. 2020;578:82–93. doi: 10.1038/s41586-020-1969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawamura-Saito M., Yamazaki Y., Kaneko K., Kawaguchi N., Kanda H., Mukai H., Gotoh T., Motoi T., Fukayama M., Aburatani H., et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum. Mol. Genet. 2006;15:2125–2137. doi: 10.1093/hmg/ddl136. [DOI] [PubMed] [Google Scholar]
- 37.Lilljebjörn H., Henningsson R., Hyrenius-Wittsten A., Olsson L., Orsmark-Pietras C., von Palffy S., Askmyr M., Rissler M., Schrappe M., Cario G., et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat. Commun. 2016;7 doi: 10.1038/ncomms11790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Musich R., Cadle-Davidson L., Osier M.V. Comparison of Short-Read Sequence Aligners Indicates Strengths and Weaknesses for Biologists to Consider. Front. Plant Sci. 2021;12 doi: 10.3389/fpls.2021.657240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tomlins S.A., Rhodes D.R., Perner S., Dhanasekaran S.M., Mehra R., Sun X.-W., Varambally S., Cao X., Tchinda J., Kuefer R., et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 40.Soda M., Choi Y.L., Enomoto M., Takada S., Yamashita Y., Ishikawa S., Fujiwara S.i., Watanabe H., Kurashina K., Hatanaka H., et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 41.Ardini E., Bosotti R., Borgia A.L., De Ponti C., Somaschini A., Cammarota R., Amboldi N., Raddrizzani L., Milani A., Magnaghi P., et al. The TPM3-NTRK1 rearrangement is a recurring event in colorectal carcinoma and is associated with tumor sensitivity to TRKA kinase inhibition. Mol. Oncol. 2014;8:1495–1507. doi: 10.1016/j.molonc.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kent W.J. BLAT—The BLAST-Like Alignment Tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hinton G., Vinyals O., Dean J. Distilling the Knowledge in a Neural Network. arXiv. 2015 doi: 10.48550/arXiv.1503.02531. Preprint at. [DOI] [Google Scholar]
- 44.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bari A.T.M.G., Reaz M.R., Choi H.-J., Jeong B.-S. In: Database Systems for Advanced Applications Lecture Notes in Computer Science. Hong B., Meng X., Chen L., Winiwarter W., Song W., editors. Springer; 2013. DNA Encoding for Splice Site Prediction in Large DNA Sequence; pp. 46–58. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
All original code has been deposited at GitHub and is publicly available as of the date of publication. URLs and DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.