

Neoantigen-based cancer vaccines have shown limited clinical efficacy. The authors find a vaccine targeting a single neoantigen can be effective when there is minimal disease burden, but multiple neoantigens and checkpoint blockade are required in the metastatic setting.
Abstract
Tumor neoantigens (nAg) represent a promising target for cancer immunotherapy. The identification of nAgs that can generate T-cell responses and have therapeutic activity has been challenging. Here, we sought to unravel the features of nAgs required to induce tumor rejection. We selected clinically validated Great Ape–derived adenoviral vectors (GAd) as a nAg delivery system for differing numbers and combinations of nAgs. We assessed their immunogenicity and efficacy in murine models of low to high disease burden, comparing multi-epitope versus mono-epitope vaccines. We demonstrated that the breadth of immune response is critical for vaccine efficacy and having multiple immunogenic nAgs encoded in a single vaccine improves efficacy. The contribution of each single neoantigen was examined, leading to the identification of 2 nAgs able to induce CD8+ T cell–mediated tumor rejection. They were both active as individual nAgs in a setting of prophylactic vaccination, although to different extents. However, the efficacy of these single nAgs was lost in a setting of therapeutic vaccination in tumor-bearing mice. The presence of CD4+ T-cell help restored the efficacy for only the most expressed of the two nAgs, demonstrating a key role for CD4+ T cells in sustaining CD8+ T-cell responses and the necessity of an efficient recognition of the targeted epitopes on cancer cells by CD8+ T cells for an effective antitumor response. This study provides insight into understanding the determinants of nAgs relevant for effective treatment and highlights features that could contribute to more effective antitumor vaccines.
Introduction
Tumor neoantigens (nAg) have emerged as promising targets for cancer immunotherapy. They are mutated peptides without preexisting central tolerance, therefore, recognized by the immune system as foreign and able to elicit tumor-specific immune responses. Neoantigen-based cancer vaccines have become of increasing interest in the immunotherapy field. Their goal is to induce effective antitumor immunity, ideally enhancing the magnitude, quality, and breadth of antigen-specific T-cell responses. Neoantigen-based vaccines can also induce epitope spreading of T-cell responses, with development of effective immune response specific for tumor antigens that were not targeted in the vaccine (1–3). Several clinical trials have tested vaccines targeting nAgs, providing compelling evidence for their safety, immunogenicity, and, in some cases, efficacy, in combination with checkpoint inhibitors (3–6).
To date, a number of nAg-based vaccines using different platforms are under clinical evaluation, including synthetic long peptides, dendritic cells, viral vectors, and nucleic acids (DNA and synthetic messenger RNA; ref. 7). Among them, genetic vaccines based on Adenoviruses (Ad) derived from non-human Great Apes–derived adenoviral vectors (GAd) have demonstrated a strong capability to elicit potent and effective T-cell responses in multiple studies in mice and humans, against infectious diseases and cancer (1, 8–12). A key aspect of the GAd platform is the ability of these Ads to encode large gene inserts so that it is possible to simultaneously target many nAgs, offering the unique opportunity to overcome the issues of tumor heterogeneity and the weakness of currently available epitope prediction methods (12). Neoantigens are largely patient-specific because only a small fraction of mutations are shared between patients. Therefore, their accurate prediction for each individual patient is a key factor for the development of a successful cancer vaccine. Current efforts are underway to improve the available algorithms used for epitope prediction. Indeed, only a minority of the tested neo-peptides elicits a T-cell response (13), suggesting that the key determinants of immunogenicity/effectiveness are not yet fully understood. Although CD8+ T cells are considered the major mediators of antitumor T-cell responses in vivo, several studies suggest that CD4+ T cells play a pivotal role during the priming phase of antigen-mediated T-cell activation (14–17). CD4+ T cells help CD8+ T cells to perform their cytotoxic effector functions and differentiate into long-lasting memory cells through a specific gene program that involves the downregulation of co-inhibitory receptors and the increase of motility and migration capacities (14). It is thus generally agreed that the inclusion of both CD8+ and CD4+ T cell–specific epitopes in vaccine design is required to maximize the therapeutic efficacy (18). In our previous work, we showed that a GAd vector encoding 31 neoantigens (GAd-31) selected from the murine CT26 colon carcinoma cell line could elicit potent T-cell antitumor immunity, with 6 out of the 31 encoded nAgs found to be immunogenic and to induce both CD8+ and CD4+ T-cell responses. The GAd-31 vaccine was effective in both prophylactic and therapeutic vaccination settings, as monotherapy and when combined with anti-PD1, respectively (12). In the present study, we generated different GAd nAg-based vaccines to determine the specific contribution of the 6 previously identified immunogenic nAgs to the vaccine efficacy and to investigate the impact of CD8+ and CD4+ T cell–mediated responses in different tumor burden settings. This study provides insights into the vaccine determinants relevant for effective antitumor responses, such as the importance of simultaneously targeting multiple nAgs and the impact of CD4+ T cells, which has the potential to improve the design and optimization of antitumor vaccines.
Materials and Methods
Mice
Six-week-old female Balb/c mice were purchased from Envigo. Daily animal care was performed by trained staff at Plaisant, Castel Romano. All in vivo experimental procedures were approved by the local animal ethics council of the Italian Ministry of Health and performed in accordance with national and international laws and policies (EEC Council Directive 86/609; Italian Legislative Decree 26/14). The ethical committee of the Italian Ministry of Health approved this research. The number of mice per experimental group was obtained by performing a power analysis.
Cell culture
CT26 cell line (Balb/c murine colon carcinoma) was purchased from the ATCC (2017). Cells were cultured in complete RPMI-1640 (Gibco) supplemented with 10% FBS (Gibco), 2 mmol/L l-glutamine (Gibco), 1% (v/v) penicillin/streptomycin (Gibco) and maintained at 37°C in 5% CO2. Cells were used on passage 7–9 after being tested for the absence of Mycoplasma contamination by PCR. Cells on passage 8 were sequenced by next-generation sequencing (NGS) to assess the presence of mutations previously described (12, 19).
Generation of #5 and #23 knockout CT26 cells
The genomic regions encoding nAg #5 (E2f8) and nAg #23 (Mtch1) were specifically targeted in CT26 cell line using CRISPR-Cas9 technology. Briefly, sgRNAs were specifically designed after PAM sequences (NGG) to include the region harboring the point mutations of epitope #5 and #23 and then cloned into a SpCas9–2A-Puro (PX459) vector (Addgene). The sequences of the 2 sgRNAs used to generate CT26#5 knockout (KO) are: sgRNA 1: 5′TCGGGCCCATCCTATGCCA 3′; sgRNA 2: 5′AACCGTCTGCCCCACCCAGC 3′. The sequences of the 2 sgRNAs used to generate CT26#23 KO are: sgRNA1: 5′ ATGACCCCGATGACACCCGGCGG 3′; sgRNA2: 5′ AGTTGGATCTCAGAATCCATAGG 3′. CT26 cells were transfected with Lipofectamine 2000 (Invitrogen) and PX459-sgRNAs plasmids (Addgene), according to the manufacturer's instructions, and selected with Puromycin (15 μg/mL; Gibco) for 3 days. Cas9 cut occurred 2–3 nucleotides after the PAM sites, as assessed by PCR, resulting in deletion of 74 nucleotides for #5 and 116 nucleotides for #23, a premature STOP codon and KO of the corresponding gene. Single-cell clones were screened for the deletions by PCR followed by genomic DNA sequencing. For #5 KO PCR, the following primers were used: 5′ GATGCAGTTGGAAGAGCAG 3′ (Forward); 5′ GCATGGTGCTAAGCATCGA 3′ (Reverse). For #23 KO PCR, the following primers were used: 5′ ATGGCTTCTGGGGCTCTATC 3′ (Forward); 5′ CTGAGTGACGTTTGCCTCTG 3′ (Reverse). Positive clones carrying homozygous deletion were selected for the subsequent in vivo experiments.
PCR for #5 and #23 epitope mutation analysis in CT26 tumors
To perform mutation analysis relative to the expression of #5 and #23 epitopes, CT26 tumors were collected for RNA extraction (RNeasy mini kit, Qiagen) following the manufacturing instruction. 1-μg of RNA was converted to cDNA (Superscript First-Strand, Invitrogen) and used for PCR (Phusion Hot Start II High Fidelity, Thermo Fisher Scientific). Primers used for E2f8 gene (encoding #5) are: Forward: 5′GATGCAGTTGGAAGAGCAG 3′; Reverse: 5′TCGATGCTTAGCACCATGC 3′. Mtch1 gene (encoding #23): Forward: 5′ ACAGTGGTGTGCTGAGTTC 3′: Reverse: 5′ TACTCCAGGGCAAAGCATG 3′. PCR products were purified using Wizard SV gel and PCR clean-up system (Promega) and analyzed by sequencing.
Production of adenoviral vectors
Adenoviral vectors were generated as previously described (12). Briefly, the coding sequences (CDS) for the transgenes encoded in Adenoviral vectors (the corresponding amino acidic sequences are listed in Supplementary Table S1) were synthesized as phosphorylated gBlock dsDNA fragments (IDT). HA tags were added at the N- and C terminus of each transgene. The CDS for all the constructs were generated by Gibson assembly (New England Biolabs) and cloned into the respective shuttle plasmid containing the CMV promoter with two TetO operator repeats and a BGH polyA. The expression cassettes were then transferred into pGAd plasmid, containing the E1/E3/E4 deleted in which the E4 is replaced with Ad5 E4 ORF6. The transgene cassettes were introduced in the E1 deletion locus of related pAdeno by homologous recombination in BJ5183 cells (Agilent). GAd vectors were then produced by transfection of adherent M9 cells (293 cells derivative) with Lipofectamine 2000 (Invitrogen) and amplification in suspension M9 cells. Vectors were then purified from infected cells by Vivapure Adenopack 20 RT (Sartorius). The titer of each vector was determined by qPCR and expressed as viral particles (vp) per mL.
In vivo tumor growth
For prophylactic experiments, a total of 2×105 CT26 or CT26#5 KO cells were subcutaneously injected into the right flank of 6-week-old female Balb/c mice, 14 days after immunization with 5×108 vp of each GAd vaccine. For experimental metastases, a total of 1×105 CT26 cells were injected intravenously into the tail vein, 3 days before vaccination with 5×108 vp of each GAd vaccine. On day 16, lungs were perfused with India Ink 15% (American Mastertech Scientific), harvested and fixed in Fekete's solution (Carlo Erba, 700 mL/L 100% ethanol, 32 mL/L 37% formalin, 40 mL/L glacial acetic acid, distilled water to 1L). Metastatic nodules on the surface of the lungs were counted using a dissecting microscope. For established tumor setting experiments, a total of 2×106 CT26 cells were injected subcutaneously After 5 days, tumor volume was measured with a digital caliper, applying the formula: 0.5 × length × width2, where the length is the longer dimension. Animals were then randomized on the basis of their tumor size (tumor size average per group 30–70 mm3) and treated (day 0) with 5×108 vp of each GAd vaccine and anti-PD1. Tumor growth was measured every 3 to 4 days and mice were euthanized as soon as signs of distress or a tumor volume above 2,000 mm3 was reached.
For second tumor challenge, tumor free mice were inoculated subcutaneously with a total of 2×106 CT26 or CT26 #5KO or CT26 #23KO cells, on day 40 after the first challenge. Tumor growth was measured every 3 to 4 days and mice were euthanized as soon as signs of distress or a tumor volume above 2,000 mm3 was reached.
In vivo treatments
Vaccines were administered via intramuscular injections in the quadriceps. A volume of 100 μL containing 5×108 vp was delivered (50 μL per side). For efficacy studies, anti-mouse PD1 (clone RMP114, BioXCell) was administered intraperitoneally, at a dosage of 200 μg twice a week, starting from day 0 until day 16.
Ex vivo immune analysis
IFNγ ELISpot assays were performed on cell suspension of spleens collected from naïve mice, 14 days after immunization with 5×108 vp of GAd vaccine. MSIP S4510 plates were coated with 10 μg/mL of anti-mouse IFNγ (U-CyTech) and incubated overnight at 4°C. After washing and blocking with RPMI medium (Gibco) and 10% FBS (Gibco), freshly isolated mouse splenocytes were plated in duplicate at two different cell densities (5×105 and 2.5×105 cells) and stimulated overnight with single 25-mer peptides (Tema Ricerca; matching the peptide sequences listed in Supplementary Table S1) or peptide pool at a final concentration of 1 μg/mL. The peptide diluent dimethyl sulfoxide (DMSO) and Concanavalin A (ConA, Sigma) were used as negative and positive controls, respectively. Plates were then incubated with biotinylated anti-mouse IFNγ (dilution: 1/100; U-CyTech), conjugated streptavidin–alkaline phosphatase (dilution, 1/2,500) and finally 5-bromo-4-chloro-3-indoyl-phosphate/nitro blue tetrazolium 1-Step solution (Thermo Fisher Scientific). An automated ELIspot assay video analysis system automated plate reader was used to analyze plates (Immuno Spot CTL). ELISpot data were expressed as IFNγ Spot Forming Colonies (SFC) per million splenocytes. ELISpot responses were considered positive if all the following conditions occurred: (i) IFNγ production present in ConA stimulated wells, (ii) the number of spots seen in positive wells was three times the number obtained in the negative control wells (DMSO), (iii) at least 30 specific spots/million splenocytes.
Cell isolation from tumor, spleen, lymph node, and blood for flow cytometry analysis of T cells
Tumors were collected on days 9 and 14 after immunization, dissociated and digested with the Miltenyi Tumor Dissociation kit, following the manufacturer's instructions. Tumor homogenates were then filtered through a 70-μm cell strainer to generate single-cell suspension, ready for staining with markers of interest. Blood was collected at different time point after immunization in heparin-containing tubes, incubated with ACK buffer (Gibco) 2 to 3 times for 10 minutes each, centrifuged at 1,500 rpm for 5 minutes and filtered through 70-μm cell strainer. Spleens were collected at days 9 and 14 after immunization. Splenocytes were isolated by manual pressing of the organ with a piston, followed by lysis of red blood cells by incubation with ACK buffer. Finally, cells were filtered through a 70-μm cell strainer, ready for the staining. Tumor-draining lymph nodes were collected at day 9 after immunization. Briefly, cells were isolated by tearing the tissue using fine forceps and incubating in HBSS + 2% FBS (Gibco) + Collagenase IV (0.4 mg/mL; Sigma) and DNase I (0.2 mg/mL; Sigma) for 30 minutes at 37°C with gentle mixing. After centrifugation at 1,500 × g, cell pellets were resuspended in FACS buffer, ready for staining. For dextramer staining: Isolated cells were incubated with 1 μg/106 cells of Fc block treatment (BD Pharmigen) then stained for the following surface markers: Dextramer#23-PE (Immudex, customized H-2-Kd dextramer); Live/Dead (Fixable Near-IR Dead Cell stain kit, Life Technologies; catalogue number L10119); CD45 PerCP-Cyanine 5.5 (clone 30-F11, eBioscience; catalogue number 45–0451–82); CD3 APC (clone 145–2C11, BD; catalogue number 553066); CD8 Brilliant Violet 510 (clone 53–6.7, BioLegend; catalogue number 100752). For Effector memory: CD8 PerCP; CD44 APC (clone IM7, BioLegend; catalogue number 103012); CD62 L FITC (clone MEL-14, BD Pharmigen; catalogue number 104406). For Exhausted (CD8+ PD1+ TIM3+ TCF1− GRZB−), stemlike (CD8+ PD1+ TIM3− TCF1+ GRZB−), Better effector (CD8+ PD1+ TIM3−, TCF1−, GRZB+): CD8 Brilliant Violet 510; PD1 Brilliant Violet 421 (clone 29F.1A12, BioLegend; catalogue number 329920); TIM3 PerCP-Cyanine 5.5 (clone B8.2C12, BioLegend; catalogue number 134012); Granzyme B FITC (clone GB11, BioLegend; catalogue number 515403); TCF1 APC (clone C63D9, Cell Signaling Technology; catalogue number 37636S). A FACS Canto was used for data acquisition and FlowJo (v.10) was used for the analysis.
Bulk RNA sequencing on tumor samples
Tumors were collected from responder (RES) and non-responder (NR) mice treated with GAd CD4+/CD8+ (Help vaccine) and from control untreated mice. RES mice were defined as animals showing at least 40% of tumor shrinkage (relative to the maximum peak of tumor volume) after treatment with vaccine and anti-PD1. Collected tumors were immediately flash-frozen in liquid nitrogen and stored at −80°C until RNA extraction performed with the RNeasy mini kit (Qiagen). Bulk RNA was sequenced as previously described (12). Briefly, RNA library construction and NGS of tumor samples was performed at Genomix4Life S.r.l (Salerno). RNA was fragmented and the sequencing library was prepared using Illumina TruSeq mRNA stranded kit. Sequencing was performed with a Hiseq2000 Genome Analyzer (Illumina) at target depth of 60 mln of paired-end reads. Quality control of sequenced reads was performed with FastQC 0.11.5 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Raw NGS reads were aligned on mm10 genome by using hisat2 software (20). Reads that aligned to more than one locus with the same mapping score were filtered using Samtools 0.1.19 (21). Reads mapped on each gene annotated in Refseq were counted with Rsubreads package (22). Reads counts were subsequently converted in transcripts per million values by using in house developed R scripts. Five and four biological replicates were used, respectively, for NR and RES groups.
Statistical analyses
Statistical significance was determined by GraphPad Prism (version 9) using the nonparametric, two-tailed Mann–Whitney U test or exact Fisher's test as stated in the figure legends.
Data availability
RNA sequencing (RNA-seq) data generated in this study have been deposited in Sequence Read Archive under the accession code PRJNA1073521 and PRJNA543001 (untreated tumors; ref. 12). All the other data of this study are available in the article and its Supplementary Files or upon request from the corresponding author.
Results
Generation of different GAd vaccine constructs to dissect nAgs contribution: in vivo immunogenicity
In our previous work (12), we demonstrated that a nAg-based GAd vaccine encoding 31 epitopes selected from CT26 tumors (GAd-31) was able to induce a potent T-cell immunity. Among the 31 predicted nAgs, 6 were found to be immunogenic, specifically 3 (here referred to as #5, #11, #23) eliciting CD8+ T-cell responses and 3 (here referred to as #4, #18, #28) eliciting CD4+ T-cell responses. To gain insights into the contribution of each single immunogenic nAg and/or different combinations of them, we generated 6 distinct GAd vaccines, shown in Fig. 1A (CD8 epitopes represented in red; CD4 epitopes in blue). These constructs were: (i) GAd-6 CD4+/CD8+, encoding all 6 previously identified immunogenic nAgs; (ii) GAd-6 CD4+/CD8− encoding only CD4 nAgs, with the 3 CD8 epitopes reverted to wild-type (WT) sequence (gray); (iii) GAd-6 CD4+/CD8#5, encoding only one CD8 nAg (#5) whereas the other 2 CD8 epitopes (#11, #23) are reverted to WT, in presence of CD4 nAgs; (iv) GAd-6 CD4+/CD8#23 encoding only one CD8 nAg (#23) whereas the other 2 CD8 epitopes (#11, #5) are reverted to WT, in presence of CD4 nAg; (v) GAd CD8#5, and (vi) CD8#23 mono-epitopes, encoding only the single CD8 epitope #5 and #23, respectively (Fig. 1A). Details and features of the 6 nAgs (12) are listed in Supplementary Table S1. Immunogenicity of each GAd construct was then tested by ex vivo IFNγ ELISpot on splenocytes isolated 14 days after immunization (Fig. 1B). Immune responses against the 6 nAgs upon GAd-31 vaccination were confirmed, as previously reported (12), with comparable levels and hierarchy of T-cell responses between GAd-31 and GAd-6 CD4+/CD8+ vaccine. The immunogenic nAgs elicited different levels of immune responses, with CD8 #5 and #23 nAgs being the most immunogenic (12), whereas #11 was weakly immunogenic and failed to elicit a positive immune response in all immunized animals and therefore, it was not investigated further. As expected, T-cell responses were never observed when the mutation was reverted to the WT sequence, providing an effective mean to assess the contribution of each nAg in the context of the different vaccine layouts. Overall, no significant changes in the immunogenicity of the nAgs were observed across the different constructs, with a trend toward lower response against the subdominant nAg #23 when encoded in presence of the stronger nAg #5 (Fig. 1B).
Figure 1.

Immunogenicity of GAd adenoviral vectors encoding CT26 nAgs. A, Schematic representation of GAd adenoviral vectors: Six immunogenic nAgs (#4, #5, #11, #18, #23, #28), including both CD4 (in blue) and CD8 (in red) reactivities, selected from the previously published GAd-31 vaccine (12), were cloned to generate different GAd-6 multi-epitopes (constructs 1–4) or GAd mono-epitope vectors (constructs 5–6). WT, wild-type. B,Ex vivo IFNγ ELISpot showing Ag-specific immune responses following naïve mice immunization with the indicated GAd vaccines. Results are expressed as mean + S.E.M. of spot forming cells (SFC) per million of splenocytes (n = 6 mice/group). Positive and negative responses are indicated with + and −, respectively, according to criteria of positivity described in the Materials and Methods section. Data are representative of three independent experiments.
Several neoantigens can drive tumor rejection in both prophylactic and early vaccination settings
In vivo antitumor efficacy of the different vaccine constructs was evaluated in a prophylactic setting (Fig. 2A). Balb/c mice were immunized with each individual GAd vaccine, followed by subcutaneously injection of CT26 tumor cells 14 days later. Tumor development was monitored over time to assess the rate of protection conferred by each GAd vaccine (Fig. 2B and C; gray bars, tumor-free mice; black bars, tumor-bearing mice). Similar to what observed with the GAd-31 vaccine (12), the GAd-6 CD4+/CD8+ vaccine led to full protection, with 100% of mice remaining tumor-free, whereas all the untreated animals developed large tumors. The antitumor protection rate decreased to 50% when the vaccine encoding only CD4 nAgs was injected (GAd-6 CD4+/CD8−), indicating the critical role played by the concomitant presence of both CD8+ and CD4+ reactivities for full vaccine effectiveness. In this setting, single CD8 nAg-encoding vaccines were able to promote tumor protection in approximately 50% and 70% of treated animals, with nAg#5 and #23, respectively. The presence of CD4 epitopes together with the single CD8 nAg#5 (CD4+/CD8#5) raised the tumor protection from 50% to 94%. CD4 epitopes also enhanced the rate of protection elicited by nAg#23, from 70% observed with the CD8#23 mono-epitope vaccine, to almost 90% with the CD4+/CD8#23 vaccine. Overall, the strongest antitumor activity was achieved when targeting multiple neoantigens in presence of both neoantigen-specific CD4+ and CD8+ T cells. The targeting of single neoantigens may lead to tumor escape. To assess whether, upon prophylactic vaccination with mono-epitope vaccines, the lack of protection in some animals was associated with the loss of the targeted mutation, we analyzed the presence of the mutation #5 and #23 in tumor tissue of NR mice. Loss of mutation #5 was found in 33% of non-responding tumors, whereas none of the tumors had lost mutation #23 (Supplementary Fig. S1). Mice that remained tumor-free after the immunization with different GAd vaccines were all protected from a second subcutaneously CT26 tumor challenge (Fig. 2D), demonstrating induction of an effective memory T-cell response.
Figure 2.

Prophylactic protection of GAd vaccines. A, Experimental scheme of the in vivo prophylactic setting. B,In vivo prophylactic efficacy of each GAd vaccine. Mice were immunized with 5×108 vp of the indicated vaccine (day 0); 2 weeks after immunization, a total of 2×105 CT26 cells were injected subcutaneously (day 14) and tumor growth was monitored over time. Graphs indicate the tumor volume (mm3) over time. Each line represents an individual animal; the percentages (%) and the number of tumor-free (TF) mice over the total number of animals are indicated. C, Graph shows the percentage of tumor-free (gray) and tumor-bearing (black) mice, 60 days after tumor cells injection. Statistical differences were calculated by using the two-tailed Fisher test, comparing treated groups versus untreated (**, P < 0.01; ***, P < 0.001; ****, P < 0.0001), CD4+/CD8#23 versus CD8#23 (ns, not significant, P > 0.99) and CD4+/CD8#5 versus CD8#5 (***, P = 0.0006). D, Tumor-free mice resulted from immunization with the indicated vaccines shown in B were subjected to a second tumor challenge with CT26 cells, 40 days after the first tumor cell inoculum; as control, untreated (No vaccine) mice were also inoculated with CT26 cells. Bars represent the percentage of tumor-bearing (black) and tumor-free (gray) mice after the second challenge. Statistical differences were calculated by using the two-tailed Fisher test, comparing treated groups versus untreated. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Data are representative of three independent experiments.
The efficacy of different GAd vaccines was further explored in the early therapeutic setting of CT26 lung metastases. In this setting, vaccination was performed 3 days after intravenous injection of tumor cells (Fig. 3A). In this setting, poly-epitope vaccines encoding CD4 and CD8 neoantigens and mono-epitope CD8 vaccines were all effective, with a significantly decreased number of tumor nodules as compared with untreated control mice (Fig. 3B). The CD4+/CD8− vaccine could only partially control lung nodule formation and protection was not significantly superior over the untreated control, suggesting a predominant role of CD8 reactivities in driving antitumor efficacy in this experimental model (Fig. 3B and C).
Figure 3.
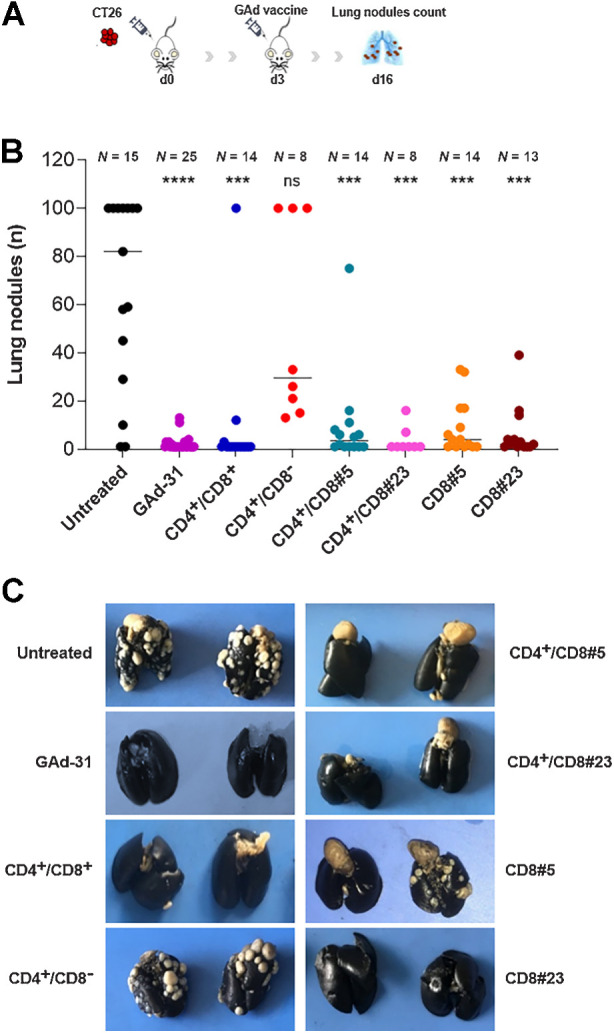
Antitumor efficacy of GAd vaccines in early metastatic setting. A, Experimental scheme for CT26 early metastatic model. Mice were inoculated intravenously with 105 CT26 cells (day 0) and vaccinated or left untreated at day 3. B, Lung nodules count at day 13 after vaccination (day 16 after tumor challenge). Dots represent the number (n) of lung nodules counted per each animal. The number (N) of mice per group is indicated. Results are shown as median. Statistical differences were calculated by using the two-tailed Mann–Whitney U test, comparing the treated groups versus untreated. ****, P < 0.0001; ***, P < 0.001; ns, not significant. C, Representative pictures of India ink-stained lungs from the indicated experimental groups. Metastatic foci are clearly visible as white dots on lung surface (black). Quantification and statistical analysis are reported in B. Data are representative of three independent experiments.
Protective vaccination results in epitope spread
To understand the mechanism of CD8#5 and CD8#23 vaccine-mediated tumor protection, we explored the ability of these vaccines to induce epitope spreading. CT26 cells knocked-out for epitope #5 or #23 (named CT26 #5KO and CT26 #23KO) were generated by CRISPR-Cas9 technology (Fig. 4A; Supplementary Fig. S2). We reasoned that, if epitope spreading was occurring, the mono-epitope vaccines would have been able to confer, through cytotoxicity against tumor cells, immunity to tumor antigens other than the nAg encoded in the vaccine, thus conferring protection also against a secondary challenge by tumors lacking the vaccine neoepitope. Mice prophylactically vaccinated with mono CD8#5 and CD8#23 vaccines that remained tumor free after a first tumor challenge with CT26 cells were subjected to a second tumor challenge with CT26 cells knocked-out for the respective epitope. The rechallenge with CT26 #5KO or CT26 #23KO resulted in 53% and 62% of protection, respectively (Fig. 4B and C), demonstrating tumor rejection also in absence of the vaccine-targeted epitope.
Figure 4.

Vaccination with GAd mono-epitope vaccines induces antigens spreading. A, Generation of CT26 cells knocked-out for #5 and #23 encoding genes, using CRISPR-Cas9 technology. Mice were immunized with CD8#5 (B) or CD8#23 (C) mono-epitope vaccine and after 14 days, mice were subcutaneously inoculated with CT26 cells (1st challenge) resulting in 40% and 75% of tumor-free (TF) mice, respectively (gray bar). After 40 days, a 2nd tumor challenge with CT26 cells knocked-out for nAg #5 (B) and #23 (C), was performed on TF mice from the 1st challenge, resulting in 50% (B) and 60% (C) of tumor protection. Bars show the percentage (%) of TF (gray) and TB (black) mice. The number (n) of animals per each group is indicated. Two-tailed Fisher test of treated groups versus untreated. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. D, Experimental scheme. Mice were immunized with CD8#5 mono-epitope vaccine and, after 14 days, they were subcutaneously inoculated with CT26 cells (1st challenge). After 40 days, a 2nd tumor challenge with CT26 cells knocked-out for nAg #5 was performed on TF mice from the 1st challenge. E, Vaccine-unrelated immune responses were evaluated by ex vivo IFNγ ELISpot in tumor-free mice derived from the 2nd challenge (black bars, n = 5) compared with CT26 tumor bearing mice, as control (gray bars, n = 4). Results are expressed as spot forming cells (SFC) per million of splenocytes. Two-tailed Mann–Whitney. *, P < 0.05. Data are representative of three independent experiments.
A control experiment of primary challenge with the CT26 KO cells was also performed, creating a setting where epitope spreading is not predicted to occur. Mice were immunized with the mono-epitope vaccine encoding nAg#5 followed by tumor challenge with CT26 cells knocked-out for #5. Under these conditions, we demonstrated that vaccination was not effective and no tumor protection was observed against the tumors lacking the vaccine targeted nAg (Supplementary Fig. S3).
To investigate the induction of T-cell responses against epitopes not targeted by vaccine, IFNγ ELISpot analysis was performed on protected mice immunized with CD8#5 and then subjected to a re-challenge with CT26 #5KO (Fig. 4D). Among the tested reactivities, the only detected T-cell responses were, as expected, against the vaccine epitope (#5) and gp70, a previously described dominant CD8+ T-cell epitope derived from an endogenous murine leukemia virus and expressed in CT26 cells eliciting spontaneous T-cell responses (ref. 23; Fig. 4E). Furthermore, CD8+ T-cell responses against gp70 were also detected in untreated, unvaccinated tumor-bearing mice, suggesting that the immunity against gp70 is not responsible for tumor rejections in vaccinated mice. Our results suggest that likely, there are other unknown epitopes responsible for a potential epitope spreading–based protection in the secondary challenge situation.
One CD8 specificity can lead to tumor rejection in an advanced therapeutic setting only in the presence of CD4+ T-cell help
Next, the in vivo therapeutic activity of GAd nAg vaccines was tested in a setting of advanced therapeutic treatment of mice bearing large, established subcutaneous CT26 tumors, in combination with anti-PD1 (Fig. 5A and B). In this setting, no vaccine monotherapy had antitumor activity, which was consistent with our previous findings (12). Combination with anti-PD1 showed that the only effective constructs were GAd-6 CD4+/CD8+ vaccine and GAd-6 CD4+/CD8#23, both curing 50% of the mice as compared with only 18% cured by anti-PD1 monotherapy (Fig. 5B and C). This was similar to what we previously reported for the GAd-31 vaccine (12). These results suggest that the therapeutic effect of GAd-6 CD4+/CD8+ is likely predominantly driven by nAg#23, with the essential contribution of the CD4 epitopes. Accordingly, the CD8#23 mono-epitope vaccine lacking CD4 epitopes could not improve the cure rate of anti-PD1 alone (18%). Interestingly, the epitope CD8#5, despite being the most immunogenic, was not effective in this advanced therapeutic setting regardless of the presence of CD4 epitopes (Fig. 5B), whereas the less immunogenic epitope CD8#23 conferred the therapeutic efficacy of GAd vaccine in the presence of CD4 epitopes (Fig. 5B and C). This apparent contradiction may be explained by the relative expression levels of the two nAgs, with #23 displaying a 5-fold higher expression in CT26 ex vivo tumors as compared with #5 (Fig. 5D). These data suggest that nAg expression levels are likely a key contributor to the effectiveness of nAg-specific T cells mediating tumor recognition and clearance.
Figure 5.

Antitumor efficacy of GAd vaccines in CT26 established tumor setting, in combination with checkpoint inhibitor anti-PD1. A, Experimental scheme. B, Five days after CT26 tumor challenge, Balb/c mice were treated with αPD1 alone or in combination with the indicated GAd vaccines. As control, a group of mice received only the tumor challenge (Untreated). Curves indicate representative experiments of the tumor volume (mm3) over time. Each line represents an individual animal; dotted lines indicate non-responders and solid lines indicate responders. Percentages of the rate of complete responses (CR) and the numbers of animals are reported within each graph. C, The percentage of tumor-free (TF, gray bars) and tumor-bearing (TB, black bars) mice is represented for the indicated groups. Two-sided Fisher's test analysis between anti-PD1 and anti-PD1 + vaccination was performed, with exact *, P = 0.02. D, CT26 tumors were collected from untreated mice for RNA-seq and the abundance of the transcripts encoding nAgs #5 and #23 is indicated as transcript per million (n = 4). Data are representative of three independent experiments.
CD4+ helper T cells promote tumor infiltration by antigen-specific CD8+ T cell
The role played by the CD4 nAgs in mediating the antitumor efficacy of the CD8#23 vaccine was further investigated. We asked whether CD4+ Th cells may affect the migratory potential of CD8+ CTLs to the tumor. Balb/c mice were subcutaneously inoculated with CT26 cells and, when tumor masses were established, mice were treated with GAd-6 CD4+/CD8#23 (here referred to as Help) and GAd CD8#23 (here referred to as No Help) along with anti-PD1, as shown in the experimental scheme in Fig. 6A and D. Tumors and spleens were collected at days 9 and 14 after vaccination and the frequency, number, and phenotype of intratumoral and peripheral CD8+ T cells and antigen-specific T cells were investigated (Fig. 6; Supplementary Figs. S4 and S5). Help and No Help vaccines induced a similar expansion in the frequency of total intratumoral CD8+ T cells compared with the untreated control group (Fig. 6B), with no differences observed in the spleen (Fig. 6E). On the subset of CD8+ antigen-specific T cells, Help vaccine induced a significantly earlier increase in the percentage of intratumoral nAg#23-specific CD8+ T cells than No Help vaccine (Fig. 6C), and the antigen-specific CD8+ T cells acquired an Effector Memory phenotype (TEM; CD44+CD62L−; Supplementary Fig. S4B). No significant differences were observed in the periphery (spleen) between Help and No Help groups in the percentage of TEM cells (Supplementary Fig. S4C). No major differences were found in the other subsets (in tumor and tumor-draining lymph node) of other immune phenotypes of T cells, except for a significant increase of stemlike CD8+ T cells (CD8+PD1+TIM3− TCF1+GRZB−) observed in the tumors of mice receiving the Help vaccine (Supplementary Fig. S6).
Figure 6.

The presence of CD4 Help increases intratumoral antigen-specific T cells. A, Schematic representation of the experiments. Mice were subcutaneously injected with CT26 cells and 5 days later, immunized with GAd vaccines CD4+CD8#23 (Help) and CD8#23 (No Help) in combination with anti-PD1. At days 9 and 14 after vaccination, tumors were collected for FACS analysis. The percentage of intratumoral CD8+ T cells (B) and nAg#23+-specific T cells (C) is shown (Untreated, n = 4; Help day 9, n = 6; no Help day 9, n = 6; Help day 14, n = 7; no Help, n = 7). Statistical differences were calculated with two-tail Mann-Whitney (**, P = 0.002). D, Schematic representation of the experiments. Mice were subcutaneously injected with CT26 cells and 5 days later, immunized with GAd vaccines CD4+CD8#23 (Help) and CD8#23 (No Help) in combination with anti-PD1. At days 9 and 14 after vaccination, spleens were collected for splenocytes isolation and FACS analysis. The percentage of CD8+ T cells (E) and nAg#23+ specific T cells (F) is shown (Untreated, n = 4; Help day 9, n = 6; no Help day 9, n = 6; Help day 14, n = 7; no Help, n = 7). G, Schematic representation of the experiments. Mice were subcutaneously injected with CT26 cells and 5 days later, immunized with GAd vaccine CD4+CD8#23 (Help) in combination with anti-PD1. At day 14 after vaccination, blood (H) and tumors (I) were collected for FACS analysis of nAg#23+-specific T cells, in responder (RES, n = 4) and nonresponders mice (NR, n = 13 for blood and n = 3 for tumors). Results are expressed as percentage (%). Statistical differences were calculated with one or two-tail Mann–Whitney (*, P< 0.05; ns, not significant). J, Heat map displaying the relative (median log2-fold change) expression of selected chemokines, chemokine receptors and adhesion genes associated with T-cell infiltration and chemotaxis, between responders (RES, n = 5) and nonresponders (NR, n = 4) tumors to Help vaccine in combination with anti-PD1, compared with the untreated group (n = 3). The color gradient indicates the normalized values (z-score) of the median gene values. K, Tumor-free mice treated with CD4+CD8#23 (Help, n = 5) and anti-PD1 were re-challenged with a second CT26 tumor inoculum at day 40 after first challenge. Untreated mice were inoculated with CT26 tumor cells as control (n = 9). Log rank test ***, P = 0.0005. Data are representative of three independent experiments.
Within the group of Help vaccine-treated mice, a higher percentage of circulating and intratumoral #23-specific CD8+ T cells was found in RES mice compared with NR mice (Fig. 6H and I), indicating a strong correlation between high frequency of antigen-specific CD8+ T cells and tumor control in the Help group. The frequency of #23-specific CD8+ T cells in blood of RES animals increased over time while remaining low in NR animals, suggestive of their recirculation upon tumor shrinkage (Supplementary Fig. S4E and S4F). In line with these results, bulk RNA-seq analysis showed an upregulation of multiple genes involved in T-cell migration, such as Ccl7, Cxcr3, Cxcr6, Ccl21, Ccl27a, Ccl6, in tumors of mice responding to the Help vaccine + anti-PD1 versus NR tumors (Fig. 6J).
The generation of a long-lasting protecting immune memory was also demonstrated by the full protection against a CT26 tumor re-challenge in animals that were subjected to a first tumor challenge and then cured by CD4+/CD8#23 (Help vaccine) + anti-PD1 treatment (Fig. 6K).
Discussion
Tumor-specific mutations are ideal targets for vaccine approaches in cancer immunotherapy, as they represent nonself antigens that can be recognized by the immune system and thus trigger antitumor responses. However, the identification of neoepitopes effective in cancer therapy remains a major challenge in the field of cancer vaccine (24).
In the present study, we sought to dissect the role played by 6 epitopes previously identified as immunogenic upon immunization with a GAd poly-epitope vaccine. We tested them within different layouts of GAd-vaccines evaluating their immunogenicity and ability to mediate antitumor protection, to identify the mechanisms underlying the therapeutic response. By using three different preclinical settings of tumor burden, we demonstrated the nAg features required for a vaccine to achieve effective outcome are strongly influenced by tumor burden, because epitopes that are effective in prophylactic or early lines of treatment become inefficacious in a high disease burden setting (Fig. 7). In prophylactic and early therapeutic settings, vaccines encoding the 6 immunogenic nAgs were fully effective and provided the maximal antitumoral activity. In these same settings, mono-epitope vaccines based on single CD8 nAgs could also significantly impact tumor growth, although at a lesser extent than poly-epitope vaccines. However, the efficacy of CD8 mono-epitope vaccines was completely lost when treating mice bearing large tumor masses, even in presence of anti-PD1. The different performance of the mono-epitope vaccines is likely due to the impact of tumor burden on the immune system. Large tumors are highly immunosuppressive and, as a consequence, they may rapidly impair vaccine-induced immune responses, leading to T-cell exhaustion and tumor progression. In contrast, in a situation of prophylactic or early intervention, when the tumor is absent or minimal, T-cell effector functions are not or are less affected. This is also in line with the need to combine vaccine and anti-PD1 treatment to achieve effective vaccination in the presence of high tumor burden.
Figure 7.

Schematic model representing the impact of tumor burden on nAg-based vaccine effectiveness. In early disease setting with minimal tumor burden, a vaccine targeting a single neoantigen can be effective. On the contrary, in advanced setting such as metastatic disease, the immunosuppressive tumor microenvironment requires multiple neoantigens and concomitant checkpoint blockade treatment for effective tumor control.
Dissection of the vaccine-encoded nAgs to find those responsible for antitumoral activity led to the identification of the CD8 epitope–driving vaccine efficacy under high disease burden. Although in the prophylactic and early vaccination settings both immunogenic CD8 nAgs could trigger tumor rejection as individual epitopes, in an advanced tumor setting, only nAg#23 could promote tumor rejection in the presence of CD4 help. The rejection-mediating antigen nAg#23 elicited a weaker immune response than nAg#5 but was expressed at higher levels, suggesting that the antitumor activity conferred by a specific epitope is not necessarily related to its immunogenicity, but that the level of expression of the mutated genes within a tumor is a key element to consider. Levels of nAg expression may correlate to the levels of nAg MHC presentation, and higher levels of presentation are likely to trigger immune responses leading to tumor recognition and clearance.
The presence of CD4 epitopes was crucial to support the cytotoxic functions of nAg#23-specific CD8+ T cells, as CTLs raised in the absence of CD4+ T-cell help were not effective in mediating tumor rejection. Several studies suggest that CD4+ T helper cells are required to generate an effective antitumor CD8+ T-cell response. In two different preclinical tumor models, antitumor activity of nAg-specific CD8+ T cells could only be observed when tumor cells expressed both MHC-I and MHC-II nAgs (18). Similarly, other studies have demonstrated that the inclusion of helper epitopes in therapeutic vaccines improved the antitumor response by increasing the expansion of CD8+ T cells, as well as reducing the expression of co-inhibitory receptors, supporting their differentiation in effector memory and increasing their migratory potential to the tumor (14, 25). In line with these findings, increased tumor infiltration of nAg#23-specific CD8+ T cells was observed in mice vaccinated with Help vaccine as compared with the ones receiving the nonhelp vaccine. Different factors may be responsible for the intratumoral increase of antigen-specific CD8+ T cells in presence of the Help, such as more efficient priming in the lymphoid organs, more robust expansion at the tumor site, or an increase in the migratory potential of the T cells. The expression of key genes known to attract T cells into the tumor (Ccl7, Cxcr3, Cxcr6, Ccl21, Ccl27, and Ccl6; ref. 26) was upregulated in tumors of mice responding to the Help vaccine and anti-PD1 versus those not responding, suggesting that the migratory phenotype may contribute to treatment effectiveness. Moreover, a higher frequency of nAg#23 CD8+ T cells was detected in the tumors of RES versus NR mice, in association with the therapeutic response. Help vaccine also induced an increase in stemlike CD8+ T cells into the tumor. Stemlike CD8+ T cells represent a subset of memory T cells, identified both in tumor and tumor-draining lymph node, with self-renewing capabilities and long-term persistence. These cells have a role in generating and sustaining effector T cells and therefore are critical for the maintenance of T-cell responses against tumors (27, 28). Although in our vaccination model we used CD4+ tumor epitopes encoded together with the CD8 nAg#23, we cannot exclude that nontumor-specific MHC-II–restricted antigens may be also effective to support the activity of CD8+ T cells against the tumor, as shown by others (14, 25). The role of CD4+ T cells was also evident in the prophylactic and early vaccination settings. In particular, prophylactic vaccination with a vaccine encoding only CD4 neoepitopes conferred protection to 50% of treated animals. Other studies have shown neoantigen-specific CD4+ T cells have antitumoral activity, which is consistent with our results (19) and with the hypothesis of a direct cytotoxic role for CD4+ T cells or, because most tumors lack MHC class II, through cross-presentation of tumor antigens by tumor stromal cells (16).
Our work also suggests that epitope spreading may occur after vaccination to neoantigens not included in a vaccine. Likely, the mechanism of epitope spreading is linked to tumor destruction mediated by vaccine-induced T cells. This event triggers a memory T-cell response reacting also against antigens not encoded in the vaccine and controlling the development of a second tumor after rechallenge. From the translational point of view, this suggests the potential impact of vaccine, when used in neoadjuvant setting, to prevent relapse even when driven by tumor cells not carrying vaccine-encoded mutations. Today, several clinical trials with neoantigen-based vaccines are ongoing in different clinical settings from advanced diseases to adjuvant treatments, including also cancer interception (NCT05078866). Our results have translational relevance for the clinical application of neoantigen vaccination by showing that in a situation of minimal disease burden, a vaccine targeting a single neoantigen can be effective. However, in the metastatic setting, the immunosuppressive tumor microenvironment raises the bar, and multiple neoantigens with concomitant checkpoint blockade treatment are required for an effective treatment. The importance of including both MHC-I- and MHC-II–restricted epitopes, as well as the relevance of parameters such as neoantigen abundance, provides insights into the optimal nAg features to select for generating a vaccine capable of mediating tumor rejection.
Supplementary Material
List of the GAd encoded CT26 neoantigens selected according to: binding predictions for MHC-I (IC50 ≤ 500nM) and MHC-II (binding score ≤ 1); tumor allele frequency (MAF≥ 25%) and RNA expression (mutated RNA reads ≥ 1). The mutated amino acid is underlined. MAF = Minor Allele Frequency.
Mutation loss in developed CT26 tumors after GAd CD8#5 and CD8#23 vaccine. A) Mice were immunized with mono-epitope GAd CD8#5 or B) CD8#23 vaccines and 14 days later, mice were inoculated s.c. with 2x10^5 CT26 cells. Mutation relative to nAg#5 (A) and nAg#23 (B) was checked by DNA sequencing in developed tumors. C) Table shows the percentage of mutation loss relative to nAg#5 (33%, E2f8 gene) and nAg#23 (0%, Mtch1 gene) in CT26 developing tumors after immunization with CD8#5 or CD8#23 vaccine.
Generation of CT26 #5KO and CT26 #23KO cells using CRISPR-Cas9 technology. A) CRISPR/Cas9 strategy used for the generation of CT26 cells lacking of nAg #5. Red arrows represent forward and reverse primers used for PCR. Blue lines 1 and 2 indicate 2 sgRNAs designed for Cas9-mediating genome editing, within exon 10, harboring #5 mutation (blue star). The position of the 2 sgRNAs is also indicated. B) Left panel: representative agarose gel of PCR screening on cDNA showing homozygous deletions of 74nt in the genomic region encoding nAg#5 (E2f8 gene). Right panel: representative sequenced region of E2f8 gene and the relative chromatogram. In red, PAM sequences are indicated. The deleted region is underlined. The mutated nucleotide of nAg#5 is in black bold. Red arrow indicates Cas9 cut site. C) CRISPR/Cas9 strategy used for the generation of CT26 cells lacking of nAg #23. Red arrows represent forward and reverse primers used for PCR. Blue lines indicate the 2 sgRNAs designed for Cas9-mediating genome editing. Exon 12 harbors #23 mutation (green star). The position of the 2 sgRNAs is also indicated. D) Left panel: representative agarose gel of PCR screening on genomic DNA showing homozygous deletions of 116nt in the genomic region encoding nAg#23 (Mtch1 gene). Right panel: representative sequenced region of Mtch1 gene and relative chromatogram. In red, PAM sequences are indicated. The deleted region is underlined. The mutated nucleotide of nAg#23 is in black bold. Red arrow indicates Cas9 cut site.
Primary tumor challenge with NeoAg #5 knocked out CT26 tumor cells. Mice were immunized with CD8#5 mono-epitope vaccine and after 14 days, mice were s.c. inoculated with CT26 cells or CT26 #5KO cells, resulting in 50% and 0% of tumor free mice, respectively. Bars show the percentage (%) of tumor free (grey) and tumor bearing (black) mice (n=8). No vaccinated mice (Untreated) were s.c. inoculated with CT26 #5KO cells as control (n=.6). One-tailed Fisher’s exact test CT26 vs CT26#5KO, p-values * p=0.03.
Characterization of neoAg#23 specific T cell subsets in the presence/ absence of T cell “Help”. A) Schematic representation of the experiment. Mice were s.c. injected with CT26 cells and 5 days later, immunized with GAd vaccines CD4+CD8#23 (Help) and CD8#23 (No Help) in combination with anti-PD1. At day 9 and day 14 post vaccination, tumors (B) and spleens (C) were collected for FACS analysis on antigen-specific nAg#23+ CD44+CD62- (T Effector memory, TEM). The percentage (%) of TEM and #23-specific T cells are shown. Two-tailed Mann-Whitney test versus CT26 tumor bearing mice, p-values * p<0.05; ns = not significant (Untreated n=4; Help day 9 n= 6; no Help day 9 n=6; Help day 14 n=7; no Help n=7). D) Schematic representation of the experiment. Mice were s.c. injected with CT26 cells and 5 days later, immunized with GAd vaccine CD4+CD8#23 (Help, n=5). E) Blood was collected at different time points post immunization from responder mice (RES) and (F) not responders (NR, n=5-14, depending on the time point) for FACS analysis of nAg#23+ specific CD8+ T-cells.
Representative numbers of total and #23+ CD8+ T cells in tumor and spleen of Help versus non Help vaccine. A) Experimental scheme, FACS gating strategy with representative FACS plots of intratumoral #23+ CD8+ T cells in Help group and untreated group. B and C) Average number of total CD8+ and #23+ T cells relative to figure 6B, 6C, 6E, 6F, per each experimental group (Untreated, Help and No Help) in tumor (B) and in spleen (C) at day 14 post immunization. Number (n) of total CD8+ (x104 cells) and Ag-#23+ specific (x102 cells). Data are shown as total cell count per tumor / organ.
Phenotype characterization of total CD8+ and nAg #23+ CD8+T cells in presence and absence of the “Help”. A) Schematic representation of the experimental set up. B) Representative FACS gating strategy for intratumoral CD8+ T cells. C-D) Phenotype characterization of CD8+ T cells infiltrating the tumors (C, n=14-17) and the tumor draining lymph nodes (D, dLN, n=5-7). The frequency of Exhausted, Stem-like and Better effector cells is shown in Help (blue bars) and No Help (red bars) groups. E) Representative FACS gating strategy for intratumoral nAg #23 specific CD8+ T cells. F-G) Phenotype characterization of intratumoral nAg #23 specific CD8+ T cells collected from mice immunized with Help and No Help vaccines at day 9 post vaccination (n=7-12). The frequency of #23+ specific CD8+ T cells (F) and Exhausted and Transitory (G) is shown in Help (blue bars) and No Help (red bars) groups. The phenotypic markers used to analyze each subtype are reported in the Material and Methods section. Results are from two independent experiments. Two-Way Anova with multiple comparison analysis or Mann Whitney test were performed between Help and No Help groups. *** p=0.0005; * p<0.05; ns = not significant.
Acknowledgments
The research project was partially supported by the FSE-REACT-EU, PON Ricerca e Innovazione 2014–2020 DM 1062/202, Cod: 31-I-14604–1 (to L. Avalle). We acknowledge the animal facility of Plaisant in Castel Romano (Rome) for maintenance and care of the mice used in this study. We would like to thank in particular Domenico Salvatori for his support with tumor calibrations for in vivo studies.
Footnotes
Note: Supplementary data for this article are available at Cancer Immunology Research Online (http://cancerimmunolres.aacrjournals.org/).
Authors' Disclosures
I. Garzia reports employment with Nouscom. L. Nocchi reports employment with Nouscom Srl. L. Avalle reports other support from Italian Research and University Ministry—MUR during the conduct of the study. F. Troise reports employment with Nouscom. G. Leoni reports employment with Nouscom Srl. Antonucci reports employment with Nouscom. G. Cotugno reports employment with Nouscom Srl. S. Allocca reports employment with Nouscom Srl. G. Romano reports employment with Nouscom Srl. V. Poli reports other support from Italian Research and University Ministry during the conduct of the study. E. Scarselli reports as a cofounder and employment with Nouscom. A.M. D'Alise reports employment with Nouscom Srl. No disclosures were reported by the other authors.
Authors' Contributions
I. Garzia: Formal analysis, investigation, methodology. L. Nocchi: Formal analysis, investigation, visualization, writing–original draft. L. Avalle: Investigation, visualization, methodology, writing–original draft. F. Troise: Validation, investigation, visualization, methodology. G. Leoni: Validation, investigation, visualization, methodology. L. Seclì: Validation, investigation, visualization, methodology. L. Antonucci: Validation, investigation, visualization, methodology. G. Cotugno: Validation, investigation, visualization, methodology. S. Allocca: Validation, investigation, visualization, methodology. G. Romano: Validation, investigation, visualization, methodology. L. Conti: Validation, investigation, visualization, methodology. C. Caiazza: Validation, investigation, visualization, methodology. M. Mallardo: Supervision, validation, investigation, visualization, methodology, writing–original draft. V. Poli: Conceptualization, supervision, validation, investigation, visualization, methodology, writing–original draft. E. Scarselli: Conceptualization, supervision, validation, investigation, visualization, methodology, writing–original draft. A.M. D'Alise: Conceptualization, supervision, validation, investigation, visualization, methodology, writing–original draft.
References
- 1. D'Alise AM, Brasu N, De Intinis C, Leoni G, Russo V, Langone F, et al. Adenoviral-based vaccine promotes neoantigen-specific CD8(+) T-cell stemness and tumor rejection. Sci Transl Med 2022;14:eabo7604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gulley JL, Madan RA, Pachynski R, Mulders P, Sheikh NA, Trager J, et al. Role of antigen spread and distinctive characteristics of immunotherapy in cancer treatment. J Natl Cancer Inst 2017;109:djw261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hu Z, Leet DE, Allesoe RL, Oliveira G, Li S, Luoma AM, et al. Personal neoantigen vaccines induce persistent memory T-cell responses and epitope spreading in patients with melanoma. Nat Med 2021;27:515–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015;348:803–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017;547:217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rojas LA, Sethna Z, Soares KC, Olcese C, Pang N, Patterson E, et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023;618:144–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. D'Alise AM, Scarselli E. Getting personal in metastatic melanoma: neoantigen-based vaccines as a new therapeutic strategy. Curr Opin Oncol 2023;35:94–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borthwick N, Ahmed T, Ondondo B, Hayes P, Rose A, Ebrahimsa U, et al. Vaccine-elicited human T cells recognizing conserved protein regions inhibit HIV-1. Mol Ther 2014;22:464–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Colloca S, Barnes E, Folgori A, Ammendola V, Capone S, Cirillo A, et al. Vaccine vectors derived from a large collection of simian adenoviruses induce potent cellular immunity across multiple species. Sci Transl Med 2012;4:115ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Capone S, Raggioli A, Gentile M, Battella S, Lahm A, Sommella A, et al. Immunogenicity of a new gorilla adenovirus vaccine candidate for COVID-19. Mol Ther 2021;29:2412–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Swadling L, Capone S, Antrobus RD, Brown A, Richardson R, Newell EW, et al. A human vaccine strategy based on chimpanzee adenoviral and MVA vectors that primes, boosts, and sustains functional HCV-specific T-cell memory. Sci Transl Med 2014;6:261ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. D'Alise AM, Leoni G, Cotugno G, Troise F, Langone F, Fichera I, et al. Adenoviral vaccine targeting multiple neoantigens as strategy to eradicate large tumors combined with checkpoint blockade. Nat Commun 2019;10:2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bjerregaard AM, Nielsen M, Jurtz V, Barra CM, Hadrup SR, Szallasi Z, et al. An analysis of natural T-cell responses to predicted tumor neoepitopes. Front Immunol 2017;8:1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ahrends T, Spanjaard A, Pilzecker B, Babala N, Bovens A, Xiao Y, et al. CD4(+) T-cell help confers a cytotoxic T-cell effector program including coinhibitory receptor downregulation and increased tissue invasiveness. Immunity 2017;47:848–61. [DOI] [PubMed] [Google Scholar]
- 15. Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4 +T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003;421:852–6. [DOI] [PubMed] [Google Scholar]
- 16. Poncette L, Bluhm J, Blankenstein T. The role of CD4 T cells in rejection of solid tumors. Curr Opin Immunol 2022;74:18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, et al. CD4(+) T-cell help is required for the formation of a cytolytic CD8(+) T-cell subset that protects against chronic infection and cancer. Immunity 2019;51:1028–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM, et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 2019;574:696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015;520:692–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 2019;37:907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. Gigascience 2021;10:giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liao Y, Smyth GK, Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res 2019;47:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Castle JC, Loewer M, Boegel S, de Graaf J, Bender C, Tadmor AD, et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics 2014;15:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brennick CA, George MM, Moussa MM, Hagymasi AT, Seesi SA, Shcheglova TV, et al. An unbiased approach to defining bona fide cancer neoepitopes that elicit immune-mediated cancer rejection. J Clin Invest 2021;131:e142823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ahrends T, Busselaar J, Severson TM, Babala N, de Vries E, Bovens A, et al. CD4(+) T-cell help creates memory CD8(+) T cells with innate and help-independent recall capacities. Nat Commun 2019;10:5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Azulay M, Shahar M, Shany E, Elbaz E, Lifshits S, Torngren M, et al. Tumor-targeted superantigens produce curative tumor immunity with induction of memory and demonstrated antigen spreading. J Transl Med 2023;21:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Y, Joe G, Hexner E, Zhu J, Emerson SG. Host-reactive CD8 +memory stem cells in graft-versus-host disease. Nat Med 2005;11:1299–305. [DOI] [PubMed] [Google Scholar]
- 28. Connolly KA, Kuchroo M, Venkat A, Khatun A, Wang J, William I, et al. A reservoir of stem-like CD8(+) T cells in the tumor-draining lymph node preserves the ongoing antitumor immune response. Sci Immunol 2021;6:eabg7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of the GAd encoded CT26 neoantigens selected according to: binding predictions for MHC-I (IC50 ≤ 500nM) and MHC-II (binding score ≤ 1); tumor allele frequency (MAF≥ 25%) and RNA expression (mutated RNA reads ≥ 1). The mutated amino acid is underlined. MAF = Minor Allele Frequency.
Mutation loss in developed CT26 tumors after GAd CD8#5 and CD8#23 vaccine. A) Mice were immunized with mono-epitope GAd CD8#5 or B) CD8#23 vaccines and 14 days later, mice were inoculated s.c. with 2x10^5 CT26 cells. Mutation relative to nAg#5 (A) and nAg#23 (B) was checked by DNA sequencing in developed tumors. C) Table shows the percentage of mutation loss relative to nAg#5 (33%, E2f8 gene) and nAg#23 (0%, Mtch1 gene) in CT26 developing tumors after immunization with CD8#5 or CD8#23 vaccine.
Generation of CT26 #5KO and CT26 #23KO cells using CRISPR-Cas9 technology. A) CRISPR/Cas9 strategy used for the generation of CT26 cells lacking of nAg #5. Red arrows represent forward and reverse primers used for PCR. Blue lines 1 and 2 indicate 2 sgRNAs designed for Cas9-mediating genome editing, within exon 10, harboring #5 mutation (blue star). The position of the 2 sgRNAs is also indicated. B) Left panel: representative agarose gel of PCR screening on cDNA showing homozygous deletions of 74nt in the genomic region encoding nAg#5 (E2f8 gene). Right panel: representative sequenced region of E2f8 gene and the relative chromatogram. In red, PAM sequences are indicated. The deleted region is underlined. The mutated nucleotide of nAg#5 is in black bold. Red arrow indicates Cas9 cut site. C) CRISPR/Cas9 strategy used for the generation of CT26 cells lacking of nAg #23. Red arrows represent forward and reverse primers used for PCR. Blue lines indicate the 2 sgRNAs designed for Cas9-mediating genome editing. Exon 12 harbors #23 mutation (green star). The position of the 2 sgRNAs is also indicated. D) Left panel: representative agarose gel of PCR screening on genomic DNA showing homozygous deletions of 116nt in the genomic region encoding nAg#23 (Mtch1 gene). Right panel: representative sequenced region of Mtch1 gene and relative chromatogram. In red, PAM sequences are indicated. The deleted region is underlined. The mutated nucleotide of nAg#23 is in black bold. Red arrow indicates Cas9 cut site.
Primary tumor challenge with NeoAg #5 knocked out CT26 tumor cells. Mice were immunized with CD8#5 mono-epitope vaccine and after 14 days, mice were s.c. inoculated with CT26 cells or CT26 #5KO cells, resulting in 50% and 0% of tumor free mice, respectively. Bars show the percentage (%) of tumor free (grey) and tumor bearing (black) mice (n=8). No vaccinated mice (Untreated) were s.c. inoculated with CT26 #5KO cells as control (n=.6). One-tailed Fisher’s exact test CT26 vs CT26#5KO, p-values * p=0.03.
Characterization of neoAg#23 specific T cell subsets in the presence/ absence of T cell “Help”. A) Schematic representation of the experiment. Mice were s.c. injected with CT26 cells and 5 days later, immunized with GAd vaccines CD4+CD8#23 (Help) and CD8#23 (No Help) in combination with anti-PD1. At day 9 and day 14 post vaccination, tumors (B) and spleens (C) were collected for FACS analysis on antigen-specific nAg#23+ CD44+CD62- (T Effector memory, TEM). The percentage (%) of TEM and #23-specific T cells are shown. Two-tailed Mann-Whitney test versus CT26 tumor bearing mice, p-values * p<0.05; ns = not significant (Untreated n=4; Help day 9 n= 6; no Help day 9 n=6; Help day 14 n=7; no Help n=7). D) Schematic representation of the experiment. Mice were s.c. injected with CT26 cells and 5 days later, immunized with GAd vaccine CD4+CD8#23 (Help, n=5). E) Blood was collected at different time points post immunization from responder mice (RES) and (F) not responders (NR, n=5-14, depending on the time point) for FACS analysis of nAg#23+ specific CD8+ T-cells.
Representative numbers of total and #23+ CD8+ T cells in tumor and spleen of Help versus non Help vaccine. A) Experimental scheme, FACS gating strategy with representative FACS plots of intratumoral #23+ CD8+ T cells in Help group and untreated group. B and C) Average number of total CD8+ and #23+ T cells relative to figure 6B, 6C, 6E, 6F, per each experimental group (Untreated, Help and No Help) in tumor (B) and in spleen (C) at day 14 post immunization. Number (n) of total CD8+ (x104 cells) and Ag-#23+ specific (x102 cells). Data are shown as total cell count per tumor / organ.
Phenotype characterization of total CD8+ and nAg #23+ CD8+T cells in presence and absence of the “Help”. A) Schematic representation of the experimental set up. B) Representative FACS gating strategy for intratumoral CD8+ T cells. C-D) Phenotype characterization of CD8+ T cells infiltrating the tumors (C, n=14-17) and the tumor draining lymph nodes (D, dLN, n=5-7). The frequency of Exhausted, Stem-like and Better effector cells is shown in Help (blue bars) and No Help (red bars) groups. E) Representative FACS gating strategy for intratumoral nAg #23 specific CD8+ T cells. F-G) Phenotype characterization of intratumoral nAg #23 specific CD8+ T cells collected from mice immunized with Help and No Help vaccines at day 9 post vaccination (n=7-12). The frequency of #23+ specific CD8+ T cells (F) and Exhausted and Transitory (G) is shown in Help (blue bars) and No Help (red bars) groups. The phenotypic markers used to analyze each subtype are reported in the Material and Methods section. Results are from two independent experiments. Two-Way Anova with multiple comparison analysis or Mann Whitney test were performed between Help and No Help groups. *** p=0.0005; * p<0.05; ns = not significant.
Data Availability Statement
RNA sequencing (RNA-seq) data generated in this study have been deposited in Sequence Read Archive under the accession code PRJNA1073521 and PRJNA543001 (untreated tumors; ref. 12). All the other data of this study are available in the article and its Supplementary Files or upon request from the corresponding author.