

ABSTRACT
Acute interstitial nephritis (AIN) is a significant contributor to acute kidney injury and can be attributed to a variety of factors, including but not limited to allergens or drugs, infections, autoimmune or systemic diseases, and idiopathic forms of the disease. In some cases, AIN requires a therapeutic action according to a single specific etiology by handling the offending agent and applying an immunosuppressant. Although AIN can be diagnosed through renal biopsy, it is not able to pinpoint the precise cause when multiple causes are suspected to be present simultaneously. Such situations arise when a patient suffering from infection develops AIN during antibiotic therapy, the exact causative factor of which becomes a challenge for the clinicians to determine. This is attributed to the different approaches employed in different etiologies, wherein clinicians are required to maintain the current antibiotic therapy or augment the dose in cases of infection as AIN etiology, without resorting to immunosuppressant therapy as the primary objective is infection killing. In contrast, antibiotics as an etiology for AIN require an alternative drug from the antibiotics group, along with an immunosuppressant. In the interim, delaying the identification of the precise cause may result in interstitial fibrosis and chronic kidney disease. This narrative review highlights certain findings that can be typical of infection-associated ATIN compared with antibiotic-associated ATIN based on clinical history and physical examination, clinical presentation of different antibiotic drug classes, histopathological features, classical and novel biomarkers, serum and urine cytokines and chemokines, cellular biomarkers, and genetic biomarkers. Although these findings cannot provide conclusive and clear recommendations that can be useful in the clinical practice, they can entice researchers to conduct original research on these features to discover clear recommendations.
Keywords: acute interstitial nephritis, antibiotics, biomarkers, chronic kidney disease, infection
INTRODUCTION
Acute tubulointerstitial nephritis (ATIN), or AIN, is an immunomediated disease that affects the tubulointerstitial area of the kidneys, accompanied by histological findings of interstitial inflammation, edema and tubulitis [1]. The tubulointerstitial area comprises 80% of the renal surface area and is composed of cellular and extracellular matrix components [2]. AIN is an important cause of acute kidney injury (AKI), with various etiologies such as allergens or drugs, infections, autoimmune or systemic diseases, and idiopathic forms of the disease [3]. It is estimated that up to 10%–27% of hospitalized AKI patients are affected by AIN, making it the third most prevalent cause of hospital-acquired AKI after acute tubular necrosis (ATN) and prerenal AKI [4–9]. In developed countries, the most common cause of AIN is an immunoallergic drug reaction, accounting for 70%–90% of cases [7, 10]. In a 2004 report based on pooled data from three large studies, antibiotics were accounted for one-third of the cases from the drug-induced etiology in 91 of the 128 cases (71.1%) [11] (Fig. 1). Among the remaining 37 cases, 20 were induced by infection, 10 were idiopathic, 6 had tubulointerstitial nephritis with uveitis (TINU) and 1 had sarcoidosis [11]. A study conducted by Valluri et al. [12] (Fig. 2) in 2015, based on 171 biopsy-confirmed cases between 2000 and 2012, revealed that antibiotics accounted for 35% of the cases, proton pump inhibitors (35%), and non-steroidal anti-inflammatory drugs (NSAIDs) (20%) were identified as the primary culprits. Therefore, a significant number of AIN cases can be caused by antibiotics and infections (Figs 1 and 2).
Figure 1:
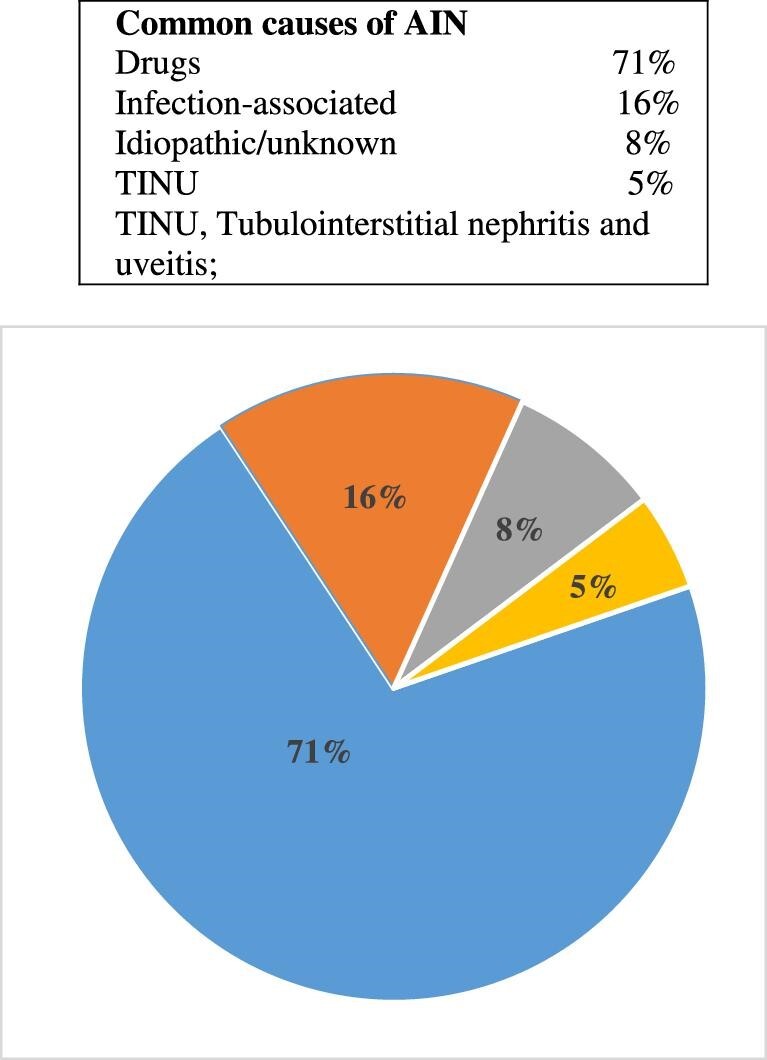
Common causes of AIN. This figure is based on data from Baker and Pusey [11].
Figure 2:
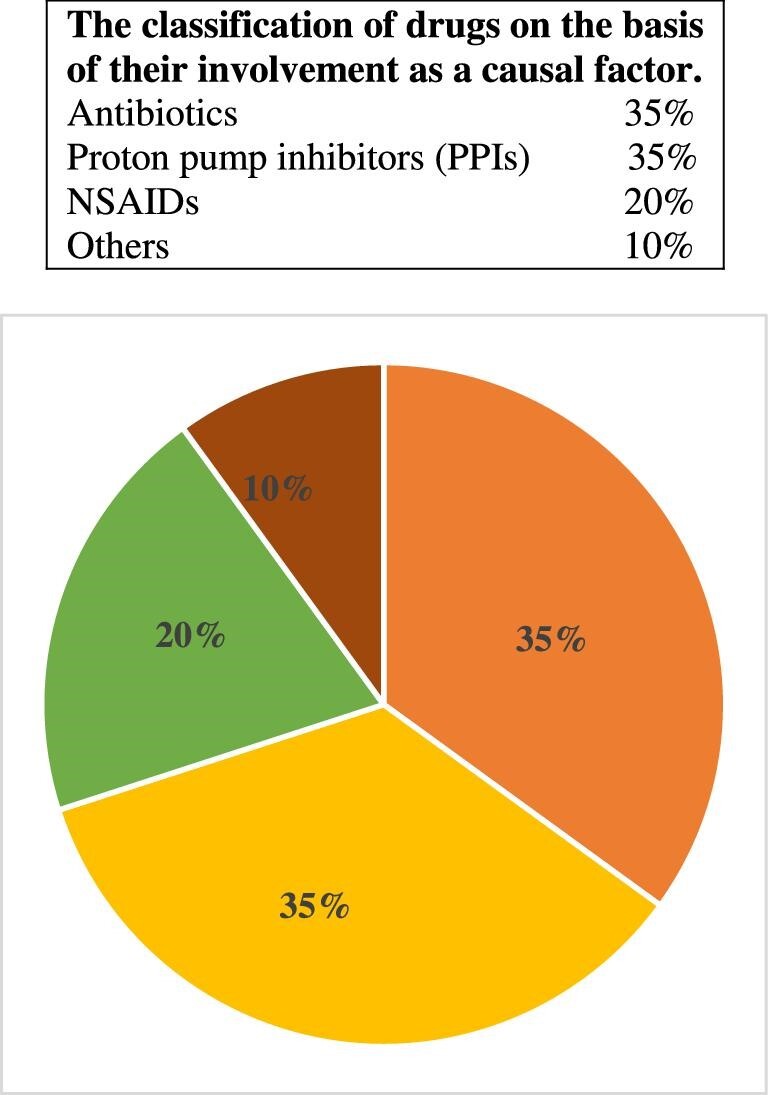
Based on data from Valluri et al. [12].
The renal biopsy procedure is widely recognized as the most reliable diagnostic method for AIN, however, it is not effective in identifying the underlying cause. Serious complications arise when a patient who is infected develops AIN during antibiotic therapy. This is due to the fact that both infection and antibiotics can be suspected as the offending agent simultaneously, resulting in misguided efforts to identify the offending agent, as both etiologies may exhibit similar histopathological or clinical characteristics. Furthermore, the two etiologies require different therapeutic actions because infection as AIN etiology requires the continuation of current antibiotic therapy or even an increase in dose without the need for immunosuppressant therapy [13–15]. Treating the underlying infection and supportive therapy is the main initial target. On the other hand, during the case of an antibiotic-related AIN, clinicians may require the use of alternative antibiotic drugs along with the application of an immunosuppressant [3, 6, 16, 17]. The delay in finding the exact cause can lead to interstitial fibrosis and chronic kidney disease (CKD) [1, 3, 11, 13, 18, 19], and research estimates that 40% to 60% of patients develop CKD after an episode of AIN [20–25].
Clinicians are required to conduct prompt diagnosis, identification and withdrawal of the offending agent, as these are crucial factors in preserving kidney function and ensuring a favorable long-term renal prognosis [26]. A recent case series published by Fernandez-Juarez et al. [27] demonstrated that the culprit drug cannot be precisely identified in nearly 30% of cases. This lack of crucial information hinders the primary therapeutic option, as it entails the withdrawal of the causative agent. Furthermore, it may increase the probability of developing recurrences and partial renal recovery [1, 11, 18]. Barreto et al. [13] demonstrated that the initial inflammatory lesions in AIN can commence to progress into irreversible interstitial fibrosis as early as 7 days after drug exposure. This may be the reason why, despite the most effective treatment options, only 40%–50% of patients with AIN experience complete recovery of kidney function [28]. AIN may also represent an earlier clinical manifestation of an underlying systemic or infectious disease with fewer extrarenal manifestations [29–31], thereby hindering the accurate diagnosis and treatment [5].
Drugs, particularly antibiotics, are known to cause AIN, which is a drug hypersensitivity reaction (DHR) that manifests itself within 7–10 days of exposure to the offending drug [20] (Tables 1 and 2 [32–49]). However, this time can be shorter if the patient is exposing to the same drug for the second time [25], thus misguiding in finding the culprit drug. AIN can also arise as a reactive, cytokine-mediated response to infection, as was first described in 1898 by Councilman for streptococcal infections [50] (Table 1 [38–48]). The exact pathomechanism of infection-induced AIN is not yet clear, though some mechanisms have been proposed. Certain microorganisms that act as planted antigens have the ability to accumulate in the interstitium, mimic a normally present antigen in the tubular basement membrane and elicit an immune response directed against this antigen (Table 1) [32–48]. Furthermore, the direct cytopathic effects of microbial antigens or cytokine-mediated inflammation may be the explanation for the renal injury [14, 51] (Table 1 [32–48]). In order to gain a more thorough comprehension of the pathophysiology and etiology associated with early diagnosis, researchers are endeavoring to identify reliable biomarkers of the disease, with a particular emphasis on urinary cytokines and chemokines that may manifest renal local inflammation (Table 3) [52–62]. There is a growing interest in finding cell-based tests that can identify the exact drug involved in hypersensitivity reactions to drugs, manifesting as AIN or ATIN [63]. Some studies also found that certain single-nucleotide polymorphisms in human leukocyte antigens (HLA) or cytokine genes confer susceptibility to some etiologies inducing ATIN [64]. Hence, it is imperative for researchers to conduct additional research on these procedures to facilitate clinicians in the timely identification of the primary aggravating factor causing ATIN, thereby aiding in diagnosis, prognosis and follow-up.
Table 1:
Literature review of antibiotics-induced AIN and infection-induced AIN.
| Offending agent | Associated clinical features | Diagnostic procedures | Pathomechanism or immunogenecity | Biopsy findings | Therapy | Outcome |
|---|---|---|---|---|---|---|
| Antibiotics | ||||||
| BLs | Fever and rash, eosinophilia, eosinophiluria and pyuria | Renal biopsy, physical examination clinical features and history | Delayed T-cell-mediated hypersensitivity reaction | Interstitial infiltration with mononuclear cells | Discontinuation of culprit drug and administration of steroid | Higher chances of recovery |
| Sulfonamides | Fever and rash, eosinophilia, eosinophiluria and pyuria | Renal biopsy, physical examination clinical features and history | Delayed T-cell-mediated hypersensitivity reaction | Interstitial infiltration with mononuclear cells | Discontinuation of sulfonamides and administration of steroid | Higher chances of recovery |
| Fluoroquinolones (Hung et al., Nephrol Dial Transplant 2006) [32] | Eosinophilia and pyuria, fever, elevated WBC count and anuria | Renal biopsy, physical examination clinical features and history | Delayed T-cell-mediated hypersensitivity reaction | Mild hypercellularity, interstitial infiltration with mononuclear cells | Discontinuation of fluoroquinolones | Recovered |
| Rifampicin (Salih et al., Saudi J Kidney Dis Transpl 2008) [33] | Fever and rash, eosinophilia, eosinophiluria, pyuria, abdominal pain, high leukocyte count, decrease in urine pH | Renal biopsy, physical examination and clinical history | Rifampicin-dependent antibodies, especially IgM | Acute tubular necrosis with mild tubulo-interstitial mononuclear cellular infiltrate | Rifampicin was discontinued, without steroid | Recovered |
| Vancomycin (Kannan et al., Front Med 2022) [34] | Inconsistent pyuria, generalized fatigue, fever, elevated WBC count, moderate hematuria | Blood culture positive for gram positive cocci, vancomycin trough was 17 µg/mL | Delayed T-cell-mediated hypersensitivity reaction | Diffuse cellular infiltrate within the interstitium with inflammatory cells including eosinophils and lymphocytes, numerous eosinophils in the interstitium | Administration of prednisone and discontinuation of vancomycin | Recovered |
| Minocycline (Sharma et al., SAGE Open Med Case Rep 2020) [35] | Generalized rash, anasarca, fever, myalgia, nausea, vomiting, sore throat and generalized body weakness, mild proteinuria | Renal biopsy, physical examination, clinical features and history | Delayed T-cell-mediated hypersensitivity reaction | Marked interstitial edema with patchy interstitial lymphoplasmacytic infiltrates | Corticosteroids, diphenhydramine, and discontinuation of minocycline | Recovered |
| Prothionamide (Asnake et al., SAGE Open Med Case Rep 2022) [36] | Fatigability, rash and intermittent fever, proteinuria, hematuria | Physical examination, clinical features and history | Delayed T-cell-mediated hypersensitivity reaction | NA | Discontinuation of prothionamide | Recovered |
| Infections | ||||||
| Staphylococcus infection [(Raina et al., J Nephropathol 2017) [37] | Nausea, vomiting, poor oral intake, and decreased urination, elevated white blood cell count with predominant neutrophils, proteinuria, sterile pyuria | Urine culture, renal biopsy, physical examination, clinical features and history | Low C3, normal C4, and positive ANA (1:80 titer) | Patchy interstitial inflammatory infiltrates with prominent eosinophilic component | CRRT, vancomycin , piperacillin + tazobactam, clindamycin, prednisone after 7th day of hospitalization | Recovered |
| Salmonella infection (Caers et al., Eur J Intern Med 2006) [38] | Fever, malaise, vomiting, diarrhea, microscopic haematuria, pyuria, subnephrotic proteinuria, and normal or mildly reduced GFR | Renal biopsy, physical examination, clinical features and history | Immunofluorescence showed the absence of immunoglobulin deposits | Interstitial edema and lymphocytic infiltrate with plasma cells and rare granulocytes | Ciprofloxacin, ceftriaxone, methylprednisolone | Recovered |
| Legionella infection (Daumas et al., J Med Case Rep 2012) [39] | Aseptic leukocyturia, hematuria, proteinuria, anuric | Legionella antigenuria was positive, renal biopsy, physical examination, clinical features and history | NA | Interstitial cell infiltrate associated with edema and few tubules lined by flattened cells, focal tubulitis with mononuclear cells that have invaded few tubules | Erythromycin and ofloxacin, HD, steroid | Recovered |
| Yersinia infection (Iijima et al., Am J Nephrol 1989) [40] | Proteinuria, granular casts of urine, fever, | Renal biopsy, physical examination, clinical features and history | NA | Extensive lymphocyte infiltration and diffuse edematous change in the interstitium | Cephalexin, acetylsalicylic acid | CKD |
| Brucella infection (Dagli et al., J Infect Dev Ctries 2011) [41] | Weakness, generalized joint pain, sweats, nausea, loss of appetite and fever, dysuria, hematuria, proteinuria, granular casts | Urine culture, physical examination, clinical features and history | Brucella agglutinins were reported at a titer of 1:160, brucella indirect hemagglutination (Rose-Bengal) test was positive, antigen-antibody reaction as the cause of the renal damage | NA | Rifampin and doxycycline | Recovered |
| Campylobacter jejuni (Rautelin et al., Scand J Urol Nephrol 1987) [42] | Fever and backache, oliguric, hematuria, proteinuria, febrile | Isolation of Campylobacter jejuni heat-stable serotype 2 from faeces, renal biopsy, physical examination, clinical features and history | Elevated titer of Campylobacter IgA class antibody was detected, antigen-antibody reaction as the cause of the renal damage | Extensive interstitial inflammation, with mononuclear cells and fewer granulocytes; infiltrates in medullar and cortical areas | NA | Recovered after 22 days from the onset of symptoms |
| Corynebacterium diphtheriae | Sore throat, weakness, fever, and swollen glands | Blood or urine culture, physical examination, clinical features and history | NA | NA | A vaccine, DTaP, effectively prevents the disease, penicillin or erythromycin | NA |
| Streptococcal infection (Chang et al., Nephrol Dial Transplant 2011) [43] | Sore throat, diarrhoea and general myalgia, erythematous rash, proteinuria | Blood and urine culture grew Group A Streptococcus pyogenes, renal biopsy, physical examination, clinical features and history | Immunohistochemical study depicted diffusely strong positive signals of anti-streptococcal pyrogenic exotoxin B antibodies in tubular epithelial cells and tubulointerstitial compartments of the cortical tissue; antigen-antibody reaction as the cause of the renal damage | Inflammatory cells infiltration and edema in the interstitium of cortex, and was infiltrated by admixed inflammatory cells, including neutrophils, lymphocytes, plasma cells and especially oeosinophils | Four sessions of haemodialysis, supportive therapy, ceftriaxone | After 23 days of hospitalization, he recovered |
| Streptococcus pneumoniae (Phillips et al., Pediatr Nephrol 2005) [44] | Fever, cough, vomiting, abdominal pain and lethargy, increase in WBCs count | Blood and urine cultures grew penicillin-sensitive S. pneumoniae, physical examination, clinical features and history | NA | NA | Intravenous ceftriaxone and maintenance hydration | Recovered post 8 weeks of hospitalization |
| Escherichia coli (Kwon et al., J Korean Med Sci 2015) [45] | Hematuria, pyuria, oliguric acute kidney injury, acute pyelonephritis | Bacteremia and megalocytic interstitial nephritis, renal biopsy, physical examination, clinical features and history | NA | Infiltration of numerous histiocytes without Michaelis-Gutmann bodies, CD68 positivity in infiltrated histiocytes, mesangial staining was positive for C1q and electron microscopy showed moderate effacement of epithelial foot processes | CRRT, HD, cefotaxime (third-generation cephalosporin) and azithromycin, high-dose steroid | Renal function was not recovered despite adequate duration of susceptible antibiotic treatment, accompanied by negative conversion of bacteremia and bacteriuria |
| Melioidosis (Prabhu et al., J Nephrol. 2021) [46] | Hyponatremia, microscopic hematuria and proteinuria, renal and lower renal tract abscesses, pneumonia, abscess formation, sepsis, and multi-organ dysfunction | Positive culture for Burkholderia pseudomallei, bacteraemia, renal biopsy, physical examination, clinical features and history | NA | Acute tubular injury, interstitial nephritis and microabscesses | HD, ceftazidime and carbapenem with or without cotrimoxazole, or chlorampheniol, doxycycline and cotrimoxazole | AKI was predicted by bacteraemia and CKD, and was associated with higher mortality and need for ICU care; but renal function recovery was observed in survivors |
| Scrub typhus (Kim et al., Kidney Res Clin Pract 2013) [47] | Proteinuria, fever, chills, myalgia, skin rash, lymphadenopathy and an eschar | Renal biopsy, physical examination, clinical features and history | Immunochromatographic antibody assay test for Orientia tsutsugamushi was positive, and the immunofluorescent antibody assay test profile showed an IgM titer 41:2048 and IgG titer 41:2048 | Diffuse lymphoplasmacytic infiltrations with scattered neutrophils in the edematous interstitium, tubulitis and acute tubular necrosis, detached tubular epithelial cells and neutrophils were detected in the tubular lumen, mild tubular atrophy | HD supportive therapy, doxycycline, azithromycin | Renal function did not improve 18 months after discharge and the patient required continuous HD; this failure to recover might be from cross reaction or superinfection from one fever type to other |
| Leptospira infection (Daher Ede et al., J Bras Nefrol 2010) [48] | Nonoliguric and hypokalemic, fever, chills, severe headache, followed by anorexia, diarrhea, nauseas, vomiting, malaise, myalgia, mild proteinuria and urinary sediment abnormalities | Blood culture, clinical findings and epidemiological data | IgM antibodies, MAT, results are considered positive when antibody titers are four times greater than the reference value | NA | Early and daily HD; low volume infusion and lung-protective strategies | Mortality in leptospirosis-associated AKI is around 22% |
NA, not available; IgM, immunoglobulin M; CRRT, continous renal replacement therapy; C3, complement 3; C4, complement 4; ANA, antinuclear antibody; GFR, glomerular filtration rate; HD, hemodialysis; IgA, immunoglobulin A; DTaP, diphtheria, tetanus and acellular pertussis; CD68, cluster of differentiation 68; C1q, complement component 1q; ICU, intensive care unit; NA, not available; MAT, microscopic agglutination test.
Table 2:
Clinical characteristics of AIN (Nussbaum et al., Clin Kidney J 2019) [49].
| Fever | Rash | Eosinophilia (blood) | Triad of fever, rash and eosinophilia | Oliguria |
|---|---|---|---|---|
| Present in 15%–36% of cases | Present in 22%–27% of cases | Present in 23%–36% of patients | Present in up to 10% of cases | Present in 50% of cases |
RBC, red blood cell.
Table 3:
A comprehensive summary of the main publications pertaining to serum and urinary biomarkers of ATIN.
| Reference | Population samples | Relevant findings | Association in distinguishing between different etiologies? |
|---|---|---|---|
| Dantas et al., Kidney Blood Press Res (2007) [52] | Glomerulopathy n = 37 | The urinary MCP-1 was correlated with the extent of tubulointerstitial infiltrate by macrophages, but not with the degree of glomerular infiltrate | Until the present research, it was not sensitive, as it was found to be significantly higher in different etiologies [63, 92, 53, 59] |
| Wu et al., Clin J Am Soc Nephrol (2010) [53] | Drug-induced ATIN n = 40Healthy controls n = 20 | ATIN patients had higher levels of MCP-1, α1-MG, NGAL and NAG compared with controls; the urinary levels of MCP-1were correlated with the extent and severity of the acute lesions | It is important to classify drugs based on their pathomechanism, since different drugs can induce ATIN through different pathomechanism which may aid in identifying the culprit drug |
| Nakashima et al., Clin Nephrol (2010) [54] | IgG4 disease-related ATIN n = 4Other cause ATIN n = 16 | The expression of IL-4, IL-10 and TGF-β RNA in kidney tissue was observed to be higher in patients with IgG4 disease, as compared with other causes of ATIN | Due to the small sample size and the presence of such biomarkers in other autoimmune diseases, these are not universally applicable as a diagnostic biomarkers in IgG4-disease-related ATIN |
| Shi et al., Am J Med Sci (2013) [55] | Drug-induced ATIN n = 51 | The rate of GFR decline was faster in patients with higher urinary levels of NAG, metalloproteinase 2 (MMP2) and MMP9 | It is important to classify drugs based on their pathomechanism, since different drugs can induce ATIN through different pathomechanism which may aid in identifying the culprit drug |
| Aoyagi et al., CEN Case Rep (2014) [56] | One case of TINU | During follow-up of an episode of TINU, serum TNF-α, IL-8 and IFN-γ levels decreased | Several studies have found that these biomarkers are higher in various etiologies, making them non-sensitive and non-specific |
| Chen et al., Braz J Med Biol Res (2018) [57] | ATIN n = 30Healthy controls n = 15 | The serum levels of IL-6, IL-10 and TNF-α were significantly higher in ATIN patients than in controls | Several studies have found that these biomarkers are higher in various etiologies, making them non-sensitive and non-specific |
| Zhao et al., Am J Physiol Renal Physiol (2019) [58] | ATIN n = 44Healthy controls n = 24 | ATIN patients had higher urinary levels of KIM-1 and C5b9 compared with healthy controls; urinary C5b9 correlated with the extent of tubulointerstitial infiltrates in kidney biopsy in ATIN patients | Several studies have found that these biomarkers are higher in various etiologies, making them non-sensitive and non-specific |
| Yun et al., BMC Nephrol (2019) [59] | ATIN n = 113Healthy controls n = 40 | Serum IL-1β, IFN-α2, TNF-α, MCP-1, IL-8, IL-17A, IL-18 and IL-23 were higher in ATIN patients compared with healthy controls; urinary IFN-α2, MCP-1, IL-6, IL-8, IL-12p70 and IL-17A were higher in ATIN patients compared with healthy controls | There are various biomarkers that may be due to the combined study of various etiologies; therefore, it is necessary to study among various etiologies in order to find specific biomarkers for each etiology |
| Moledina et al., JCI Insight (2019) [60] | ATIN n = 32Other kidney diseases n = 186 | Urinary TNF-α and IL-9 were higher in ATIN patients compared with other kidney diseases; urinary IL-5 was higher among ATIN patients with prominent eosinophil infiltrates | ATIN vs other kidney diseases, cannot analyze such findings among various ATIN etiologies from this study |
| Moledina et al., Nephron (2019) [61] | ATIN n = 32ATN n = 41 | Urinary TNF-α and IL-9 were higher in ATIN patients | ATIN vs ATN, cannot analyze such findings among various ATIN etiologies from this study |
| Moledina et al., JCI (2023) [62] | AIN n = 31Healthy controls n = 57 | CXCL9 was significantly higher in AIN patients compared with healthy controls | It is therefore necessary to further study to determine the correlation between various etiologies of AIN |
NAG: N-acetyl-neuraminidase; α1-MG: α1-microglobulin; NGAL: neutrophil gelatinase-associated lipocalin; TGF-β: transforming growth factor-β; GFR: glomerular filtration rate; KIM-1: kidney injury molecule.
FEATURES OF INFECTION IN CORRELATION WITH AIN
Infection-induced AIN usually results in a sterile infiltrate, indicating that immunological disturbances may be the cause of AIN [65]. Therefore, in most instances, blood or urine culture may not be effective in identifying infection as an etiology of AIN (Table 1) [32–48]. An early diagnosis of the cause is essential to eliminate and prevent the responsible causes from occurring in the future [65]. Sonoda et al. [66] also found that local infection may induce an autoimmune response against autoantigens in the infected kidney, which could trigger organ-specific autoimmune disease. Therefore, it leads to confusion in determining whether infection is the root cause or autoimmune disease. Previously, infections were the primary causes of AIN; however, in recent times, an immuno-allergic mechanism triggered by medications such as antibiotics, NSAIDs and numerous others has emerged as the predominant cause [1].
Since infections and antibiotics induce AIN through autoimmune and immuno-allergic pathomechanisms, respectively, it is possible that their histopathological and clinical characteristics may overlap. According to some studies, the leading cause of AIN is bacterial pyelonephritis, which can be unilateral and typically localized to the renal pyramid [67]. In this instance, a healthcare professional can utilize positive uroculture, or, in most instances, blood cultures as a diagnostic tool for infection-associated AKI, particularly when AKl occurs in the absence of septic shock [67]. The presence of a large number of neutrophils [1], particularly as micro-abscesses, should alert one to the possibility of pyelonephritis [68]. The study conducted by Raghavan et al. [6] revealed a significant amount of neutrophilic infiltration, which tends to be negative on immunofluorescence microscopy. Such cases can present with both oliguric and non-oliguric renal insufficiency, without the classical clinical triad of AIN (fever, rash and arthralgia), usually with reversible renal function if infection is treated in a timely manner [37] (Tables 2 and 4 [49]). In patients with infection-induced AIN, steroids may increase the risk of immunosuppression, which could worsen the infection. Therefore, steroids should be delayed until the active infection is completely controlled [13–15] (Table 1 [38–48]).
Table 4:
Urinalysis in AIN (Nussbaum et al., Clin Kidney J 2019) [49].
| Basic urinalysis | Urine microscopy | Urine eosinophils | Urine chemistries |
|---|---|---|---|
| Proteinuria: present in 90% of cases | WBC casts: present in 3%–14% of cases | Sensitivity: 31% | FENa: can be >1% or <1% |
| Nephrotic-range proteinuria rare | RBC casts: present in up to 29% cases | Specificity: 68% | FEUrea: can be >35% or <35% |
| Hematuria: present in 50% of cases | RTE and granular casts: present in up to 86% cases | Also present in ATN, GN and other renal diseases | |
| Pyuria: present in 50%–80% of cases | Bland urine sediment: present in 20% cases |
RTE: renal tubular epithelial; GN: glomerulonephritis; FENa: fractional excretion of Na; FEUrea: fractional excretion of urea.
FEATURES OF ANTIBIOTICS ASSOCIATED WITH AIN
The capacity to elicit an immune response to β-lactams (BLs) can exhibit varying degrees of variability: some patients exhibit a specific response to a specific BL and demonstrate tolerance to others, whereas other patients exhibit a universal response to all BLs [69] (Table 1 [32–38]). This phenomenon may be attributable to the fact that distinct patients may possess distinct HLA alleles that exhibit distinct responses to these BLs. Therefore, further investigation is necessary to identify specific HLA alleles that are more susceptible to AIN [64]. Perazella et al. [1] observed relatively short duration of exposure to the causative BL antibiotic, ranging from a few days to a few weeks, with the classical clinical triad of fever, rash or eosinophilia in >75% of patients, and the occurrence of proteinuria, leukocyturia or hematuria in approximately 75% of affected patients. AIN can also occur in association with the administration of non-BL antibiotics, such as rifampicin, when administered intermittently to treat mycobacterial infections (Table 1 [32–48]). The occurrence of AIN caused by sulfonamide antibiotics may be associated with typical hypersensitivity reactions, including fever, rash and eosinophilia [70]. Patients who have been infected with HIV, transplant recipients and patients who have pre-existing renal disease are at a higher risk of developing sulfonamide-induced AIN [71, 72]. The fluoroquinolone antibiotics, especially ciprofloxacin, can induce AIN without hypersensitivity syndrome [1].
Renal pathologists suspect the drug as an offending cause when a significant eosinophilic infiltrate (>10 eosinophils per 20× field) is present [1]. Immunofluorescence microscopy is typically negative in patients with AIN, although a very few cases of methicillin-induced AIN with tubular basement membrane deposits of immunoglobulin have been reported [73]. Immune complex deposits are relatively uncommon in drug-induced AIN—they have been reported with drugs such as methicillin and rifampin [73–75], which exhibit linear or granular staining for immunoglobulin G and C3 on tubular basement membrane (anti–tubular basement membrane antibodies). For example, the presence of hypersensitivity may be due to drugs such as BLs or sulfonamides. Asim et al. [76] presented a case of hemorrhagic tubulointerstitial nephritis caused by amoxicillin–clavulanate, characterized by systemic eosinophilia, a high concentration of eosinophils and plasma cells in the interstitial infiltrate, indicating a delayed hypersensitivity immune response mediated by antigen-reactive T cells. This case demonstrates an idiosyncratic immune response, with no correlation between the dose or the duration of amoxicillin–clavulanate therapy [6, 14, 77]. Although the alternative medication is typically recommended in cases of medication hypersensitivity associated AIN [3], it is important to note that there may be cross-reactivity between the two drugs. For instance, a case of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome was reported, wherein cross-reactivity was observed between vancomycin and subsequent teicoplanin administration [78], thereby enhancing the difficulty in determining the precise etiology. Similarly, several studies have found that patients with penicillin allergy have a higher risk of developing cephalosporin hypersensitivity associated AIN, but this finding is not universal [25, 79, 80]. The structural features of BLs could be the reason for this, as all BLs contain a five- or six-member ring, with the exception of monobactams (e.g. aztreonam), which have no reported cross-reactivity with penicillins [20].
CLINICAL HISTORY AND PHYSICAL EXAMINATION IN RELATION TO INFECTION AND ANTIBIOTICS
A careful history can provide a clue about the offending agent but not a definitive confirmation, as previously described (Tables 1, 2 and 4 [32–48, 49]). For instance, the onset of either a mild or intermittently spiking fever typically occurs within a span of 2 weeks following the administration of a medication [1]. However, in the event that the patient has been previously exposed to the aforementioned drug, the duration of fever may be limited to a few hours or days [25]. The occurrence of fever as a systemic feature of AIN is variable, and the rate of variation is contingent upon the class of drug involved [1] (Table 1 [32–38]). Fever may be absent in AIN caused by many drugs, such as NSAIDs and non-BL medications, but it has been manifested in as many as 50%–100% of patients with AIN caused by penicillin derivatives, particularly methicillin [25, 77, 80–82]. The occurrence of AIN-associated rash is observed in 15%–50% of patients with drug-induced AIN, particularly with agents that can trigger a hypersensitivity reaction, such as penicillin derivatives, sulfonamides, allopurinol and phenytoin [11, 16, 80]. Patients receiving rifampicin frequently experience symptoms of flank pain and tenderness, resulting from the distention of the renal capsule caused by inflammation and parenchymal swelling [83, 84].
CLASSICAL BIOMARKERS OF AIN IN RELATION TO INFECTION AND ANTIBIOTIC
Commonly ordered tests
A series of urine and serum tests is frequently ordered to assess and distinguish the precise cause of AIN. According to a recently published study, dipstick pyuria was observed in 60–80% of AIN cases caused by antibiotics [10] (Tables 2 and 4 [49]). However, the absence of pyuria does not exclude the diagnosis [63]. Numerous studies on AIN have reported the presence of mild to moderate proteinuria, which is incapable of distinguishing between infection or antibiotic as the causative agent, or among other AIN etiologies [49]. However, nephrotic-range proteinuria is rare [4, 5, 10, 28, 85, 86], and when found in the setting of AIN, it should raise suspicion for NSAID-induced nephrotoxicity or underlying glomerular diseases [5, 10, 87, 88]. Hematuria is also a nonspecific and insensitive laboratory test, despite several studies indicating a mean of 50% (with a range of 20%–80%) in cases of AIN [10, 85]. It is more common with some drugs, especially methicillin and the BL class of antibiotics, which can cause hematuria in up to 90% of patients with BL antibiotic-induced AIN [25]. The presence of white blood cell (WBC) casts has been demonstrated in the context of intrarenal infection, such as pyelonephritis, or in conjunction with inflammatory renal lesions, such as proliferative glomerulonephritis and AIN, rendering them non-specific and insensitive for AIN [49]. Numerous studies have revealed that eosinophilia was observed in 80% of cases of drug-induced AIN caused by BL antibiotics, such as methicillin, while it was only observed in less than one-third of cases of drug-induced AIN caused by non-BL antibiotics [16, 80, 89]. Eosinophiluria cannot be used in diagnosing AIN because it is detected in many renal and nonrenal diseases such as pyelonephritis, cystitis, prostatitis, atheroembolic disease, acute tubular nephritis, rapidly progressive glomerulonephritis and bladder malignancies, among others [50, 73, 90, 91].
Novel biomarkers
In recent times, a variety of novel biomarkers have been evaluated for the identification of the distinct characteristics of interstitial infiltrates and the cytokines produced by this infiltrate, in relation to other inflammatory renal diseases (Table 3). Their detection in serum or urine has been identified as potential diagnostic and prognostic tools for ATIN. The presence of specific chemokines and cytokines can provide a clue towards a potential causative agent, as the pathophysiologic process may vary depending on the etiology. For instance, Moledina et al. [62] observed a higher level of the major eosinophilic attractant C-C motif ligand 3 (CCL3) in patients with AIN compared with the control group. This finding may serve as a means to distinguish patients with AIN due to antibiotics, as they are known to have a higher number of interstitial eosinophils. On the contrary, it is possible that the level of monocyte chemoattractant protein-1 (MCP-1) is elevated in infection-induced AIN due to its significant role in the recruitment of monocytes, neutrophils and lymphocytes in tissue inflammation processes [63]. Furthermore, several studies have reported a higher level of neutrophils in infection-induced AIN [6]. However, it is not feasible to assess its sensitiveness or specificity as it has been identified as a chemokine implicated in numerous autoimmune diseases [92]. It is noteworthy that Muhammad et al. [93] hypothesized that drugs may be able to induce autoimmune diseases that induce AIN. Therefore, researchers are uncertain about the causative agent that triggers the MCP-1 level in such a scenario. Wu et al. [53] also observed a higher level of MCP-1 in a cohort of 40 patients with drug-induced AIN in comparison with controls. Similarly, Yun et al. [59] found a higher level of MCP-1 in serum and urine in 113 patients with AIN, resulting from different causes, through a bead-based multiplex assay. Despite the findings of Moledina et al. [60, 62, 94], who observed a higher level of urinary C-X-C motif ligand 9 (CXCL9), interleukin-9 (IL-9) and tumor necrosis factor-α (TNF-α) levels in patients with AIN, these biomarkers are unable to definitively identify the causative agent among various etiologies. As IL-9 and IL-5 are implicated in allergic responses and promotes mast cell accumulation, which in turn is a source of TNF-α [60, 61, 95–98], it is plausible that these cytokines, along with CCL3, may provide a clue in identifying the culprit drug in hypersensitivity-associated AIN. In a similar context, elevated levels of IL-6 and IL-8 may indicate infection as the causative agent of AIN [59, 98–101]. However, there are no published studies that specifically demonstrate the significance of these cytokines as indicators of the offending agent.
Cellular biomarkers
The binding of T cells to a drug is a complicated process that can be measured through cellular assays based on the demonstration of the lymphocyte proliferation response and the cytokine secretion response when exposed to the suspected drug [63]. Fluorescence cytometry techniques can be employed to assess the activation phenotype of lymphocytes subsequent to drug exposure. Several studies have demonstrated the detection of activation markers on the surface of lymphocytes subsequent to incubation with the offending drug, such as CD25, CD69 or HLA drug reaction (DR), indicating hypersensitivity [95, 101].
A sensitive test, the enzyme-linked immunospot (ELISpot) assay, has been reported to be useful in evaluating drug hypersensitivity reactions. It allows the ex vivo measurement of the release of cytokines by lymphocytes in response to a certain stimulus [102]. There is, however, limited evidence regarding the usefulness of this assays specifically in patients with AIN. Punrin et al. [103] reported that 50% of patients with drug-induced ATIN had a positive interferon (IFN)-γ ELISpot assay in a cohort study.
Lymphocyte transformation test
It is possible to use the lymphocyte transformation test (LTT) to measure the proliferation of lymphocytes in response to the pure form of a suspicious drug. Koda et al. [104] reported the utilization of LTT in identifying the culprit drug in ATIN among lansoprazole and nivolumab, and it demonstrated reactivity against lansoprazole, but not against nivolumab. Positive LTT has also been reported in the context of ATIN induced by BLs and NSAIDs [98]. Nonetheless, the LTT concept can yield accurate results only if drugs are able to directly interact with the T-cell receptor and act as hapten, without any prior metabolism or binding to proteins [98]. Beta-lactams are known to be able to act as haptens and bind to amino groups of amino acids, such as lysine [105]. However, clinicians should understand that sometimes the reaction is not caused by the drug itself, but by a component within the drug or a metabolite which transforms this so-called pro-hapten to hapten [106], which may result in negative LTT. Sulfamethoxazole is a typical example of a drug that acts as prohapten, because it is transformed to sulfamethoxazole–hydroxylamine and further oxidized to sulfamethoxazole–nitroso [107–109]. Drugs that exhibit pseudoallergic responses in nonimmune-mediated hypersensitivity reactions by affecting the innate immune system and/or effector cells, such as basophils directly, may also be tested negative, without any evidence of involvement of the specific immune system [98]. The possible drawback of this method could be the long time it takes for the procedure to display the result, because in the acute setting, the LTT has a low specificity because sensitization does not always associate disease [110]. Chung et al. [111] confirmed through LTT that vancomycin, but not other glycopeptide antibiotics, specifically induced T-cell proliferation in the reported case.
Genetic biomarkers
Some studies have described the association of AIN, mainly in the setting of TINU, with certain HLA or single-nucleotide polymorphisms as markers of disease susceptibility. According to a study conducted in a cohort of 154 Chinese patients with different causes of AIN, HLA-DQA1*0104/DQB1*0503/DRB1*1405 are risk haplotypes for the development of AIN [64]. These studies suggest that these variants may enhance antigen presentation and facilitate renal interstitial inflammation. Therefore, it is important to determine which specific HLA alleles are more susceptible to which etiology of AIN. We may consider combining this HLA testing for diverse etiologies of AIN with adjunctive testing, such as IFN-γ ELISpot, to obtain a more favorable outcome.
DISCUSSION
This narrative review demonstrates the relevant features of ATIN in the setting of infection, as opposed to the situation of a drug-induced immune allergic reaction, with a special focus on the antibiotic-associated ATIN. The main purpose of the study was to analyze the peculiar findings that are typical of an infection-associated ATIN compared with an antibiotic-associated ATIN caused by an allergic reaction. Although these findings cannot provide conclusive and clear recommendations that can be useful in the clinical practice, they might encourage researchers to conduct original research on these features to find clear recommendations.
Hospital-acquired AKI affects up to 10%–27% of patients affected by AIN, which has various etiologies, among which antibiotics and infections account for up to 35% and 16% of cases, respectively [4–9]. Both etiologies necessitate distinct therapeutic measures in a timely manner, as previously described [13–15]. The delay in identifying the precise culprit agent can impede therapeutic measures, ultimately leading to renal fibrosis and CKD [1, 3, 11, 13, 18, 19]. Previous studies have demonstrated that, in up to 30% of cases, the culprit drug cannot be precisely identified, and this type of misdiagnosis can lead to partial renal recovery, recurrent disease or CKD in up to 40%–60% of cases [20–25]. Therefore, a timely and thorough investigation is required for complete recovery. Currently, there is no universal procedure that can be utilized to determine the precise cause, despite the fact that certain clinical and histopathological features may provide a clue towards the potential suspected cause, as previously described. Therefore, it is imperative for clinicians to exercise caution in selecting the appropriate therapeutic measures, particularly when both infections and antibiotic-related causes are suspected; however, they treat patients on the basis of clinical history, physical examinations and biopsy findings, without caring much about the exact etiology.
We have highlighted various characteristics associated with antibiotics and infections that give clues, but cannot be deemed universal findings. For example, an increase in neutrophil levels points towards infection, while an increase in eosinophils points towards hypersensitivity to drugs [6]. In contrast, an increase in neutrophils can also be caused by urinary tract infections. Similarly, different antibiotics exhibit different hypersensitive reactions in different patients, with some individuals exhibiting tolerability to certain drugs while others exhibit hypersensitivity to other drugs. This can be challenging to discern when taking multiple antibiotics. Clinicians also follow the typical triad of fever, rash and arthralgia, but it is not always helpful. The time period from drug exposure, which typically ranges from 7–10 days, is utilized as a diagnostic indicator [20]. However, this method is not universally accurate for all drugs, particularly when a patient is exposed to the same drug for the second time, where the hypersensitivity reaction can be more rapid, as previously described. In cases of suspected infection, some clinicians also rely on blood or urine culture, but this may not be effective because it usually results in a sterile infiltrate, as infection induces AIN primarily through immunological disturbances. Thus, both etiologies may have similar histopathological and clinical characteristics due to the same immunological pathomechanism. Clinicians may also prescribe alternative medications in case of hypersensitivity to one drug, but there may be cross-reactivity between the previous drug and the alternative, which may be misleading in finding the offending agent.
Various serum and urine tests, including dipstick pyuria, proteinuria, hematuria and WBCs, are routinely ordered and relied upon to distinguish between the two etiologies, however they are neither sensitive nor specific, as previously described. Numerous novel biomarkers including CCL3, CXCL9, MCP-1, TNF-α, IL-9, IL-5, IL-6 and IL-8 have been evaluated for the identification of the distinct characteristics of interstitial infiltrates in different etiologies. However, they are not routinely performed and are not recommended at universal guidelines. There are some reports that indicate the utility of the ELISpot assay in evaluating drug hypersensitivity reactions [64]. However, there is limited evidence regarding its efficacy in patients with AIN. The LTT possesses several drawbacks, including the inability to display results promptly and its inability to be utilized across all classes of drugs due to the variability of drug metabolism, rendering it unsuitable for routine use. Several studies have published associations between ATIN and certain HLA or single-nucleotide polymorphisms as markers of disease susceptibility, mainly in the setting of TINU [64]. However, there is currently no further investigation into the identification of HLA risk alleles in other causes of AIN, which is crucial for determining the prognosis of AIN.
In brief, our study does not provide conclusive and clear recommendations that can be useful in the clinical practice, and further research is required regarding the features described above.
Contributor Information
Amir Muhammad, Department of Nephrology, Xiangya Hospital, Central South University, Changsha, China.
Yingli Zhang, Department of Nephrology, Third Hospital of Changsha, Changsha, China.
Ling Huang, Department of Nephrology, Xiangya Hospital, Central South University, Changsha, China.
Qiongjing Yuan, Department of Nephrology, Xiangya Hospital, Central South University, Changsha, China.
Wei Wang, Department of Nephrology, Xiangya Hospital, Central South University, Changsha, China.
Jiaxi Pu, Department of Nephrology, Xiangya Hospital, Central South University, Changsha, China.
Wei Lin, Department of Pathology, Xiangya Hospital, Central South University, Changsha, China.
Rong Tang, Department of Nephrology, Xiangya Hospital, Central South University, Changsha, China.
Xiangcheng Xiao, Department of Nephrology, Xiangya Hospital, Central South University, Changsha, China.
STATEMENT OF ETHICS
The study was approved by the Medical Ethics Committee of Xiangya Hospital of Central South University and followed the Declaration of Helsinki.
FUNDING
This work was supported by National Key Research and Development Program of China, grant number 2020YFC2005000; Natural Science Foundation of Hunan Province, grant number 2022JJ30070.
AUTHORS’ CONTRIBUTIONS
Formal analysis and writing—original draft preparation: A.M. Conception, supervision and writing—review and editing: R.T., X.X. Revision: Y.Z., L.H., Q.Y., W.W., J.P. Pathology guidance: W.L. All authors have read and approved the final manuscript.
DATA AVAILABILITY STATEMENT
All data included in this study are available upon request from the corresponding author.
CONFLICT OF INTEREST STATEMENT
The authors declare that there were no conflicts of interest.
REFERENCES
- 1. Perazella MA, Markowitz GS. Drug-induced acute interstitial nephritis. Nat Rev Nephrol 2010;6:461–70. 10.1038/nrneph.2010.71 [DOI] [PubMed] [Google Scholar]
- 2. Tanaka T, Nangaku M. Pathogenesis of tubular interstitial nephritis. Contrib Nephrol 2011;169:297–310. 10.1159/000314577 [DOI] [PubMed] [Google Scholar]
- 3. Krishnan N, Perazella MA. Drug-induced acute interstitial nephritis: pathology, pathogenesis, and treatment. Iran J Kidney Dis 2015;9:3–13. [PubMed] [Google Scholar]
- 4. Perazella MA. Clinical approach to diagnosing acute and chronic tubulointerstitial disease. Adv Chronic Kidney Dis 2017;24:57–63. 10.1053/j.ackd.2016.08.003 [DOI] [PubMed] [Google Scholar]
- 5. Perazella MA. Diagnosing drug-induced AIN in the hospitalized patient: a challenge for the clinician. Clin Nephrol 2014;81:381–8. 10.5414/CN108301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Raghavan R, Eknoyan G. Acute interstitial nephritis—a reappraisal and update. Clin Nephrol 2014;82:149–62. 10.5414/cn108386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nast CC. Medication-induced interstitial nephritis in the 21st century. Adv Chronic Kidney Dis 2017;24:72–9. 10.1053/j.ackd.2016.11.016 [DOI] [PubMed] [Google Scholar]
- 8. Gilbert SJ, Weiner DE, Bomback AS et al. National Kidney Foundation's Primer on Kidney Diseases, 7th edn. Philadelphia, PA: Elsevier, 2018, 299–326. [Google Scholar]
- 9. Liano F, Pascual J. Epidemiology of acute renal failure: a prospective multicenter, community-based study. Kidney Int 1996;50:811–8. 10.1038/ki.1996.380 [DOI] [PubMed] [Google Scholar]
- 10. Moledina DG, Perazella MA. Drug-induced acute interstitial Nephritis. Clin J Am Soc Nephrol 2017;12:2046–9. 10.2215/CJN.07630717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baker RJ, Pusey CD. The changing profile of acute tubulointerstitial nephritis. Nephrol Dial Transplant 2004;19:8–11. 10.1093/ndt/gfg464 [DOI] [PubMed] [Google Scholar]
- 12. Valluri A, Hetherington L, Mcquarrie E et al. Acute tubulointerstitial nephritis in Scotland. QJM 2015;108:527–32. 10.1093/qjmed/hcu236 [DOI] [PubMed] [Google Scholar]
- 13. Barreto EF, Rule AD. Management of drug-associated acute Interstitial Nephritis. Kidney360 2020;1:62–4. 10.34067/KID.0000042019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Praga M, González E. Acute interstitial nephritis. Kidney Int 2010;77:956–61. 10.1038/ki.2010.89 [DOI] [PubMed] [Google Scholar]
- 15. Perazella MA, Rosner MH. Drug-induced acute kidney injury. Clin J Am Soc Nephrol 2022;17:1220–33. 10.2215/CJN.11290821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clarkson MR, Giblin L, O'Connell FP et al. Acute interstitial nephritis: clinical features and response to corticosteroid therapy. Nephrol Dial Transplant 2004;19:2778–83. 10.1093/ndt/gfh485 [DOI] [PubMed] [Google Scholar]
- 17. González E, Gutiérrez E, Galeano C et al. Early steroid treatment improves the recovery of renal function in patients with drug-induced acute interstitial nephritis. Kidney Int 2008;73:940–6. 10.1038/sj.ki.5002776 [DOI] [PubMed] [Google Scholar]
- 18. Caravaca-Fontán F, Shabaka A, Sánchez-Álamo B et al. Recurrent acute interstitial nephritis: what lies beneath. Clin Kidney J 2020;14:197–204. 10.1093/ckj/sfaa018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Robles NR, Lopez-Gomez J, Garcia-Pino G et al. Use of α1-microglobulin for diagnosing chronic interstitial nephropathy. Clin Exp Med 2014;14:315–20. 10.1007/s10238-013-0242-9 [DOI] [PubMed] [Google Scholar]
- 20. Raghavan R, Shawar S. Mechanisms of drug-induced interstitial nephritis. Adv Chronic Kidney Dis 2017;24:64–71. 10.1053/j.ackd.2016.11.004 [DOI] [PubMed] [Google Scholar]
- 21. Verghese PS, Luckritz KE, Eddy AA. Interstitial nephritis in children. In: Denis F. Geary and Franz Schaefer (eds.). Pediatric Kidney Disease. Division of Nephrology. Springer, 2016, 1013–36. 10.1007/978-3-662-52972-0_38 [DOI] [Google Scholar]
- 22. Dibek Misirlioglu E, Guvenir H, Bahceci S et al. Severe cutaneous adverse drug reactions in pediatric patients: a multicenter study. J Allergy Clin Immunol Pract 2017;5:757–63. 10.1016/j.jaip.2017.02.013 [DOI] [PubMed] [Google Scholar]
- 23. Geevasinga N, Coleman PL, Webster AC et al. Proton pump inhibitors and acute interstitial nephritis. Clin Gastroenterol Hepatol 2006;4:597–604. 10.1016/j.cgh.2005.11.004 [DOI] [PubMed] [Google Scholar]
- 24. Buysen JG, Houthoff HJ, Krediet RT et al. Acute interstitial nephritis: a clinical and morphological study in 27 patients. Nephrol Dial Transplant 1990;5:94–9. 10.1093/ndt/5.2.94 [DOI] [PubMed] [Google Scholar]
- 25. Rossert J. Drug-induced acute interstitial nephritis. Kidney Int 2001;60:804–17. 10.1046/j.1523-1755.2001.060002804.x [DOI] [PubMed] [Google Scholar]
- 26. Aziz A, Yaqub S, Awan S et al. Clinicopathological characteristics of drug-induced acute interstitial nephritis and role of steroids in management: a single-center observational study. World J Nephrol Urol 2022;11:24–30. 10.14740/wjnu427 [DOI] [Google Scholar]
- 27. Fernandez-Juarez G, Perez JV, Caravaca-Fontán F et al. Duration of treatment with corticosteroids and recovery of kidney function in acute interstitial nephritis. Clin J Am Soc Nephrol 2018;13:1851–8. 10.2215/CJN.01390118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Muriithi AK, Leung N, Valeri AM et al. Biopsy-proven acute interstitial nephritis, 1993-2011: a case series. Am J Kidney Dis 2014;64:558–66. 10.1053/j.ajkd.2014.04.027 [DOI] [PubMed] [Google Scholar]
- 29. Praga M, Sevillano A, Aunon P et al. Changes in the aetiology, clinical presentation and management of acute interstitial nephritis, an increasingly common cause of acute kidney injury. Nephrol Dial Transplant 2015;30:1472–9. 10.1093/ndt/gfu326 [DOI] [PubMed] [Google Scholar]
- 30. Oliva-Damaso N, Oliva-Damaso E, Payan J. Acute and chronic tubulointerstitial nephritis of rheumatic causes. Rheum Dis Clin North Am 2018;44:619–33. 10.1016/j.rdc.2018.06.009 [DOI] [PubMed] [Google Scholar]
- 31. Su T, Gu Y, Sun P et al. Etiology and renal outcomes of acute tubulointerstitial nephritis: a single-center prospective cohort study in China. Nephrol Dial Transplant 2018;33:1180–8. 10.1093/ndt/gfx247 [DOI] [PubMed] [Google Scholar]
- 32. Hung CC, Kuo MC, Chang JM et al. Fluoroquinolone-induced acute interstitial nephritis in immunocompromised patients: two case reports. Nephrol Dial Transplant 2006;21:237–8. 10.1093/ndt/gfi178. [DOI] [PubMed] [Google Scholar]
- 33. Salih SB, Kharal M, Qahtani M et al. Acute interstitial nephritis induced by intermittent use of rifampicin in patient with brucellosis. Saudi Journal of Kidney Diseases and Transplantation 2008;19:450–2. [PubMed] [Google Scholar]
- 34. Kannan L, Raj R. Case report: vancomycin-associated tubulointerstitial nephritis in clinical practice-case report and review of literature. Frontiers in Medicine 2022;9:899886. 10.3389/fmed.2022.899886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sharma K, Geagan N, Tengsupakul S. Severe acute interstitial nephritis secondary to minocycline use in an adolescent girl. SAGE Open Medical Case Reports 2020;8:2050313X20943069. 10.1177/2050313X20943069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Asnake M, Henock A, Abayneh M et al. Acute interstitial nephritis with prothionamide. SAGE Open Medical Case Reports 2022;10:2050313X221094076. 10.1177/2050313X221094076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Raina R, Ale S, Chaturvedi T et al. Infection associated acute interstitial nephritis; a case report. J Nephropathol 2017;6:53–57. 10.15171/jnp.2017.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Caers J, Peeters P, Houte KV et al. Acute interstitial nephritis associated with salmonellosis. European Journal of Internal Medicine 2006;17:217–9. 10.1016/j.ejim.2005.09.026. [DOI] [PubMed] [Google Scholar]
- 39. Daumas A, El-Mekaoui F, Bataille S et al. Acute tubulointerstitial nephritis complicating Legionnaires' disease: a case report. Journal of Medical Case Reports 2012;6:1–6. 10.1186/1752-1947-6-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Iijima K, Yoshikawa N, Sato K et al. Acute interstitial nephritis associated with Yersinia pseudotuberculosis infection. Am J Nephrol 1989;9:236–40. 10.1159/000167971. [DOI] [PubMed] [Google Scholar]
- 41. Dagli O, Dokur M, Guzeldag G et al. Acute renal failure due to Brucella melitensis. The Journal of Infection in Developing Countries 2011;5:893–5. 10.3855/jidc.1442. [DOI] [PubMed] [Google Scholar]
- 42. Rautelin HI, Outinen AV, Kosunen TU et al. Tubulointerstitial nephritis as a complication of Campylobacter jejuni enteritis: a case report. Scand J Urol Nephrol 1987;21:151–2. 10.3109/00365598709180313. [DOI] [PubMed] [Google Scholar]
- 43. Chang JF, Peng YS, Tsai CC et al. A possible rare cause of renal failure in streptococcal infection. Nephrol Dial Transplant 2011;26:368–71. 10.1093/ndt/gfq569. [DOI] [PubMed] [Google Scholar]
- 44. Phillips J, Palmer A, Baliga R. Glomerulonephritis associated with acute pneumococcal pneumonia: a case report. Pediatr Nephrol 2005;20:1494–5. 10.1007/s00467-005-1994-6. [DOI] [PubMed] [Google Scholar]
- 45. Kwon HJ, Yoo KH, Kim IY et al. Megalocytic interstitial nephritis following acute pyelonephritis with Escherichia coli bacteremia: a case report. J Korean Med Sci 2015;30:110. 10.3346/jkms.2015.30.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Prabhu RA, Shaw T, Rao IR et al. Acute kidney injury and its outcomes in melioidosis. J Nephrol 2021;1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim DY, Park HS, Han DJ et al. A case of scrub typhus requiring maintenance hemodialysis. Kidney Research and Clinical Practice 2013;32:190–3. 10.1016/j.krcp.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Daher ED, Abreu KL, Silva Junior GB. Leptospirosis-associated acute kidney injury. Brazilian Journal of Nephrology 2010;32:408–15. 10.1590/S0101-28002010000400010.21541456 [DOI] [Google Scholar]
- 49. Nussbaum EZ, Perazella MA. Diagnosing acute interstitial nephritis: considerations for clinicians. Clin Kidney J 2019;12:808–13. [Google Scholar]
- 50. Councilman WT. Acute interstitial nephritis. J Exp Med 1898;3:393–420. 10.1084/jem.3.4-5.393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Caravaca-Fontán F, Fernández-Juárez G, Praga M. Acute kidney injury in interstitial nephritis. Curr Opin Crit Care 2019;25:558–64. 10.1097/MCC.0000000000000654 [DOI] [PubMed] [Google Scholar]
- 52. Dantas M, Almeida Romão E, Silva Costa R et al. Urinary excretion of monocyte chemoattractant protein-1: a biomarker of active tubulointerstitial damage in patients with glomerulopathies. Kidney and Blood Pressure Research 2007;30:306–13. 10.1159/000107806. [DOI] [PubMed] [Google Scholar]
- 53. Wu Y, Yang L, Su T et al. Pathological significance of a panel of urinary biomarkers in patients with drug-induced tubulointerstitial nephritis. Clinical Journal of the American Society of Nephrology 2010;5:1954–9. 10.2215/CJN.02370310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nakashima H, Miyake K, Moriyama M et al. An amplification of IL-10 and TGF-β in patients with IgG4-related tubulointerstitial nephritis. Clin Nephrol 2010;73:385. 10.5414/CNP73385. [DOI] [PubMed] [Google Scholar]
- 55. Shi Y, Su T, Qu L et al. Evaluation of urinary biomarkers for the prognosis of drug-associated chronic tubulointerstitial nephritis. Am J Med Sci 2013;346:283–8. 10.1097/MAJ.0b013e318271f910. [DOI] [PubMed] [Google Scholar]
- 56. Aoyagi J, Kanai T, Ito T et al. Cytokine dynamics in a 14-year-old girl with tubulointerstitial nephritis and uveitis syndrome. CEN Case Reports 2014;3:49–52. 10.1007/s13730-013-0084-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen Y, Zheng Y, Zhou Z et al. Baicalein alleviates tubular-interstitial nephritis in vivo and in vitro by down-regulating NF-κb and MAPK pathways. Braz J Med Biol Res 2018;51:e7476. Published 2018 Aug 6. 10.1590/1414-431x20187476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhao WT, Huang JW, Sun PP et al. Diagnostic roles of urinary kidney injury molecule 1 and soluble C5b-9 in acute tubulointerstitial nephritis. Am J Physiol Renal Physiol 2019;317:F584–92. 10.1152/ajprenal.00176.2019. [DOI] [PubMed] [Google Scholar]
- 59. Yun D, Jang MJ, An JN et al. Effect of steroids and relevant cytokine analysis in acute tubulointerstitial nephritis. BMC nephrology 2019;20:1–0. 10.1186/s12882-019-1277-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Moledina DG, Wilson FP, Pober JS et al. Urine TNF-α and IL-9 for clinical diagnosis of acute interstitial nephritis. JCI insight 2019;4. 10.1172/jci.insight.127456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Moledina DG, Parikh CR. Differentiating acute interstitial nephritis from acute tubular injury: a challenge for clinicians. Nephron 2019;143:211–6. 10.1159/000501207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Moledina DG, Obeid W, Smith RN et al. Identification and validation of urinary CXCL9 as a biomarker for diagnosis of acute interstitial nephritis. J Clin Invest 2023;133. 10.1172/JCI168950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Martinez Valenzuela L, Draibe J, Fulladosa X et al. New biomarkers in acute tubulointerstitial nephritis: a novel approach to a classic condition. Int J Mol Sci 2020;21:4690. 10.3390/ijms21134690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jia Y, Su T, Gu Y et al. HLA-DQA1, -DQB1, and -DRB1 alleles associated with acute tubulointerstitial nephritis in a Chinese population: a single-center cohort study. J Immunol 2018;201:423–31. 10.4049/jimmunol.1800237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ulinski T, Sellier-Leclerc AL, Tudorache E et al. Acute tubulointerstitial nephritis. Pediatr Nephrol 2012;27:1051–7. 10.1007/s00467-011-1915-9 [DOI] [PubMed] [Google Scholar]
- 66. Sonoda K, Mukasa A, Matsuzaki G et al. The induction of renal autoantigen-specific T cells by a local Listeria monocytogenes infection. Immunology 1995;86:190–8. [PMC free article] [PubMed] [Google Scholar]
- 67. Schurder J, Buob D, Perrin P et al. Acute interstitial nephritis: aetiology and management. Nephrol Dial Transplant 2021;36:1799–802. 10.1093/ndt/gfz262 [DOI] [PubMed] [Google Scholar]
- 68. John R, Herzenberg AM. Renal toxicity of therapeutic drugs. J Clin Pathol 2009;62:505–15. 10.1136/jcp.2008.058271 [DOI] [PubMed] [Google Scholar]
- 69. Gruchalla RS, Pirmohamed M. Clinical practice. Antibiotic allergy. N Engl J Med 2006;354:601–9. 10.1056/NEJMcp043986 [DOI] [PubMed] [Google Scholar]
- 70. Kleinknecht D, Vanhille P, Morel-Maroger L et al. Acute interstitial nephritis due to drug hypersensitivity. An up-to-date review with a report of 19 cases. Adv Nephrol Necker Hosp 1983;12:277–308. [PubMed] [Google Scholar]
- 71. Perazella MA. Acute renal failure in HIV-infected patients: a brief review of common causes. Am J Med Sci 2000;319:385–91. 10.1097/00000441-200006000-00008 [DOI] [PubMed] [Google Scholar]
- 72. Perazella MA. Drug-induced renal failure: update on new medications and unique mechanisms of nephrotoxicity. Am J Med Sci 2003;325:349–62. 10.1097/00000441-200306000-00006 [DOI] [PubMed] [Google Scholar]
- 73. Border WA, Lehman DH, Egan JD et al. Antitubular basement-membrane antibodies in methicillin-associated interstitial nephritis. N Engl J Med 1974;291:381–4. 10.1056/NEJM197408222910803 [DOI] [PubMed] [Google Scholar]
- 74. Gabow PA, Lacher JW, Neff TA. Tubulointerstitial and glomerular nephritis associated with rifampin. Report of a case. JAMA 1976;235:2517–8. 10.1001/jama.235.23.2517 [DOI] [PubMed] [Google Scholar]
- 75. De Vriese AS, Robbrecht DL, Vanholder RC et al. Rifampicin-associated acute renal failure: pathophysiologic, immunologic, and clinical features. Am J Kidney Dis 1998;31:108–15. 10.1053/ajkd.1998.v31.pm9428460 [DOI] [PubMed] [Google Scholar]
- 76. Asim M, Ahmad F, Akhtar M. Florid interstitial hemorrhages: a novel feature of amoxicillin-clavulanate-induced acute tubulointerstitial nephritis. Am J Case Rep 2021;22:e928989. 10.12659/AJCR.928989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kodner CM, Kudrimoti A. Diagnosis and management of acute interstitial nephritis. Am Fam Physician 2003;67:2527–34. [PubMed] [Google Scholar]
- 78. Miyazu D, Kodama N, Yamashita D et al. DRESS syndrome caused by cross-reactivity between vancomycin and subsequent teicoplanin administration: a case report. Am J Case Rep 2016;17:625–31. 10.12659/ajcr.899149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Alexopoulos E. Drug-induced acute interstitial nephritis. Ren Fail 1998;20:809–19. 10.3109/08860229809045178 [DOI] [PubMed] [Google Scholar]
- 80. Toto RD. Acute tubulointerstitial nephritis. Am J Med Sci 1990;299:392–410. 10.1097/00000441-199006000-00007 [DOI] [PubMed] [Google Scholar]
- 81. Bhaumik SK, Kher V, Arora P et al. Evaluation of clinical and histological prognostic markers in drug-induced acute interstitial nephritis. Ren Fail 1996;18:97–104. 10.3109/08860229609052779 [DOI] [PubMed] [Google Scholar]
- 82. Ooi BS, Jao W, First MR et al. Acute interstitial nephritis. A clinical and pathologic study based on renal biopsies. Am J Med 1975;59:614–28. 10.1016/0002-9343(75)90223-5 [DOI] [PubMed] [Google Scholar]
- 83. Simenhoff ML, Guild WR, Dammin GJ. Acute diffuse interstitial nephritis. Review of the literature and case report. Am J Med 1968;44:618–25. 10.1016/0002-9343(68)90063-6 [DOI] [PubMed] [Google Scholar]
- 84. Baker SB, Williams RT. Acute interstitial nephritis due to drug sensitivity. Br Med J 1963;1:1655–8. 10.1136/bmj.1.5346.1678-b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Fogazzi GB, Ferrari B, Garigali G et al. Urinary sediment findings in acute interstitial nephritis. Am J Kidney Dis 2012;60:330–2. 10.1053/j.ajkd.2012.05.002 [DOI] [PubMed] [Google Scholar]
- 86. Wilson GJ, Kark AL, Francis LP et al. The increasing rates of acute interstitial nephritis in Australia: a single centre case series. BMC Nephrol 2017;18:329. 10.1186/s12882-017-0747-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Glassock RJ. Secondary minimal change disease. Nephrol Dial Transplant 2003;18 Suppl 6:vi52–8. 10.1093/ndt/gfg1060 [DOI] [PubMed] [Google Scholar]
- 88. Kleinknecht D. Interstitial nephritis, the nephrotic syndrome, and chronic renal failure secondary to nonsteroidal anti-inflammatory drugs. Semin Nephrol 1995;15:228–35. [PubMed] [Google Scholar]
- 89. Corwin HL, Korbet SM, Schwartz MM. Clinical correlates of eosinophiluria. Arch Intern Med 1985;145:1097–9. 10.1001/archinte.1985.00360060165025 [DOI] [PubMed] [Google Scholar]
- 90. Cornell LD, Chicano SL, Deshpande V et al. Pseudotumors due to IgG4 immune-complex tubulointerstitial nephritis associated with autoimmune pancreatocentric disease. Am J Surg Pathol 2007;31:1586–97. 10.1097/PAS.0b013e318059b87c [DOI] [PubMed] [Google Scholar]
- 91. D'Agati VD, Theise ND, Pirani CL et al. Interstitial nephritis related to nonsteroidal anti-inflammatory agents and beta-lactam antibiotics: a comparative study of the interstitial infiltrates using monoclonal antibodies. Mod Pathol 1989;2:390–6. [PubMed] [Google Scholar]
- 92. Deshmane SL, Kremlev S, Amini S et al. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 2009;29:313–26. 10.1089/jir.2008.0027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Muhammad A, Xiao Z, Lin W et al. Acute interstitial nephritis caused by ANCA-associated vasculitis: a case based review. Clin Rheumatol 2024;43:1227–44. 10.1007/s10067-023-06798-z [DOI] [PubMed] [Google Scholar]
- 94. Moledina DG, Wilson FP, Kukova L et al. Urine interleukin-9 and tumor necrosis factor-α for prognosis of human acute interstitial nephritis. Nephrol Dial Transplant 2021;36:1851–8. 10.1093/ndt/gfaa169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Wu Y, Farrell J, Pirmohamed M et al. Generation and characterization of antigen-specific CD4+, CD8+, and CD4+CD8+ T-cell clones from patients with carbamazepine hypersensitivity. J Allergy Clin Immunol 2007;119:973–81. 10.1016/j.jaci.2006.12.617 [DOI] [PubMed] [Google Scholar]
- 96. Zanni MP, Mauri-Hellweg D, Brander C et al. Characterization of lidocaine-specific T cells. J Immunol 1997;158:1139–48. 10.4049/jimmunol.158.3.1139 [DOI] [PubMed] [Google Scholar]
- 97. Sachs B, Erdmann S, Malte Baron J et al. Determination of interleukin-5 secretion from drug-specific activated ex vivo peripheral blood mononuclear cells as a test system for the in vitro detection of drug sensitization. Clin Exp Allergy 2002;32:736–44. 10.1046/j.1365-2222.2002.01382.x [DOI] [PubMed] [Google Scholar]
- 98. Pichler WJ, Tilch J. The lymphocyte transformation test in the diagnosis of drug hypersensitivity. Allergy 2004;59:809–20. 10.1111/j.1398-9995.2004.00547.x [DOI] [PubMed] [Google Scholar]
- 99. Kesmez Can F, Özkurt Z, Öztürk N et al. Effect of IL-6, IL-8/CXCL8, IP-10/CXCL 10 levels on the severity in COVID 19 infection. Int J Clin Pract 2021;75:e14970. 10.1111/ijcp.14970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wu Y, Su T, Yang L et al. Correlation between urinary biomarkers and pathological lesions in drug-induced tubulointerstitial nephritis. Zhonghua Nei Ke Za Zhi 2010;49:568–71. [PubMed] [Google Scholar]
- 101. Koponen M, Pichler WJ, de Weck AL. T cell reactivity to penicillin: phenotypic analysis of in vitro activated cell subsets. J Allergy Clin Immunol 1986;78:645–52. 10.1016/0091-6749(86)90083-7 [DOI] [PubMed] [Google Scholar]
- 102. Tanvarasethee B, Buranapraditkun S, Klaewsongkram J. The potential of using enzyme-linked immunospot to diagnose cephalosporin-induced maculopapular exanthems. Acta Derm Venereol 2013;93:66–9. 10.2340/00015555-1386 [DOI] [PubMed] [Google Scholar]
- 103. Punrin S, Thantiworasit P, Mongkolpathumrat P et al. Evaluated the diagnostic utility of interferon-gamma enzyme-linked immunospot (ELISPOT) assays in 117 patients with non-immediate drug hypersensitivity reactions. J Allergy Clin Immunol 2016;137:AB36. 10.1016/j.jaci.2015.12.117 [DOI] [Google Scholar]
- 104. Koda R, Watanabe H, Tsuchida M et al. Immune checkpoint inhibitor (nivolumab)-associated kidney injury and the importance of recognizing concomitant medications known to cause acute tubulointerstitial nephritis: a case report. BMC Nephrol 2018;19:48. 10.1186/s12882-018-0848-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Schneider CH, de Weck AL. Chemical aspects of penicillin allergy. The direct penicilloylation of epsilon-amino groups by penicillin at pH 7.4. Helv Chim Acta 1966;49:1695–706. 10.1002/hlca.19660490532 [DOI] [PubMed] [Google Scholar]
- 106. Park BK, Pirmohamed M, Kitteringham NR. Role of drug disposition in drug hypersensitivity: a chemical, molecular, and clinical perspective. Chem Res Toxicol 1998;11:969–88. 10.1021/tx980058f [DOI] [PubMed] [Google Scholar]
- 107. Schnyder B, Mauri-Hellweg D, Zanni M et al. Direct, MHC-dependent presentation of the drug sulfamethoxazole to human alphabeta T cell clones. J Clin Invest 1997;100:136–41. 10.1172/JCI119505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Naisbitt DJ, Gordon SF, Pirmohamed M et al. Antigenicity and immunogenicity of sulphamethoxazole: demonstration of metabolism-dependent haptenation and T-cell proliferation in vivo. Br J Pharmacol 2001;133:295–305. 10.1038/sj.bjp.0704074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Naisbitt DJ, Farrell J, Gordon SF et al. Covalent binding of the nitroso metabolite of sulfamethoxazole leads to toxicity and major histocompatibility complex-restricted antigen presentation. Mol Pharmacol 2002;62:628–37. 10.1124/mol.62.3.628 [DOI] [PubMed] [Google Scholar]
- 110. Spanou Z, Keller M, Britschgi M et al. Involvement of drug-specific T-cells in acute drug-induced interstitial nephritis. J Am Soc Nephrol 2006;17:2919–27. 10.1681/ASN.2006050418 [DOI] [PubMed] [Google Scholar]
- 111. Chung KB, Hwang JH, Kim D. A case of vancomycin-induced drug reaction with eosinophilia, systemic symptoms and multiorgan involvement proven using lymphocyte transformation test. Ann Dermatol 2023;35:140. 10.5021/ad.20.341 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data included in this study are available upon request from the corresponding author.