

ABSTRACT
The incidence of isoniazid (INH) resistant Mycobacterium tuberculosis is increasing globally. This study aimed to identify the molecular mechanisms behind the development of INH resistance in M. tuberculosis strains collected from the same patients during the standard course of treatment. Three M. tuberculosis strains were collected from a patient before and during antituberculosis (anti-TB) therapy. The strains were characterized using phenotypic drug susceptibility tests, Mycobacterial Interspersed Repeated Unit-Variable-Number Tandem Repeats (MIRU-VNTR), and whole-genome sequencing (WGS) to identify mutations associated with INH resistance. To validate the role of the novel mutations in INH resistance, the mutated katG genes were electroporated into a KatG-deleted M. tuberculosis strain (GA03). Three-dimensional structures of mutated KatG were modeled to predict their impact on INH binding. The pre-treatment strain was susceptible to INH. However, two INH-resistant strains were isolated from the patient after anti-TB therapy. MIRU-VNTR and WGS revealed that the three strains were clonally identical. A missense mutation (P232L) and a nonsense mutation (Q461Stop) were identified in the katG of the two post-treatment strains, respectively. Transformation experiments showed that katG of the pre-treatment strain restored INH susceptibility in GA03, whereas the mutated katG genes from the post-treatment strains rendered negative catalase activity and INH resistance. The protein model indicated that P232L reduced INH-KatG binding affinity while Q461Stop truncated gene transcription. Our results showed that the two katG mutations, P232L and Q461Stop, accounted for the co-emergence of INH-resistant clones during anti-TB therapy. The inclusion of these mutations in the design of molecular assays could increase the diagnostic performance.
IMPORTANCE
The evolution of drug-resistant strains of Mycobacterium tuberculosis within the lung lesions of a patient has a detrimental impact on treatment outcomes. This is particularly concerning for isoniazid (INH), which is the most potent first-line antimycobacterial drug. However, the precise genetic factors responsible for drug resistance in patients have not been fully elucidated, with approximately 15% of INH-resistant strains harboring unknown genetic factors. This raises concerns about the emergence of drug-resistant clones within patients, further contributing to the global epidemic of resistance. In this study, we revealed the presence of two novel katG mutations, which emerged independently due to the stress exerted by antituberculosis (anti-TB) treatment on a parental strain. Importantly, we experimentally demonstrated the functional significance of both mutations in conferring resistance to INH. Overall, this research sheds light on the genetic mechanisms underlying the evolution of INH resistance within patients and provides valuable insights for improving diagnostic performance by targeting specific mutations.
KEYWORDS: isoniazid resistance, Mycobacterium tuberculosis, whole-genome sequencing, transformation, antituberculosis treatment, protein modeling
INTRODUCTION
The standard treatment for tuberculosis (TB) involves a combination of multiple antituberculosis (anti-TB) drugs. The current 6-month regimen for drug-susceptible TB includes 2 months of intensive phase treatment with isoniazid (INH), rifampicin (RIF), pyrazinamide (PZA), and ethambutol (EMB) followed by 4 months of continuation phases with INH and RIF (1). However, prolonged treatment has led to poor adherence to the regimen, which increases the likelihood of the emergence of drug-resistant Mycobacterium tuberculosis strains (2).
Recent studies have reported the emergence of drug-resistant conferring mutations within a patient under standard treatment (3–5). Trauner et al. (3) suggested that the emergence and fixation of drug-resistant conferring mutations depends on the pressure exerted by the drugs. They noted that an accurate dosage of anti-TB drugs substantially clears the bacteria and limits the emergence of drug-resistant strains. In contrast, when the drug pressure declines owing to host-related or programmatic failures, drug-resistant clones would gradually emerge. Strikingly, Liu et al. reported that the microevolution of drug-resistant clones within lung lesions of a patient adversely affects treatment responses reinforcing the above observation (5). The emergence of genetic determinants of drug resistance within patients is becoming a serious concern, particularly for INH due to its complex mechanisms of action and resistance (6).
INH is one of the most powerful first-line antimycobacterial drugs (7). It is a prodrug that requires activation by the bacterial catalase-peroxidase enzyme KatG. The activated INH radicals combine with nicotinamide adenine dinucleotide species (NADH/NAD+) to form INH-NAD adducts that inhibit the biosynthesis of mycobacterial mycolic acid. The drug has pleiotropic effects on both the growing and dormant form of mycobacteria (8). Despite the essential role of INH in anti-TB therapy, the emergence and spreading of INH-resistant M. tuberculosis strains become the major challenge for the treatment of TB. The increasing incidence of INH resistance might be attributed to the higher spontaneous mutation rate of M. tuberculosis toward INH compared to other drugs (9).
While most of the known INH resistance mechanisms occur through the alteration of genes encoding the activating enzyme or through mutations of other genes that encode the targets of the activated INH, the mechanism of resistance to INH is complex and involves multiple genetic loci (10). Arrays of genetic polymorphisms in genes via mabA-inhA promoter, oxyR-ahpC intergenic region, mshA, nat, ndh, kasA, ahp, inhA, and katG have been implicated in INH resistance (11–14). A single point mutation of S315T in katG accounts for approximately 60% of INH resistance, while mutations in the regulator and a structural segment of inhA contribute to 19% of INH resistance (6). Yet the resistance mechanisms remain elusive with an estimated 15% of INH-resistant strains harboring unknown genetic determinants (15, 16).
Furthermore, with the advancement of deep sequencing technology, it is now possible to identify minor subpopulations of drug-resistant strains in patient specimens. This has raised significant concerns about the emergence of drug-resistant clones in patients who are undergoing standard treatment (17).
This study investigated three M. tuberculosis isolates collected from a patient before and during the standard course of TB treatment. The pre-treatment isolate was found to be susceptible to INH, while the two isolates obtained during anti-TB treatment were INH-resistant without any known mutation. Mycobacterial Interspersed Repeated Unit-Variable-Number Tandem Repeats (MIRU-VNTR) typing revealed that all three isolates were clonal. Comparative whole-genome sequencing (WGS) identified two novel mutations in the katG gene of the two post-treatment isolates. The functional study of these mutations confirmed their causative role. These findings underscore the need for further studies to elucidate the mechanisms of the emergence of INH resistance within a patient and determine the role of novel mutations through functional genetics.
MATERIALS AND METHODS
M. tuberculosis clinical isolates
The M. tuberculosis isolates were obtained from a 60-year-old male patient diagnosed with pulmonary TB. The initial isolate, labeled as M_11806, was obtained on 28 October 2015, before treatment. Two subsequent isolates, named D1_12327 and D2_12328, were obtained on 11 January and 14 January 2016, respectively, while the patient was undergoing standard anti-TB therapy. Full panel first-line phenotypic drug susceptibility tests (pDST) for the three strains were conducted retrospectively at The University of Hong Kong, while total nucleic acid extracts were then sent to The Hong Kong Polytechnic University for genomic characterization.
In addition, a catalase-negative M. tuberculosis isolate (GA03) which exhibited mono-INH resistance (minimum inhibitory concentration = 256 mg/L) and lacked catalase activity, was used as the recipient for cloned katG genes in a transformation experiment. Sequencing analysis revealed a truncated katG gene with a nonsense mutation (Q434Stop) in the GA03 isolate.
Phenotypic drug susceptibility tests
Agar proportion susceptibility tests for RIF and INH were performed prospectively in the Public Health Laboratory Centre, the reference laboratory in Hong Kong, according to CSLI guidelines (18). The Bactec MGIT 960 SIRE kit and PZA kit (Becton, Dickinson, Baltimore, MD, USA) were then retrospectively done to determine the first-line drug susceptibilities of the three M. tuberculosis isolates according to the previously described protocol (19, 20). The critical concentration values used were 0.1 mg/L and 0.4 mg/mL for INH resistance, 1.0 mg/L for RIF resistance, 5.0 mg/L for EMB resistance, 1.0 mg/L and 4.0 mg/mL for streptomycin (STR) resistance, and 100 mg/L for PZA resistance.
MIRU-VNTR typing
DNA isolation was conducted using the Roche Cobas Amplicor extraction kit (Roche Diagnostics, USA) as previously described (21). The PCR amplification of the 24 MIRU-VNTR loci was conducted using the primers and PCR conditions provided in MIRU-VNTR (22).
Whole-genome sequence and bioinformatic analysis
The M. tuberculosis strains were initially isolated from respiratory specimens and cultured on either Stonebrink agar (D1_12327) or Lowenstein Jensen slants (M_11896 and D2_12328). Sub-cultures were then performed on 7H11 agar to obtain single colonies. Each strain was individually picked and inoculated into 15 mL of 7H9 broth supplemented with OADC. The cultures were incubated at 37°C with agitation at 120 rpm for 14 days prior to DNA extraction. The phenol-chloroform method was used for isolating genomic DNA as described previously (23–25). The purity and integrity of the genomic DNA were examined using the Qubit HS assay (Thermo Scientific, USA) and 0.8% agarose gel electrophoresis, respectively. The isolated DNA was subjected to QIAseq FX DNA Library Preparation Kit (Qiaen, Hilden, Germany) according to the manufacturer’s instructions, and was sequenced on the Illumina MiSeq platform using 2 × 150 bp version 2 sequencing chemistry (Illumina, San Diego, USA).
Core-genome single nucleotide polymorphism (SNP) phylogeny was constructed to confirm the clonality and the global phylogenetic placement of the three M. tuberculosis strains. The detailed methodology was described in Supplemental Materials and Methods.
In addition, a comparative genome analysis was conducted, in which the M. tuberculosis H37Rv genome (NC_000962.3) was used as the reference, to identify genetic variants responsible for INH resistance in the three isolates. Particularly, previously reported resistance genes or loci, including katG, furA-katG, mabA-inhA promoter, inhA, ahpC, oxyR-ahpC intergenic, iniCAB, Rv0340, fabD, kasA, fbpC, srmR, accD6, efpA, fadE24, ndh, Rv1592c, Rv1772, and nhoA were selected and analyzed (26). Variants were called when the coverage of a specific position was higher than 50× and variant frequency was >20% as previously described (27). The detailed method for comparative genome analysis was described in Supplemental Materials and Methods.
Construction of cloning plasmid and transformation
A pOLYG plasmid with a hygromycin resistance marker was introduced into Escherichia coli DH5α, and the resulting cells were streaked on hygromycin-containing Luria-Bertani (LB) agar. A single colony was then transferred to LB broth and incubated for 16 hours. The plasmid was isolated using the QIAprep spin miniprep kit (QIAGEN). The entire furA-katG operon, including the promoter region, was amplified from the three clinical isolates. The vector backbone and furA-katG amplicon were subjected to restriction digestion with XbaI and HindIII, followed by ligation to produce the cloned plasmids listed in Table S1. The plasmids containing the insert were then electroporated into a catalase-negative INH-resistant M. tuberculosis strain GA03 using an Eporator (Eppendorf, Germany) with the setting of 2.5 kV, 25 µF, and resistance at 1,000 Ω. The presence of mutations of interest in the insert was confirmed by Sanger sequencing using additional forward and reverse primers (Table S2) to cover the entire furA-katG operon as previously described (28).
Minimal inhibitory concentration
The minimal inhibitory concentrations (MICs) of INH for M. tuberculosis H37Rv, GA03 (INH-resistant), GA03 harboring pOLYG (vector control), and GA03 harboring pOLYG furA-katG insert from strains M_11806, D1_12327, and D2_12328 were determined using agar proportion method as previously described (29). All experiments were conducted in triplicate (detailed method is described in Supplemental Materials and Methods).
Semi‐quantitative catalase assay
The catalase test was conducted for M. tuberculosis transformants harboring the furA-katG operon from three strains (M_11806, D1_12327, and D2_12328), M. tuberculosis H37Rv (positive control), and strain GA03 without the plasmid (negative control). All experiments were conducted in triplicate as described previously (28).
Modeling of INH binding in wild and mutant katG
The three-dimensional structure of the wild type and mutated katG proteins from three experimental strains was built by protein homology modeling with the crystal structure of the katG protein of M. tuberculosis (1SJ2) (30) using the server SWISS-MODEL with default parameters. The model was further analyzed using the Discovery Studio visualizer (Accelrys), and the Ramachandran plot was examined to ensure that the structure of the model was not in any unfavorable region. INH-heme binding model in the constructed was created by superimposing the katG structure with the drug.
Furthermore, the Protein Variation Effect Analyzer (PROVEAN), a web server tool used to calculate pairwise alignment scores between a protein sequence and a large protein database with precomputed scores (31), was utilized to predict the functional consequences of the novel amino acid substitutions on the KatG protein.
RESULTS
The clinical case
On 5 October 2015, a 60-year-old male patient with presumptive symptoms of TB visited a chest clinic for medical consultation. A sputum sample was collected for acid-fast bacilli (AFB) culture, and on 28 October 2015, pulmonary TB was confirmed when the first M. tuberculosis strain, M_11806 (mother strain), was isolated. Standard anti-TB therapy (2HRZE) was initiated on 4 November 2015. However, as the symptoms persisted, two follow-up consultations on 7 December and 14 December 2015, resulted in the collection of two additional sputum samples. On 11 January and 14 January 2016, respectively, two M. tuberculosis strains, D1_12327 and D2_12328 (daughter strains), were isolated from these samples. Drug susceptibility tests for RIF and INH were requested for all three strains on 27 March 2016. The results, obtained on 9 May 2016, revealed that the M_11806 strain was susceptible to INH but resistant to RIF. Interestingly, the two daughter strains, D1_12327 and D2_12328, were found to be INH-resistant, resulting in a multidrug-resistant TB (MDR-TB) case. On 17 May 2016, the therapy was adjusted to include levofloxacin, para-aminosalicylic acid, and kanamycin. Unfortunately, the patient defaulted on treatment twice afterward due to travel outside of Hong Kong. The detailed treatment timeline is presented in Fig. 1.
Fig 1.

Treatment history of the clinical case. A 60-year-old patient presenting with presumptive symptoms of TB visited a chest clinic for medical consultation on 5 October 2015. A sputum sample was collected for AFB culture. On 28 October 2015, pulmonary TB was confirmed when the first M. tuberculosis strain, M_11806 (mother strain), was isolated. Standard anti-TB therapy (2HRZE) was initiated on 4 November 2015. However, despite treatment, the patient’s symptoms persisted. Two follow-up consultations took place on 7 December and 14 December 2015, during which two additional sputum samples were collected. Subsequently, on 11 January and 14 January 2016, two M. tuberculosis strains, D1_12327 and D2_12328 (daughter strains), were isolated from these samples. Consequently, the patient missed several medical appointments in February and March 2016. The patient sought medical consultation again on 27 March 2016. Phenotypic DSTs for INH and RIF were requested for all M. tuberculosis isolates (including the mother strain and the two daughter strains) collected from this case. On 9 May 2016, MDR-TB was confirmed based on the pDST results. Consequently, RIF and INH were discontinued and replaced with levofloxacin, para-aminosalicylic acid, and kanamycin on 17 May 2016. However, the patient defaulted the treatment in August 2016 due to unknown reasons. He returned to treatment in late November 2016. Disappointedly, he defaulted again 2 weeks later as he emigrated from Hong Kong.
All three strains were retrospectively sent to the University of Hong Kong for repeat pDST using the Bactec MGIT 960 SIRE kit and PZA kit. The results confirmed that the mother strain was susceptible to INH and EMB but resistant to RIF, PZA, and STR. Both daughter strains exhibited conversion to INH resistance at concentrations of 0.1 mg/L and 0.4 mg/L (Table 1).
TABLE 1.
Phenotypic and genotypic characteristics of M. tuberculosis strains
| M. tuberculosis strains isolated from the same patient | ||||
|---|---|---|---|---|
| M_11806 | D1_12327 | D2_12328 | ||
| Phenotypic drug susceptibility patterna | ||||
| INH-S, RIF-R, PZA-R, EMB-S, STR-R | INH-R, RIF-R, PZA-R, EMB-S, STR-R (MDR-TB) | INH-R, RIF-R, PZA-R, EMB-S, STR-R (MDR-TB) | ||
| Genotypic drug susceptibility pattern | ||||
| Related TB drugs | Resistance genes/loci | Mutations b (Allele frequency) | ||
| Isoniazid (INH) | katG | Arg463Leu (100.0%) | Arg463Leu (99.8%) Pro232Pro (33.0%) Pro232Leu (67.0%) |
Arg463Leu (99.8%) Pro232Pro (91.0%) Pro232Leu (9.0%) Gln461Stop (97.0%) |
| furA-katG | WTc | WT | WT | |
| inhA | WT | WT | WT | |
| mabA-inhA | WT | WT | WT | |
| oxyR-ahpC | WT | WT | WT | |
| ahpC | WT | WT | WT | |
| Rv0340 | WT | WT | WT | |
| iniA | Ser501Ser (99.0%) | Ser501Ser (99.0%) | Ser501Ser (99.0%) | |
| iniB | WT | WT | WT | |
| iniC | WT | WT | WT | |
| srmR | WT | WT | WT | |
| fabD | WT | WT | WT | |
| kasA | WT | WT | WT | |
| accD6 | Asp200Asp (98.0%) Asp229Gly (98.0%) |
Asp200Asp (97.0%) Asp229Gly (98.0%) |
Asp200Asp (98.0%) Asp229Gly (98.0%) |
|
| fbpC | WT | WT | WT | |
| fadE24 | WT | WT | WT | |
| efpA | WT | WT | WT | |
| Rv1772 | WT | WT | WT | |
| Rv1592c | Ile322Gly (99.0%) | Ile322Gly (94.0%) | Ile322Gly (95.0%) | |
| ndh | WT | WT | WT | |
| nhoA/nat | WT | WT | WT | |
| Rifampicin (RIF) | rpoB | His526Leu (98.0%) | His526Leu (97.0%) | His526Leu (96.0%) |
| Pyrazinamide (PZA) | pncA | −11 A > C (98.0%) | −11 A > C (93.0%) | −11 A > C (98.0%) |
| rpsA | WT | WT | WT | |
| Ethambutol (EMB) | embB | WT | WT | WT |
| Streptomycin (STR) | rpsL | WT | WT | WT |
| rrs | WT | WT | WT | |
Phenotypic drug susceptibility pattern was determined by Bactec MGIT 960 SIRE kit and PZA kit.
Variants are called based on H37Rv reference genome NC_000962.3.
WT: Wildtype.
Clonality of the three M. tuberculosis strains
The 24-loci MIRU-VNTR analysis and the in silico spoligotyping revealed that M_11806, D1_12327, and D2_12328 shared the same genetic patterns, suggesting that they originated from the same clonal population and were classified as belonging to the Beijing lineage (Fig. 2). The clonality of the three strains was validated by core-genome SNP phylogeny, which showed that they shared 100% identical core genome, which refers to the genome excluding the accessory genes like drug resistance genes and virulence genes (genomes highlighted in red grid in Fig. 3). In addition, the phylogenetic placement in the tree demonstrated that the three isolates were clustered with M. tuberculosis strains of lineage (L) 2.2.2., supporting that these isolates belong to Beijing/Asia Ancestral 1 sub-lineage.
Fig 2.

24-loci MIRU-VNTR and spoligotyping of the three M. tuberculosis strains from the same patient.
Fig 3.

Core-genome SNP phylogeny of the three M. tuberculosis strains in this study. Strains M_11806, D1_12327, and D2_12328 (indicated inside the red grid) had identical core genomes (i.e., no core genome SNP difference), suggesting that they belong to the same clone. Regarding the phylogenetic placement, these strains were clustered with other M. tuberculosis strains of lineage 2.2.2., which were also classified as Beijing/Asia Ancestral 1 sub-lineage.
Comparative genome analysis and INH resistance-conferring mutations
Each isolate was sequenced and yielded nearly 6 million sequence reads with an average read length of 147.3 bp and a depth of coverage of 186.4× (Table S3). The reads provided uniform coverage across the entire reference genome NC_000962.3. After removing polymorphisms in the pe/ppe gene families and large deletions, which are markers of the Beijing genotype, and fixing the allelic frequency to ≥20×, we discovered a total of 1,621 mutations shared by the three isolates when compared to M. tuberculosis H37Rv. A total of 4 and 16 unique single nucleotide polymorphisms were found only in isolates D1_12327 and D2_12328, respectively (Fig. S1). A comprehensive comparative analysis of WGS was performed to detect INH resistance-conferring mutations in 19 structural and 2 regulatory regions (Table 1). Two novel mutations in katG were discovered in on-treatment strains, Pro232Leu substitution (CCG→CTG, chromosomal position 2155417) at an allele frequency of 67% and Gln461Stop termination (CCG→CTG, chromosomal position 2155417) at an allele frequency of 97% in the D1_12327 and D2_12328 strains, respectively (Table 1). Notably, Pro232Leu substitution was also detected in the D2_12328 strain with an allele frequency of 9.0% although Sanger sequencing confirmed only the Gln461Stop mutation in this strain.
Moreover, all three strains exhibited mutations that were consistent with pDST. Specifically, a missense mutation, His526Leu, was identified in rpoB, which confers resistance to RIF. Additionally, a nucleotide change, −11A > C, was found in the promoter region of pncA, which confers resistance to PZA (Table 1).
Functional validation of novel katG mutations
The entire furA-katG operon, along with the upstream promoter region, from M_11806, D1_12327, and D2_12328 strains were successfully transformed into the catalase-negative strain, GA03 (Fig. S2). The MICs of INH were determined for M. tuberculosis H37Rv, strain GA03, GA03 containing the vector alone and the vector cloned with the furA-katG operon from M_11806, D1_12327, and D2_12328 strains, respectively.
Strain GA03 carried a nonsense mutation, Q434Stop, in katG, which resulted in the complete loss of catalase activity and high INH MICs of 256 mg/L. As depicted in Fig. 4 and Table 2, the introduction of the furA-katG operon of M_11806 successfully reduced the MIC to 32 mg/L, partially restoring susceptibility to INH. However, the transformants carrying the constructs pOLYG::katG Q461Stop and pOLYG::katG P232L remained highly resistant to INH, with MICs of 128 mg/L and 256 mg/L, respectively. We also evaluated the impact of these mutations by measuring the functional activity of catalase produced by the transformants carrying the constructs with novel katG mutations. The results showed that the complementation of the furA-katG operon of M_11806 had recovered the catalase activity of GA03. However, constructs with novel mutations substantially lost catalase activity, with a bubble height of ≤3 mm, which was ≥eightfold lower than the construct carrying pOLYG::M_11806 and M. tuberculosis H37Rv. Remarkably, catalase production was lower for the construct that had a termination mutation than for the one with the amino acid substitution.
Fig 4.

MIC and catalase activity using the agar proportion method and semi-quantitative catalase activity of the katG transformants and H37Rv. The label on the plate indicates the serial dilution of INH in mg/L; N denoted drug-free well inoculated with neat bacterial concentration, i.e., 10,000 CFU/well; 10–2 denoted drug-free well inoculated with 1:99 diluted bacteria, i.e., 100 CFU/well. (a) H37Rv reference strain (wild; control) with MIC = 0.2 mg/L, catalase = 30 mm; (b)GA03 (pOLYG::M_11806) with MIC = 32 mg/L, catalase = 25 mm; (c) GA03 (pOLYG::D1_12327) with MIC = 256 mg/m, catalase = 3 mm; and (d) GA03 (pOLYG::D2_12328) with MIC = 256 mg/L, catalase = 1 mm.
TABLE 2.
The minimal inhibitory concentration results of transformed mutants and reference strains
| Strain | Characteristics | MIC (mg/L) | Catalase activity |
|---|---|---|---|
| GA03 | A clinical isolate of M. tuberculosis with nonsense mutation, Q434Stop, in katG, resulting in negative catalase and high-level INH resistance | 256 | 0 |
| GA03 (pOLYG) | M. tuberculosis GA03 that was transformed with intact mycobacterial expression vector, pOLYG | 256 | 0 |
| GA03 (pOLYG::M_11806) | M. tuberculosis GA03 that was transformed with pOLYG carrying entire furA-katG operon from M_11806, pOLYG::Wild katG | 32 | 25 |
| GA03 (pOLYG::D1_12327) | M. tuberculosis GA03 that was transformed with pOLYG carrying entire furA-katG operon from D1_12327, pOLYG::katG P232L (CCG→CTG) | 256 | 3 |
| GA03 (pOLYG::D2_12328) | M. tuberculosis GA03 that was transformed with pOLYG carrying entire furA-katG operon from D2_12328, pOLYG::katG Q461Stop (CAG→TAG) | 256 | 1 |
| H37Rv | Reference strain used in MIC test | 0.2 | 30 |
Protein model for prediction of the effect of novel mutations on INH binding
To explain the impact of the mutations on the binding with INH, we modeled a three-dimensional (3D) structure of katG (mutant and wild) with INH and heme (Fig. 5). The natural polymorphism, R463L, in all three isolates is located far away from the active binding site and does not alter any structural properties of the protein. However, the other two mutations (Pro232Leu and Gln461Stop) in D1_12327 and D2_12328, respectively, have a significant effect on the catalytic activity of INH. The proline residue in wild-type katG at codon 232 is crucial for the binding of INH/Heme, as previously reported (32). The substitution of proline with leucine in D1_12327 (Pro232Leu) interfered with the binding of INH to the enzyme in the active site, limiting the access of INH to the iron atom, where catalysis takes place. The mutation in D2_12328 (Gln461Stop) results in a truncated protein from 461 residues to 741 residues in the C-terminus (280 total residues), causing the loss of enzymatic activity, as the protein cannot fold correctly. The deleterious impact of these two novel mutations on the catalase-peroxidase protein was further evaluated using PROVEAN (31). The PROVEAN scores of −9.818 and −10.943 were obtained for Pro232Leu and Gln461Stop, respectively, which are considered deleterious to the function of the catalase-peroxidase enzyme.
Fig 5.
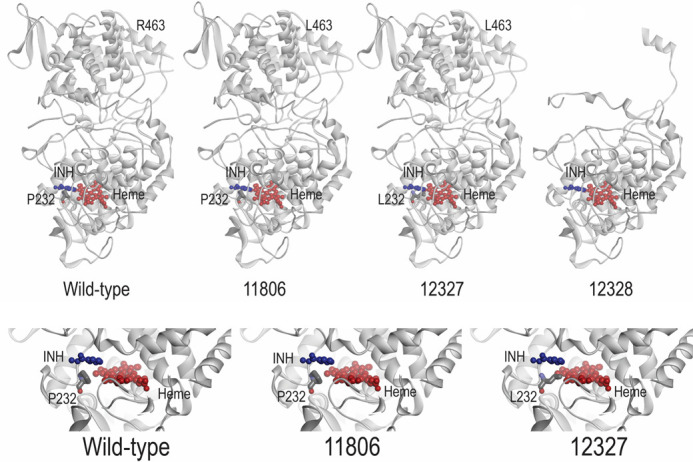
KatG protein model of H37Rv (wildtype) and M. tuberculosis strains M_11806, D1_12327, and D3_12328 interacted with INH and heme.
DISCUSSION
INH remains the backbone of the standard treatment regimen for TB. Resistance to INH is often associated with a variety of polymorphisms, including complete loss of katG (26), single amino acid substitutions, termination mutations (33), deletions, and frameshift insertions in katG (34). These mutations can have varying effects on katG enzymatic activity and the level of resistance to INH (35).
In this study, we documented the co-emergence of MDR-TB clones in a patient during the standard course of treatment and demonstrated the causal role of emerging katG mutations in the development of INH resistance. The patient was initially infected with a strain of M. tuberculosis that was resistant to RIF and PZA but susceptible to INH and EMB. Without the guidance of pDST, the standard short-course 6-month regimen was administered to the patient. However, no clinical improvement was observed during the treatment. After 2 months of treatment, two consecutive M. tuberculosis strains were collected, both of which were found to be INH-resistant. Standard 24-MIRU-VNTR and core-genome phylogeny confirmed that the strains were clonally related and belonged to the Beijing lineage. Minimal genetic variations were observed across the whole genome of the three isolates (<15 SNPs identified in accessory genes at ≥20% allelic frequency, and no SNP difference identified in the core genome), supporting the clonal relatedness of the strains. These findings suggest that the two INH-resistant strains evolved from the pre-treatment INH-susceptible strain during treatment.
Comparative WGS analysis of the entire furA-katG and mabA-inhA genetic regions as well as previously reported genetic stretches associated with INH resistance was performed (26). We found the absence of INH resistance-conferring genetic polymorphisms in all loci except for two novel mutations in katG, P232L, and Q461Stop, in the strains isolated during the treatment. M. tuberculosis has a natural propensity of spontaneous mutations that confer INH resistance in approximately 1 out of 106–108 bacterial populations (9). Suboptimal drug pressure, either due to patient-related factors or programmatic mismanagement, could facilitate the selection of mutant (drug-resistant) clones. The M. tuberculosis lineage has also been shown to affect the speed of acquisition of genetic variants. The Beijing lineage, in particular, is hypermutable compared to other lineages (36), which may have increased the chance of the emergence of INH resistance while the patient was undergoing standard treatment.
The emergence of drug-resistant clones in a single patient has caught substantial attention in the last several decades. While previous studies have produced conflicting results regarding the speed of emergence and fixation of drug-resistant clones in vivo, recent research utilizing highly sensitive next-generation sequencing technologies has shown that there is a high level of diversity of mycobacterial strains within individual patients (37, 38). Studies that have examined serial sputum samples from single or multiple patients under standard treatment regimens have demonstrated the emergence and coexistence of multiple drug-resistant-conferring alleles. The ultimate fixation of these alleles is determined by their fitness cost and level of resistance. Generally, mutations that confer high levels of resistance but have low fitness costs are fixed in patients under therapy and subsequently transmitted. For example, Sun et al. studied three serial sputum samples collected from a single patient and identified four INH-resistant associated mutations (three in katG and one in inhA). After 23 months, only one variant with a high resistance level and low fitness cost was fixed (4). A similar study examining RIF and STR-associated mutations in sputum collected from the same patient found that high drug resistance-conferring and low fitness cost genetic mutations were fixed (39, 40). Furthermore, previous studies have revealed that M. tuberculosis employs various compensatory mechanisms to counteract the loss of catalase activity caused by mutations in katG. These mechanisms include the upregulation of alkylhydroperoxidase (AhpC) expression, which has been shown to restore catalase activity in the presence of katG mutations (41, 42). Sherman et al. demonstrated that ahpC and katG share reactive nitrogen intermediates and organic peroxides, which contribute to enhancing the compensatory mechanism when mycobacteria experience a loss of catalase function due to katG mutations (43). However, it is important to note that only a small proportion (~9%) of katG mutants were found to carry mutations in the oxyR-ahpC region, resulting in the overexpression of AhpC (44). Similarly, in our study, no variants were identified in the oxyR-ahpC region in both daughter strains. Therefore, it is speculated that both strains may compensate for the reduced catalase activity through epigenetic mechanisms (45).
In this study, although we did not obtain data supporting the role of two novel mutations in relation to the fitness cost, we demonstrated that both mutations confer high INH resistance through functional genetics. To determine the impact of the mutations, a katG-deleted M. tuberculosis strain was transformed with a mycobacterial vector (pOLYG) carrying the furA-katG operon from the two strains (Table 2). The introduction of mutants katG P232L and Q461Stop resulted in high INH resistance with MICs of 256 mg/L and 128 mg/L, respectively, indicating that katG genes with these mutations are defective. These MIC results are consistent with a range of 0.19 to 256 mg/L obtained for single missense mutations of katG in clinical isolates by Ramaswamy et al. (26). However, it is worth noting that Ramaswamy et al. (26) did not conduct a transformation experiment to confirm the causal role of the identified substitutions. Similarly, Kandler et al. (34) and Torres et al. (6) also identified and cloned several novel mutations in katG that increased the MIC of the transformants. Nonetheless, the MIC results in our study were higher than those reported in these two studies, which could be due to the difference in the identified mutations and the models (H37Rv and Mycobacterium smegmatis) used for the functional assay in the respective studies.
Catalase-peroxidase, encoded by katG, plays a crucial role in converting pro-INH to an active form that has pleiotropic effects on the bacilli (46). Mutations in the coding stretch of katG may reduce the catalytic activity of the gene product, potentially leading to decreased drug susceptibility (26). In vitro evaluation of the catalase activity of the transformants can enable the assessment of the impact of novel mutations in katG (26). As determined by catalase activity, our study also revealed that the M. tuberculosis strain, GA03, which harbored mutated katG (P232L and Q461Stop), has reduced catalase activity by more than eightfold compared to the transformants carrying the gene from the pre-treatment strain, suggesting that the mutations resulted in a defective protein. Previous studies have shown that mutants associated with high MIC were characterized by a drastic reduction in catalase activities (35, 47).
To consolidate the role of the two novel mutations in INH resistance and understand the effect of amino acid substitution and termination, 3D protein-INH binding models were constructed. The P232L substitution is located very close to the INH binding site and could potentially obstruct the binding of INH. As expected, the crystallographic analysis showed that the P232L substitution reduces INH-KatG binding affinity, affecting the conversion of the prodrug to the active form, which is a critical step in the mechanism of action of INH. Transformants carrying this mutation were associated with high resistance to INH, as demonstrated by the MIC study. On the other hand, the termination mutation (Q461Stop) generates a truncated protein at 461 residues without 280 residues in the C-terminal domain. The Q461Stop mutation truncates gene transcription, limiting the activity of the enzyme and causing high-level INH resistance.
In conclusion, our results showed that the two katG mutations, P232L and Q461Stop, contributed to the co-emergence of INH-resistant clones during the standard course of anti-TB therapy. We confirmed the presence of two independent mutations that evolved due to anti-TB stress from a parental strain and experimentally demonstrated the functional role of both mutations in conferring INH resistance. The inclusion of these mutations in the design of molecular assays can improve diagnostic performance.
ACKNOWLEDGMENTS
In loving memory of Dr. Kenneth Siu-Sing Leung, whose talent and presence will forever be remembered and cherished.
We would also like to express our appreciation to the Department of Microbiology at the University of Hong Kong for allowing us to conduct parts of this work in their mycobacteriology laboratory.
This work was supported by the General Research Fund (grant number 15102220), which is administrated under the Research Grant Council of the University Grants Committee, the Government of Hong Kong Special Administrative Region.
Contributor Information
Gilman Kit-Hang Siu, Email: gilman.siu@polyu.edu.hk.
Sophia B. Georghiou, Foundation for Innovative New Diagnostics, Geneva, Switzerland
DATA AVAILABILITY
The genomes of the three M. tuberculosis strains were submitted to the NCBI GenBank and were assigned the accession numbers SAMN34123194, SAMN34123195, and SAMN34123196, respectively.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/spectrum.02133-23.
Supplemental methods, supplemental tables, and Supplemental figures.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Horsburgh CR, Barry CE, Lange C. 2015. Treatment of tuberculosis. N Engl J Med 373:2149–2160. doi: 10.1056/NEJMra1413919 [DOI] [PubMed] [Google Scholar]
- 2. Caminero J, Van A, Fujiwara P. 2013. Guidelines for clinical and operational management of drug-resistant tuberculosis, p 18–19. International Union Against Tuberculosis and Lung Disease. [Google Scholar]
- 3. Trauner A, Liu Q, Via LE, Liu X, Ruan X, Liang L, Shi H, Chen Y, Wang Z, Liang R, Zhang W, Wei W, Gao J, Sun G, Brites D, England K, Zhang G, Gagneux S, Barry CE, Gao Q. 2017. The within-host population dynamics of Mycobacterium tuberculosis vary with treatment efficacy. Genome Biol 18:71. doi: 10.1186/s13059-017-1196-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sun G, Luo T, Yang C, Dong X, Li J, Zhu Y, Zheng H, Tian W, Wang S, Barry CE, Mei J, Gao Q. 2012. Dynamic population changes in Mycobacterium tuberculosis during acquisition and fixation of drug resistance in patients. J Infect Dis 206:1724–1733. doi: 10.1093/infdis/jis601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu Q, Via LE, Luo T, Liang L, Liu X, Wu S, Shen Q, Wei W, Ruan X, Yuan X, Zhang G, Barry CE, Gao Q. 2015. Within patient microevolution of Mycobacterium tuberculosis correlates with heterogeneous responses to treatment. Sci Rep 5:17507. doi: 10.1038/srep17507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Torres JN, Paul LV, Rodwell TC, Victor TC, Amallraja AM, Elghraoui A, Goodmanson AP, Ramirez-Busby SM, Chawla A, Zadorozhny V, Streicher EM, Sirgel FA, Catanzaro D, Rodrigues C, Gler MT, Crudu V, Catanzaro A, Valafar F. 2015. Novel katG mutations causing isoniazid resistance in clinical M. tuberculosis isolates. Emerg Microbes Infect 4:e42. doi: 10.1038/emi.2015.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Almeida Da Silva PEA, Palomino JC. 2011. Molecular basis and mechanisms of drug resistance in Mycobacterium tuberculosis: classical and new drugs. J Antimicrob Chemother 66:1417–1430. doi: 10.1093/jac/dkr173 [DOI] [PubMed] [Google Scholar]
- 8. Zhang Y, Yew W-W. 2015. Mechanisms of drug resistance in Mycobacterium tuberculosis: update 2015. Int J Tuberc Lung Dis 19:1276–1289. doi: 10.5588/ijtld.15.0389 [DOI] [PubMed] [Google Scholar]
- 9. McGrath M, Gey van Pittius NC, van Helden PD, Warren RM, Warner DF. 2014. Mutation rate and the emergence of drug resistance in Mycobacterium tuberculosis. J Antimicrob Chemother 69:292–302. doi: 10.1093/jac/dkt364 [DOI] [PubMed] [Google Scholar]
- 10. Seifert M, Catanzaro D, Catanzaro A, Rodwell TC. 2015. Genetic mutations associated with isoniazid resistance in Mycobacterium tuberculosis: a systematic review. PLoS One 10:e0119628. doi: 10.1371/journal.pone.0119628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Timmins GS, Deretic V. 2006. Mechanisms of action of isoniazid. Mol Microbiol 62:1220–1227. doi: 10.1111/j.1365-2958.2006.05467.x [DOI] [PubMed] [Google Scholar]
- 12. Huyen MNT, Cobelens FGJ, Buu TN, Lan NTN, Dung NH, Kremer K, Tiemersma EW, van Soolingen D. 2013. Epidemiology of isoniazid resistance mutations and their effect on tuberculosis treatment outcomes. Antimicrob Agents Chemother 57:3620–3627. doi: 10.1128/AAC.00077-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dalla Costa ER, Ribeiro MO, Silva MSN, Arnold LS, Rostirolla DC, Cafrune PI, Espinoza RC, Palaci M, Telles MA, Ritacco V, Suffys PN, Lopes ML, Campelo CL, Miranda SS, Kremer K, da Silva PEA, Fonseca L de S, Ho JL, Kritski AL, Rossetti MLR. 2009. Correlations of mutations in katG, oxyR-ahpC and inhA genes and in vitro susceptibility in Mycobacterium tuberculosis clinical strains segregated by spoligotype families from tuberculosis prevalent countries in South America. BMC Microbiol 9:39. doi: 10.1186/1471-2180-9-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jagielski T, Bakuła Z, Roeske K, Kamiński M, Napiórkowska A, Augustynowicz-Kopeć E, Zwolska Z, Bielecki J. 2014. Detection of mutations associated with isoniazid resistance in multidrug-resistant Mycobacterium tuberculosis clinical isolates. J Antimicrob Chemother 69:2369–2375. doi: 10.1093/jac/dku161 [DOI] [PubMed] [Google Scholar]
- 15. Hazbón MH, Brimacombe M, Bobadilla del Valle M, Cavatore M, Guerrero MI, Varma-Basil M, Billman-Jacobe H, Lavender C, Fyfe J, García-García L, León CI, Bose M, Chaves F, Murray M, Eisenach KD, Sifuentes-Osornio J, Cave MD, Ponce de León A, Alland D. 2006. Population genetics study of isoniazid resistance mutations and evolution of multidrug-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemother 50:2640–2649. doi: 10.1128/AAC.00112-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Projahn M, Köser CU, Homolka S, Summers DK, Archer JAC, Niemann S. 2011. Polymorphisms in isoniazid and prothionamide resistance genes of the Mycobacterium tuberculosis complex. Antimicrob Agents Chemother 55:4408–4411. doi: 10.1128/AAC.00555-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cohen KA, Manson AL, Desjardins CA, Abeel T, Earl AM. 2019. Deciphering drug resistance in Mycobacterium tuberculosis using whole-genome sequencing: progress, promise, and challenges. Genome Med 11:45. doi: 10.1186/s13073-019-0660-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Woods GL, Brown-Elliott BA, Conville PS, Desmond EP, Hall GS, Lin G, Pfyffer GE, Ridderhof JC, Siddiqi SH, Wallace RJ, Warren NG, FG W. 2011. Susceptibility testing of mycobacteria, nocardiae, and other aerobic actinomycetes. Vol. M24-A2. Clinical and Laboratory Standards Institute, Wayne (PA). [PubMed] [Google Scholar]
- 19. Tam K-G, Leung K-S, Siu G-H, Chang K-C, Wong S-Y, Ho P-L, Leung E-C, Yam W-C. 2019. Direct detection of pyrazinamide resistance in Mycobacterium tuberculosis by use of pncA PCR sequencing. J Clin Microbiol 57:e00145-19. doi: 10.1128/JCM.00145-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tam K-G, Leung K-S, To S-C, Siu G-H, Lau T-K, Shek V-M, Tse C-S, Wong S-Y, Ho P-L, Yam W-C. 2017. Direct detection of Mycobacterium tuberculosis and drug resistance in respiratory specimen using Abbott Realtime MTB detection and RIF/INH resistance assay. Diagn Microbiol Infect Dis 89:118–124. doi: 10.1016/j.diagmicrobio.2017.06.018 [DOI] [PubMed] [Google Scholar]
- 21. Yam WC, Tam CM, Leung CC, Tong HL, Chan KH, Leung ETY, Wong KC, Yew WW, Seto WH, Yuen KY, Ho PL. 2004. Direct detection of rifampin-resistant Mycobacterium tuberculosis in respiratory specimens by PCR-DNA sequencing. J Clin Microbiol 42:4438–4443. doi: 10.1128/JCM.42.10.4438-4443.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tafess K, Beyen TK, Girma S, Girma A, Siu G. 2021. Spatial clustering and genetic diversity of Mycobacterium tuberculosis isolate among pulmonary tuberculosis suspected patients, Arsi Zone, Ethiopia. BMC Pulm Med 21:206. doi: 10.1186/s12890-021-01567-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Benjak A, Sala C, Hartkoorn RC. 2015. Whole-genome sequencing for comparative genomics and de novo genome assembly. Methods Mol Biol 1285:1–16. doi: 10.1007/978-1-4939-2450-9_1 [DOI] [PubMed] [Google Scholar]
- 24. Rajwani R, Yam WC, Zhang Y, Kang Y, Wong BKC, Leung KSS, Tam KKG, Tulu KT, Zhu L, Siu GKH. 2017. Comparative whole-genomic analysis of an ancient L2 lineage Mycobacterium tuberculosis reveals a novel phylogenetic clade and common genetic determinants of hypervirulent strains. Front Cell Infect Microbiol 7:539. doi: 10.3389/fcimb.2017.00539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Leung KS-S, Siu GK-H, Tam KK-G, To SW-C, Rajwani R, Ho P-L, Wong SS-Y, Zhao WW, Ma OC-K, Yam W-C. 2017. Comparative genomic analysis of two clonally related multidrug resistant Mycobacterium tuberculosis by single molecule real time sequencing. Front Cell Infect Microbiol 7:478. doi: 10.3389/fcimb.2017.00478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ramaswamy SV, Reich R, Dou S-J, Jasperse L, Pan X, Wanger A, Quitugua T, Graviss EA. 2003. Single nucleotide polymorphisms in genes associated with isoniazid resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 47:1241–1250. doi: 10.1128/AAC.47.4.1241-1250.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eldholm V, Norheim G, von der Lippe B, Kinander W, Dahle UR, Caugant DA, Mannsåker T, Mengshoel AT, Dyrhol-Riise AM, Balloux F. 2014. Evolution of extensively drug-resistant Mycobacterium tuberculosis from a susceptible ancestor in a single patient. Genome Biol 15:490. doi: 10.1186/s13059-014-0490-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Siu GKH, Yam WC, Zhang Y, Kao RYT. 2014. An upstream truncation of the furA-katG operon confers high-level isoniazid resistance in a Mycobacterium tuberculosis clinical isolate with no known resistance-associated mutations. Antimicrob Agents Chemother 58:6093–6100. doi: 10.1128/AAC.03277-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Blanco-Ruano D, Roberts DM, Gonzalez-Del-Rio R, Álvarez D, Rebollo MJ, Pérez-Herrán E, Mendoza A. 2015. Antimicrobial susceptibility testing for Mycobacterium sp. Methods Mol Biol 1285:257–268. doi: 10.1007/978-1-4939-2450-9_15 [DOI] [PubMed] [Google Scholar]
- 30. Bertrand T, Eady NAJ, Jones JN, Jesmin, Nagy JM, Jamart-Grégoire B, Raven EL, Brown KA. 2004. Crystal structure of Mycobacterium tuberculosis catalase-peroxidase. J Biol Chem 279:38991–38999. doi: 10.1074/jbc.M402382200 [DOI] [PubMed] [Google Scholar]
- 31. Choi Y, Chan AP. 2015. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31:2745–2747. doi: 10.1093/bioinformatics/btv195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kamachi S, Hirabayashi K, Tamoi M, Shigeoka S, Tada T, Wada K. 2015. The crystal structure of isoniazid-bound KatG catalase-peroxidase from Synechococcus elongatus PCC7942. FEBS J 282:54–64. doi: 10.1111/febs.13102 [DOI] [PubMed] [Google Scholar]
- 33. Bollela VR, Namburete EI, Feliciano CS, Macheque D, Harrison LH, Caminero JA. 2016. Detection of katG and inhA mutations to guide isoniazid and ethionamide use for drug-resistant tuberculosis. Int J Tuberc Lung Dis 20:1099–1104. doi: 10.5588/ijtld.15.0864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kandler JL, Mercante AD, Dalton TL, Ezewudo MN, Cowan LS, Burns SP, Metchock B, Cegielski P, Posey JE, Global PETTS Investigators . 2018. Validation of novel Mycobacterium tuberculosis isoniazid resistance mutations not detectable by common molecular tests. Antimicrob Agents Chemother 62:e00974-18. doi: 10.1128/AAC.00974-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brossier F, Boudinet M, Jarlier V, Petrella S, Sougakoff W. 2016. Comparative study of enzymatic activities of new KatG mutants from low- and high-level isoniazid-resistant clinical isolates of Mycobacterium tuberculosis. Tuberculosis (Edinb) 100:15–24. doi: 10.1016/j.tube.2016.06.002 [DOI] [PubMed] [Google Scholar]
- 36. Merker M, Blin C, Mona S, Duforet-Frebourg N, Lecher S, Willery E, Blum MGB, Rüsch-Gerdes S, Mokrousov I, Aleksic E, et al. 2015. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat Genet 47:242–249. doi: 10.1038/ng.3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pérez-Lago L, Comas I, Navarro Y, González-Candelas F, Herranz M, Bouza E, García-de-Viedma D. 2014. Whole genome sequencing analysis of intrapatient microevolution in Mycobacterium tuberculosis: potential impact on the inference of tuberculosis transmission. J Infect Dis 209:98–108. doi: 10.1093/infdis/jit439 [DOI] [PubMed] [Google Scholar]
- 38. Cohen T, Chindelevitch L, Misra R, Kempner ME, Galea J, Moodley P, Wilson D. 2016. Within-host heterogeneity of Mycobacterium tuberculosis infection is associated with poor early treatment response: a prospective cohort study. J Infect Dis 213:1796–1799. doi: 10.1093/infdis/jiw014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meacci F, Orrù G, Iona E, Giannoni F, Piersimoni C, Pozzi G, Fattorini L, Oggioni MR. 2005. Drug resistance evolution of a Mycobacterium tuberculosis strain from a noncompliant patient. J Clin Microbiol 43:3114–3120. doi: 10.1128/JCM.43.7.3114-3120.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sander P, Springer B, Prammananan T, Sturmfels A, Kappler M, Pletschette M, Böttger EC. 2002. Fitness cost of chromosomal drug resistance-conferring mutations. Antimicrob Agents Chemother 46:1204–1211. doi: 10.1128/AAC.46.5.1204-1211.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hsu LY, Lai LY, Hsieh PF, Lin TL, Lin WH, Tasi HY, Lee WT, Jou R, Wang JT. 2020. Two novel katG mutations conferring isoniazid resistance in Mycobacterium tuberculosis. Front Microbiol 11:1644. doi: 10.3389/fmicb.2020.01644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vilchèze C, Jacobs WR. 2007. The mechanism of isoniazid killing: clarity through the scope of genetics. Annu Rev Microbiol 61:35–50. doi: 10.1146/annurev.micro.61.111606.122346 [DOI] [PubMed] [Google Scholar]
- 43. Sherman DR, Mdluli K, Hickey MJ, Arain TM, Morris SL, Barry CE, Stover CK. 1996. Compensatory ahpC gene expression in isoniazid-resistant Mycobacterium tuberculosis. Science 272:1641–1643. doi: 10.1126/science.272.5268.1641 [DOI] [PubMed] [Google Scholar]
- 44. Haratiasl AA, Hamzelou G, Amini S, Kardan-Yamchi J, Haeili M, Heidari F, Feizabadi MM. 2020. Molecular identification of mutations conferring resistance to rifampin, isoniazid and pyrazinamide among Mycobacterium tuberculosis isolates from Iran. J Chemother 32:75–82. doi: 10.1080/1120009X.2020.1716479 [DOI] [PubMed] [Google Scholar]
- 45. Wilson T, de Lisle GW, Marcinkeviciene JA, Blanchardand JS, Collins DM. 1998. Antisense RNA to ahpC, an oxidative stress defence gene involved in isoniazid resistance, indicates that AhpC of Mycobacterium bovis has virulence properties. Microbiology (Reading) 144 ( Pt 10):2687–2695. doi: 10.1099/00221287-144-10-2687 [DOI] [PubMed] [Google Scholar]
- 46. Middlebrook G. 1954. Isoniazid-resistance and catalase activity of tubercle bacilli; a preliminary report. Am Rev Tuberc 69:471–472. doi: 10.1164/art.1954.69.3.471 [DOI] [PubMed] [Google Scholar]
- 47. Ando H, Kondo Y, Suetake T, Toyota E, Kato S, Mori T, Kirikae T. 2010. Identification of katG mutations associated with high-level isoniazid resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 54:1793–1799. doi: 10.1128/AAC.01691-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental methods, supplemental tables, and Supplemental figures.
Data Availability Statement
The genomes of the three M. tuberculosis strains were submitted to the NCBI GenBank and were assigned the accession numbers SAMN34123194, SAMN34123195, and SAMN34123196, respectively.