

Abstract
The association between coronary artery disease (CAD) and posttraumatic stress disorder (PTSD) contributes to the high morbidity and mortality observed for these conditions. To understand the dynamics underlying PTSD-CAD comorbidity, we investigated large-scale genome-wide association (GWA) statistics from the Million Veteran Program (MVP), the UK Biobank (UKB), the Psychiatric Genomics Consortium, and the CARDIoGRAMplusC4D Consortium. We observed a genetic correlation of CAD with PTSD case-control and quantitative outcomes, ranging from 0.18 to 0.32. To investigate possible cause-effect relationships underlying these genetic correlations, we performed a two-sample Mendelian randomization (MR) analysis, observing a significant bidirectional relationship between CAD and PTSD symptom severity. Genetically-determined PCL-17 (PTSD 17-item Checklist) total score was associated with increased CAD risk (odds ratio = 1.04; 95% confidence interval, 95% CI = 1.01–1.06). Conversely, CAD genetic liability was associated with reduced PCL-17 total score (beta =−0.42; 95% CI =−0.04 to −0.81). Because of these opposite-direction associations, we conducted a pleiotropic meta-analysis to investigate loci with concordant vs. discordant effects on PCL17 and CAD, observing that concordant-effect loci were enriched for molecular pathways related to platelet amyloid precursor protein (beta = 1.53, p = 2.97 × 10−7) and astrocyte activation regulation (beta = 1.51, p = 2.48 × 10−6) while discordant-effect loci were enriched for biological processes related to lipid metabolism (e.g., triglyceride-rich lipoprotein particle clearance, beta = 2.32, p = 1.61 × 10−10). To follow up these results, we leveraged MVP and UKB electronic health records (EHR) to assess longitudinal changes in the association between CAD and posttraumatic stress severity. This EHR-based analysis highlighted that earlier CAD diagnosis is associated with increased PCL-total score later in life, while lower PCL total score was associated with increased risk of a later CAD diagnosis (Mann–Kendall trend test: MVP tau = 0.932, p < 2 × 10−16; UKB tau = 0.376, p = 0.005). In conclusion, both our genetically-informed analyses and our EHR-based follow-up investigation highlighted a bidirectional relationship between PTSD and CAD where multiple pleiotropic mechanisms are likely to be involved.
INTRODUCTION
Coronary artery disease (CAD) is a major contributor to premature mortality among individuals affected by posttraumatic stress disorder (PTSD). Several studies applied different approaches to investigate the underlying dynamics linking these illnesses [1–3], but there is no clear understanding of whether PTSD-CAD associations are due to causal effects or to shared pathogenic processes. A major challenge to disentangle this complex relationship is to model the longitudinal changes that lead to PTSD-CAD comorbidity. Previous prospective investigations reported that PTSD is associated with reduced coronary microcirculatory function and greater deterioration over time, and that differences in blood pressure and heart rate among trauma-exposed individuals could have implications for cardiovascular disease risk [4, 5].
Genetic associations can be used as instrumental variables in Mendelian randomization (MR) analyses to test the causal effect of exposures on outcomes of interest [6]. Genetically-informed analyses can be used to investigate cause-effect hypotheses that may otherwise not be feasible to test in randomized trials due to practical or ethical considerations. For PTSD, MR has been used to investigate causal hypotheses with respect to psychiatric comorbidities [7, 8], obesity-related traits [9, 10], respiratory outcomes [11], tobacco smoking [12], inflammatory biomarkers [13], sleeping patterns [14], and socioeconomic factors [15–17]. These MR studies support a complex network of cause-effect relationships involving PTSD. To our knowledge, no genetically-informed causal inference analysis was conducted to explain the comorbidity between PTSD and CAD. Previously, a trans-ancestry meta-analysis observed that PTSD was associated with decreased systolic and increased diastolic blood pressure, but these effects were not moderated by polygenic risk [18]. More recently, a multi-omics study showed that transcriptomic and proteomic regulatory mechanisms are partially shared between PTSD symptoms and hematologic and cardiometabolic traits [19], suggesting that certain pathogenic processes may be shared between these health outcomes.
Previous studies [20–22] explored three main mechanisms to explain the observed PTSD-CAD comorbidity: (i) PTSD could have a causal effect on CAD as an effect of posttraumatic stress on the cardiovascular system; (ii) CAD could change PTSD vulnerability because of associated life-threatening and life-altering events; and (iii) certain pathogenic processes may affect both PTSD and CAD risk. To test these hypotheses using a causal inference framework, we leveraged genome-wide information from the Million Veteran Program (MVP) [23], the Psychiatric Genomics Consortium (PGC) [24], the CARDIoGRAMplusC4D Consortium [25], and the UK Biobank (UKB) [26]. Additionally, we also investigated the electronic health records (EHR) available for 319,036 MVP participants and 155,817 UKB participants who were also assessed with PTSD Checklist instruments (PCL; for MVP the 17-item checklist, PCL-17; for UKB the six-item checklist, PCL-6) [27].
METHODS
This study was conducted using genome-wide association (GWA) statistics generated from MVP, UKB, PGC, and the CARDIoGRAMplusC4D Consortium and individual-level information from MVP and UKB EHRs and PCL assessments. The use of GWA statistics (i.e., previously collected, de-identified, aggregated data) did not require institutional review board approval. The use of UKB individual-level data has been conducted through the application reference no. 58146. UKB has approval from the Northwest Multi-center Research Ethics Committee (MREC) as a Research Tissue Bank (RTB) approval. This approval means that researchers do not require separate ethical clearance and can operate under the RTB approval. The use of MVP individual-level data was conducted under project CSP575b. MVP is approved by the VA Central IRB and project CSP575b was also approved by local VA IRBs in Boston, San Diego, and West Haven.
Cohorts
Million Veteran Program.
In our study, we investigated MVP genome-wide information and EHRs related to PTSD and CAD. We used GWA statistics from a recent PTSD GWAS conducted in MVP [28] that analyzed an HER-validated algorithmic PTSD diagnosis [29] and PCL-17-derived quantitative phenotypes related to PTSD symptom clusters (re-experiencing, REX; avoidance, AVOID; hyperarousal, HYPER; and overall severity score, PCL-17) [28]. We included GWA statistics generated from 36,301 cases and 178,107 controls of European descent for the PTSD diagnosis and from 186,689 individuals of European descent for the PCL-17-derived quantitative phenotypes. For CAD, we used GWA statistics from a CAD GWAS conducted on 293,470 MVP participants of European descent. In our follow-up analysis, we investigated PTSD information derived from the PCL-17 assessment and CAD information derived from VHA EHRs. As part of the MVP lifestyle survey, MVP participants completed a PCL-17 assessment, which asks respondents to report the extent to which they had been affected in the previous month by symptoms in response to stressful life experiences. To derive CAD diagnosis from VHA EHR, we used the ICD (International Classification of Diseases) codes used by the CARDIoGRAMplusC4D Consortium [25]. Briefly, CAD was defined as myocardial infarction, chronic ischemic heart disease, or angina (ICD9: 410–413, 414.0, 414.8, 414.9; ICD10: I20, I21–I24, I25.1, I25.2, I25.5–I25.9). Exclusion criteria were aneurysm and atherosclerotic cardiovascular disease (ICD9 414.1 and ICD 10 I25.0, I25.3, I25.4). These exclusion criteria were defined in accordance with those proposed by the CARDIoGRAMplusC4D Consortium to refine CAD cases through ICD codes [25]. Through this CAD definition, we identified 73,074 cases and 245,962 controls that completed the PCL-17 assessment.
UK Biobank.
UKB contributed to the PGC and CARDIoGRAMplusC4D GWAS meta-analyses described in the subsequent paragraphs. As part of the online-mental health questionnaire [30], 157,366 participants were also assessed for items included in the PCL-6 assessment. Similar to the CARDIoGRAMplusC4D Consortium [25], we defined CAD cases leveraging UKB information derived from questionnaires and EHRs. Based on the questionnaires, we defined CAD cases as UKB participants having ‘vascular/heart problems diagnosed by doctor’, ‘non-cancer illnesses that self-reported as angina or heart attack’, or self-reported surgery including ‘percutaneous transluminal coronary angioplasty’, ‘coronary artery bypass grafting’, or ‘triple heart bypass’. Through UKB EHRs, we defined CAD cases as participants reporting codes related to infarction, percutaneous transluminal coronary angioplasty, coronary artery bypass grafting, chronic ischemic heart disease, or angina (ICD9: 410–413, 414.0, 414.8, 414.9; ICD10: I20, I21–I24, I25.1, I25.2, I25.5–I25.9; OPCS-4: K40–K46; K49, K50.1, K75). Exclusion criteria were aneurysm and atherosclerotic cardiovascular disease (ICD9 414.1 and ICD 10 I25.0, I25.3, I25.4). These exclusion criteria were defined in accordance with those proposed by the CARDIoGRAMplusC4D Consortium to refine CAD cases through ICD codes [25]. Through this CAD definition, we identified 8942 cases and 146,875 controls that completed the PCL-6 assessment.
Psychiatric Genomics Consortium.
We used GWA statistics generated by the PGC-PTSD workgroup, which conducted a GWAS meta-analysis across 60 different PTSD studies, ranging from clinically deeply characterized, small patient groups to large cohorts with self-reported PTSD symptoms [31]. We analyzed two datasets: PGC-PTSD version 1.5 including 12,823 cases and 35,648 controls of European descent; PGC-PTSD version 2 (i.e., PGC-PTSD version 1.5 plus UKB) including 23,212 cases 151,447 controls of European descent.
CARDIoGRAMplusC4D Consortium.
This collaborative initiative combined data from multiple large-scale genetic studies to identify risk loci for CAD and myocardial infarction. We analyzed two CARDIoGRAMplusC4D datasets: a GWAS meta-analysis of 48 studies including 60,801 cases and 123,504 controls [32] and a follow-up GWAS meta-analysis combining these data with UKB reaching a total of 79,602 cases and 261,418 controls [25].
Other GWA datasets.
We also investigated additional cardiovascular traits, including blood pressure, heart failure, PR interval, and resting heart rate. For blood pressure, we analyzed GWA statistics related to systolic blood pressure (N = 738,170) [33], diastolic blood pressure (N = 757,601) [33], pulse pressure (N = 738,170) [33], and self-reported high blood pressure (144,793 case and 313,761 controls) [34]. For heart failure, the GWA statistics used in the present study were generated from 47,309 cases and 930,014 controls [35]. For resting heart rate, we used GWA statistics derived from 458,969 individuals [34].
Data analysis
Effect size distribution of genetic effects in PTSD and CAD phenotypes.
To investigate the genetic architecture of PTSD and CAD traits, we applied GENESIS (GENetic Effect-Size distribution Inference from Summary-level data) package [36]. This likelihood-based framework permitted us to estimate the number of underlying susceptibility SNPs and their expected effect sizes with respect to the PTSD and CAD phenotypes investigated.
Genetic correlation analysis.
We assessed SNP-based heritability and genetic correlation among GWA datasets analyzed using the linkage disequilibrium (LD) score regression method [37]. The SNP-based heritability is the proportion of phenotypic variance that could be explained by the aggregated effect of all SNPs. The genetic correlation estimates represent the genetic covariation between the two traits based on all polygenic effects captured by SNPs. The LD score regression analysis was performed using pre-computed LD scores based on 1000 Genomes Project populations of European descent (available at https://github.com/bulik/ldsc).
Polygenic risk scoring.
We used the gtx R package incorporated in PRSice software [38] to calculate an approximate estimate of the explained variance from a multivariate regression model [39]. PRSice is based on the clumping-thresholding method, where the clumping procedure is used to define LD-independent associations and thresholding is used to define the variants to include in the PRS calculation [40]. Because the PRS analysis was conducted to define instrumental variables for the subsequent MR analysis, we selected the clumping-thresholding method, which is also the method to define instrumental variables in a two-sample MR analysis. The PRS were defined considering a genome-wide significance threshold (P < 5 × 10−8) and applying a clumping procedure with an LD cutoff of R2= 0.001 within a 10,000-kilobase window. The European samples from the 1000 Genomes Project were used as the LD reference panel [41].
Genetically-informed causal inference analysis.
MR approaches allow inference of putative causal relationships between an exposure and an outcome of interest using genetic variants (SNPs) as instrumental variables [42]. We followed the STROBE (STrengthening the Reporting of Observational studies in Epidemiology) guidelines [43] to design and report our MR analyses and the related sensitivity tests. Considering genetic instruments that survived multiple testing correction in the PRS analysis, we conducted a two-sample MR analysis using the TwoSampleMR R package [44]. The primary analysis was conducted considering a random-effects inverse-variance weighted (IVW) method, because of its high statistical power [44]. The secondary MR methods included MR Egger [45], simple mode [46], weighted median [47], and weighted mode [46]. These additional MR methods permitted us to assess the model considering different assumptions: all variants valid or balanced pleiotropy for IVW; InSIDE (Instrument Strength Independent of Direct Effect) for MR Egger; majority valid for weighted median; plurality valid for mode-based estimation [48]. We also conducted multiple MR sensitivity analyses to exclude possible biases (horizontal pleiotropy, i.e. the variants included in the genetic instrument have an effect on disease outside their effects on the exposure in MR [49]) under different scenarios in the MR estimates. These included the IVW-heterogeneity test [50], the MR-Egger intercept [45], the MR-Robust Adjusted Profile Score (MR-RAPS) overdispersion test [51], and the MR–Pleiotropy Residual Sum and Outlier (MR-PRESSO) global test [52]. Finally, a leave-one-out analysis was conducted to identify potential outliers among the variants included in the genetic instruments tested. The outlier variants were defined as those affecting the results of the IVW heterogeneity test outside the range observed for the other variants. To test the MR findings by applying another genetically-informed causal inference method based on different assumptions, we used the latent causal variable (LCV) approach [53]. This distinguishes causation from genetic correlation by estimating whether a latent variable has a causal effect on the traits investigated using GWA statistics [53].
Pleiotropic meta-analysis.
We leveraged the ASSET (Association Analysis based on Subsets) approach [54] to identify loci with concordant and discordant effect directions between PCL-17 (MVP) and CAD (UKB-CARDIoGRAMplusC4D). The ASSET method allows the meta-analysis of subsets of loci based on their different effect directions, and applies a tail-probability approximation to account for correlation among different test statistics [54]. The ASSET results were annotated using Multi-marker Analysis of GenoMic Annotation (MAGMA) [55] implemented in FUMA [56] and tested for enrichment with respect to gene sets available from the Molecular Signatures Database (MSigDB) [57]. The enrichment analysis was performed using the hypergeometric test to estimate whether the genes prioritized are statistically overrepresented in certain molecular pathways and biological processes [56].
EHR-based follow-up analysis.
We used individual-level EHR and PCL data available from MVP and UKB cohorts to explore further the association between CAD and PTSD symptom severity. Specifically, we assessed the association of CAD with PCL total score, stratifying the analysis by the difference between the time of the PCL assessment with respect to the time of the first CAD diagnosis. As described in the section Cohorts, MVP participants were screened using the PCL-17 assessment and CAD diagnosis was based on the ICD codes defined by the CARDIoGRAMplusC4D Consortium. UKB participants were screened using the PCL-6 assessment and CAD diagnosis was based on the ICD and OPCS-4 codes and on self-reported information using the same approach previously applied for this cohort by the CARDIoGRAMplusC4D Consortium [25]. A linear regression analysis was conducted to test the association of CAD with PCL total score. In MVP analysis, we included age, sex, income, self-reported ethnicity, and self-reported racial background as covariates. In UKB analysis, we included age, sex, Townsend deprivation index (a measure of material deprivation [58]), and self-reported ethnic background. Within CAD cases, we also tested the association of PCL total score with the difference between the time of the PCL assessment and the time of the first CAD diagnosis. This analysis was conducted considering a linear regression model using the same sets of covariates described above. We used the Mann-Kendall trend test to verify whether there is a statistically significant trend in the changes in PCL scores across CAD cases depending on the difference in time between CAD diagnosis and PCL assessment.
RESULTS
Genetic correlation analysis
Among the genome-wide datasets investigated, we observed a high genetic correlation among PTSD/PCL phenotypes (e.g., PGC PTSD v2 – MVP PCL-17 rg = 0.96, p = 3.99 × 10−247; Supplementary Table 1A) and among CAD datasets (e.g., MVP CAD – UKB-CARDIoGRAMplusC4D rg = 0.97, p = 1.01 × 10−299; Supplementary Table 1B). Comparing their genetic architectures, PTSD traits had a higher degree of polygenicity (i.e., higher number of variants with small individual effect) than CAD (Supplementary Fig. 1). Similar to other brain-related phenotypes [59], PTSD quantitative phenotypes appear to model better the continuum of the genetic liability than PTSD case-control definitions. With respect to CAD, we observed slightly different patterns of polygenic architecture between CARDIoGRAMplusC4D GWAS meta-analysis and UKB-CARDIoGRAMplusC4D GWAS meta-analysis (Supplementary Fig. 1). This may be due to the characteristics of the UKB cohort (e.g., age, sex, and comorbidities).
For three CAD datasets investigated (i.e., CARDIoGRAMplusC4D, UKB-CARDIoGRAMplusC4D, and MVP), the genetic correlation between CAD and PTSD traits ranged from 0.18 to 0.32 (Fig. 1-left; Supplementary Table 2). Most of the variation observed among the genetic correlation estimates appears to be due to a higher degree of uncertainty in the less powered PTSD datasets. The statistically more powerful PTSD datasets converged around a 20% genetic correlation with CAD (e.g., MVP PCL-17 rg = 0.20, p = 6.71 × 10−7; Fig. 1-left; Supplementary Table 2).
Fig. 1. Genetic correlation analysis.
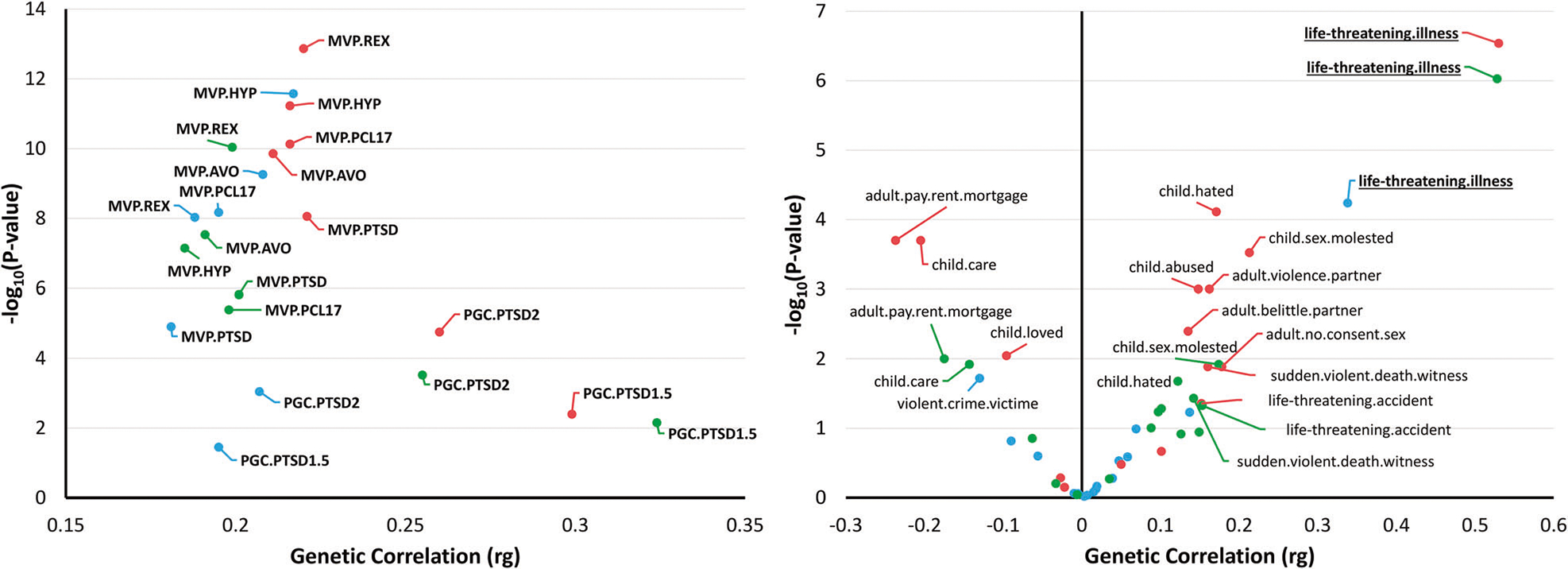
Left. Genetic correlation (rg) between CAD (coronary artery disease) and PTSD (posttraumatic stress disorder) traits. Details are reported in Supplementary Table 2. Right. Genetic correlation of CAD with traits related to traumatic events and social support. Details are reported in Supplementary Table 3. Labels are reported for genetic correlation surviving multiple testing correction. Underlined bolded labels are reported for genetic correlation surviving multiple testing correction across the three CAD datasets tested. CAD datasets are color-coded as light blue for MVP (Million Veteran Program), red for UKB-CardioGRAMplusC4D meta-analysis, and green for CardioGRAMplusC4D meta-analysis. MVP.PTSD: MVP PTSD GWAS; MVP.PCL17: MVP GWAS of PTSD 17-item checklist total score; MVP.AVO: MVP GWAS of avoidance symptom severity score; MVP.HYP: MVP GWAS of hyperarousal symptom severity score; MVP.REX: MVP GWAS of reexperiencing symptom severity score; PGC.PTSD2: Psychiatric Genomic Consortium (PGC) GWAS of PTSD version 2; PGC.PTSD1.5: Psychiatric Genomic Consortium (PGC) GWAS of PTSD version 1.5.
We also investigated the genetic correlation of CAD with 16 UKB-derived traits related to traumatic events and social support (Fig. 1-right; Supplementary Table 3). Considering a Bonferroni correction accounting for the number of traits tested, only “Been diagnosed with a life-threatening illness” (UKB Data-Field: 20528) showed statistically significant genetic correlation with the CAD datasets investigated (CARDIoGRAMplusC4D: rg = 0.53, p = 9.37 × 10−7; UKB-CARDIoGRAMplusC4D: rg = 0.53, p = 2.89 × 10−7; MVP: rg = 0.34, p = 5.78 × 10−5). Considering the UKB-CARDIoGRAMplusC4D dataset, we identified significant genetic correlations of CAD with six other phenotypes, but none of these survived multiple testing correction in the other two CAD datasets. For the variable “Been diagnosed with a life-threatening illness”, PTSD datasets showed genetic correlation estimates similar to the one observed with respect to CAD (e.g., MVP PCL-17 rg = 0.5, p = 1.28 × 10−6; MVP AVOID rg = 0.51, p = 1.02 × 10−6; MVP HYPER rg = 0.45, p = 4.77 × 10−6; MVP REX rg = 0.44, p = 7.62 × 10−6; Supplementary Table 4).
Genetically-informed causal inference analysis
Similar to previous studies [17], we defined informative genetic instruments conducting a cross-phenotype PRS analysis based on variants reaching genome-wide significance (p < 5 × 10−8) in the exposure GWAS. Because the two-sample MR approach can be biased by sample overlap [60], we selected non-overlapping exposure and outcome datasets (see Cohorts description) and prioritized the largest sample size when multiple datasets were available. Applying a Bonferroni multiple testing correction (p = 0.005), we identified seven significant PRS associations, observing bidirectional relationships between CAD and PTSD traits (top-results: MVP PCL-17 PRS on UKB-CARDIoGRAMplusC4D p = 1.3 × 10−5; UKB-CARDIoGRAMplusC4D PRS on MVP PCL 17 p = 4.29 × 10−4; Supplementary Table 5). Considering the strength of these PRS associations, we considered the cause-effect relationship between CAD (UKB-CARDIoGRAMplusC4D) and PCL-17 total score (MVP) as the primary test in our two-sample MR analysis. While genetically-determined PCL-17 total score was associated with increased CAD risk (IVW odds ratio = 1.04, p = 2.56 × 10−3; Fig. 2-left), genetically-determined CAD was associated with decreased PCL-17 total score (IVW beta =−0.42, p = 0.029). These estimates were consistent across different MR methods and no evidence of horizontal pleiotropy was detected via the MR-Egger intercept (Supplementary Table 6). However, the CAD → PCL-17 association was characterized by strong heterogeneity (IVW heterogeneity test: Q = 89.4, p = 2.45 × 10−7; MR-RAPS global test: RSSobs = 110.9, p < 10−4). We conducted a leave-one-out (LOO) analysis to identify the variants contributing to the heterogeneity within the CAD instrumental variable (Supplementary Table 7). After removing the outliers identified in the LOO analysis, we confirmed that increased CAD genetic liability is associated with reduced PCL-17 score (IVW beta = −0.50, p = 2.61 × 10−4; Fig. 2-right, Supplementary Table 8) in the absence of heterogeneity (IVW heterogeneity test: Q = 36.8, p = 0.121; MR-RAPS global test: RSSobs = 39.6, p = 0.137). We observed similar results when testing the effect of genetically-determined CAD on PTSD symptom cluster severity (i.e., AVOID, HYPER, and REX; Supplementary Table 9). These estimates were also characterized by strong heterogeneity, but the effects were confirmed after the removal of outlier variants from the instrumental variables (Supplementary Table 10).
Fig. 2. Bidirectional Mendelian randomization analysis.
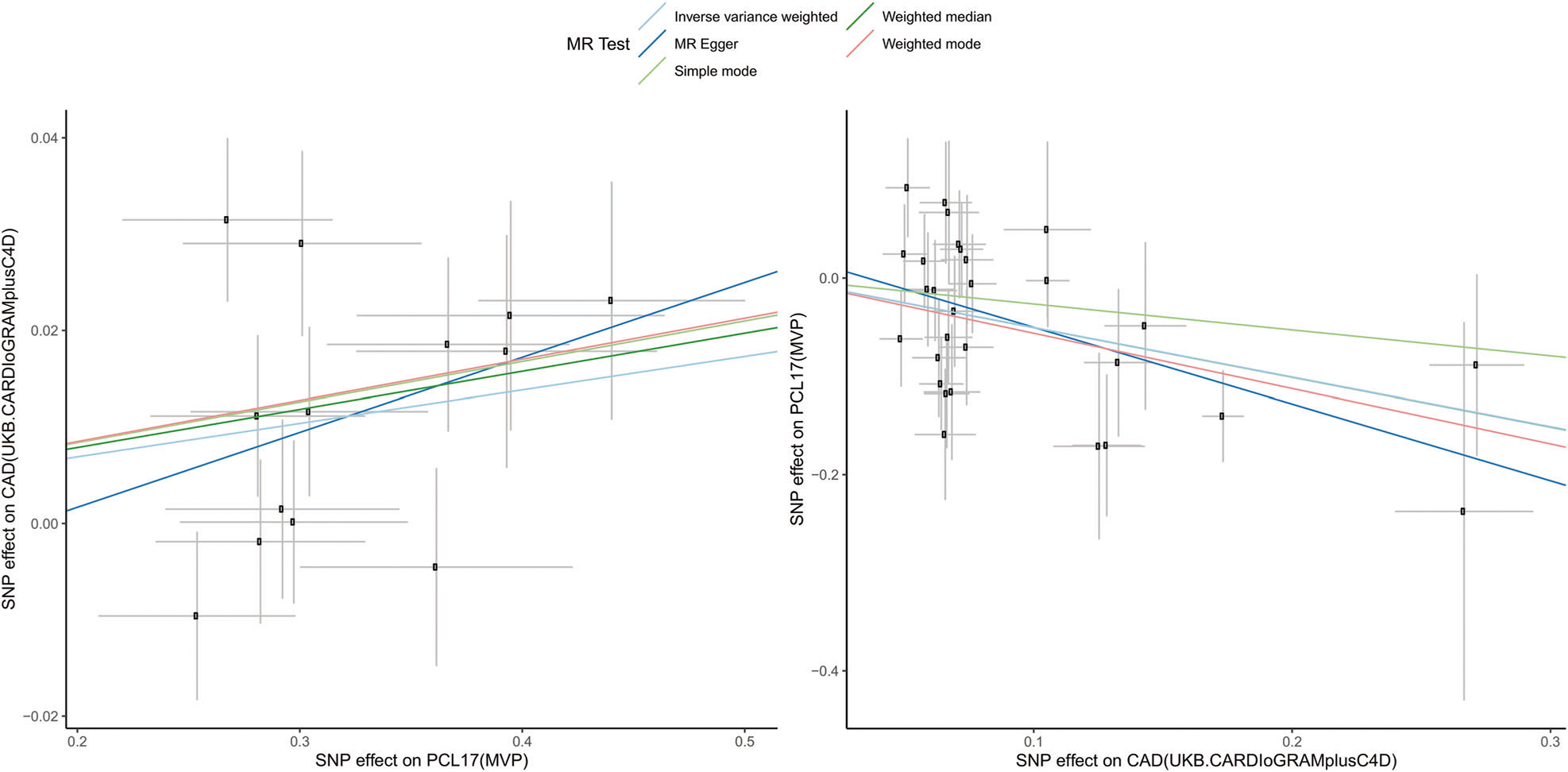
Left. SNP-exposure (coronary artery disease, CAD associations, logOR) and SNP-outcome (PTSD 17-item checklist total score, PCL17 associations, beta) coefficients. Right. SNP-exposure (PCL17 associations, beta) and SNP-outcome (CAD associations, logOR) coefficients. Error bars (95% CIs) are reported for each association. Slopes are reported for multiple Mendelian randomization (MR) methods.
To evaluate whether the CAD → PCL-17 effect was due to characteristics of the datasets investigated (UKB-CARDIoGRAMplusC4D CAD GWAS and MVP PCL-17 GWAS), we re-tested the association using the other datasets available. Using the CARDIoGRAMplusC4D GWAS (not including UKB) as the exposure dataset, we observed the same pattern of associations across the PCL-17-derived traits (i.e., total score, AVOID, HYPER, and REX; Supplementary Table 11). Similar to the analyses conducted using the UKB-CARDIoGRAMplusC4D GWAS, there was evidence of significant heterogeneity in these associations (Supplementary Table 11), but the estimates were confirmed after the removal of the variant outliers (Supplementary Table 12). With regard to PTSD severity, there are two GWAS datasets not including MVP: a PCL-6 GWAS conducted in the UKB cohort and a PGC GWAS of a PTSD quantitative phenotype combining UKB PCL-6 with PTSD severity information from other cohorts [61]. To avoid sample overlap bias, the MR analyses were conducted testing CARDIoGRAMplusC4D CAD → UKB PCL-6 and ii) MVP CAD → PGC PTSD quantitative phenotype. The effects observed from these analyses were null with inconsistent effect direction across MR methods in both cases (Supplementary Table 13). This null result could be due to a lack of statistical power due to the limited genetic correlation of MVP PCL-17 with UKB PCL-6 (rg = 0.81, p = 6.26 × 10−75) and PGC-PTSD quantitative phenotype (rg = 0.84, p = 2.5 × 10−75). These estimates are lower than the genetic correlations observed among PTSD traits assessed in different cohorts (Supplementary Table 1).
To evaluate whether the effect observed was specific to CAD, we tested other cardiovascular outcomes and risk factors (see Methods). We identified significant genetic correlation of MVP PCL-17 total score with heart failure (rg = 0.24, p = 9.45 × 10−7), self-reported high blood pressure (rg = 0.23, p = 5.86 × 10−16), and resting heart rate (rg = 0.15, p = 1.1 × 10−10). Among these, we observed significant PRS association with MVP PCL-17 only for heart failure (p = 0.001; Supplementary Table 14). Our MR analysis showed that increased risk of genetically-determined heart failure is associated with lower PCL-17 (beta =−1.35, p = 1.71 × 10−4). This effect was consistent across different MR methods and no evidence of heterogeneity and horizontal pleiotropy was observed (Supplementary Table 15).
To explore further the putative cause-effect relationships identified in our MR analysis, we applied the LCV approach. This is an alternative method to perform causal inference analyses modeling genetic effects [53]. LCV analysis did not identify a statistically significant genetic causality proportion between these traits (e.g., MVP PCL-17 → UKB-CARDIoGRAMplusC4D CAD: genetic causal proportion 0.46 ± 0.36; Supplementary Table 16). This null result is likely due to the fact that the LCV approach is not designed to model bidirectional associations as causal [53].
Pleiotropic meta-analysis
Testing for concordant and discordant genetic associations between PCL-17 (MVP) and CAD (UKB-CARDIoGRAMplusC4D), we identified seven variants with concordant effects and ten variants with discordant effects that reached genome-wide significance (p < 5 × 10−8) in the pleiotropic meta-analysis (Supplementary Table 17). To investigate the underlying mechanisms of concordant and discordant genetic effects, we conducted an enrichment analysis for molecular pathways and gene ontologies (GO). After applying a Bonferroni multiple testing correction (p = 3.25 × 10−6), we identified two processes that were enriched for loci with concordant effects between PCL-17 and CAD: “Platelet Amyloid Precursor Protein (APP) Pathway” (MSigDB: M6487; beta = 1.53, p = 2.97 × 10−7) and “Negative Regulation of Astrocyte Activation” (GO:0061889; beta = 1.51, p = 2.48 × 10−6). Seven enrichments were identified with loci with discordant effect: “Triglyceride-Rich Lipoprotein Particle Clearance” (GO:0071830; beta = 2.32, p = 1.61 × 10−10), “Chylomicron Clearance” (MSigDB: M27845; beta = 2.99, p = 8.92 × 10−9); “Low-Density Lipoprotein (LDL) Pathway during Atherogenesis” (MSigDB: M22051; beta = 2.45, p = 3.18 × 10−8); “Protein-Lipid Complex Assembly” (GO:0065005; beta = 0.94, p = 7.48 × 10−8), “Phosphatidylcholine Catabolic Process” (GO:0034638; beta = 1.58, p = 2 × 10−6); “Tumor Suppressor Arf Inhibits Ribosomal Biogenesis” (MSigDB: M11358; beta = 1.08, p = 1.62 × 10−6); “Plasma Lipoprotein Assembly” (MSigDB: M27841; beta = 1.31; p = 3.06 × 10−6).
EHR-based follow-up analysis
To complement the results obtained from the genetically-informed analyses, we used individual-level EHR and PCL information from MVP and UKB. Specifically, we considered the temporal relationship between CAD (status and time of the diagnosis) and PCL (total score and time of the assessment). The time of the assessment is relevant, because PCL allows assessing the extent to which the participants had been affected in the previous month by symptoms in response to stressful life experiences. Stratifying CAD cases for whether the disease diagnosis was before, after, or the same year of the PCL assessment, we observed positive associations of CAD with PCL total score across all comparison groups in both MVP and UKB (Table 1). Within CAD cases, we also investigated whether the time difference between CAD onset and PCL assessment (Supplementary Figs. 2 and 3) was associated with PCL total score. In both MVP and UKB, there was a significant pattern where CAD cases with a larger time difference between the disease onset and the PCL assessment had higher PCL total scores (MVP: Mann–Kendall Trend Test tau = 0.932, p < 2 × 10−16; UKB: Mann–Kendall Trend Test tau = 0.376, p = 0.005; Fig. 3). Stratifying CAD cases depending on whether the disease onset was before or after the PCL assessment, this relationship appears to be consistent with the possible negative mental health consequences associated with CAD and its symptoms (Table 1). Indeed, CAD cases with PCL assessment after their cardiac disease onset had higher PCL total scores when the time difference between disease onset and PCL assessment was longer (MVP: beta = 0.113, p = 6.95 × 10−16; UKB: beta = 0.011, p = 0.027). Conversely, CAD cases with PCL assessment before cardiac disease onset had PCL-17 total scores lower when the time difference between the disease diagnosis and PCL assessment was larger (MVP: beta = 0.192, p = 0.011).
Table 1.
Association of CAD (status and time of the diagnosis) with PCL total scores in MVP and UKB.
| CAD | MVP (PCL-17; control N = 245,962) | UKB (PCL-6, control N = 146,875) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| case N | Case-Control | Case-only | case N | Case-Control | Case-only | |||||||||
| Beta | SE | P | Beta | SE | P | Beta | SE | P | Beta | SE | P | |||
| Total sample | 73,074 | 2.89 | 0.06 | < 10−300 | 0.112 | 0.01 | 4.08 × 10−29 | 8942 | 0.58 | 0.034 | 3.98 × 10−66 | 0.01 | 0.004 | 0.001 |
| Onset after PCL assessment | 12,267 | 2.19 | 0.13 | 1.12 × 10−63 | 0.192 | 0.075 | 0.011 | 167 | 0.25 | 0.234 | 0.391 | −0.57 | 0.533 | 0.287 |
| Onset same-year PCL assessment | 4646 | 2.71 | 0.21 | 4.23 × 10−38 | − | − | − | 590 | 0.44 | 0.125 | 3.94 × 10−4 | − | − | − |
| Onset before PCL assessment | 56,161 | 3.03 | 0.07 | < 10−300 | 0.11 | 0.014 | 6.95 × 10−16 | 8185 | 0.6 | 0.035 | 7.11 × 10−66 | 0.011 | 0.005 | 0.027 |
In the case-control analysis, the PCL total scores of controls were compared with those of CAD cases (total sample and stratified by the timing of their cardiac disease onset with respect to PCL assessment). In the case-only analysis, we tested the association of PCL total scores with the temporal difference (years) between the time of CAD diagnosis and the time of PCL assessment. In MVP, the analyses were corrected for age, sex, income, self-reported ethnicity, and self-reported racial background. In UKB, the analyses were corrected for age, sex, Townsend deprivation index, and self-reported ethnic background.
Fig. 3. EHR-based follow-up analysis.
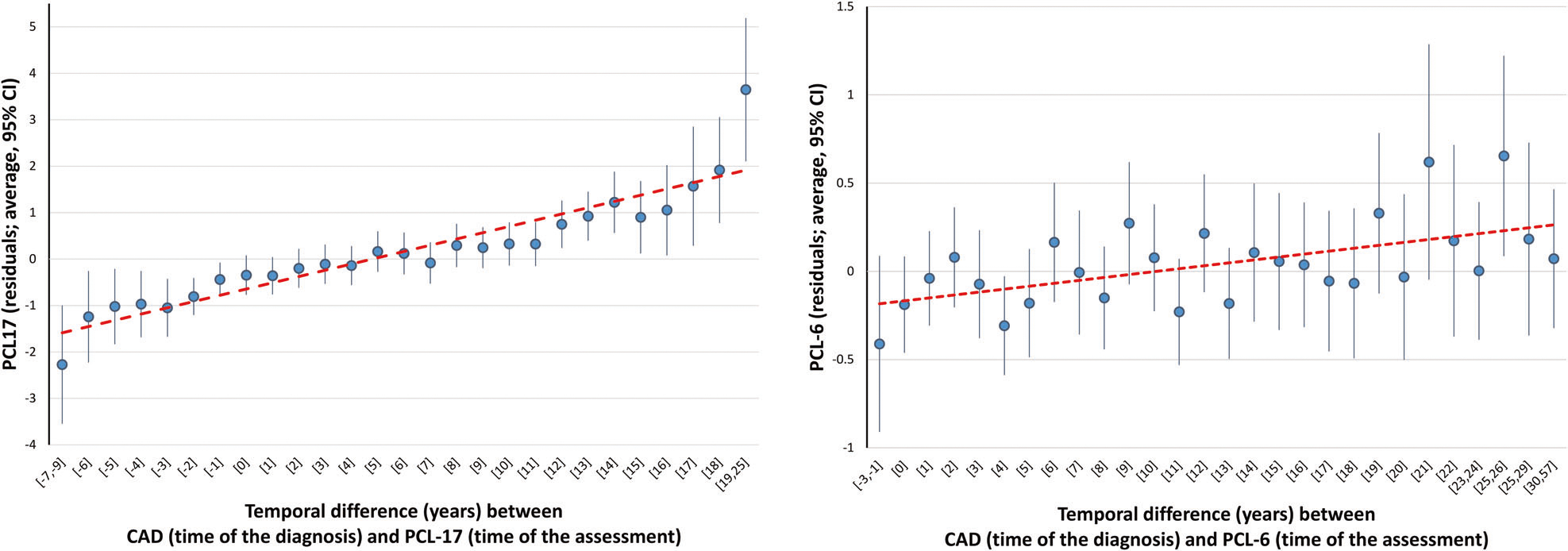
Relationship of PCL (PTSD checklist) total scores with the temporal difference between CAD (time of the diagnosis) and PCL (time of the assessment) in Million Veteran Program (MVP; left) and UK Biobank (UKB; right). In MVP, 17-item PCL (PCL-17) residuals were obtained regressing PCL-17 total score on age, sex, income, self-reported ethnicity, and self-reported racial background. In UKB, PCL-6 residuals were obtained regressing PCL-6 total score on age, sex, Townsend deprivation index, and self-reported ethnic background.
DISCUSSION
Leveraging GWA statistics generated by consortium meta-analyses and biobanks, we assessed the genetic correlation of CAD with PTSD, defining PTSD via case-control or quantitative measures. We observed consistent results across datasets converging on ~20% CAD-PTSD genetic correlation. Conversely, when testing traits related to traumatic experience and social support, there was a lack of consistent pleiotropy with CAD across the datasets investigated. This supports the notion that the PTSD-CAD comorbidity can be partially due to pleiotropic mechanisms by which some risk loci contribute to both diseases, but these shared genetic effects did not appear to be due to the association of PTSD with traumatic events and social support. The only exception was the shared genetic correlation (rg = ~0.5) of PTSD and CAD with “Been diagnosed with life-threatening disease”. This likely reflects CAD prevalence among life-threatening diseases [62]. CAD diagnosis itself and the medical encounters during which it is diagnosed may be experienced as traumatic experiences leading to posttraumatic stress [63].
In our genetically-informed causal inference analysis, we observed a significant bidirectional relationship between CAD and PCL-17 total score. The effect of genetically-determined PCL-17 total score on CAD was in line with the positive genetic correlation observed between these two traits: an increase of one standard deviation in the PCL-17 total score was associated with a 3.5% increase in the odds of CAD risk. Conversely, we observed that genetically-determined CAD was associated with a reduction in PCL-17 total scores. Following the STROBE-MR guidelines, we verified the consistency of the estimates across MR methods, data sources, and PTSD outcomes and also evaluated the effect of possible biases on the MR estimates (i.e., heterogeneity and horizontal pleiotropy within the instrumental variables). Our MR results were consistent across the sensitivity analyses performed. Nevertheless, we observed a null effect of CAD on PCL-6 total scores assessed in UKB cohort. Because MVP is the only large-scale GWAS of PCL-17 total score available to date, we cannot exclude that the effect of CAD on PCL-17 total score is due to characteristics specific to the MVP cohort or to patients within the VA healthcare system. Another possible bias that we could not test in our study is the possible survival bias in CAD genetic associations. However, a previous study indicated this should be very limited [64].
Our pleiotropic meta-analysis to investigate loci with concordant and discordant effects between PCL-17 and CAD highlighted different biological processes. Loci with concordant PCL-17 and CAD associations showed enrichment for molecular pathways related to platelet APP and astrocyte activation regulation. APP is a major inhibitor of platelet aggregation and is released from activated platelets in patients with acute coronary syndrome [65, 66]. APP demonstrated neuroprotective properties following traumatic brain injuries, including long-term neuronal survival and neuronal extension [67]. In this context, the regulation of astrocyte activation suggests that loci with concordant effects between PCL17 and CAD are involved in pathways related to neuroprotective functions. These may contribute to the comorbidity of trauma and PTSD with dementias [68, 69]. Conversely, the loci with discordant effects between PCL17 and CAD were enriched for multiple pathways related to lipid metabolism. Increased lipid levels (triglycerides and LDL) are associated with adverse cardiac outcomes [70]. With respect to PTSD, altered lipid levels are observed among affected patients in the context of interrelated somatic features including a proinflammatory milieu, metabolomic changes, and metabolic dysregulation [71].
In our EHR-based follow-up analysis, CAD was associated with increased PCL total score in both MVP and UKB cohorts, also when stratifying cases by whether the CAD diagnosis was received before, after, or in the same year as the PCL assessment. We also conducted a case-only analysis investigating whether there is a relationship between the time difference between PCL assessment and CAD diagnosis. Among CAD cases diagnosed before the PCL assessment, an earlier CAD diagnosis was associated with a higher PCL total score later in life in both MVP and UKB cohorts. A growing literature regarding cardiac-disease-induced PTSD highlights the negative mental outcomes associated with acute coronary syndrome and related medical procedures [72]. Additionally, the enduring somatic threat model has been proposed to address PTSD due to acute manifestations of chronic diseases that are enduring/internal in nature [63]. However, these reports suggest that the onset of CAD is followed closely in time with PTSD. Conversely, our EHR-based findings highlight the potential long-term effect of CAD on PTSD severity.
With respect to CAD subjects who received their CAD diagnosis after the PCL assessment, MVP participants with lower PCL-17 total score had received a CAD diagnosis later in time than individuals with higher PCL-17 total score. This association is in line with the positive association of genetically-determined PCL-17 on CAD identified in our two-sample MR analysis. However, we cannot exclude the possibility that individuals receiving a CAD diagnosis closer to the time of their PCL assessment may at the time be experiencing cardiac symptoms that increase their psychological stress and PTSD symptoms.
Although our EHR-based follow-up analysis did not find direct evidence supporting a causal effect between CAD and PTSD severity, we found the relationship between CAD and PTSD severity is affected by the timing of CAD onset. Earlier health-event occurrence is genetically correlated with a higher polygenic risk of disease susceptibility, but the age of first occurrence has specific genetic risk factors not associated with susceptibility to the disease [73]. With respect to cardiovascular outcomes, the genetic correlation between age of first occurrence and disease susceptibility was only partial. Because of the effect of CAD onset on PTSD severity, we speculate that the association of higher CAD genetic liability with lower PCL-17 score could be due to the unaccounted effect of the genetic predisposition to CAD onset. Unfortunately, there is no large-scale GWAS of CAD onset to test this hypothesis using genetically-informed causal inference methods.
Although the present study provides novel insights into the complex relationship between PTSD and CAD, we acknowledge several limitations. Genetically-informed causal inference analysis was conducted using genome-wide association statistics generated from cohorts including individuals of European descent because of the unavailability of large-scale PTSD and CAD GWAS in other ancestry groups. In the EHR-based follow-up analysis, we investigate UKB and MVP participants of different racial and ethnic backgrounds considering the latter as a covariate in the regression analysis. Due to the limited sample size available, we did not explore possible differences across racial and ethnic backgrounds. Accordingly, further studies will be needed to investigate genetic and phenotype differences in PTSD-CAD relationships across worldwide populations. Because of the higher statistical power of the PCL-17 GWAS, our primary analysis tested this cross-sectional assessment with respect to CAD (a lifetime diagnosis). Although PCL-17 assessment was not performed in the year of the index trauma or at the onset of PTSD symptoms, the high genetic correlation observed (rg > 0.95) strongly supports that last-month PCL-17 cross-sectional assessment is a very good proxy of PTSD lifetime diagnosis. Nevertheless, we cannot exclude that the timing of the PCL-17 assessment may have affected the results of our analyses. Finally, we did not explore the effect of possible mediators and moderators in the PTSD-CAD relationship, because our main goal was to perform a comprehensive investigation of the bidirectional associations between PTSD and CAD, testing multiple datasets and methods. Psychiatric and somatic comorbidities of PTSD can contribute to the bidirectional associations observed. Indeed, PTSD cases are often accompanied by other mental illnesses including major depression, anxiety disorders, and substance use disorders [74]. Additionally, somatic comorbidities of PTSD include several cardiovascular outcomes, such as angina pectoris, tachycardia, and hypertension [75]. Although our genetically-informed analyses demonstrated that the type of traumatic experience and social support should not have affected the observed PTSD-CAD associations, future multivariable analyses will be needed to investigate whether the effects detected are due to risk factors and comorbid conditions shared between PTSD and CAD. Another important aspect that is likely to play a role in PTSD-CAD comorbidity is treatment. Accordingly, the extraction of treatment information from EHR and the modeling of the longitudinal changes of drug combinations, doses, and treatment cessations will help to disentangle further the complex associations between psychiatric disorders and cardiovascular diseases.
In conclusion, our study highlights that PTSD-CAD comorbidity is likely due to mechanisms that can act on the disease risk through multiple paths. Leveraging multiple complementary analytic approaches, we confirmed that PTSD can have a causal effect on CAD and that these disorders also share some molecular pathways. In addition to supporting these previous hypotheses with large-scale genome-wide data, we also identified a possible effect of CAD reducing PTSD risk, but further studies will be needed to verify whether this association is affected by the timing of CAD onset or other possible confounders.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the veterans who participated in this study, and the members of the VA CSP and MVP study teams, without whom this work would not have been possible. This research has been also conducted using the UK Biobank Resource (application reference no. 58146).
FUNDING
This research is based on data from the MVP, Office of Research and Development, Veterans Health Administration and was supported by funding from the VA Cooperative Studies Program (CSP, no. CSP575B) and the Veterans Affairs Office of Research and Development MVP (grant nos. MVP000 and VA Merit MVP025). The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the United States government.
Footnotes
COMPETING INTERESTS
RP received a research grant from Alkermes. RP and JG received personal fees from Karger Publishers for their editorial work for Complex Psychiatry. JG is named as co-inventor on Patent Cooperation Treaty application no. 15/878,640 titled ‘Genotype-guided dosing of opioid agonists’, filed on January 24, 2018. CJO. received payment for editorial work for UpToDate and JAMA Cardiology and is currently employed by Novartis Institute of Biomedical Research. MBS received consulting fees from Acadia Pharmaceuticals, Aptinyx, Bionomics, Boehringer Ingelheim, Clexio Biosciences, EmpowerPharm, Engrail Therapeutics, Epivario, GW Pharmaceuticals, Janssen, and Jazz Pharmaceuticals; and payment for editorial work for UpToDate and the journals Biological Psychiatry and Depression and Anxiety. The other authors declare no competing financial interests.
Reprints and permission information is available at http://www.nature.com/reprints
ADDITIONAL INFORMATION
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41380-022-01735-z.
DATA AVAILABILITY
All data used discussed in this study are provided as Supplementary Material.
REFERENCES
- 1.O’Donnell CJ, Schwartz Longacre L, Cohen BE, Fayad ZA, Gillespie CF, Liberzon I, et al. Posttraumatic stress disorder and cardiovascular disease: state of the science, knowledge gaps, and research opportunities. JAMA Cardiol. 2021;6:1207–16. [DOI] [PubMed] [Google Scholar]
- 2.De Hert M, Detraux J, Vancampfort D. The intriguing relationship between coronary heart disease and mental disorders. Dialogues Clin Neurosci. 2018;20:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coughlin SS. Post-traumatic stress disorder and cardiovascular disease. Open Cardiovasc Med J. 2011;5:164–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seligowski AV, Steuber ER, Hinrichs R, Reda MH, Wiltshire CN, Wanna CP, et al. A prospective examination of sex differences in posttraumatic autonomic functioning. Neurobiol Stress. 2021;15:100384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vaccarino V, Shah AJ, Moncayo V, Nye J, Piccinelli M, Ko YA, et al. Posttraumatic stress disorder, myocardial perfusion, and myocardial blood flow: A Longitudinal Twin Study. Biol Psychiatry. 2022;91:615–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bountress KE, Wendt F, Bustamante D, Agrawal A, Webb B, Gillespie N, et al. Potential causal effect of posttraumatic stress disorder on alcohol use disorder and alcohol consumption in individuals of European descent: A Mendelian Randomization Study. Alcohol Clin Exp Res. 2021;45:1616–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang F, Rao S, Cao H, Zhang X, Wang Q, Xu Y, et al. Genetic evidence suggests posttraumatic stress disorder as a subtype of major depressive disorder. J Clin Invest. 2022;132:e145942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hubel C, Gaspar HA, Coleman JRI, Hanscombe KB, Purves K, Prokopenko I, et al. Genetic correlations of psychiatric traits with body composition and glycemic traits are sex- and age-dependent. Nat Commun. 2019;10:5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polimanti R, Amstadter AB, Stein MB, Almli LM, Baker DG, Bierut LJ, et al. A putative causal relationship between genetically determined female body shape and posttraumatic stress disorder. Genome Med. 2017;9:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu Z, Zhu X, Liu CL, Shi H, Shen S, Yang Y, et al. Shared genetics of asthma and mental health disorders: a large-scale genome-wide cross-trait analysis. Eur Respir J. 2019;54:1901507. [DOI] [PubMed] [Google Scholar]
- 12.Yuan S, Yao H, Larsson SC. Associations of cigarette smoking with psychiatric disorders: evidence from a two-sample Mendelian randomization study. Sci Rep. 2020;10:13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muniz Carvalho C, Wendt FR, Maihofer AX, Stein DJ, Stein MB, Sumner JA, et al. Dissecting the genetic association of C-reactive protein with PTSD, traumatic events, and social support. Neuropsychopharmacology 2021;46:1071–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lind MJ, Brick LA, Gehrman PR, Duncan LE, Gelaye B, Maihofer AX, et al. Molecular genetic overlap between posttraumatic stress disorder and sleep phenotypes. Sleep. 2020;43:zsz257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orri M, Pingault JB, Turecki G, Nuyt AM, Tremblay RE, Cote SM, et al. Contribution of birth weight to mental health, cognitive and socioeconomic outcomes: two-sample Mendelian randomisation. Br J Psychiatry. 2021:219:507–14. [DOI] [PubMed] [Google Scholar]
- 16.Tamman AJF, Wendt FR, Pathak GA, Krystal JH, Montalvo-Ortiz JL, Southwick SM, et al. Attachment Style Moderates Polygenic Risk for Posttraumatic Stress in United States Military Veterans: Results From the National Health and Resilience in Veterans Study. Biol Psychiatry. 2021;89:878–87. [DOI] [PubMed] [Google Scholar]
- 17.Polimanti R, Ratanatharathorn A, Maihofer AX, Choi KW, Stein MB, Morey RA, et al. Association of Economic Status and Educational Attainment With Posttraumatic Stress Disorder: A Mendelian Randomization Study. JAMA Netw Open. 2019;2:e193447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sumner JA, Maihofer AX, Michopoulos V, Rothbaum AO, Almli LM, Andreassen OA, et al. Examining Individual and Synergistic Contributions of PTSD and Genetics to Blood Pressure: A Trans-Ethnic Meta-Analysis. Front Neurosci. 2021;15:678503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pathak GA, Singh K, Wendt FR, Fleming TW, Overstreet C, Koller D, et al. Genetically regulated multi-omics study for symptom clusters of posttraumatic stress disorder highlights pleiotropy with hematologic and cardio-metabolic traits. Mol Psychiatry. 2022;27:1394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fu Q Autonomic dysfunction and cardiovascular risk in post-traumatic stress disorder. Auton Neurosci. 2022;237:102923. [DOI] [PubMed] [Google Scholar]
- 21.Edmondson D, von Kanel R. Post-traumatic stress disorder and cardiovascular disease. Lancet Psychiatry. 2017;4:320–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koenen KC, Sumner JA, Gilsanz P, Glymour MM, Ratanatharathorn A, Rimm EB, et al. Post-traumatic stress disorder and cardiometabolic disease: improving causal inference to inform practice. Psychol Med. 2017;47:209–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hunter-Zinck H, Shi Y, Li M, Gorman BR, Ji SG, Sun N, et al. Genotyping Array Design and Data Quality Control in the Million Veteran Program. Am J Hum Genet. 2020;106:535–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sullivan PF, Agrawal A, Bulik CM, Andreassen OA, Borglum AD, Breen G, et al. Psychiatric Genomics: An Update and an Agenda. Am J Psychiatry. 2018;175:15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson CP, Goel A, Butterworth AS, Kanoni S, Webb TR, Marouli E, et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet. 2017;49:1385–91. [DOI] [PubMed] [Google Scholar]
- 26.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018;562:203–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilkins KC, Lang AJ, Norman SB. Synthesis of the psychometric properties of the PTSD checklist (PCL) military, civilian, and specific versions. Depress Anxiety. 2011;28:596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stein MB, Levey DF, Cheng Z, Wendt FR, Harrington K, Pathak GA, et al. Genome wide association analyses of post-traumatic stress disorder and its symptom subdomains in the Million Veteran Program. Nat Genet. 2021;53:174–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harrington KM, Quaden R, Stein MB, Honerlaw JP, Cissell S, Pietrzak RH, et al. Validation of an Electronic Medical Record-Based Algorithm for Identifying Posttraumatic Stress Disorder in U.S. Veterans. J Trauma Stress. 2019;32:226–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis KAS, Coleman JRI, Adams M, Allen N, Breen G, Cullen B, et al. Mental health in UK Biobank - development, implementation and results from an online questionnaire completed by 157 366 participants: a reanalysis. BJPsych Open. 2020;6:e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nievergelt CM, Maihofer AX, Klengel T, Atkinson EG, Chen CY, Choi KW, et al. International meta-analysis of PTSD genome-wide association studies identifies sex- and ancestry-specific genetic risk loci. Nat Commun. 2019;10:4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evangelou E, Warren HR, Mosen-Ansorena D, Mifsud B, Pazoki R, Gao H, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet. 2018;50:1412–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu Z, Wang X, Li X, Lin Y, Shen S, Liu CL, et al. Genetic overlap of chronic obstructive pulmonary disease and cardiovascular disease-related traits: a large-scale genome-wide cross-trait analysis. Respir Res. 2019;20:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shah S, Henry A, Roselli C, Lin H, Sveinbjornsson G, Fatemifar G, et al. Genome wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat Commun. 2020;11:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Qi G, Park JH, Chatterjee N. Estimation of complex effect-size distributions using summary-level statistics from genome-wide association studies across 32 complex traits. Nat Genet. 2018;50:1318–26. [DOI] [PubMed] [Google Scholar]
- 37.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47:1236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Euesden J, Lewis CM, O’Reilly PF. PRSice: Polygenic Risk Score software. Bioinformatics 2015;31:1466–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dastani Z, Hivert MF, Timpson N, Perry JR, Yuan X, Scott RA, et al. Novel loci for adiponectin levels and their influence on type 2 diabetes and metabolic traits: a multi-ethnic meta-analysis of 45,891 individuals. PLoS Genet. 2012;8:e1002607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi SW, Mak TS, O’Reilly PF. Tutorial: a guide to performing polygenic risk score analyses. Nat Protoc. 2020;15:2759–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanderson E, Glymour MM, Holmes MV, Kang H, Morrison J, Munafò MR, et al. Mendelian randomization. Nat Rev Methods Prim. 2022;2:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skrivankova VW, Richmond RC, Woolf BAR, Yarmolinsky J, Davies NM, Swanson SA, et al. Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization: The STROBE-MR Statement. JAMA. 2021;326:1614–21. [DOI] [PubMed] [Google Scholar]
- 44.Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 2018;7:e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44:512–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46:1985–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bowden J, Davey, Smith G, Haycock PC, Burgess S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet Epidemiol. 2016;40:304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burgess S, Davey Smith G, Davies NM, Dudbridge F, Gill D, Glymour MM, et al. Guidelines for performing Mendelian randomization investigations. Wellcome Open Res. 2019;4:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hemani G, Bowden J, Davey Smith G. Evaluating the potential role of pleiotropy in Mendelian randomization studies. Hum Mol Genet. 2018;27:R195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity Analyses for Robust Causal Inference from Mendelian Randomization Analyses with Multiple Genetic Variants. Epidemiology 2017;28:30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qingyuan Z, Jingshu W, Gibran H, Jack B, Dylan SS. Statistical inference in two-sample summary-data Mendelian randomization using robust adjusted profile score. Ann Stat. 2020;48:1742–69. [Google Scholar]
- 52.Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50:693–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O’Connor LJ, Price AL. Distinguishing genetic correlation from causation across 52 diseases and complex traits. Nat Genet. 2018;50:1728–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bhattacharjee S, Rajaraman P, Jacobs KB, Wheeler WA, Melin BS, Hartge P, et al. A subset-based approach improves power and interpretation for the combined analysis of genetic association studies of heterogeneous traits. Am J Hum Genet. 2012;90:821–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11:e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Townsend P, Phillimore P, Beattie A. Health and Deprivation: Inequality and the North. Croom Helm. 1988. [Google Scholar]
- 59.Wendt FR, Pathak GA, Overstreet C, Tylee DS, Gelernter J, Atkinson EG, et al. Characterizing the effect of background selection on the polygenicity of brain-related traits. Genomics 2021;113:111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two-sample Mendelian randomization. Genet Epidemiol. 2016;40:597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maihofer AX, Choi KW, Coleman JRI, Daskalakis NP, Denckla CA, Ketema E, et al. Enhancing Discovery of Genetic Variants for Posttraumatic Stress Disorder Through Integration of Quantitative Phenotypes and Trauma Exposure Information. Biol Psychiatry. 2022;91:626–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ralapanawa U, Sivakanesan R. Epidemiology and the Magnitude of Coronary Artery Disease and Acute Coronary Syndrome: A Narrative Review. J Epidemiol Glob Health. 2021;11:169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Edmondson D An Enduring Somatic Threat Model of Posttraumatic Stress Disorder Due to Acute Life-Threatening Medical Events. Soc Personal Psychol Compass 2014;8:118–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hu YJ, Schmidt AF, Dudbridge F, Holmes MV, Brophy JM, Tragante V, et al. Impact of Selection Bias on Estimation of Subsequent Event Risk. Circ Cardiovasc Genet. 2017;10:e001616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Evin G, Li QX. Platelets and Alzheimer’s disease: Potential of APP as a biomarker. World J Psychiatry. 2012;2:102–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kitazume S, Yoshihisa A, Yamaki T, Oikawa M, Tachida Y, Ogawa K, et al. Soluble amyloid precursor protein 770 is released from inflamed endothelial cells and activated platelets: a novel biomarker for acute coronary syndrome. J Biol Chem. 2012;287:40817–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Plummer S, Van den Heuvel C, Thornton E, Corrigan F, Cappai R. The Neuroprotective Properties of the Amyloid Precursor Protein Following Traumatic Brain Injury. Aging Dis. 2016;7:163–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Meziab O, Kirby KA, Williams B, Yaffe K, Byers AL, Barnes DE. Prisoner of war status, posttraumatic stress disorder, and dementia in older veterans. Alzheimers Dement. 2014;10:S236–241. [DOI] [PubMed] [Google Scholar]
- 69.Weiner MW, Veitch DP, Hayes J, Neylan T, Grafman J, Aisen PS, et al. Effects of traumatic brain injury and posttraumatic stress disorder on Alzheimer’s disease in veterans, using the Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement. 2014;10:S226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anroedh S, Hilvo M, Akkerhuis KM, Kauhanen D, Koistinen K, Oemrawsingh R, et al. Plasma concentrations of molecular lipid species predict long-term clinical outcome in coronary artery disease patients. J Lipid Res. 2018;59:1729–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mellon SH, Gautam A, Hammamieh R, Jett M, Wolkowitz OM. Metabolism, Metabolomics, and Inflammation in Posttraumatic Stress Disorder. Biol Psychiatry. 2018;83:866–75. [DOI] [PubMed] [Google Scholar]
- 72.Vilchinsky N, Ginzburg K, Fait K, Foa EB. Cardiac-disease-induced PTSD (CDIPTSD): A systematic review. Clin Psychol Rev. 2017;55:92–106. [DOI] [PubMed] [Google Scholar]
- 73.Feng Y-CA, Ge T, Cordioli M, FinnGen, Ganna A, Smoller JW, et al. Findings and insights from the genetic investigation of age of first reported occurrence for complex disorders in the UK Biobank and FinnGen. medRxiv. 2020: 2020.2011.2020.20234302. [Google Scholar]
- 74.Pietrzak RH, Goldstein RB, Southwick SM, Grant BF. Prevalence and Axis I comorbidity of full and partial posttraumatic stress disorder in the United States: results from Wave 2 of the National Epidemiologic Survey on Alcohol and Related Conditions. J Anxiety Disord. 2011;25:456–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pietrzak RH, Goldstein RB, Southwick SM, Grant BF. Medical comorbidity of full and partial posttraumatic stress disorder in US adults: results from Wave 2 of the National Epidemiologic Survey on Alcohol and Related Conditions. Psychosom Med. 2011;73:697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used discussed in this study are provided as Supplementary Material.