

Abstract
Diabetic nephropathy (DN) is a significant complication of diabetes and the leading cause of end-stage renal disease. Hyperglycemia-induced dysfunction of the glomerular podocytes is a major contributor to the deterioration of renal function in DN. Previously, we demonstrated that podocyte-specific disruption of the Src homology phosphatase 2 (Shp2) ameliorated lipopolysaccharide-induced renal injury. This study aims to evaluate the contribution of Shp2 to podocyte function under hyperglycemia and explore the molecular underpinnings. We report elevated Shp2 in the E11 podocyte cell line under high glucose and the kidney under streptozotocin- and high fat diet-induced hyperglycemia. Consistently, Shp2 disruption in podocytes was associated with partial renoprotective effects under hyperglycemia, as evidenced by the preserved renal function. At the molecular level, Shp2 deficiency was associated with altered renal insulin signaling and diminished hyperglycemia-induced renal endoplasmic reticulum stress, inflammation, and fibrosis. Additionally, Shp2 knockdown in E11 podocytes mimicked the in vivo deficiency of this phosphatase and ameliorated the deleterious impact of high glucose, whereas Shp2 reconstitution reversed these effects. Moreover, Shp2 deficiency attenuated high glucose-induced E11 podocyte migration. Further, we identified the protein tyrosine kinase FYN as a putative mediator of Shp2 signaling in podocytes under high glucose. Collectively, these findings suggest that Shp2 inactivation may afford protection to podocytes under hyperglycemia and highlight this phosphatase as a potential target to ameliorate glomerular dysfunction in DN.
Keywords: Src homology phosphatase 2, Hyperglycemia, Podocyte, Diabetic nephropathy, Endoplasmic reticulum stress, Inflammation, Fibrosis, FYN
Introduction
Diabetic nephropathy (DN) is a major microvascular complication of diabetes affecting about 20–40% of patients with diabetes and the leading cause of end-stage renal disease (ESRD) in Europe and the United States [1–3]. Additionally, the increasing prevalence of type 2 diabetes may escalate the number of patients with ESRD [4, 5]. The effective treatments of ESRD include dialysis, with a high mortality rate within five years [6], and kidney transplantation, which is limited by organ availability. Glomerular injury and dysfunction are apparent in DN, and a clinical hallmark of the disease is the leakage of albumin through the glomerular filtration barrier (GFB) [7]. GFB is formed of three major components: the podocyte, glomerular basement membrane, and endothelium. The podocytes are terminally differentiated, highly specialized cells that consist of the cell body, major processes, and foot processes [7]. The foot processes of neighboring podocytes interdigitate to form the intercellular junction slit diaphragm, which is an essential part of the GFB to prevent urinary protein loss [8, 9]. Podocyte injury and loss are common early features in several glomerulopathies, including DN [10]. Indeed, podocyte density is a predictor of DN progression, and in experimental animal models, a loss of more than 20% leads to glomerular impairment [11]. Notably, studies using transgenic mouse models establish that podocyte-specific interventions are sufficient to halt DN progression and suggest podocentric therapeutic strategies to combat this deadly malady [12].
A convergence of multiple injurious pathways contributes to podocyte loss and the development of DN (reviewed in [13]). In particular, it is increasingly recognized that dysregulated insulin signaling contributes to podocyte dysfunction and DN (reviewed in [14]). The podocytes are insulin-responsive cells that can uptake glucose following insulin stimulation via the glucose transporters GLUT1 and GLUT4 [15]. Additionally, podocytes may become insulin resistant preceding the development of DN [16]. Insulin signaling is crucial for podocyte function, as evidenced by mice with podocyte-specific disruption of the insulin receptor (IR) that exhibit features of DN despite normal blood glucose concentrations [17]. Moreover, insulin signaling may impact podocyte function by modulating key pathways, including the endoplasmic reticulum (ER) stress [18], actin dynamics [19], and calcium mobilization [20, 21].
Compelling evidence establish protein tyrosine phosphatases (PTPs) as regulators of podocyte function (reviewed in [22]). The prototypical protein tyrosine phosphatase 1B (PTP1B) is upregulated in murine models of renal injury (membranous nephropathy and puromycin aminonucleoside) and dephosphorylates the essential podocyte protein, Nephrin [23]. Additionally, podocyte PTP1B expression is elevated under hyperglycemia, and the deficiency of this phosphatase mitigates hyperglycemia-induced podocyte injury [24]. Similarly, the Src homology domain-containin phosphatase 1 (Shp1) can dephosphorylate Nephrin [25] and is elevated in murine models of DN [25–27].
Src homology phosphatase 2 (Shp2; encoded by Ptpn11) is a ubiquitously expressed non-transmembrane phosphatase [28] that plays an essential role in most receptor tyrosine kinase signaling where it is required for normal activation of the extracellular signal-regulated kinase pathways [29, 30]. Additionally, Shp2 is an established modulator of insulin signaling, engaging targets in the proximal cascade [31–35] with cell/tissue-specific outcomes [36–38]. We and others report that podocyte-specific Shp2 ablation ameliorates acute podocyte and renal injury induced by protamine sulfate, nephrotoxic serum, and lipopolysaccharide [39, 40]. However, the contribution of Shp2 in podocytes to DN is not clear. In the present study, we test the hypothesis that podocyte Shp2 deficiency may be protective against the adverse effects of hyperglycemia and explore the underlying mechanisms.
Materials and methods
Reagents.
Reagents were purchased from MilliporeSigma unless otherwise indicated. HRP-conjugated secondary antibodies were purchased from Bio-Rad Laboratories. The pharmacological inhibitor of ROCK (Y-27632, NC1052806) was purchased from Thermo Fisher Scientific.
Mouse studies.
Mice with podocyte-specific disruption of Shp2 (Ptpn11fl/fl; Pod-Cre) were generated and genotyped as we previously described [40]. Briefly, Shp2 floxed (Ptpn11fl/fl) mice (129Sv/J x C57Bl/6J background) [41] were bred to podocin-Cre transgenic mice (C57Bl/6J background) (Jackson laboratories). Mice were maintained at a 12-hour light-dark cycle with access to water and rodent laboratory chow (Purina lab chow; # 5001). For high-fat-diet (HFD)-induced hyperglycemia, eight weeks old male Ptpn11fl/fl and Ptpn11fl/fl; Pod-Cre littermates were fed a HFD (Research Diets; # D12492, 60% kcal from fat) for 24 weeks. For streptozotocin (STZ)-induced hyperglycemia, 9–12 weeks old male mice were intraperitoneally injected with STZ (in 100 mM sodium citrate buffer) at a single high dose (160 μg/g body weight) or five day multiple low doses (50 μg/g body weight). Mice were sacrificed 20 weeks after STZ treatment. Fed parameters were measured in samples collected between 7–9 a.m., and the fasted samples were assayed from overnight-fasted mice. We determined the albumin, creatinine, and blood urea nitrogen (BUN) concentrations using the corresponding kits according to the manufacturer’s instructions (Sigma; MAK124, MAK080, MAK006). Blood glucose concentration was determined using an Easy Plus II blood glucose monitor (Home Aide Diagnostics). Diastolic and systolic blood pressures were measured by the noninvasive CODA high throughput mouse rat tail-cuff system (Kent Scientific Corporation). For renal insulin signaling studies, overnight-fasted male mice (10 weeks old) were intraperitoneally injected with insulin (Lilly; Humulin R U-100, 10 U/kg bodyweight) and then sacrificed after 5 and 15 minutes as we described [24, 36]. At sacrifice, the kidneys were harvested and stored for further biochemical and histological analyses. For immunohistochemistry, the kidney sections were fixed in 4% paraformaldehyde, embedded in paraffin, and deparaffinized in xylene. Sections were stained using antibodies for Shp2 (sc-280), Nephrin (sc-19000) (both from Santa Cruz Biotechnology), Synaptopodin (Novus Biologicals; NBP2–33357), pAKT Ser473 (4060), pERK1/2 Tyr202/Thr204 (4370) (both from Cell Signaling Technology), and Collagen III (Abcam; ab7778) at 4°C overnight. Then sections were incubated with the appropriate fluorescent dye-conjugated secondary antibodies (Thermo Fisher Scientific; A-21206: Alexa Fluor 488 anti-rabbit, A-31570: Alexa Fluor 555 anti-mouse, A-21432: Alexa Fluor 555 anti-goat) for an hour at room temperature and visualized using an Olympus FV1000 confocal microscope. In addition, the Periodic acid–Schiff (PAS) staining was done by a commercial kit according to the instruction from Sigma (395B-1KT), and images were captured using an Olympus BX51 microscope. All animal studies were approved by the Institutional Animal Care and Use Committee at the University of California Davis.
Cell experiments.
The murine kidney podocyte cell line E11 was purchased from Cell Lines Service GmbH (400494). E11 cell lines with Shp2 knockdown (KD) and reconstitution (KD-R) with wild-type human Shp2 were previously generated as we described [40]. Additionally, the Shp2 knockdown cells were reconstituted with the substrate-trapping double mutant human Shp2 (KD-DM; D425A and C459S) [42]. Shp2 KD cells were selected with puromycin (2 μg/ml), and Shp2 KD-R and KD-DM cells were maintained under the selection of puromycin (2 μg/ml) and Geneticin (G418, 400 μg/ml). All E11 cell lines were maintained at 33°C in RPMI-1640 medium (which contains 5.6 mM glucose and supplemented with 10% FBS and 2 mM glutamine) and then induced to differentiate by culture for two weeks at 37°C [24, 40]. To investigate the insulin signaling cascade, differentiated Shp2 KD and KD-R podocytes were cultured without and with high glucose (HG, 25 mM) for 72 hours. Subsequently, overnight-starved cells were stimulated without and with insulin (10 nM) for 10 and 20 minutes. For the wound healing assay, differentiated Shp2 KD and KD-R podocytes were starved 24 hours, and then straight scratches were created using 200 μl pipette tips. Cells were cultured without and with HG for an additional 48 hours. For pharmacological inhibition of ROCK, differentiated Shp2 KD and KD-R podocytes were starved 24 hours then the wound was generated as described earlier. Cells were further cultured under HG for 48 hours without and with the ROCK inhibitor (Y-27632, 10 μM). Bright-field images were captured using an Olympus BX51 microscope, and the number of cell migration to the wound was quantitated manually with enlarged pictures from four independent experiments. For the substrate-trapping experiment, differentiated Shp2 KD-DM podocytes were cultured without and with HG for 72 hours, and proteins were extracted using NP40 (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 5 mM EDTA, 20 mM NaF, 10 mM Na4P2O7, 1% Igepal CA-630, and proteases inhibitors) and RIPA (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.1% SDS, 5 mM EDTA, 20 mM NaF, 10 mM Na4P2O7, 1% Triton X-100, 1% sodium deoxycholate and proteases inhibitors). Protein lysates (500 μg) were incubated with antibodies for Shp2 (10 μg) for 4 hours at 4°C. The immunocomplexes were pulled down by 30 μl protein A/G magnetic beads (MilliporeSigma; LSKMAGAG10) for 2 hours at 4°C. Then proteins were eluted by sample buffer (60 mM Tris-Cl pH 6.8, 1% SDS, 10% glycerol, 0.01% bromophenol blue, 2.5% 2-mercaptoethanol) for 5 minutes at 95°C. For immunoprecipitation of FYN, differentiated Shp2 KD and KD-R cells were cultured without and with HG for 72 hours, and total proteins were extracted using RIPA buffer with 2 mM sodium orthovanadate. Protein lysates (200 μg) were incubated with antibodies for FYN (5 μg) for 4 hours at 4°C, then immunoprecipitated as described above.
Biochemical analyses.
Total proteins were extracted from cells and kidneys using RIPA buffer. Lysates were centrifuged at 12,000 x g for 10 minutes, and protein concentrations were determined using a bicinchoninic acid protein assay kit (Pierce Chemical). Extracted proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to PVDF membranes. Immunoblotting was performed using antibodies for pNF-κBp65 Ser536 (3033), NF-κBp65 (8242), pAKT Ser473 (4060), pERK1/2 Tyr202/Thr204 (4370), ERK1/2 (4695), pSrc Tyr416 (2101), pSrc Tyr527 (2105), PERK (3192), peIF2α Ser51 (9721), IRE1α (3294), GAPDH (3683) (all from Cell Signaling Technology), AKT (sc-81434), Shp2 (sc-280), pPERK Thr981 (sc-32577), eIF2α (sc-133132), TGFβRII (sc-17792), FYN (sc-73388), Actin (sc-47778), Tubulin (sc-8035), Nephrin (sc-19000) (all from Santa Cruz Biotechnology), pNephrin Tyr1176/Tyr1193 (Abcam; ab80299), and Synaptopodin and pIRE1α Ser724 (NB100–2323) from Novus Biologicals at 4°C overnight. Then membranes were incubated with the appropriate HRP-conjugated secondary antibodies for an hour at room temperature. Protein bands were visualized using HyGLO enhanced chemiluminescence (Denville Scientific) and further quantitated using FluorChem 9900 program (Alpha Innotech). For quantitative real-time PCR, total RNA was extracted from the kidney samples by TRIzol (Invitrogen), and cDNA was generated using a high-capacity cDNA synthesis Kit (Applied Biosystems; 4368814). Then samples were subjected to CFX96 Touch Real-Time PCR Detection System (Bio-Rad) with a mixing of SsoAdvanced Universal SYBR Green Supermix (Thermo Fisher Scientific) and relevant primer pairs. Relative gene expression was normalized with TATA-box binding protein (TBP) and determined using the ΔCT method. The primers used were listed as forward and reverse: IL1β, 5’-TAGCTTCAGGCAGGCAGTATC and 5’-TAAGGTCCACGGGAAAGACAC; IL6, 5’-ACAACCACGGCCTTCCCTACTT and 5’-CACGATTTCCCAGAGAACATGTG; MCP1, 5’-CCCAATGAGTAGGCTGGAGA and 5’-TCTGGACCCATTCCTTCTTG; TNFα, 5’-TGACGTGGAACTGGCAGAAGAG and 5’-TTGCCACAAGCAGGAATGAGA; pro-Collagen, 5’-AGAGGCGAAGGCAACAGTCG and 5’-GCAGGGCCAATGTCTAGTCC; αSMA, 5’-TCAGCGCCTCCAGTTCCT and 5’-AAAAAAAACCACGAGTAACAAATCAA; TBP, 5’-TTGGCTAGGTTTCTGCGGTC and 5’-GCCCTGAGCATAAGGTGGAA.
Statistical analyses.
Data are expressed as means ± standard error of the mean (SEM). Statistical analyses were performed by the SPSS program (IBM) using Dunnett’s test (Fig. 1) and one-way ANOVA with Tukey’s honest significance test. Differences were considered significant at p ≤ 0.05.
Fig. 1:
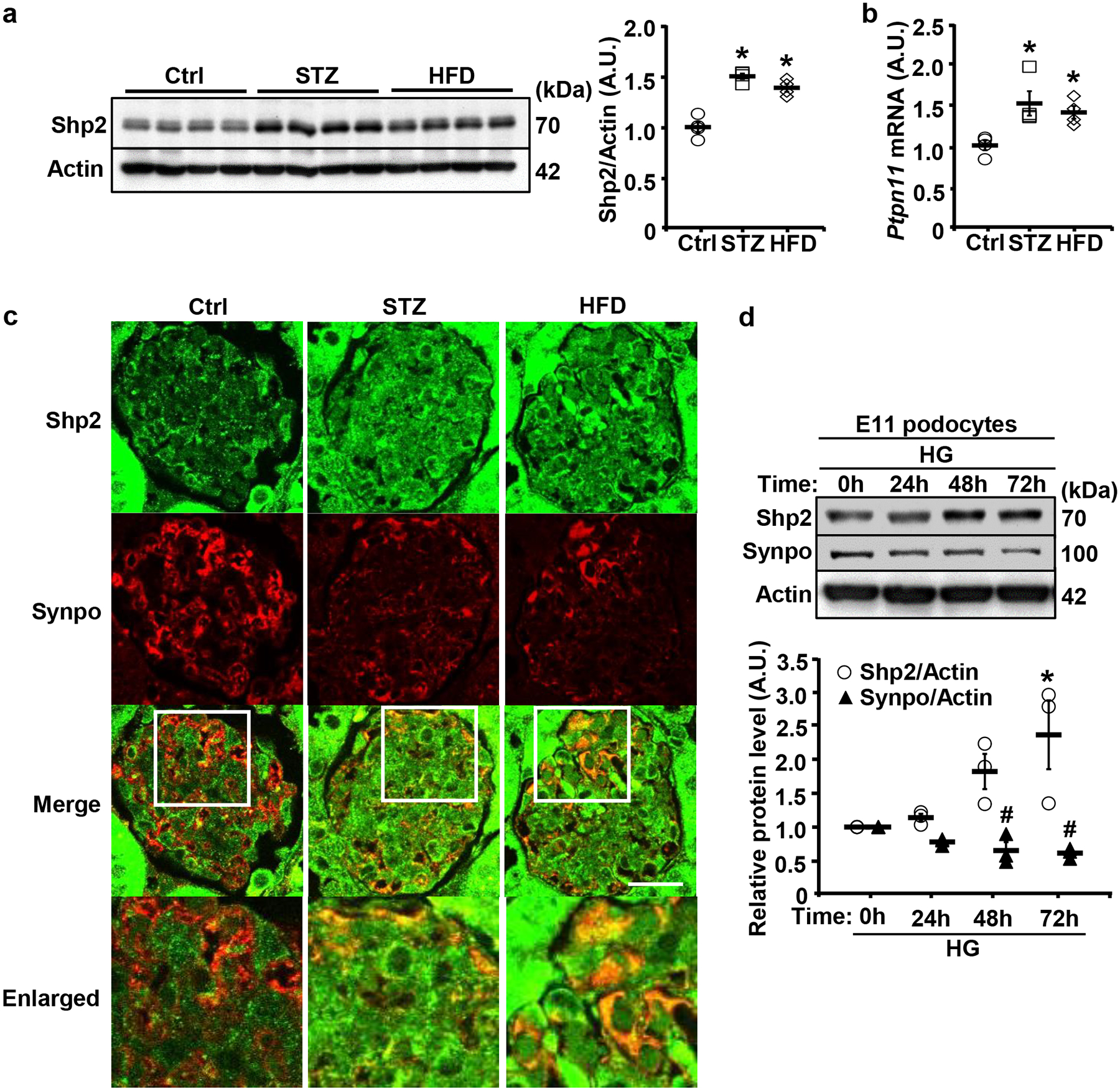
Upregulation of Shp2 in the kidney and E11 podocyte cell line under hyperglycemia. a,b Immunoblots of Shp2 and Actin (a), and Ptpn11 mRNA expression (b) in kidneys from male mice fed with regular chow (Ctrl), HFD (24 weeks), or challenged with STZ (160 μg/g body weight, 20 weeks). Actin served as a protein loading control for immunoblotting (n=4 per group), and mRNA expression was normalized to Tbp (n=4 per group). Each lane in immunoblot represents a sample from an individual mouse. *p ≤ 0.05 indicates a significant difference between Ctrl versus STZ-treated and HFD-fed mice. c Confocal images of kidney sections from Ctrl, HFD-fed (24 weeks), and STZ-treated (160 μg/g body weight, 20 weeks) wild-type male mice co-immunostained for Shp2 (green) and Synaptopodin (Synpo, red). Scale bar: 20 μm. The boxed areas in the Merge are enlarged in the lower panel. d Differentiated E11 podocytes were treated with high glucose (25 mM; HG) for 0, 24, 48, and 72 hours. Cell lysates were immunoblotted for Shp2, Synpo, and Actin. The protein expression level was normalized to Actin from three independent experiments. Statistical significance in (d) was considered *p ≤ 0.05 for Shp2 and # p ≤ 0.05 for Synpo between 0h and 24h, 48h, and 72 h. A.U.: arbitrary unit.
Results
Elevated Shp2 in the kidney under hyperglycemia and the E11 podocyte cell line under high glucose.
Previously, we demonstrated an increased expression of renal Shp2 in a mouse model of lipopolysaccharide-induced renal injury [40]. Additionally, Shp2 tyrosine phosphorylation (Tyr542) is elevated in a subgroup of human glomerular diseases suggestive of increased phosphatase activity [39]. In the present study, we evaluated Shp2 expression in the kidney of wild-type (WT) male mice under STZ- and HFD-induced hyperglycemia. Renal Shp2 protein and mRNA were significantly elevated under hyperglycemia compared with normoglycemia (Fig. 1a, b). Moreover, co-immunostaining Shp2 and Synaptopodin suggested upregulation of this phosphatase in the kidney, including podocytes under hyperglycemia (Fig. 1c). Correspondingly, Shp2 protein expression significantly increased in differentiated E11 podocytes cultured in high glucose compared with normal glucose (Fig. 1d). On the other hand, Synaptopodin expression decreased under hyperglycemia in mice (Fig. 1c) and E11 podocytes cultured in high glucose (Fig. 1d). Collectively, these observations suggest the upregulation of Shp2 in the kidney and podocytes under hyperglycemia and support the notion that deregulated signaling of this phosphatase may contribute to hyperglycemia-induced podocyte dysfunction.
Shp2 ablation in podocytes is associated with attenuated hyperglycemia-induced renal dysfunction in mice.
To investigate the potential contribution of Shp2 in podocytes to renal function under hyperglycemia, we used mice with podocyte-specific Shp2 disruption (Ptpn11fl/fl; Pod-Cre) [40]. Hyperglycemia was induced in control (Ptpn11fl/fl) and Ptpn11fl/fl; Pod-Cre mice by a single high dose and multiple low doses of STZ and HFD feeding. Both genotypes exhibited comparable alterations in body weight and kidney weight/body weight ratio (Supplementary Fig. 1). STZ-induced hyperglycemia significantly elevated the urine albumin/creatinine ratio (ACR), blood urea nitrogen (BUN), fed and fasted glucose concentrations, and systolic and diastolic blood pressure in Ptpn11fl/fl but to a significantly lower level in Ptpn11fl/fl; Pod-Cre mice (Fig. 2a). Similarly, hyperglycemia induced by multiple low doses of STZ significantly increased ACR, BUN, and fed and fasted glucose in Ptpn11fl/fl but was diminished in Ptpn11fl/fl; Pod-Cre mice (Fig. 2b). In keeping with these observations, in HFD-induced hyperglycemia, the elevation in ACR, fasted glucose, and systolic blood pressure was significantly lower in Ptpn11fl/fl; Pod-Cre compared with Ptpn11fl/fl mice (Fig. 2c). Altogether, these findings demonstrate that Shp2 disruption in podocytes is associated with partial renoprotective effects under hyperglycemia, as evidenced by the preserved renal function, blood pressure and glucose control in the various murine models used herein.
Fig. 2:
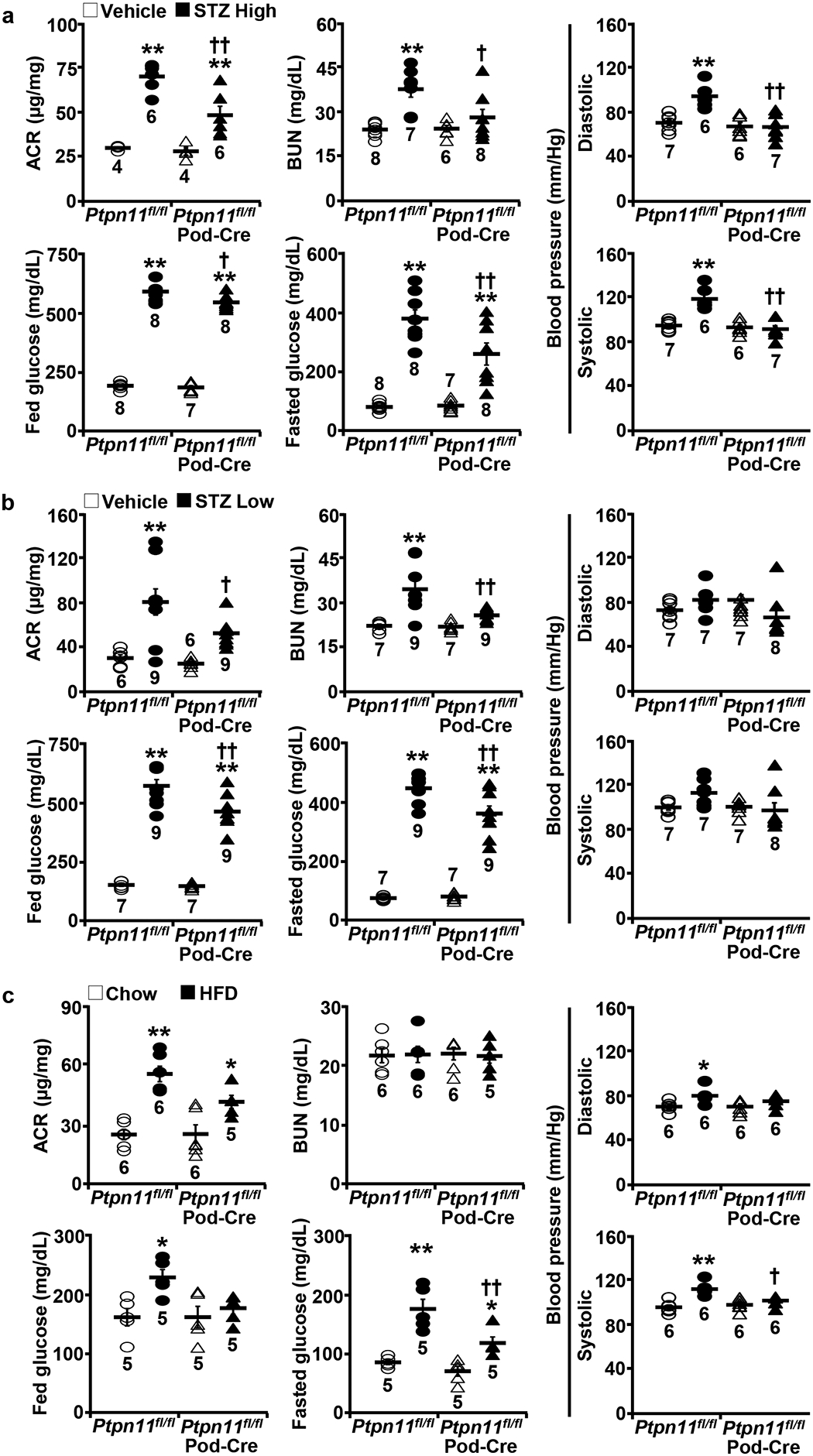
Podocyte Shp2 ablation is associated with improved renal function under hyperglycemia. a−c Urinary albumin to creatinine ratio (ACR), blood urea nitrogen (BUN), fed and fasted serum glucose concentrations, and systolic and diastolic blood pressure of Ptpn11fl/fl and Ptpn11fl/fl; Pod-Cre mice treated with vehicle and two STZ models including (a) High (160 μg/g body weight x1) and (b) Low (50 μg/g body weight x5) doses, and fed a HFD (c). The sample number for each group was indicated in dot plots. *p ≤ 0.05, **p ≤ 0.01 vehicle versus STZ (a, b), and chow versus HFD (c) of mice with the same genotype. †p ≤ 0.05, ††p ≤ 0.01 Ptpn11fl/fl versus Ptpn11fl/fl; Pod-Cre under the same treatment.
Diminished hyperglycemia-evoked renal ER stress, inflammation, and fibrosis in mice with Shp2 disruption.
Having demonstrated the salutary effects of Shp2 disruption in podocytes under hyperglycemia, next, we investigated the mechanisms that may mediate Shp2 action. Protein misfolding and ER stress are apparent in distinct renal diseases, including DN [43]. Accordingly, we monitored the activation of critical sensors in the unfolded protein response (UPR), namely protein kinase R-like ER kinase (PERK) and inositol-requiring enzyme 1α (IRE1α). Immunoblotting of total kidney lysates demonstrated a hyperglycemia-induced increase in the phosphorylation of PERK and its downstream target eukaryotic translation initiation factor 2α (eIF2α), as well as IRE1α and that, was significantly lower in Ptpn11fl/fl; Pod-Cre compared with Ptpn11fl/fl mice (Fig. 3a, and Supplementary Fig. 2a). In keeping with the diminished ER stress, the hyperglycemia-induced inflammation was mitigated upon Shp2 disruption, as evidenced by lower nuclear factor-kappa B (NF-κB) phosphorylation (Fig. 3a and Supplementary Fig. 2a) and renal mRNA of interleukin 1 beta (IL1β), monocyte chemoattractant protein 1 (MCP1), interleukin 6 (IL6), and tumor necrosis factor-alpha (TNFα) (Fig. 3b and Supplementary Fig. 2b). Moreover, Ptpn11fl/fl; Pod-Cre mice exhibited lower fibrosis under hyperglycemia compared with control animals given the decreased expression of the renal transforming growth factor-beta receptor type II (TGFβRII), mRNA of pro-Collagen and alpha-smooth muscle actin (αSMA) and Periodic acid-Schiff (Fig. 4a–c) and Collagen III staining (Supplementary Fig. 2c). Collectively, these observations establish the amelioration of hyperglycemia-induced renal ER stress, inflammation, and fibrosis upon Shp2 disruption in podocytes.
Fig. 3:
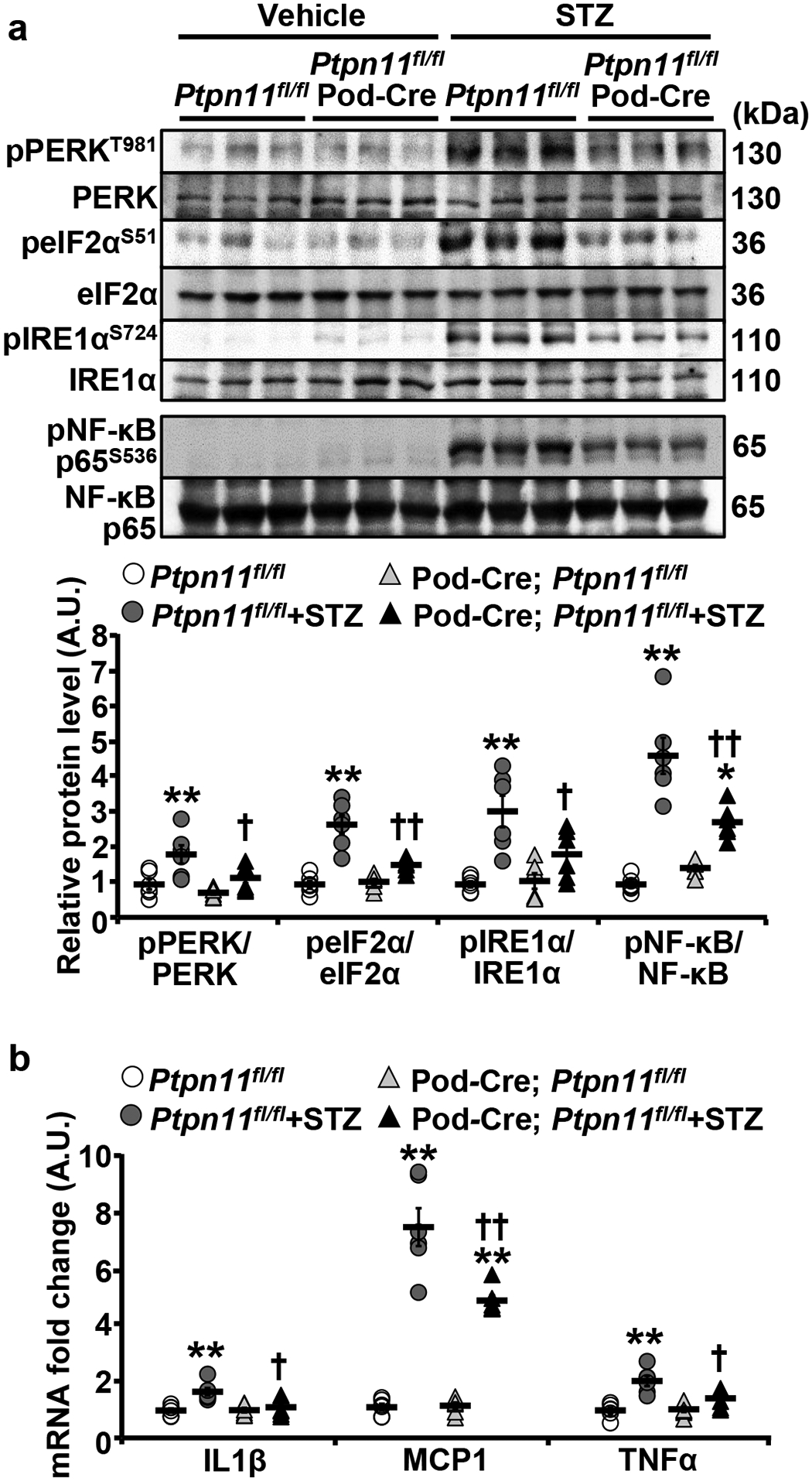
Podocyte Shp2 ablation mitigates hyperglycemia-induced renal ER stress, and inflammation. a, b Immunoblots of pPERK T981, PERK, peIF2α S51, eIF2α, pIRE1α S724, IRE1α, pNF-κB p65 S536, and NF-κB p65 (a), and mRNA expression of inflammatory cytokines IL1β, MCP1, and TNFα (b) in kidneys from Ptpn11fl/fl and Ptpn11fl/fl; Pod-Cre mice treated without (vehicle) and with STZ (160 μg/g body weight, 20 weeks). Each lane represents lysate from an individual animal. Phosphorylation levels of PERK, eIF2α, IRE1α, , and NF-κB p65 were normalized with their respective protein (n=6 each group), and mRNA expression was normalized to Tbp (n=6 in each group). *p ≤ 0.05, **p ≤ 0.01 vehicle versus STZ of mice with the same genotype. †p ≤ 0.05, ††p ≤ 0.01 Ptpn11fl/fl versus Ptpn11fl/fl; Pod-Cre under the same treatment. A.U.: arbitrary unit.
Fig. 4:
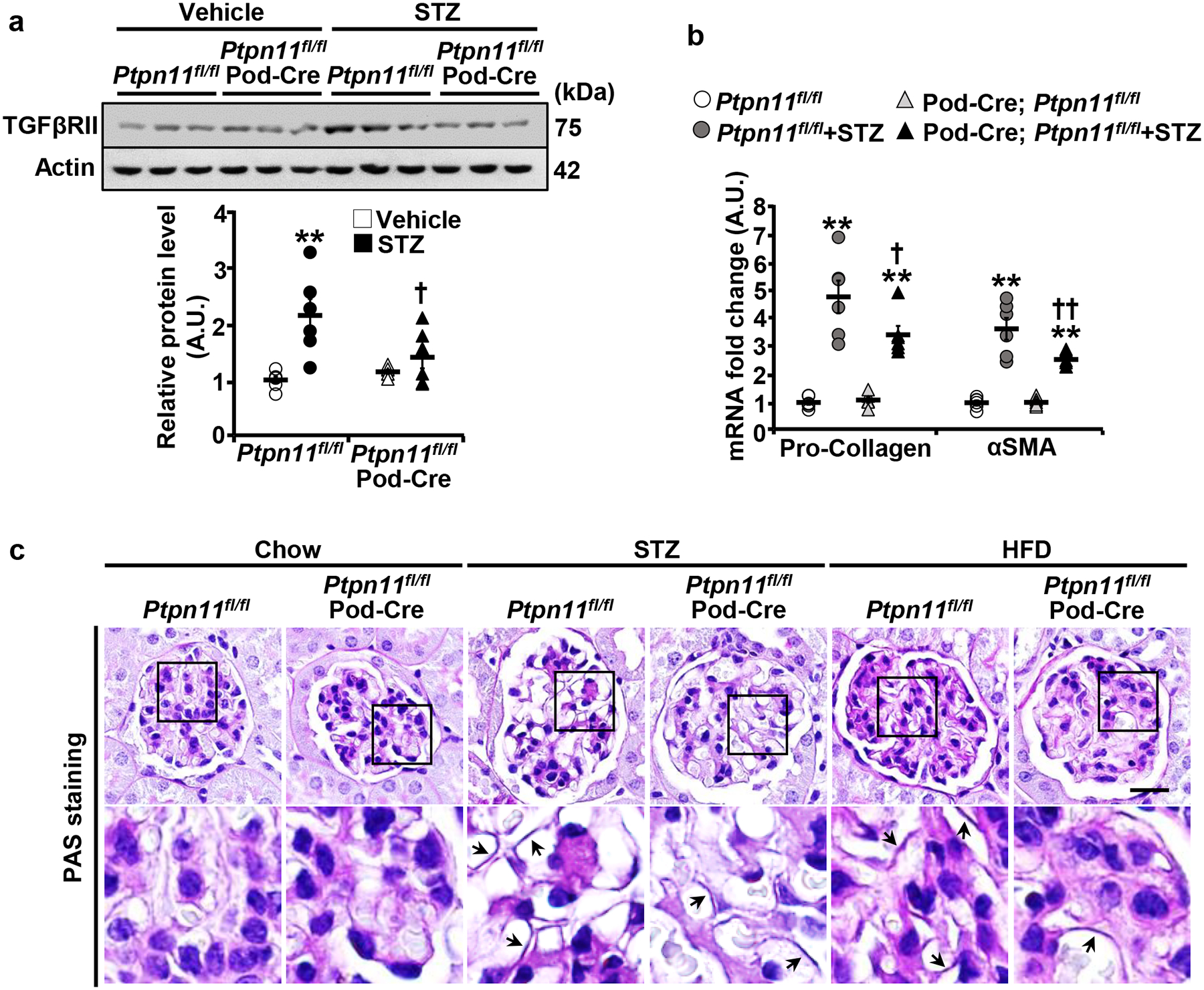
Podocyte Shp2 deficiency attenuates hyperglycemia-induced fibrosis in mice. a,b Immunoblots of TGFβRII and Actin (a), and mRNA expression of fibrosis markers pro-Collagen and αSMA (b) in kidneys from Ptpn11fl/fl and Ptpn11fl/fl; Pod-Cre mice treated without (vehicle) and with STZ (160 μg/g body weight, 20 weeks). Each lane in the immunoblot represents lysate from an individual animal. Actin served as a protein loading control for immunoblot (n=6 in each group), and mRNA expression was normalized to Tbp (n=6 in each group). *p ≤ 0.05, **p ≤ 0.01 vehicle versus STZ of mice with the same genotype. †p ≤ 0.05, ††p ≤ 0.01 Ptpn11fl/fl versus Ptpn11fl/fl; Pod-Cre under the same treatment. A.U.: arbitrary unit. c PAS staining of kidney sections from chow, STZ-treated (160 μg/g body weight, 20 weeks), and HFD-fed (24 weeks) Ptpn11fl/fl and Ptpn11fl/fl; Pod-Cre mice. Lower panel images are enlarged areas as highlighted by boxes. Arrows indicate glomerular basement membrane thickening. Scale bar: 20 μm.
Altered renal insulin signaling in mice with Shp2 disruption.
Normal insulin signaling in podocytes is critical for maintaining renal function [14, 17]. Additionally, Shp2 is an established physiological regulator of insulin signaling, exhibiting tissue-specific modulation of this pathway [35]. Accordingly, we determined the impact of podocyte Shp2 disruption on renal insulin signaling. In total kidney lysates, the insulin-induced AKT phosphorylation was increased in Ptpn11fl/fl; Pod-Cre compared with Ptpn11fl/fl mice (Fig. 5a). Notably, immunostaining of kidney sections revealed elevated insulin-induced AKT phosphorylation in the glomerular area of Ptpn11fl/fl; Pod-Cre mice (Fig. 5b). On the other hand, immunoblotting of total kidney lysates and immunostaining of kidney sections suggested diminished insulin-induced ERK phosphorylation upon Shp2 disruption in keeping with the established role of this phosphatase [36, 37] (Fig. 5a, c). Therefore, podocyte Shp2 disruption affects renal insulin signaling and differentially influences insulin-induced AKT and ERK activation.
Fig. 5:
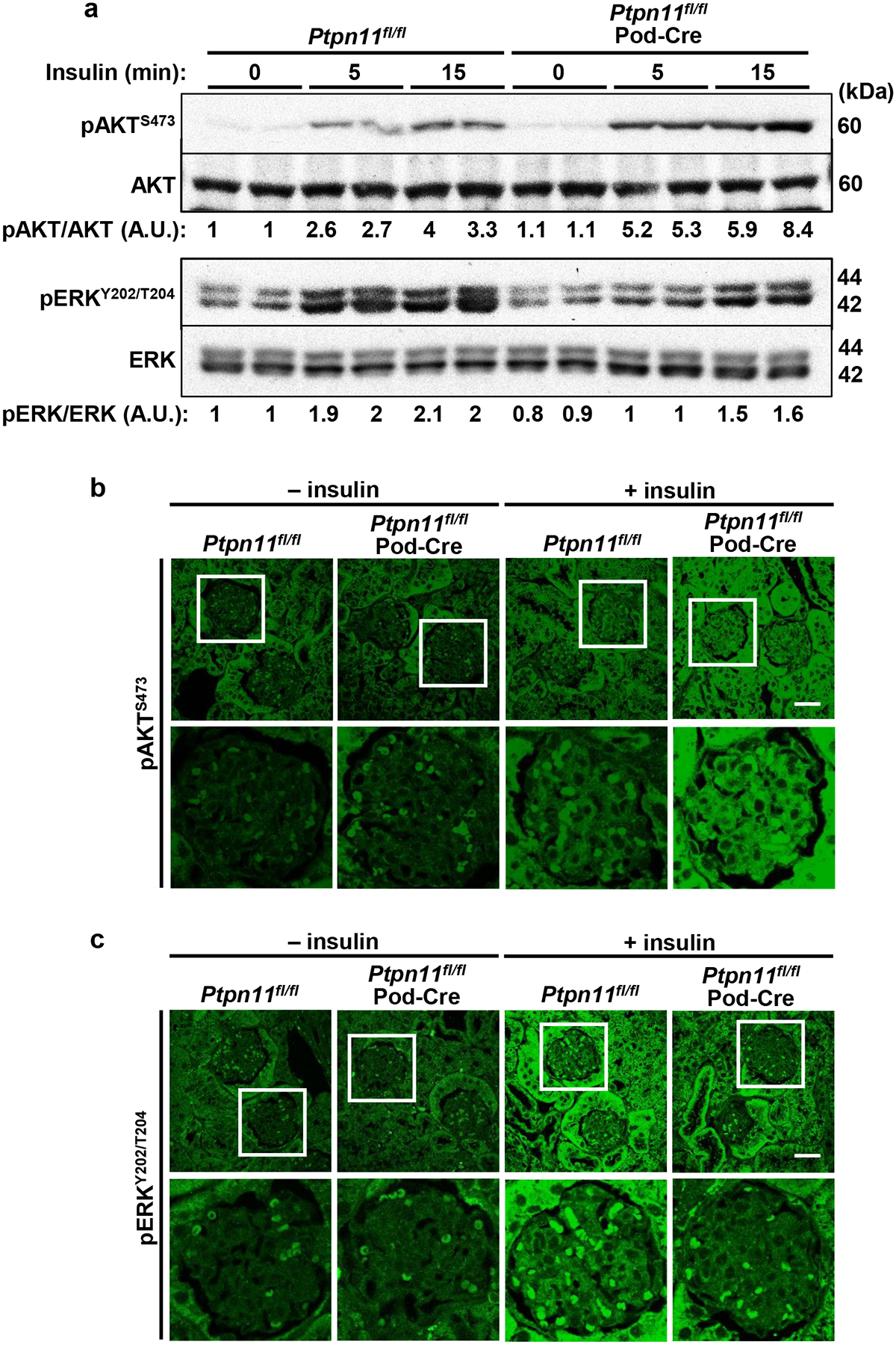
Podocyte Shp2 ablation enhances insulin-induced renal AKT signaling while attenuating ERK activation. a Immunoblots of pAKT S473, AKT, pERK Y202/T204, and ERK in kidney lysates from Ptpn11fl/fl and Ptpn11fl/fl; Pod-Cre mice without (0) and with insulin administration (10 U/kg body weight, for 5 and 15 minutes). Each lane represents lysate from an individual animal. Phosphorylation levels of AKT and ERK were normalized to the expression of the respective protein. A.U.: arbitrary unit. b, c Confocal images of kidney sections from Ptpn11fl/fl and Ptpn11fl/fl; Pod-Cre mice without (−) and with (+) insulin (15 minutes) immunostained for (b) pAKT S473 and (c) pERK Y202/T204. Boxed areas are enlarged in the lower panel. Scale bar: 25 μm.
The deficiency of Shp2 in E11 podocytes mimics the disruption in vivo.
We sought to determine if Shp2 deficiency ex vivo simulates the disruption of this phosphatase in vivo and ameliorates the deleterious effects of high glucose. To this end, we cultured the differentiated Shp2 knockdown (KD) and reconstituted (KD-R) podocytes [40] in normal and high glucose and then monitored alterations in insulin signaling, ER stress, and inflammation. In line with the observations in vivo, Shp2 knockdown in E11 podocytes enhanced insulin-induced AKT phosphorylation under high glucose while abrogating insulin-induced ERK phosphorylation (Fig. 6a). Additionally, high glucose-evoked ER stress was mitigated in Shp2 deficient podocytes as evidenced by the diminished phosphorylation of PERK, eIF2α, and IRE1α (Fig. 6b). In keeping with this observation, NF-κB phosphorylation was diminished upon Shp2 knockdown in podocytes under high glucose (Fig. 6b). The amelioration of the adverse effects of high glucose by Shp2 deficiency and reversal by reconstitution of this phosphatase in E11 podocytes are consistent with the in vivo findings. These observations suggest potential cell-autonomous effects, although the contribution of other cell types cannot be ruled out.
Fig. 6:
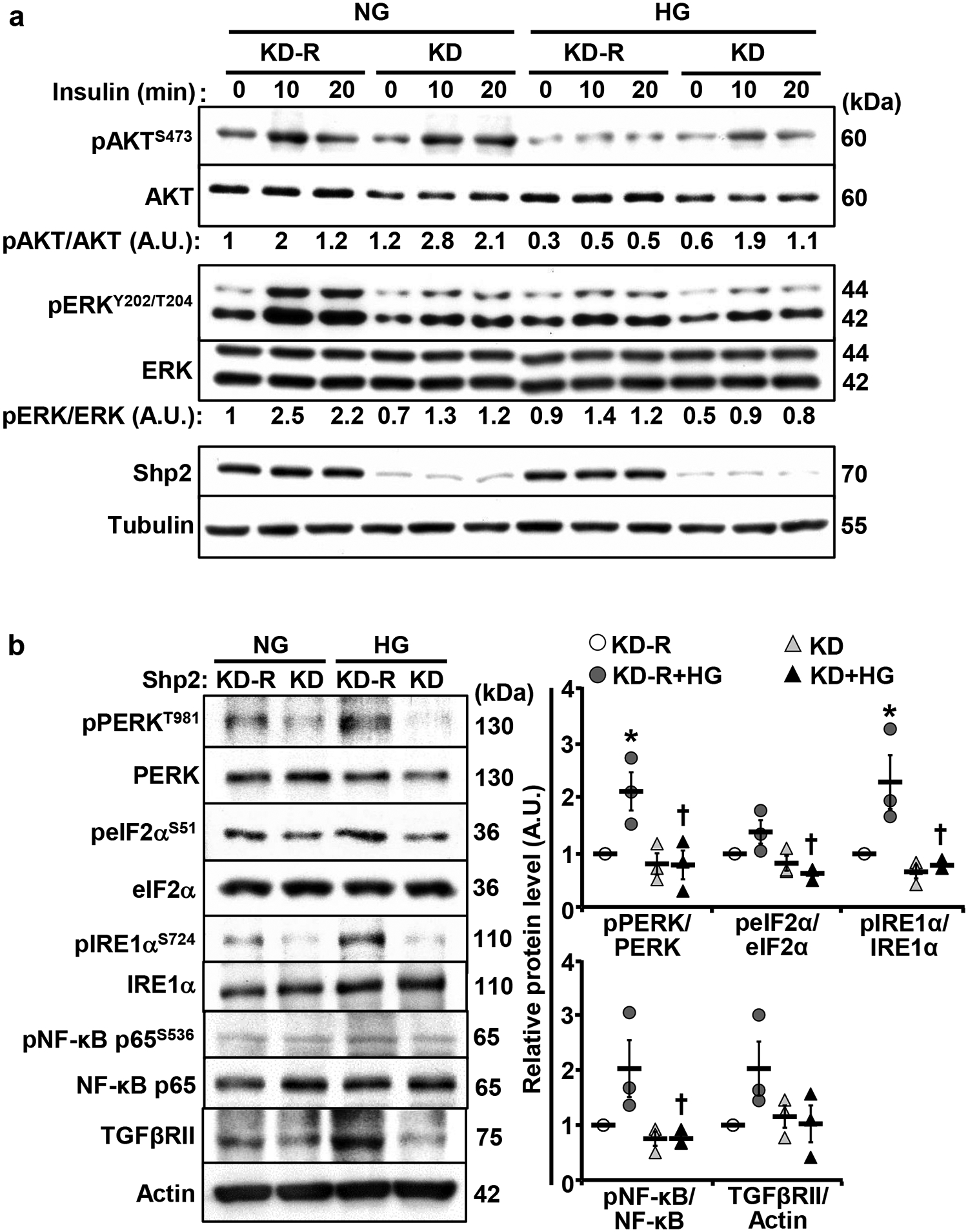
Shp2 knockdown in E11 podocytes enhances insulin-induced AKT, attenuates insulin-induced ERK activation, and mitigates high glucose-evoked ER stress and inflammation. Differentiated E11 podocytes with Shp2 knockdown (KD) and reconstitution (KD-R) were cultured under normal glucose (NG, 5.6 mM) and high glucose (HG, 25mM) for 72 hours. a Overnight starved cells were stimulated without (0) and with insulin (10 nM; for 10 and 20 minutes). Total cell lysates were immunoblotted with antibodies for pAKT S473, AKT, pERK Y202/T204, ERK, Shp2, and Tubulin. Phosphorylation levels of AKT and ERK were normalized with their respective protein. b Differentiated Shp2 KD and KD-R podocytes were lysed and immunoblotted with antibodies for pPERK T981, PERK, peIF2α S51, eIF2α, pIRE1α S724, IRE1α, pNF-κB p65 S536, NF-κB p65, TGFβRII, and Actin. Phosphorylation levels of PERK, eIF2α, IRE1α, and NF-κB p65 were normalized with their respective protein, and TGFβRII expression was normalized with Actin. Data presented in dot plots were quantified from three independent experiments. *p ≤ 0.05 NG versus HG of cells with the same background. †p ≤ 0.05 KD-R versus KD under the same treatment. A.U.: arbitrary unit.
Shp2 deficiency attenuates high glucose-induced E11 podocyte migration in culture.
Hyperglycemia causes rearrangements in the actin cytoskeleton that promote podocyte motility and contribute to glomerular injury [7]. Shp2 deficiency attenuates LPS-induced podocyte motility, and Shp2 enhances Nephrin tyrosine phosphorylation in models of podocyte injury [39, 40]. In keeping with these observations, hyperglycemia-evoked decrease in Nephrin protein expression and phosphorylation at Tyr1176/Tyr1193 were mitigated upon Shp2 disruption (Supplementary Fig. 3). Additionally, we evaluated the contribution of Shp2 to glucose-induced podocyte migration in the knockdown and reconstituted E11 podocytes using the wound healing assay. As expected, high glucose increased podocyte migration, as indicated by the high number of cells in the wound, and that was significantly decreased upon Shp2 deficiency (Fig. 7a). To gain insights into the molecular underpinning of Shp2 modulation of podocyte motility, we reconstituted the knockdown podocytes with the substrate-trapping Shp2 mutant (KD-DM) then performed substrate-trapping as described in methods. Several putative Shp2 substrates were identified, including the FYN tyrosine kinase that is implicated in remodeling the actin cytoskeleton in podocytes under high glucose [44]. FYN activity is modulated by intermolecular interactions and phosphorylation of the key tyrosine residues, Tyr527 and Tyr416 [45, 46]. Additionally, Shp2 dephosphorylates FYN Tyr527 to promote its Tyr416 autophosphorylation for full kinase activation [47, 48]. We detected co-association of FYN and the Shp2 substrate-trapping mutant under normal glucose that was enhanced under high glucose and disrupted upon lysis in the stringent RIPA buffer (Fig. 7b). Moreover, to monitor the activation of FYN, we immunoprecipitated it from the lysates of Shp2 knockdown and reconstituted podocytes, then immunoblotted using pTyr416 and pTyr527 antibodies. The high glucose-induced increase in FYN activation was abrogated in podocytes with Shp2 deficiency, as evidenced by the elevated Tyr416 and diminished Tyr527 phosphorylation (Fig. 7c). FYN regulates podocyte migration under high glucose via promoting the activation of Rho-associated coiled-coil forming protein kinase (ROCK) [44]. To further evaluate the role of Shp2-mediated FYN interaction in podocyte mobility, we performed ROCK pharmacological inhibition. High glucose-induced podocyte migration was significantly attenuated by ROCK inhibition in reconstituted podocytes but not significantly altered in podocytes with Shp2 knockdown (Fig. 7d). Collectively, these observations suggest that Shp2 deficiency attenuates high glucose-induced podocyte migration, at least in part, through modulating the FYN/ROCK signaling axis.
Fig. 7:
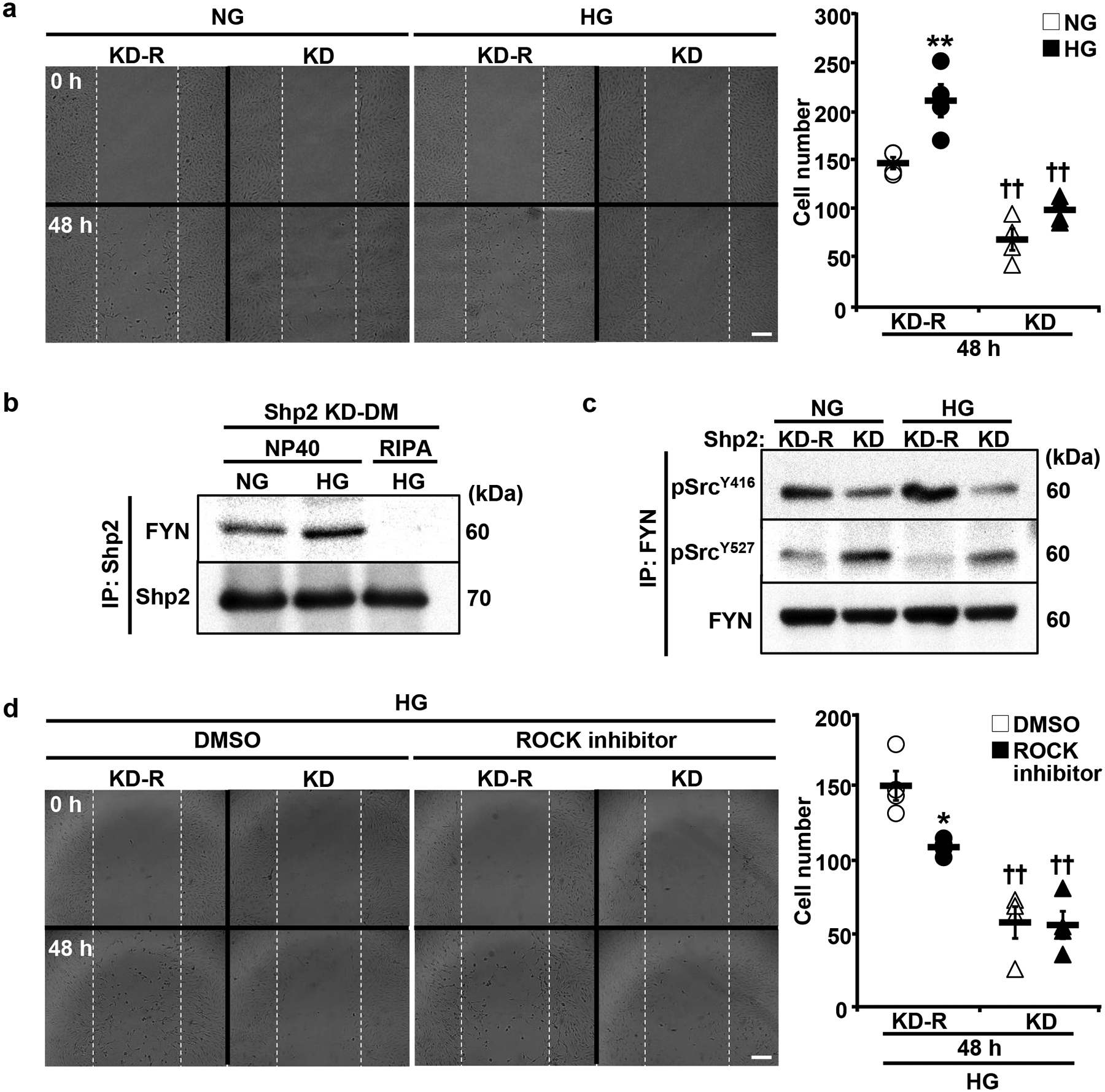
Shp2 disruption attenuates high glucose-induced podocyte migration. a Differentiated E11 podocytes with Shp2 knockdown (KD) and reconstitution (KD-R) were subjected to migration assay under normal glucose (NG, 5.6 mM) and high glucose (HG, 25 mM) conditions. Images were captured at 0 and 48 hours post conditioning. Cells migrated into the wound area were quantified from four independent experiments. **p ≤ 0.01 NG versus HG of cells with the same background. ††p ≤ 0.01 KD-R versus KD under the same treatment. b Differentiated E11 podocytes with Shp2 substrate-trapping mutant reconstitution (KD-DM) were cultured with NG and HG for 72 hours, and total proteins were extracted by NP40 and RIPA buffers as indicated. The Shp2 immunoprecipitated fractions were immunoblotted to detect FYN and Shp2 levels. c Differentiated Shp2 KD, and KD-R podocytes were treated with NG and HG for 72 hours. The FYN immunoprecipitated fractions were subjected to immunoblotting of pSrc Y416, pSrc Y527, and FYN. d Differentiated Shp2 KD and KD-R podocytes were subjected to migration assay without (DMSO) and with ROCK inhibition (Y27632, 10 μM). Images were captured at 0 and 48 hours under HG culture. *p ≤ 0.05 DMSO versus ROCK inhibitor of cells with the same background. ††p ≤ 0.01 KD-R versus KD under the same treatment. Scale bars: 200 μm.
Discussion
The present study implicates Shp2 in podocyte function under hyperglycemia and suggests that the inactivation of this phosphatase may ameliorate glomerular dysfunction in DN, a devastating complication of diabetes. We report elevated Shp2 in the kidney, including podocytes under STZ- and HFD-induced hyperglycemia in mice and E11 podocytes cultured in high glucose. However, if the increased expression of Shp2 translates to corresponding changes in the enzyme activity and if that simulates the disease state in humans is yet to be established. Additionally, the factor(s) modulating Shp2 expression in podocytes, and other kidney cell types, under hyperglycemia is currently unknown. A recent study highlights Shp2 as a target of miR-204–5p and demonstrates that inhibition or disruption of Mir204 upregulates Shp2 in models of hypertensive renal injury [49]. The current observations are consistent with elevated Shp2 in models of renal injury and the putative increase in its activity in some human glomerular diseases (minimal change nephrosis and membranous nephropathy) [39, 40]. A growing body of evidence demonstrates the upregulation of other phosphatases, including PTP1B [23, 24, 50] and Shp1 [25–27], in experimental models of podocyte injury and suggests that deregulation of phosphotyrosine signaling may contribute to DN. Indeed, overexpression of PTP1B in podocytes leads to proteinuria and foot process effacement [50], whereas PTP1B deficiency mitigates hyperglycemia-induced renal injury [24]. It remains to be determined if elevated Shp2 contributes to DN. However, findings herein indicate that the deficiency of this phosphatase in podocytes ameliorates some of the deleterious effects of hyperglycemia in mice.
Podocyte Shp2 disruption was associated with beneficial renal and systemic outcomes in mouse models of hyperglycemia as evidenced by decreased albuminuria and BUN, preserved glucose control, and reduced hypertension. A limitation is the potential renal toxicity caused by a high dose of STZ, which is countered by the use of additional models of hyperglycemia that incorporate multiple low doses of STZ and high-fat feeding. Notably, the effects of podocyte Shp2 deficiency in vivo were qualitatively comparable in the various models used. While no animal model recapitulates all the disease features in humans, the complementary models herein enabled evaluating the role of Shp2 in podocytes under hyperglycemia. The renal protective effects in mice with podocyte disruption are in keeping with the amelioration of renal injury upon Shp2 disruption in different injury models (e.g. protamine sulfate-, nephrotoxic serum- [39], and LPS-induced challenge [40]), as well as suppression of crescentic glomerulonephritis and attenuation of acute kidney injury in lupus-prone mice [51, 52]. Additionally, mice with podocyte Shp2 disruption exhibited lower blood glucose than control animals under STZ and HFD challenges. Potential contributors to the preserved glucose control are enhanced insulin signaling, increased glucose clearance in the urine, improved renal gluconeogenesis and/or altered cross-talk between the kidney and other insulin-responsive tissue(s). Moreover, the mitigation of hyperglycemia-induced hypertension in mice with Shp2 disruption is in line with the observation that Shp2 signaling in proopiomelanocortin neurons contributes to the leptin-induced chronic hypertensive effect in mice [53]. However, the current studies cannot discern the precise contribution of lower glycemia and blood pressure to the preserved renal function in mice with podocyte Shp2 disruption, and additional investigation is warranted to delineate the renal versus systemic effects.
Shp2 deficiency in podocytes was associated with the amelioration of hyperglycemia-induced renal ER stress, inflammation, fibrosis, and enhanced insulin signaling that likely contributed to the salutary effects of the disruption. A limitation is the use of total kidney lysates in the biochemical studies to monitor changes in key signaling pathways, and podocytes constitute a small fraction of the cells in the kidney. However, complementary immunohistochemical approaches that assessed fibrosis and insulin signaling support alterations of these pathways in podocytes. Additionally, the observations were further validated using the isogenic E11 podocytes with Shp2 knockdown and reconstitution. Indeed, Shp2 knockdown mimicked the in vivo deficiency of this phosphatase and ameliorated the deleterious impact of high glucose. These findings are consistent with cell-autonomous effects, although we cannot rule out the contribution of other cell-type(s). On the other hand, Shp2 reconstitution reversed the effects of the knockdown in podocytes in support of these being due to the phosphatase deficiency. Hyperglycemia and other metabolic alterations in diabetes trigger ER stress in glomerular cells [54], and pharmacological approaches that normalize stress may constitute a mechanism-based therapy [43]. The attenuation of hyperglycemia-induced PERK and IRE-1α activation upon Shp2 disruption is in keeping with the reported role of this phosphatase [40, 55, 56]. Moreover, the amelioration of hyperglycemia-induced inflammation is in line with Shp2 silencing in tubular epithelial cells leading to reduced apoptosis and inflammatory cytokines by attenuating NF-κB signaling [57]. Consistent with the diminished renal ER stress and inflammation, mice with podocyte Shp2 disruption presented with reduced hyperglycemia-induced renal fibrosis. This finding is consistent with the genetic and pharmacological inactivation of Shp2 promoting JAK2 (Tyr570) phosphorylation and diminishing JAK2/STAT3 signaling to ameliorate fibrosis [58]. Of note, communication between the various cell types in the kidney influences the cumulative integrated outcome. For example, podocyte injury contributes to tubular epithelial cell dysfunction in DN via paracrine mediators, including TNFα, IL6, vascular endothelial growth factor (VEGF), and TGFβ, among others [59]. Additionally, podocyte-specific pyruvate kinase M2 transgenic mice are protected from STZ-induced diabetic glomerular pathology, partly by maintaining podocyte VEGF expression to improve mitochondrial metabolism in glomerular endothelial cells [60]. Further investigation is needed to delineate the impact of Shp2 disruption in podocytes on cell-cell communication and its contributions to renal function.
Shp2 podocyte disruption modulated renal insulin signaling, enhancing AKT while attenuating ERK activation, in keeping with the renoprotective effects in vivo. These observations are consistent with elevated insulin-induced AKT and attenuated ERK activation in mice with hepatic Shp2 disruption [36]. The enhanced AKT activation in podocytes is associated with protection against renal injury. Indeed, podocyte AKT2 disruption enhances disease progression in mouse models of subtotal nephrectomy and aging nephropathy [61], and the insulin-induced AKT phosphorylation is attenuated in podocytes of diabetic animals to promote apoptosis [62]. Conversely, the mitigation of insulin-stimulated ERK phosphorylation is in keeping with the established role of Shp2 as a positive modulator of ERK signaling [36, 37]. Additionally, abnormal activation of ERK is associated with chronic kidney injury, including DN [63]. Indeed, the elevated ERK activation in glomeruli and cultured mesangial cells is abrogated by troglitazone to prevent glomerular dysfunction in diabetic rats [64]. Moreover, pretreatment of the ERK pharmacological inhibitor (U0126) suppresses puromycin aminonucleoside-induced apoptosis in cultured murine podocytes [65]. Shp2 can conceivably modulate insulin signaling in podocytes by directly engaging the IR and IR substrates [66]. Moreover, Shp2 may indirectly influence insulin signaling via the regulation of Nephrin [39], which interacts with the IR [67]. Further, the Shp2-MAPK axis is implicated in IR endocytosis and the regulation of insulin signaling [38]. Taken together, the current findings and the reported modulation of insulin signaling in podocytes by other phosphatases such as PTP1B [24] and Shp1 [26] highlight these enzymes as emerging orchestrators of this vital signaling pathway.
Shp2-mediated signaling in podocytes is complex, multifactorial, and likely mediated through the interactions of this phosphatase with several substrates. In support of this notion, substrate trapping studies identified numerous putative substrates of Shp2 in E11 podocytes, including, but not limited to, FYN (Hsu and Haj, data not shown). Findings herein are consistent with FYN being an Shp2 substrate in podocytes, given the co-association of the Shp2 substrate-trapping mutant with endogenous FYN, which was enhanced upon culture in high glucose. However, we cannot rule out an indirect association, as both proteins may be components of the same complex. Additionally, FYN tyrosine phosphorylation status was modulated by Shp2 in podocytes. Shp2 deficiency elevated Tyr527 while decreasing Tyr416 phosphorylation corresponding to FYN inactivation, whereas Shp2 reconstitution reversed the phosphorylation, suggesting activation of this kinase. Notably, Shp2 dephosphorylation of FYN is required to release the intramolecular inhibition and enhance its activity on Nephrin [39]. Moreover, we cannot rule out the regulation of FYN phosphorylation in podocytes by other PTP(s). Indeed, Nephrin phosphorylation is concomitantly modulated by PTP1B [23], PTP-PEST [23], Shp1 [25], and Shp2 [39]. Interestingly, the high glucose induced activation of the FYN/ROCK axis and enhanced motility in cultured podocytes are reversed by FYN knockdown and ROCK inhibition [44]. In line with these findings, we observed attenuation of high glucose-induced E11 podocyte migration upon Shp2 knockdown (inactivated FYN) and ROCK inhibition. Although the E11 podocytes are an imperfect surrogate, the alterations in cytoskeleton and mobility may model the in vivo setting and help decipher the contribution of Shp2 and its substrates. Collectively, the current findings establish that Shp2 deficiency attenuates high glucose-induced podocyte migration, and highlight FYN as one of the putative mediator of Shp2 action in podocytes.
Despite advancements in understanding the pathomechanisms underlying DN, effective treatments that halt the progression to ESRD remain an unmet medical need. Findings herein suggest that Shp2 inactivation might afford protection to podocytes from high glucose evoked dysfunction and highlight this phosphatase as a pharmacologically tractable candidate. Noteworthy, Shp2 is a therapeutic target for receptor tyrosine kinase-driven cancers [68], and an allosteric inhibitor of Shp2 shows promise in experimental animal models [69]. Additional studies are warranted to delineate the therapeutic potential of Shp2 pharmacological inhibition in combating DN.
Supplementary Material
Acknowledgments.
Research in the Haj laboratory was funded by the National Institute of Diabetes and Digestive and Kidney Diseases grants RO1DK095359 and R01DK090492, the National Institute of Environmental Health Sciences grant P42ES04699, and NIFA grant CA-D*-NTR-7836H. Dr. Haj is a Co-Leader of the Endocrinology and Metabolism Core of UC Davis Mouse Metabolic Phenotyping Center, which is funded by U24DK092993. Dr. Hsu was supported by National Institute on Alcohol Abuse and Alcoholism grant R21AA027633. Dr. Afkarian was supported by the grant R01-DK-104706 from the National Institute of Diabetes and Digestive and Kidney Diseases. The Light Microscopy Imaging Facility (UC Davis) is supported by National Institutes of Health grant 1S10RR019266.
Footnotes
Conflicts of interest: The authors disclose no conflicts associated with the manuscript.
References
- [1].Kramer A, Pippias M, Noordzij M, Stel VS, Andrusev AM, Aparicio-Madre MI, Arribas Monzon FE, Asberg A, Barbullushi M, Beltran P, Bonthuis M, Caskey FJ, Castro de la Nuez P, Cernevskis H, De Meester J, Finne P, Golan E, Heaf JG, Hemmelder MH, Ioannou K, Kantaria N, Komissarov K, Korejwo G, Kramar R, Lassalle M, Lopot F, Macario F, Mackinnon B, Palsson R, Pechter U, Pinera VC, Santiuste de Pablos C, Segarra-Medrano A, Seyahi N, Slon Roblero MF, Stojceva-Taneva O, Vazelov E, Winzeler R, Ziginskiene E, Massy Z, Jager KJ, The European Renal Association - European Dialysis and Transplant Association (ERA-EDTA) Registry Annual Report 2016: a summary, Clin Kidney J 12(5) (2019) 702–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Umanath K, Lewis JB, Update on Diabetic Nephropathy: Core Curriculum 2018, Am J Kidney Dis 71(6) (2018) 884–895. [DOI] [PubMed] [Google Scholar]
- [3].Jitraknatee J, Ruengorn C, Nochaiwong S, Prevalence and Risk Factors of Chronic Kidney Disease among Type 2 Diabetes Patients: A Cross-Sectional Study in Primary Care Practice, Sci Rep 10(1) (2020) 6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kainz A, Hronsky M, Stel VS, Jager KJ, Geroldinger A, Dunkler D, Heinze G, Tripepi G, Oberbauer R, Prediction of prevalence of chronic kidney disease in diabetic patients in countries of the European Union up to 2025, Nephrol Dial Transplant 30 Suppl 4 (2015) iv113–8. [DOI] [PubMed] [Google Scholar]
- [5].McCullough KP, Morgenstern H, Saran R, Herman WH, Robinson BM, Projecting ESRD Incidence and Prevalence in the United States through 2030, J Am Soc Nephrol 30(1) (2019) 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Saran R, Robinson B, Abbott KC, Agodoa LY, Albertus P, Ayanian J, Balkrishnan R, Bragg-Gresham J, Cao J, Chen JL, Cope E, Dharmarajan S, Dietrich X, Eckard A, Eggers PW, Gaber C, Gillen D, Gipson D, Gu H, Hailpern SM, Hall YN, Han Y, He K, Hebert H, Helmuth M, Herman W, Heung M, Hutton D, Jacobsen SJ, Ji N, Jin Y, Kalantar-Zadeh K, Kapke A, Katz R, Kovesdy CP, Kurtz V, Lavalee D, Li Y, Lu Y, McCullough K, Molnar MZ, Montez-Rath M, Morgenstern H, Mu Q, Mukhopadhyay P, Nallamothu B, Nguyen DV, Norris KC, O’Hare AM, Obi Y, Pearson J, Pisoni R, Plattner B, Port FK, Potukuchi P, Rao P, Ratkowiak K, Ravel V, Ray D, Rhee CM, Schaubel DE, Selewski DT, Shaw S, Shi J, Shieu M, Sim JJ, Song P, Soohoo M, Steffick D, Streja E, Tamura MK, Tentori F, Tilea A, Tong L, Turf M, Wang D, Wang M, Woodside K, Wyncott A, Xin X, Zang W, Zepel L, Zhang S, Zho H, Hirth RA, Shahinian V, US Renal Data System 2016 Annual Data Report: Epidemiology of Kidney Disease in the United States, Am J Kidney Dis 69(3 Suppl 1) (2017) A7–A8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lin JS, Susztak K, Podocytes: the Weakest Link in Diabetic Kidney Disease?, Curr Diab Rep 16(5) (2016) 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Greka A, Mundel P, Cell biology and pathology of podocytes, Annu Rev Physiol 74 (2012) 299–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gil CL, Hooker E, Larrivee B, Diabetic Kidney Disease, Endothelial Damage, and Podocyte-Endothelial Crosstalk, Kidney Med 3(1) (2021) 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wolf G, Chen S, Ziyadeh FN, From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy, Diabetes 54(6) (2005) 1626–34. [DOI] [PubMed] [Google Scholar]
- [11].Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC, Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene, J Am Soc Nephrol 16(10) (2005) 2941–52. [DOI] [PubMed] [Google Scholar]
- [12].Kravets I, Mallipattu SK, The Role of Podocytes and Podocyte-Associated Biomarkers in Diagnosis and Treatment of Diabetic Kidney Disease, J Endocr Soc 4(4) (2020) bvaa029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lu CC, Wang GH, Lu J, Chen PP, Zhang Y, Hu ZB, Ma KL, Role of Podocyte Injury in Glomerulosclerosis, Adv Exp Med Biol 1165 (2019) 195–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lay AC, Coward RJM, The Evolving Importance of Insulin Signaling in Podocyte Health and Disease, Front Endocrinol (Lausanne) 9 (2018) 693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Coward RJ, Welsh GI, Yang J, Tasman C, Lennon R, Koziell A, Satchell S, Holman GD, Kerjaschki D, Tavare JM, Mathieson PW, Saleem MA, The human glomerular podocyte is a novel target for insulin action, Diabetes 54(11) (2005) 3095–102. [DOI] [PubMed] [Google Scholar]
- [16].Rask-Madsen C, King GL, Diabetes: Podocytes lose their footing, Nature 468(7320) (2010) 42–4. [DOI] [PubMed] [Google Scholar]
- [17].Welsh GI, Hale LJ, Eremina V, Jeansson M, Maezawa Y, Lennon R, Pons DA, Owen RJ, Satchell SC, Miles MJ, Caunt CJ, McArdle CA, Pavenstadt H, Tavare JM, Herzenberg AM, Kahn CR, Mathieson PW, Quaggin SE, Saleem MA, Coward RJ, Insulin signaling to the glomerular podocyte is critical for normal kidney function, Cell Metab 12(4) (2010) 329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Madhusudhan T, Wang H, Dong W, Ghosh S, Bock F, Thangapandi VR, Ranjan S, Wolter J, Kohli S, Shahzad K, Heidel F, Krueger M, Schwenger V, Moeller MJ, Kalinski T, Reiser J, Chavakis T, Isermann B, Defective podocyte insulin signalling through p85-XBP1 promotes ATF6-dependent maladaptive ER-stress response in diabetic nephropathy, Nat Commun 6 (2015) 6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lay AC, Hurcombe JA, Betin VMS, Barrington F, Rollason R, Ni L, Gillam L, Pearson GME, Ostergaard MV, Hamidi H, Lennon R, Welsh GI, Coward RJM, Prolonged exposure of mouse and human podocytes to insulin induces insulin resistance through lysosomal and proteasomal degradation of the insulin receptor, Diabetologia 60(11) (2017) 2299–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kim EY, Anderson M, Dryer SE, Insulin increases surface expression of TRPC6 channels in podocytes: role of NADPH oxidases and reactive oxygen species, Am J Physiol Renal Physiol 302(3) (2012) F298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Xia S, Liu Y, Li X, Thilo F, Tepel M, Insulin Increases Expression of TRPC6 Channels in Podocytes by a Calcineurin-Dependent Pathway, Cell Physiol Biochem 38(2) (2016) 659–69. [DOI] [PubMed] [Google Scholar]
- [22].Geraldes P, Protein phosphatases and podocyte function, Curr Opin Nephrol Hypertens 27(1) (2018) 49–55. [DOI] [PubMed] [Google Scholar]
- [23].Aoudjit L, Jiang R, Lee TH, New LA, Jones N, Takano T, Podocyte Protein, Nephrin, Is a Substrate of Protein Tyrosine Phosphatase 1B, J Signal Transduct 2011 (2011) 376543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ito Y, Hsu MF, Bettaieb A, Koike S, Mello A, Calvo-Rubio M, Villalba JM, Haj FG, Protein tyrosine phosphatase 1B deficiency in podocytes mitigates hyperglycemia-induced renal injury, Metabolism 76 (2017) 56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Denhez B, Lizotte F, Guimond MO, Jones N, Takano T, Geraldes P, Increased SHP-1 protein expression by high glucose levels reduces nephrin phosphorylation in podocytes, J Biol Chem 290(1) (2015) 350–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Drapeau N, Lizotte F, Denhez B, Guay A, Kennedy CR, Geraldes P, Expression of SHP-1 induced by hyperglycemia prevents insulin actions in podocytes, Am J Physiol Endocrinol Metab 304(11) (2013) E1188–98. [DOI] [PubMed] [Google Scholar]
- [27].Lizotte F, Denhez B, Guay A, Gevry N, Cote AM, Geraldes P, Persistent Insulin Resistance in Podocytes Caused by Epigenetic Changes of SHP-1 in Diabetes, Diabetes 65(12) (2016) 3705–3717. [DOI] [PubMed] [Google Scholar]
- [28].Sugimoto S, Lechleider RJ, Shoelson SE, Neel BG, Walsh CT, Expression, purification, and characterization of SH2-containing protein tyrosine phosphatase, SH-PTP2, J Biol Chem 268(30) (1993) 22771–6. [PubMed] [Google Scholar]
- [29].Feng GS, Shp2-mediated molecular signaling in control of embryonic stem cell self-renewal and differentiation, Cell Res 17(1) (2007) 37–41. [DOI] [PubMed] [Google Scholar]
- [30].Chan G, Kalaitzidis D, Neel BG, The tyrosine phosphatase Shp2 (PTPN11) in cancer, Cancer Metastasis Rev 27(2) (2008) 179–92. [DOI] [PubMed] [Google Scholar]
- [31].Maile LA, Clemmons DR, Regulation of insulin-like growth factor I receptor dephosphorylation by SHPS-1 and the tyrosine phosphatase SHP-2, J Biol Chem 277(11) (2002) 8955–60. [DOI] [PubMed] [Google Scholar]
- [32].Ling Y, Maile LA, Clemmons DR, Tyrosine phosphorylation of the beta3-subunit of the alphaVbeta3 integrin is required for embrane association of the tyrosine phosphatase SHP-2 and its further recruitment to the insulin-like growth factor I receptor, Mol Endocrinol 17(9) (2003) 1824–33. [DOI] [PubMed] [Google Scholar]
- [33].Dixit M, Zhuang D, Ceacareanu B, Hassid A, Treatment with insulin uncovers the motogenic capacity of nitric oxide in aortic smooth muscle cells: dependence on Gab1 and Gab1-SHP2 association, Circ Res 93(10) (2003) e113–23. [DOI] [PubMed] [Google Scholar]
- [34].Mussig K, Staiger H, Fiedler H, Moeschel K, Beck A, Kellerer M, Haring HU, Shp2 is required for protein kinase C-dependent phosphorylation of serine 307 in insulin receptor substrate-1, J Biol Chem 280(38) (2005) 32693–9. [DOI] [PubMed] [Google Scholar]
- [35].Hall C, Yu H, Choi E, Insulin receptor endocytosis in the pathophysiology of insulin resistance, Exp Mol Med 52(6) (2020) 911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Matsuo K, Delibegovic M, Matsuo I, Nagata N, Liu S, Bettaieb A, Xi Y, Araki K, Yang W, Kahn BB, Neel BG, Haj FG, Altered glucose homeostasis in mice with liver-specific deletion of Src homology phosphatase 2, J Biol Chem 285(51) (2010) 39750–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Princen F, Bard E, Sheikh F, Zhang SS, Wang J, Zago WM, Wu D, Trelles RD, Bailly-Maitre B, Kahn CR, Chen Y, Reed JC, Tong GG, Mercola M, Chen J, Feng GS, Deletion of Shp2 tyrosine phosphatase in muscle leads to dilated cardiomyopathy, insulin resistance, and premature death, Mol Cell Biol 29(2) (2009) 378–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Choi E, Kikuchi S, Gao H, Brodzik K, Nassour I, Yopp A, Singal AG, Zhu H, Yu H, Mitotic regulators and the SHP2-MAPK pathway promote IR endocytosis and feedback regulation of insulin signaling, Nat Commun 10(1) (2019) 1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Verma R, Venkatareddy M, Kalinowski A, Patel SR, Salant DJ, Garg P, Shp2 Associates with and Enhances Nephrin Tyrosine Phosphorylation and Is Necessary for Foot Process Spreading in Mouse Models of Podocyte Injury, Mol Cell Biol 36(4) (2015) 596–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hsu MF, Bettaieb A, Ito Y, Graham J, Havel PJ, Haj FG, Protein tyrosine phosphatase Shp2 deficiency in podocytes attenuates lipopolysaccharide-induced proteinuria, Sci Rep 7(1) (2017) 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhang SQ, Yang W, Kontaridis MI, Bivona TG, Wen G, Araki T, Luo J, Thompson JA, Schraven BL, Philips MR, Neel BG, Shp2 regulates SRC family kinase activity and Ras/Erk activation by controlling Csk recruitment, Mol Cell 13(3) (2004) 341–55. [DOI] [PubMed] [Google Scholar]
- [42].Agazie YM, Hayman MJ, Development of an efficient “substrate-trapping” mutant of Src homology phosphotyrosine phosphatase 2 and identification of the epidermal growth factor receptor, Gab1, and three other proteins as target substrates, J Biol Chem 278(16) (2003) 13952–8. [DOI] [PubMed] [Google Scholar]
- [43].Cybulsky AV, Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases, Nat Rev Nephrol 13(11) (2017) 681–696. [DOI] [PubMed] [Google Scholar]
- [44].Lv Z, Hu M, Ren X, Fan M, Zhen J, Chen L, Lin J, Ding N, Wang Q, Wang R, Fyn Mediates High Glucose-Induced Actin Cytoskeleton Reorganization of Podocytes via Promoting ROCK Activation In Vitro, J Diabetes Res 2016 (2016) 5671803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Saito YD, Jensen AR, Salgia R, Posadas EM, Fyn: a novel molecular target in cancer, Cancer 116(7) (2010) 1629–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kumar A, Jaggi AS, Singh N, Pharmacology of Src family kinases and therapeutic implications of their modulators, Fundam Clin Pharmacol 29(2) (2015) 115–30. [DOI] [PubMed] [Google Scholar]
- [47].Roskoski R Jr., Src kinase regulation by phosphorylation and dephosphorylation, Biochem Biophys Res Commun 331(1) (2005) 1–14. [DOI] [PubMed] [Google Scholar]
- [48].Fang X, Lang Y, Wang Y, Mo W, Wei H, Xie J, Yu M, Shp2 activates Fyn and Ras to regulate RBL-2H3 mast cell activation following FcepsilonRI aggregation, PLoS One 7(7) (2012) e40566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cheng Y, Wang D, Wang F, Liu J, Huang B, Baker MA, Yin J, Wu R, Liu X, Regner KR, Usa K, Liu Y, Zhang C, Dong L, Geurts AM, Wang N, Miller SS, He Y, Liang M, Endogenous miR-204 Protects the Kidney against Chronic Injury in Hypertension and Diabetes, J Am Soc Nephrol 31(7) (2020) 1539–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kumagai T, Baldwin C, Aoudjit L, Nezvitsky L, Robins R, Jiang R, Takano T, Protein tyrosine phosphatase 1B inhibition protects against podocyte injury and proteinuria, Am J Pathol 184(8) (2014) 2211–24. [DOI] [PubMed] [Google Scholar]
- [51].Wang J, Mizui M, Zeng LF, Bronson R, Finnell M, Terhorst C, Kyttaris VC, Tsokos GC, Zhang ZY, Kontaridis MI, Inhibition of SHP2 ameliorates the pathogenesis of systemic lupus erythematosus, J Clin Invest 126(6) (2016) 2077–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Jiang J, Hu B, Chung CS, Chen Y, Zhang Y, Tindal EW, Li J, Ayala A, SHP2 inhibitor PHPS1 ameliorates acute kidney injury by Erk1/2-STAT3 signaling in a combined murine hemorrhage followed by septic challenge model, Mol Med 26(1) (2020) 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].do Carmo JM, da Silva AA, Ebaady SE, Sessums PO, Abraham RS, Elmquist JK, Lowell BB, Hall JE, Shp2 signaling in POMC neurons is important for leptin’s actions on blood pressure, energy balance, and glucose regulation, Am J Physiol Regul Integr Comp Physiol 307(12) (2014) R1438–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wang Z, do Carmo JM, da Silva AA, Fu Y, Hall JE, Mechanisms of Synergistic Interactions of Diabetes and Hypertension in Chronic Kidney Disease: Role of Mitochondrial Dysfunction and ER Stress, Curr Hypertens Rep 22(2) (2020) 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Nagata N, Matsuo K, Bettaieb A, Bakke J, Matsuo I, Graham J, Xi Y, Liu S, Tomilov A, Tomilova N, Gray S, Jung DY, Ramsey JJ, Kim JK, Cortopassi G, Havel PJ, Haj FG, Hepatic Src homology phosphatase 2 regulates energy balance in mice, Endocrinology 153(7) (2012) 3158–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wang HC, Zhou Y, Huang SK, SHP-2 phosphatase controls aryl hydrocarbon receptor-mediated ER stress response in mast cells, Arch Toxicol 91(4) (2017) 1739–1748. [DOI] [PubMed] [Google Scholar]
- [57].Teng JF, Wang K, Jia ZM, Guo YJ, Guan YW, Li ZH, Ai X, Lentivirus-Mediated Silencing of Src Homology 2 Domain-Containing Protein Tyrosine Phosphatase 2 Inhibits Release of Inflammatory Cytokines and Apoptosis in Renal Tubular Epithelial Cells Via Inhibition of the TLR4/NF-kB Pathway in Renal Ischemia-Reperfusion Injury, Kidney Blood Press Res 43(4) (2018) 1084–1103. [DOI] [PubMed] [Google Scholar]
- [58].Zehender A, Huang J, Gyorfi AH, Matei AE, Trinh-Minh T, Xu X, Li YN, Chen CW, Lin J, Dees C, Beyer C, Gelse K, Zhang ZY, Bergmann C, Ramming A, Birchmeier W, Distler O, Schett G, Distler JHW, The tyrosine phosphatase SHP2 controls TGFbeta-induced STAT3 signaling to regulate fibroblast activation and fibrosis, Nat Commun 9(1) (2018) 3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Leung JC, Lai KN, Tang SCW, Crosstalk between Podocytes and Tubular Epithelial Cells, Podocytopathy 183 (2014) 54–63. [Google Scholar]
- [60].Fu J, Shinjo T, Li Q, St-Louis R, Park K, Yu MG, Yokomizo H, Simao F, Huang Q, Wu IH, King GL, Regeneration of glomerular metabolism and function by podocyte pyruvate kinase M2 in diabetic nephropathy, JCI Insight 7(5) (2022) e155260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Canaud G, Bienaime F, Viau A, Treins C, Baron W, Nguyen C, Burtin M, Berissi S, Giannakakis K, Muda AO, Zschiedrich S, Huber TB, Friedlander G, Legendre C, Pontoglio M, Pende M, Terzi F, AKT2 is essential to maintain podocyte viability and function during chronic kidney disease, Nat Med 19(10) (2013) 1288–96. [DOI] [PubMed] [Google Scholar]
- [62].Tejada T, Catanuto P, Ijaz A, Santos JV, Xia X, Sanchez P, Sanabria N, Lenz O, Elliot SJ, Fornoni A, Failure to phosphorylate AKT in podocytes from mice with early diabetic nephropathy promotes cell death, Kidney Int 73(12) (2008) 1385–93. [DOI] [PubMed] [Google Scholar]
- [63].Feliers D, Kasinath BS, Erk in kidney diseases, J Signal Transduct 2011 (2011) 768512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Isshiki K, Haneda M, Koya D, Maeda S, Sugimoto T, Kikkawa R, Thiazolidinedione compounds ameliorate glomerular dysfunction independent of their insulin-sensitizing action in diabetic rats, Diabetes 49(6) (2000) 1022–32. [DOI] [PubMed] [Google Scholar]
- [65].Liu S, Ding J, Fan Q, Zhang H, The activation of extracellular signal-regulated kinase is responsible for podocyte injury, Mol Biol Rep 37(5) (2010) 2477–84. [DOI] [PubMed] [Google Scholar]
- [66].Xu E, Schwab M, Marette A, Role of protein tyrosine phosphatases in the modulation of insulin signaling and their implication in the pathogenesis of obesity-linked insulin resistance, Rev Endocr Metab Disord 15(1) (2014) 79–97. [DOI] [PubMed] [Google Scholar]
- [67].Villarreal R, Mitrofanova A, Maiguel D, Morales X, Jeon J, Grahammer F, Leibiger IB, Guzman J, Fachado A, Yoo TH, Busher Katin A, Gellermann J, Merscher S, Burke GW, Berggren PO, Oh J, Huber TB, Fornoni A, Nephrin Contributes to Insulin Secretion and Affects Mammalian Target of Rapamycin Signaling Independently of Insulin Receptor, J Am Soc Nephrol 27(4) (2016) 1029–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Liu Q, Qu J, Zhao M, Xu Q, Sun Y, Targeting SHP2 as a promising strategy for cancer immunotherapy, Pharmacol Res 152 (2020) 104595. [DOI] [PubMed] [Google Scholar]
- [69].Yuan X, Bu H, Zhou J, Yang CY, Zhang H, Recent Advances of SHP2 Inhibitors in Cancer Therapy: Current Development and Clinical Application, J Med Chem 63(20) (2020) 11368–11396. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.