

Abstract
Although immunotherapies of tumors have demonstrated promise for altering the progression of malignancies, immunotherapies have been limited by an immunosuppressive tumor microenvironment (TME) that prevents infiltrating immune cells from performing their anti-cancer functions. Prominent among immunosuppressive cells are myeloid-derived suppressor cells (MDSC) and tumor-associated macrophages (TAM) that inhibit T cells via release of immunosuppressive cytokines and engagement of checkpoint receptors. Here we explore the properties of MDSC and TAM from freshly isolated mouse and human tumors and find that an immunosuppressive subset of these cells can be distinguished from the non-immunosuppressive population by its upregulation of folate receptor beta (FRβ) within the TME and its restriction to the TME. This FRβ+ subpopulation could be selectively targeted with folate-linked drugs. Delivery of a folate-targeted TLR7 agonist to these cells i) reduced their immunosuppressive function, ii) increased CD8+ T cell infiltration, iii) enhanced M1/M2 macrophage ratios, iv) inhibited tumor growth, v) blocked tumor metastasis, and vi) improved overall survival without demonstrable toxicity. These data reveal a broadly applicable strategy across tumor types for reprogramming MDSC and TAM into anti-tumorigenic immune cells using a drug that would otherwise be too toxic to administer systemically. The data also establish FRβ as the first marker that distinguishes immunosuppressive from non-immunosuppressive subsets of MDSC and TAM. Because all solid tumors accumulate MDSC and TAM, a general strategy to both identify and reprogram these cells should be broadly applied in the characterization and treatment of multiple tumors.
Introduction
Myeloid derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) are well known for their abilities to suppress anti-tumor immunity (1), release tumor growth factors (2), promote tumor angiogenesis (3) and enhance tumor metastasis (4). Because both cell types are present in the tumor microenvironment (TME) of virtually all tumor types, they pose a major hurdle to developing immunotherapies for almost all solid tumors. Strategies to inhibit the functions of MDSCs and TAMs have focused primarily on nonspecific drugs like phosphatidylinositol-3-kinase inhibitors and anti-CCL2 antibodies, however such drugs have generally displayed adequate efficacy only at concentrations that were compromised by off-target toxicity (5). While current immunotherapeutic strategies have shown considerable promise in treating cancer, approaches to reprogram immunoregulatory myeloid cells are still needed to enable most immunotherapies of solid tumors to achieve their full potentials (6).
Despite significant advances in MDSC and TAM biology (1,7,8), both populations are still primarily defined functionally, since antigenic markers that segregate with MDSCs and TAMs are also expressed on non-immunosuppressive myeloid populations (8). In fact, to date there has been no diagnostic marker has been identified that distinguishes immunosuppressive from non-immunosuppressive MDSC or TAM. Previous studies have shown that folate receptor beta (FRβ) constitutes a marker for TAMs (9) and other forms of activated macrophages (10), however, use of this marker has focused to date on imaging of autoimmune diseases (11,12), with little effort devoted to exploiting it for therapeutic applications or evaluating its association with function (13). Ongoing research by our group has shown the immediate suppressive function of MDSCs to be located within inflammatory sites and the TME (14). Given that FRβ is also localized to inflammatory sites and the TME, we initiated studies to define the functional properties of FRβ+ TME-derived myeloid cells. Herein we demonstrate that FRβ+ MDSCs and TAMs freshly isolated from the TME are the dominant immunosuppressive myeloid populations and that highly specific delivery of both imaging and therapeutic agents to these cells in the TME can be achieved with folate-targeting. We further show that a potent folate-targeted TLR7 agonist that is too toxic to administer in nontargeted form can be safely and specifically targeted to myeloid cells within the TME by conjugation to folate, resulting in reprogramming of tumor myeloid cells, abrogation of MDSC/TAM immunosuppressive activities, enhancement of T cell infiltration, induction of antitumor activity and significant improvement in overall survival.
Methods
Animals
BALB/cJ, FVB/NJ, and C57BL/6J mice were purchased from the Jackson Laboratory and housed in accordance with Purdue Animal Use and Care Committee guidelines. Mice were used at 8–12 weeks of age for tumor implantation studies. Unless otherwise specified mice were housed on corn cob bedding and fed complete chow. 4T1 tumors were implanted in the mammary fat pad of female BALB/cJ at a dose of 1×106 cells/mouse. EMT6, MB49, RM-1 TRAMP-C2, and CT26 were implanted on the flank of the animal and implanted at a dose of 1×106 cells/mouse. Myc-Cap tumors were implanted orthotopically in the prostate of male FVB mice at a dose of 1×105 cells per mouse. RM-1 were additionally implanted as an intraperitoneal tumor (IP) tumor by injecting 1×106 cells into the IP space and allowing to grow for 7 days. Outgrowth of the tumor was monitored by length vs width measurements for solid flank tumors. Tumors were utilized for studies starting at 100 mm3 for the start of treatment studies and harvested at a size of 600–800 mm3 for non-treatment studies unless otherwise specified
Cell Lines
4T1 and CT26 cells were purchased from ATCC (CRL-2539 and CRL-2638) and maintained in RPMI-1640 supplemented with 10% FBS and 1% Penn/Strep (10,000 units/mL stock). EMT6 cells were purchased from ATCC (CRL-2755) and were cultured in Waymouth’s MB 752/1 media supplemented with 15% FBS and 2 mM L-glutamine. MB49, Myc-Cap, and RM-1 cell lines were cultured in DMEM supplemented with 10% FBS and 1% Penn/strep. TRAMP-C2 cells were a generous gift from James Allison (MD Anderson) and maintained in DMEM supplemented with 5% FBS, 5% NuSerum IV, 10 mM HEPES, 4 mM L-glutamine, 5 μg/mL insulin, 10 μM dehydroisoandrosterone, 55 μM 2-mercaptoethanol, and 1% Penn/Strep. Cells were maintained in 10 cm2 tissue culture treated plastic dishes. All studies were done within 10 passages of thawing the cells from frozen stocks. No cell line verification or testing was conducted.
Human Studies
Human bladder tissue samples were acquired at IU Health Methodist Hospital through the Indiana University Health Biorepository (Indianapolis, IN) under Institutional Review Board number 1408862191.
Lung carcinoma studies were conducted on tissue samples from 6 patients with stage I-II lung cancer who underwent surgical resection. Tissue was collected under IRB 813004. All samples were histologically confirmed to be adenocarcinomas. Tumor samples were digested to single cell suspensions as previously described (15). Tumor digests were compared to PBMC preps from the same patients and analyzed for FRβ and CD14.
MDSC and TAM isolation for live cell sorting and flow cytometry analysis
MDSCs and TAMs were isolated from all tumor models by generating a single cell suspension from the tumor by collagenase I (1mg/ml) and DNase (100 ug/ml) for 1 hr. with shaking at 37°C. Isolation of IP tumors was carried out by washing the IP cavity of a tumor bearing animal with 10 mL of phosphate buffered saline (PBS). After digestion or collection from IP space, samples were filtered through a 70 μm mesh and treated with ACK to remove red blood cells. MDSCs were labeled with antibodies against CD11b (M1/70 Biolegend), Ly-6C (HK1.4 Biolegend), Ly-6G (1A8 Biolegend). M-MDSCs were identified as CD11b+ Ly-6C+ and G-MDSC were identified as CD11b+ Ly-6G+. TAMs were labeled with the same set of antibodies as MDSC with the addition of F4/80 (Clone BM8 Biolegend). TAMs were isolated as CD11b+ Ly-6C− F4/80+. MDSCs and TAMs were also isolated from the spleens of tumor bearing animals by removal of the spleen followed by mechanical disruption to generate a single cell suspension. Red blood cell lysis was then performed using ACK lysis buffer. Samples were then washed in PBS, centrifuged, and resuspended in RPMI-1640 for sorting or 10% formalin and stored in the dark at 4°C for analysis by flow cytometry. Additional analysis of MDSCs and TAMs included staining with anti-PD-L1 antibody (Clone 10F.9G2 BioLegend), CD86 (Clone GL-1 BioLegend), and CD206 (Clone C068C2 BioLegend).
Isolation for both MDSCs and TAMs was done using a BD FACSAria III with the assistance of the Purdue University Flow Cytometry and Cell Separation Facility. An anti-mouse folate receptor beta antibody was generously contributed by Dr. Dimiter Dimitrov.
All flow cytometry sample collection was carried out on a BD LSRFortessa analyzer with data analysis performed using FlowJo v10 (Treestar Software) analysis software.
in vivo and in vitro folate staining
Folate conjugates were provided through OnTarget Laboratories (West Lafayette, IN). A near infrared dye bound to folate (OTL38) (16) was used for all in vitro and in vivo labeling experiments.
For in vivo labeling experiments, male C57Bl/6j mice at 8 weeks of age were placed on a folate deficient chow (Teklad Envigo) for 2 weeks prior to use in experiments. Once tumors reached a size of approximately 0.5 cm (width or length) mice were injected with 100 nmols of OTL38. Tumors were harvested 2 hours later and digested using 1 mg/ml collagenase type I (Sigma Aldrich) with 10 μg/mL DNase I (Sigma Aldrich) for 1 hour at 37°C with shaking. Digested tumors were filtered through a 70 μm filter and labeled for MDSC and TAM markers. For in vitro labeling, 7 day IP RM-1 tumors were isolated as described and treated as previously described (10).
For in vivo imaging, 4T1 tumors were implanted on balb/c mice on folate deficient chow and monitored until the tumor reached 600 mm3. 10 nmols of OTL38 with and without 200-fold excess folic acid competition (folate-glucosamine mixed with the OTL38 as a single injection) were IV injected. 2 hours later, the mice were imaged with an AMI live imager (Spectral Imaging). The same mice were then euthanized and tumors harvested for flow cytometric quantification.
Suppression assay
MDSC and TAM T cell suppression assays were carried out in an antigen specific manner utilizing the OT-1 ovalbumin T cell model system as previously described (14). OT-1 splenocytes were treated with SIINFEKL peptide (Bachem 4033142) by plating 2×106 splenocytes per well in a 24 well plate in 2 mL of complete RPMI-1640 for 24 hrs. with 1 μg/mL of SIINFEKL peptide prior to the start of the suppression assay. Activated splenocytes were harvested by gradient centrifugation with Ficoll/Lite-LM (Atlanta Biologicals 140650) to isolate activated T cells. T cells were plated at a 1:2 MDSC to T cell ratio (5×104:1×105) for 18hrs under hypoxic or normoxic culture conditions in a 96 well plate. Antigen non-specific splenic assays were carried out by incubating splenic isolated MDSCs from a tumor bearing animal with splenocytes from a non-tumor bearing animal and plate bound CD3 (clone 145–2C11 ThermoFisher 14–0031-82) and soluble CD28 (clone CD82.2 ThermoFisher 16–0289-81) for 48 hrs. under hypoxic tension at 1:2 MDSC to splenocyte ratio in a 96 well plate. Antigen specific splenic suppression assays were performed by treating OT-1 splenocytes with 1 μg/mL SIINFEKL and co-culturing them at a 1:2 MDSC to splenocyte ratio with tumor isolated MDSCs for 48 hrs. under hypoxic tension. Suppression was determined by measuring T cell proliferation by EdU by following manufacturer instructions (ThermoFisher C10337). Percent suppression was calculated as (1 − proliferation with MDSC proliferation without MDSC) × 100.
FA-TLR7a and FA-PI3Ki therapy
Balb/c mice (6–8 weeks old) were implanted with 0.05 million 4T1 tumor (S.Q.) after two weeks of folate deficient diet. On day 4 post tumor implantation, animals were treated with folate-conjugated TLR7 agonist or folate-conjugated PI3K inhibitor (10 nmol/mouse for both compounds) (17). Folate conjugated treatments were compared with non-targeted TLR7 agonist or non-targeted PI3K inhibitor (10 nmol/mouse). Additionally, specificity of the folate targeting was tested by competition with (200x free folate-glucosamine) in combination with folate-conjugated TLR7 agonist or folate-conjugated PI3Ki (10 nmol/mouse). Treatments were carried out daily for 5 days per week (12 total days). The tumor growth and animal weight change were monitored during the study. For a survival study, treatment was extended to 5 weeks.
Metastatic colony assay
4T1 mammary fat pad tumors were treated starting on day 6 post implantation and continued for 14 days daily by IV injection of FA-TLR7 agonist, FA-PI3K inhibitor, TLR7 agonist, PI3K inhibitor (10 nmol/mouse). At the end of treatment, metastasis in lung was evaluated in disease control and treatment groups by co-culturing lung digested cells (digested using Collagenase IV, 10mL per lung with concentration of 1mg/mL to obtain single cell suspension) with 6-thioguanine (60 μM) in complete RPMI-1640 media for 10 days in petri plates. Metastatic colonies were visualized by fixing the plates with 5 mL of methanol followed by a 5 mL deionized water wash and staining with 0.03% methylene blue. Total blue colonies from each plate were then counted on a microscope.
Statistics
Statistical analysis was performed using a student’s t-test (paired, single tail analysis) with data represented as mean +/− SEM. Prism 6.0 (GraphPad) was used for all statistical analyses and graph generation. Survival studies were analyzed as a Kaplan-Meier plot with both a Mantel-Cox test and Gehan-Breslow-Wilcoxon test used. Significance is shown as ns P> 0.05, *P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, **** P ≤ 0.0001.
Results
Only MDSCs within a tumor mass express folate receptor beta
Previous studies from our lab demonstrated a functional difference between spleen and tumor resident MDSCs, with only the tumor resident MDSCs exhibiting immunosuppressive properties when immediately examined ex vivo (14). Based on this functional difference, we proceeded to compare gene expression patterns using a microarray comparing tumor resident MDSC subsets and splenic MDSCs (GSE116596). Over 3,000 genes were found to be differentially expressed between the two MDSCs subtypes, with folr2, the gene for FRβ among the most prominently upregulated in the tumor MDSCs. Since Folr2 was previously identified as a marker of activated macrophages (9,10), we hypothesized that it might also be a marker of immunosuppressive MDSCs and macrophages. We first verified the upregulation of folr2 in tumor-derived MDSCs compared to spleen MDSCs using qPCR and then also analyzed expression of the other isoforms of the folate receptor. As shown in Fig. 1A, folr2 was upregulated in tumor resident MDSCs of both subtypes, with minimal to no expression of other isoforms of the folate receptor.
Figure 1: Folate receptor beta is only expressed on tumor resident MDSCs and TAMs.

(A) qPCR analysis of tumor and spleen isolated MDSCs subsets, CD11b+ Ly-6G+ (G-MDSCs) and CD11b+ Ly-6C+ (M-MDSCs), from 7-day IP RM-1 tumor bearing animals. folr1, folr2, and folr4 were analyzed. Data from 5 mice pooled. (B) Prevalence of FRβ expression in MDSCs and TAMs isolated from various tumor cell lines. Solid tumor digests were stained in vitro with OTL38 (50 nM) to determine the proportion of the tumor comprised of FRβ expressing MDSCs (CD11b+ GR-1+) and TAMs (CD11b+ Ly-6C− F4/80+). Data are represented as percent FRβ+ MDSCs and TAMs within the total tumor mass.(n=2–5 mice per group) (C) Analysis of OTL38 delivery to a solid tumor. 4T1 tumors were grown to approximately 600 mm3 in size. OTL38 (50 nM) was injected IV with or without 200x folic acid (mixed with OTL38) to compete binding. Mice were then imaged after 2 hrs. to analyze uptake. (D) MDSCs and TAMs subset analysis from mice bearing solid 800 mm3 RM-1 tumors and treated by IV delivery of OTL38 (50 nM). OTL38 was delivered as described in panel C. Tumor-derived and spleen MDSCs subsets and TAMs were isolated and analyzed for uptake of OTL38. Red histogram is tumor OTL38 uptake, blue histogram is PBS injected control, and green histogram is spleen OTL38 uptake. (n=5 mice per group) (E) Quantification of flow data shown in panel D with percent uptake indicating percentage M-MDSCs and TAMs that labeled positively for OTL38. (F) Expression of FRβ in CD14+ cells in both cancerous lung tissue and patient matched peripheral blood samples. Mann-Whitney non-parametric test was used for quantification. (G) FRβ expression in renal carcinoma tissue. Determination of FRβ expression in human cancer. Formalin fixed, paraffin embedded tissue sections were processed for antigen retrieval and stained with anti-FRβ antibody (m909). Bladder cancer sections were stained with anti-CD11b antibody (brown stain, black arrows) and co-stained with anti-FRβ antibody (red stain, white arrows).
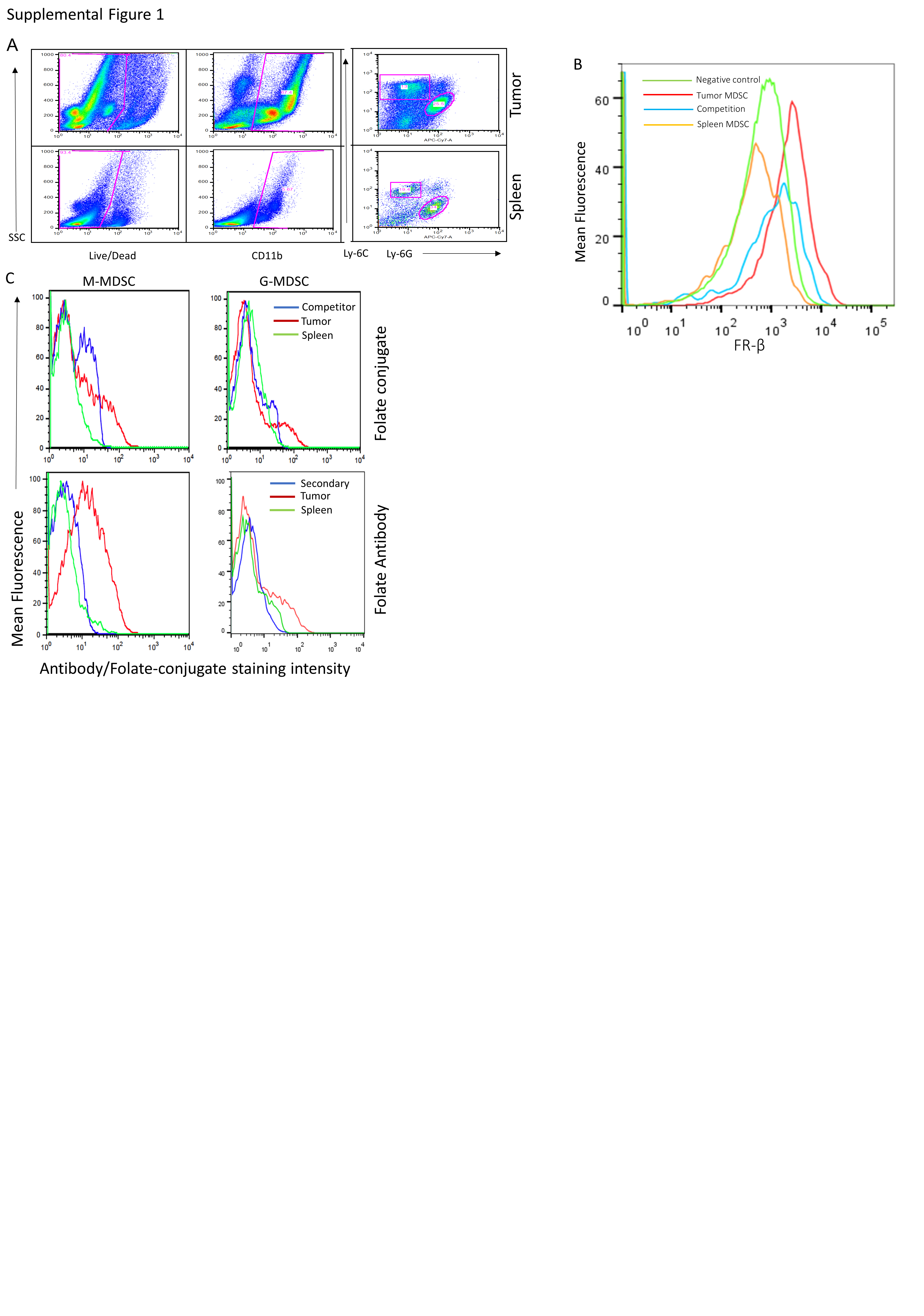
We next examined expression of FRβ in MDSCs and TAMs isolated from solid tumor grafts derived from a variety of murine tumor cell lines, including 4T1, EMT6, MB49, RM1, TRAMP-C2, Myc-CaP, and CT26. Previous studies demonstrated that folate-linked fluorescent dyes could be exploited to label FRβ-expressing myeloid cells in vivo that accumulate at sites of nonmalignant inflammation (10), suggesting that the same strategy might be used to identify tumor resident FRβ+ MDSCs and TAMs. For this purpose, subcutaneous murine tumors were digested to single cell suspensions and labeled with both OTL38 (a folate-targeted fluorescent dye (16)) and various markers for MDSCs and TAMs (SFig. 1A). 4T1 and CT26 tumors were found to contain the highest percentages of MDSCs (CD11b+ Ly-6C+ Ly-6G+ (GR-1)) expressing a functional FRβ, with 10–20% of all cells in the tumor mass staining positive for both OTL38 and MDSC markers (Fig. 1B). In the case of TAMs, the percentage of all tumor-derived cells that stained positive for both OTL38 and TAM markers (CD11b+, Ly-6C−, and F4/80+) was lower at 1–6%, with the highest observed in EMT6 and CT26 tumors (Fig 1B). Based on these data, we conclude that a substantial fraction of cells (up to 11–26% of total tumor cells) in murine tumor models constitute FRβ-expressing MDSCs and TAMs.
To characterize in vivo targeting of FRβ+ myeloid populations, we injected mice bearing an FR negative tumor (4T1) intravenously with OTL38 and examined the fluorescence of tumor resident MDSCs/TAMS. As seen in Fig. 1C, accumulation of OTL38 was only observed in solid tumors, with no uptake seen in peripheral tissues. Moreover, when folate receptors were blocked by injection of excess unlabeled folic acid (Fig. 1C), OTL38 uptake was prevented, demonstrating that accumulation of the folate-dye conjugate in the tumor was folate receptor-mediated. Moreover, following digestion of both tumors and spleens to release associated myeloid cells, FACS analysis revealed that OTL38 uptake was detected only in the tumor-derived MDSCs/TAMS and not the splenic-derived MDSCs or macrophages (SFig. 1B). These data demonstrate folate receptor expressing murine myeloid cells are localized to tumor tissue and not present in related myeloid cells in the spleen.
To further characterize the MDSC and TAM subsets that express a functional FRβ, folate receptor negative RM-1 tumor-bearing mice were treated with OTL38, after which tumors were harvested and analyzed for uptake of the folate-targeted dye by different subpopulations of MDSCs and TAMs. As shown in Fig. 1 panels D and E, although both M-and G-MDSCs displayed OTL38 uptake, OTL 38 accumulation was more prominent in M- than G-MDSCs. Expression of FRβ was then confirmed in the same cells by in vitro labeling with an anti-FRβ monoclonal antibody (SFig. 1C). Together these data demonstrate a dominant association of FRβ with murine the monocyte/macrophage-derived subtype of MDSCs and a subset of tumor-derived TAMs.
We next studied human myeloid cells for expression of FRβ. Previous work had demonstrated widespread distribution of FRβ-expressing histologically identified macrophages in human neoplastic tissues, however, no cell surface marker analysis was conducted to establish that the FRβ-positive cells were TAMs or MDSCs (18). To more clearly define the level of FRβ expression in human peripheral blood cells and solid tumors, a monocyte population defined by CD14 expression from peripheral blood and tumor tissue of lung adenocarcinoma patients was analyzed for FRβ expression. We observed a significant increase in FRβ expression in CD14+ cells isolated from cancerous lung tissue compared to CD14+ cells in the peripheral blood from the same patient (Fig. 1F). Previous reports on lung adenocarcinomas have indicated that these CD14+ cells are predominantly TAM like (19). These data taken together show a clear segregation of FRβ with MDSC/TAM populations that reside in tumor tissues and a decrement in FRβ expression among CD14+ cells found outside of the tumor microenvironment. To further characterize the presence of FRβ expression in TME myeloid cells, we co-stained tissue sections from human bladder cancers with antibodies to both human FRβ (20) and CD11b (a pan-myeloid marker) and examined their co-expression by immunohistochemistry and immunofluorescence. As shown in a comparison of the panels in Fig. 1G, cells that stained positive for FRβ also labeled prominently with anti-CD11b, demonstrating that the FRβ-expressing cells in bladder cancer also are members of the tumor-associated myeloid cell population.
Immunosuppressive activity resides in the FRβ+ subpopulations of M-MDSCs and TAMs under hypoxic conditions
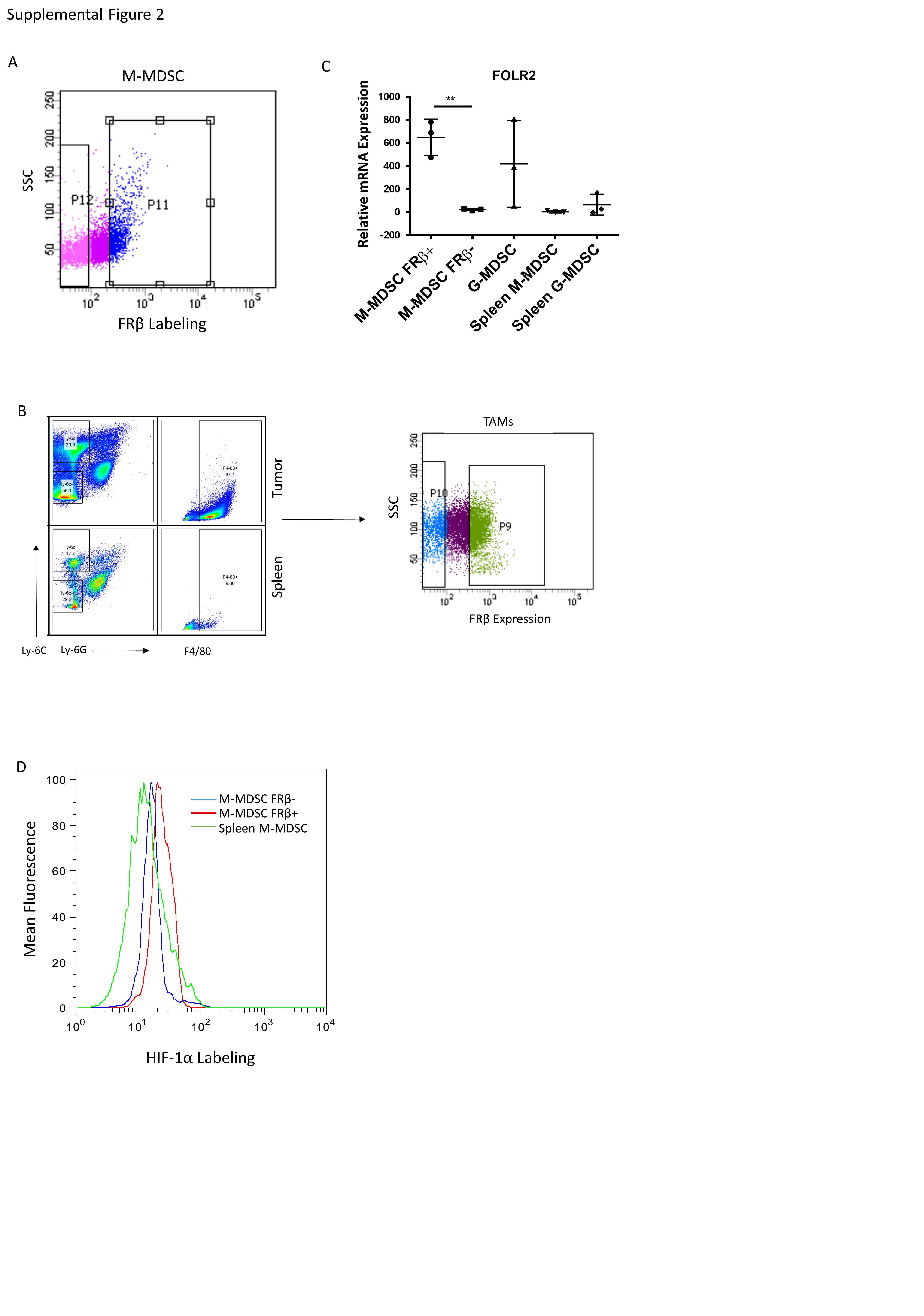
Since MDSCs and TAMs from healthy tissues (e.g. spleen, peripheral blood, etc.) express little or no FRβ (Figure 1) and because these same MDSCs and TAMs are not immunosuppressive (14), the question naturally arose whether FRβ might define the immunosuppressive subset of MDSCs and TAMs. To address this question, we set up an assay to compare the immunosuppressive properties of murine FRβ+ and FRβ− MDSCs and TAMs obtained from freshly isolated solid RM1 tumors (Sfig. 2 A and B). Moreover, because studies from other labs had shown that solid tumors are largely hypoxic (21) and that hypoxia is linked to the immunosuppressive properties of MDSCs and TAMs (22), we performed the comparative studies in the presence and absence of hypoxia. As shown in SFig. 2C, folr2 mRNA analysis confirmed a clean separation of FRβ+ from FRβ− populations (SFig. 2C). Importantly, under normoxic conditions, no discernible difference was observed between FRβ+ and FRβ− M-MDSC populations. However, under hypoxic conditions, the immunosuppressive function was observed to segregate entirely with the FRβ+ M-MDSC subset (Fig. 2A). FRβ+ TAMs were also found to be the highly immunosuppressive population under hypoxic conditions (Fig. 2B). These data suggest that FRβ identifies the critical immunosuppressive myeloid subpopulations within the hypoxic TME.
Figure 2: Suppressive function segregates with the FRβ+ populations.

(A and B) Analysis of suppression within FRβ positive and negative M-MDSCs and TAMs. M-MDSCs (A) and TAMs (B) were divided into FRβ+/− subsets from IP RM-1 tumors and tested in an 18 hr. suppression assay as described in panel A under either normoxic or hypoxic (1% O2) conditions. (n=5 mice per group pooled and data is representative of 4 independent experiments) (C and D) Determination of the hypoxic status of tumor-derived and spleen MDSCs and TAMs in tumor bearing mice. IP RM-1 tumor-bearing mice were treated with pimonidizole (60 mg/Kg of body mass by IP injection) 1.5 hrs. prior to harvest. Tumors were isolated and labeled with antibodies against MDSCs and TAM markers along with an antibody against pimonidizole. Under hypoxic conditions pimonidizole forms adducts in protein that can be detected via antibody. Red histogram is FRβ+ population, blue histogram is FRβ− population, and green histogram is spleen isolated population. (data representative of 5 mice)
To explore a possible association between hypoxia and FRβ expression in vivo, mice were injected with pimonidazole, a molecule that forms covalent adducts with proteins under highly hypoxic conditions (<1% O2) (23). Levels of FRβ and pimonidazole-protein adducts were compared by flow cytometry in freshly isolated M-MDSCs and TAMs. As shown in Figs. 2C and 2D, pimonidazole reactivity in vivo segregated with FRβ expression in both TAMs and M-MDSCs, with no hypoxia (or FRβ expression) detected in either splenic MDSCs or macrophages. Additionally, HIF-1α, which has been shown by others to be essential to the development of immunosuppressive MDSCs (22), segregated with FRβ expression in M-MDSCs (SFig. 2D). These data demonstrate that FRβ constitutes a unique marker that identifies the immunosuppressive state in MDSCs and TAMs under hypoxic tumor conditions.
Because PD-L1 expression has been implicated in the immunosuppressive properties of MDSCs and TAMs (24,25), particularly under hypoxia (25), we next undertook to investigate whether PD-L1 might correlate with FRβ expression. We first compared PD-L1 and FRβ expression by flow cytometry. As shown in Fig. 3A, we observed a clear co-segregation of PD-L1 expression with FRβ expression in both M-MDSCs and TAMs. PD-L1 and FRβ co-segregation was also prominent across all tumor types tested. (SFig. 3A). We then investigated the contribution of PD-L1 to the immunosuppressive activity of the tumor resident myeloid cells. For this purpose, OT-1 T cells (which we demonstrated express PD-1; SFig. 3B) were incubated with either FRβ+ or FRβ− M-MDSCs and TAMs and examined for immunosuppression in the presence and absence of an anti-PD-L1 antibody to block any contribution made by the PD-L1. As shown in Fig. 3B, the anti-PD-L1 antibody demonstrated no effect on tumor-derived TAM or MDSC suppression of OT-1 cell proliferation when compared to isotype antibody control. To verify the functionality of our reagents, conditions described in the original characterization of PD-L1 function in MDSCs were replicated (25). In these studies, spleen-derived MDSCs were tested for their ability to suppress anti-CD3/anti-CD28 activated T cells in an antigen non-specific 3 day assay. Although some PD-L1 mediated T cell suppression was observed, as evidenced by partial abrogation of suppression by anti-PD-L1 antibody, an equivalent reduction in suppression was also seen when treating with a nitric oxide inhibitor; i.e. another mechanism of MDSC suppression (SFig. 3C). This differential suppressive activity between antigen specific T cell activation and anti-CD3/anti-CD28 stimulation is consistent with previous reports (26).
Figure 3: M-MDSCs and TAMs depend on nitric oxide production for immunosuppression.

(A) Expression of PD-L1 in tumor-derived FRβ+/− M-MDSCs and TAMs. IP RM-1 tumor exudates were stained with MDSC markers, anti-FRβ antibody, and an anti-PD-L1 antibody. FRβ+ M-MDSCs (red), FRβ− (blue) M-MDSCs, and spleen CD11b+ Ly-6C+ cells (green) were analyzed. (Data representative of 5 mice) (B) Impact of anti-PD-L1 blocking of PD-L1 on suppression of CD8 T cell proliferation mediated by tumor-derived MDSCs and TAMs (calculated as described in Materials and Methods). M-MDSCs and TAMs were isolated from IP RM-1 tumor bearing mice and divided into FRβ+/− subsets. Those cells were then tested in an 18 hr. suppression assay with antigen activated (SIINFEKL peptide 1 μg/mL) activated OT-1 T cells as described in Materials and Methods. Samples were treated with anti-PD-L1 antibody (10F.9G2) and suppression was compared to untreated control. (n=5 mice per group pooled with data representative of 2 experiments) (C) Impact of inhibiting nitric oxide production by tumor-derived MDSCs and TAMs on suppression. L-NMMA (50 μM), an inhibitor of nitric oxide production, was tested in the same assay as described in panel B. (n=5 mice per group pooled with data representative of 2 experiments) (D) Nitric oxide production in FRβ positive and negative M-MDSCs and TAMs. Tumor-derived and spleen M-MDSCs and TAMs were treated with IFNγ for 24 hrs. after isolation from IP RM-1 tumor bearing mice. Cells were lysed by freezing and lysates were tested with for nitric oxide production by Griess reagent. (n=5 mice per group pooled with data representative of 2 experiments)
Based on previous observations that i) TAM/MDSC-induced suppression of T cell activity can derive from their release of NO and the consequent nitrosylation of the T cell receptor (27), ii) HIF-1α expression is required for induction of iNOS and its synthesis of NO in MDSCs (22), and iii) immunosuppressive mechanisms vary based on location within the tumor and hypoxia (28), we explored whether TAM/MDSC-derived NO might constitute a prominent mediator of tumor myeloid cell immunosuppression. For this purpose, we performed the suppression assay described above in the presence of L-NMMA, a potent inhibitor of iNOS (29) to directly assess whether FRβ+ M-MDSCs or TAMs might rely on NO production for their immunosuppressive functions. Remarkably, inhibition with L-NMMA under hypoxic conditions completely abrogated M-MDSC and TAM immunosuppression of T cells (Fig. 3C), suggesting that NO production constitutes a major mechanism through which MDSCs and TAMs exert their immunosuppressive activities. Not surprisingly, under both hypoxic and normoxic conditions, NO production was significantly higher in FRβ+ than FRβ− M-MDSCs and TAMs (Fig. 3D). These data document the use of at least two distinct immunosuppressive mechanisms by MDSCs and help explain why immune checkpoint inhibitors alone can be insufficient to control them.
Developing Therapeutic Approaches by Targeting FRβ+ Myeloid Cells.
Preliminary studies showed that FRβ was also expressed on MDSCs and TAMs (Fig. 4A) from FR− MB49 solid tumors (SFig. 4) and that depletion of these FRβ+ MDSCs and TAMs using a folate-targeted photodynamic therapy agent eliminated all FRβ+ myeloid cells, but that full replenishment of FRβ+ MDSCs and TAMs was observed within 3 days (Fig. 4B) and that a rebound of tumor growth then followed (Fig. 4C). Finally, no other significant difference was detected in tumor resident immune populations (Fig. 4D). Because of this rapid replenishment of tumor-infiltrating myeloid cells, we concluded that eradication of tumor-associated myeloid cells was not likely to provide a sustained alteration of the TME. Thus, alternative approaches were pursued.
Figure 4: Targeting M-MDSCs and TAMs via FRβ depletes tumor resident populations.

(A) Characterization of FRβ expression in MB49 solid tumors. MB49 IP tumors were generated by implanting 1×106 MB49 cells in the IP cavity and harvesting 7 days later. IP and splenic MDSCs and TAMs were analyzed for FRβ expression via antibody. Red histogram is tumor resident populations, green histogram is unstained, and blue histogram is splenic cells. (n=5 mice per group) (B) Depletion of M-MDSCs and TAMs from solid tumors by FRβ targeted photodynamic therapy. Intradermal MB49 tumors (2.5×106 cells per mouse) were grown for 10–14 days. At this point, 40 nmol of PG05132 photodynamic therapy compound was delivered to tumor bearing animals via IV injection. The compound was allowed to circulate for 1.5 hrs. at which time the mice were treated at the tumor site with a 400 mA 750 nm laser for 30 min. Tumors were harvested at 48 and 72 hrs. and the presence of analyze M-MDSCs and TAMs were analyzed as described in Materials and Methods. Treated mice were compared to PBS injected mice that were treated with the laser. (n=3 mice per group) (C) MB49 tumors were grown treated with PDT compound and laser as described in panel B. After an initial treatment, tumors were measured for 8 days post treatment with PDT treated mice compared to PBS injected mice treated with the laser. (n=8–9 mice per group). (D) Flow cytometric quantification of tumor resident CD8+, CD4+, and B220+ from PDT treated and control tumors. Populations were gated under viable cell and CD45+ markers in addition to specific subset markers.
Antitumor activity of myeloid specific delivery of TLR7 agonist and PI3K inhibitor.
To test the antitumor activities of the above drugs, an orthotopic 4T1 FR− tumor model in BALB/c mice was employed because it allowed determination of therapeutic activity on both primary and metastatic tumors. Additionally, all cell lines used in these studies were characterized for uptake of OTL38 which will only bind a functional FR uptake of folate conjugates by OTL38 which will only bind a functional FR (30). All cell lines showed little to no uptake of folate conjugates compared to LPS activated RAW 264.7 macrophages (SFig. 4). As shown in Fig. 5A, the folate-TLR7a conjugate was indeed able to retard tumor growth in a sustained manner. In contrast, the non-targeted TLR7a was unable to control tumor growth and was furthermore highly toxic compared to FA-TLR7a (Fig. 5A and B) and accompanied by a substantial increase in systemic cytokine production (SFig. 5A-C). The absence of toxicity of FA-TLR7 was consistent with the ability of folate to concentrate its attached cargo in FRβ+ cells and thereby avoid uptake by FR negative cell types, resulting in no detectable increase in systemic inflammatory cytokines (SFig. 5A-C). Moreover, when TAM and MDSC folate receptors were blocked with excess folic acid, the anti-tumor activity of folate-TLR7a was abrogated, confirming that the potency of folate-TLR7a conjugate was dependent on FRβ-mediated uptake (Fig. 5A). These data are consistent with previous reports that systemic TLR7 administration, while effective at controlling tumor growth through immune modulation, is ultimately toxic and not appropriate for systemic therapy (31,32).
Figure 5: M-MDSCs and TAMs targeted by folate delivered toll-like receptor 7 agonist and phosphoinositide 3-kinase inhibitor controls tumor growth and metastatic development.

(A and B) Inhibition of tumor growth by folate mediated delivery of TLR7a (FA-TLR7a) and PI3Ki (FA-PI3Ki) to MDSCs and TAMs. Balb/c mice (6–8 weeks old) were implanted with 0.05 million 4T1 tumor (Subcutaneous). On day 4 post tumor implantation, animals were treated with folate-conjugated TLR7 agonist (10 nmol/mouse), or non-targeted TLR7 agonist (10 nmol/mouse), or competition (200x free folate) with folate-conjugated TLR7 agonist (10 nmol/mouse) daily for 5 days per week. The tumor growth (A) and animal weight change (B) were monitored during the study. (C and D) Control of tumor growth by FRβ targeted reprogramming of MDSCs and TAMs by FA-PI3Ki. Balb/c mice (6–8 weeks old) were implanted with 0.05 million 4T1 tumor (subcutaneous). On day 4 post tumor implantation, animals were treated with folate-conjugated PI3K inhibitor (10 nmol/mouse), or non-targeted PI3K inhibitor (10 nmol/mouse), or competition (200x free folate) with Folate-conjugated PI3K inhibitor (10 nmol/mouse) daily for 5 days per week. The tumor growth (C) and animal weight change (D) were monitored during the study. (E and F) Impact of FA-TLR7a and FA-PI3Ki metastasis in 4T1 tumor bearing mice. 0.05 million 4T1 cells were implanted in the fat pad in 6–8-week-old balb/c mice (n=3). Treatment was started at day 6 post implantation and was continued for 14 days daily (IV). FA-TLR7 agonist, FA-PI3K inhibitor, TLR7 agonist, PI3K inhibitor (10 nmol/mouse) (defined in panels A and B). At the end of in vivo study, metastasis to the lung was evaluated in disease control and treatment groups by co-culture lung digested cells with 6-thioguanine (60 μM) for 14 days as described in methods (E and F). Data are compiled from two independent experiments with 4–5 mice per group.
We next tested FA-PI3Ki for antitumor activity. As seen in Figs. 5C and D, while some suppression of tumor growth was observed following FA-PI3Ki administration, the anti-tumor activity of the conjugate was not as potent as that seen with FA-TLR7a, suggesting that conversion of TAMs/MDSCs to a more pro-inflammatory phenotype may be more effective than simply inhibiting their immunosuppressive activities with a PI3K inhibitor.
Finally, because many publications have reported that TAMs and MDSCs participate intimately in the metastatic process (33,34), we tested whether FA-TLR7a or FA-PI3Ki might inhibit the development of lung metastasis of orthotopic 4T1 tumors. For this purpose, 4T1 syngeneic breast cancer cells were implanted into mammary fat pads of BALB/c mice (35) and beginning on day 4 post-implantation, mice were injected intravenously 5x/week with either FA-TLR7a, FA-PI3Ki or saline. When analyzed on day 18, few if any lung metastases were detectable in mice treated with either FA-PI3Ki or FA-TLR7a (Fig. 5E), whereas lung metastases in mice treated with non-targeted PI3Ki or TLR7a were similar in number to saline-treated controls (Fig. 5F). Because folate targeting is specific for TAMs and MDSCs, these data support the hypothesis that FRβ+ TAMs and/or MDSCs play a pivotal role in the mechanism of metastasis. Moreover, because neither FA-TLR7a nor FA-PI3Ki has any effect on the growth of 4T1 cells in culture, even at concentrations 1000x higher than those used this study (SFig. 5D and E), and since the 4T1 cancer cells express no folate receptors, one can conclude that these anti-metastatic effects do not stem from a direct effect on cancer cells, but rather derive from modulation of FRβ-expressing MDSCs and TAMs.
Repolarization of M-MDSCs and TAMs generates a pro-inflammatory tumoricidal phenotype
In an effort to find a more durable approach for suppressing FRβ+ MDSC/TAM activity, we explored the use of folic acid (FA) to target either an attached toll-like receptor 7 agonist (TLR7a) or a PI3K inhibitor (PI3Ki) selectively to the FRβ+ myeloid cells in the TME to determine whether either drug could reprogram the TAMs/MDSCs from a tissue regenerating/immunosuppressive phenotype to a tumoricidal M1-like phenotype (36). For this purpose, we examined the effects of systemic administration of the above FR-targeted drugs on the numbers and phenotypes of the FRβ+ MDSCs and TAMs isolated from FR negative 4T1 solid tumors 2 weeks following initiation of therapy. As shown in Fig. 6A, treatment with FA-TLR7a reduced the percentage of cells with TAM markers by more than half, while administration of FA-PI3Ki decreased TAM numbers by 40%. Reduction in MDSC numbers was also >50% upon exposure to TLR7a, but surprisingly MDSC numbers were not significantly affected by PI3Ki (Fig. 6B). Analysis of the pro- and anti-inflammatory markers within the TAM population (Fig. 6C) revealed a significant shift from an M2 towards M1 phenotype following treatment with either FA-TLR7a or FA-PI3Ki, and quantitation of CD4 and 8+ T cell numbers displayed a nearly tenfold increase after treatment with either drug conjugate (Fig. 6D and E).
Figure 6: Repolarization of tumor associated M-MDSCs and TAMs observed after treatment TLR7a and PI3KI.

Determination of changes in intratumoral myeloid populations by in Balb/c mice (6–8 weeks old) that were implanted with 0.05 million 4T1 tumor (subcutaneous). On day 4 post tumor implantation, animals were treated with folate-conjugated (FA-TLR7A or FA-PI3KI) (10 nmol/mouse) daily for 2 weeks. After the animals were euthanized, the tumor was digested to analyze MDSCs, TAMs, and T cell populations. (A and B) MDSCs (A) and TAMs (B) percentage in tumor digests. (C) The ratio of M1 and M2 macrophages in tumor digests as determined by the ratio of CD86+ (M1) and CD206 (M2) percentages within total F4/80+ macrophages. (D) Percentage of CD4+ T cells in total CD3 T cell population from digested tumor cells. (E) Percentage of CD8+ T cells in total CD3 T cell population from digested tumor cells. (F) G-CSF levels in plasma at day 12 post tumor implantation.
Phenotypic changes in MDSCs and TAMs after in vitro treatment with reprogramming compounds. MDSCs/TAMs isolated from ascites of Balb/c mice (6–8 weeks old) implanted with 10 million 4T1 tumor (IP) were incubated with 1 μM TLR7 agonist or 1 μM PI3K inhibitor for 48 hours. (G) TGF-β concentrations. (H) CD86 expression on MDSCs measured by flow cytometry. (I) T cell suppression represented by percent positive CD8+IFNγ as compared to control T cells.
Data are compiled from two independent experiments with 4–5 mice per group.
In a separate series of experiments, measurement of plasma cytokine levels in the tumor-bearing mice demonstrated that both FA-TLR7a and FA-PI3Ki significantly reduced plasma concentrations of G-CSF (Fig. 6F). Moreover, when MDSCs and TAMs were isolated from the ascites fluid of mice bearing untreated 4T1 peritoneal tumors and subsequently incubated in vitro with TLR7a or PI3Ki, TGFβ production by MDSCs was found to decrease upon treatment with TLR7a but not PI3Ki (Fig. 6G). Moreover, an increase in CD86 expression on the tumor MDSC population treated with FA-TLR7a suggested that the therapy may have induced maturation of this immature population of myeloid cells (Fig. 6H). Finally, analysis of the abilities of the aforementioned drugs to inhibit the suppression of CD8 T cell expression of IFNγ by MDSCs (Fig. 6I) revealed that the FA-TLR7a was able to reverse their inhibitory effects, whereas PI3Ki had no significant effect (Fig. 6I). Collectively, these data demonstrate the ability of a folate-targeted TLR7 agonist or PI3K inhibitor to repolarize the FRβ+ myeloid cells towards a more pro-inflammatory phenotype.
Finally, combining the observations that FA-TLR7 therapy was able to control tumor growth, prevent metastatic development, and drive a pro-inflammatory phenotype in both MDSCs and TAMs, we tested the impact of FA-TRL7a in a longitudinal survival study using the above 4T1 mammary carcinoma model. Orthotopic 4T1 tumors were implanted as described in Fig. 5, but treatment was continued for 5 weeks using the same dosing schedule. Mice were euthanized when tumor size exceeded 2 cm in any direction or became ulcerated. As shown in Fig. 7A, median survival of mice treated with FA-TLR7a was 37 days whereas median survival for saline-treated mice was only 27 days. Increased CD8+ T-cell infiltration into the tumor mass was also observed in treated mice (Fig. 7B). Collectively, these data demonstrate that selective treatment of FRβ+ MDSCs and TAMs with folate-targeted repolarizing/inactivating drugs can reprogram the TME leading to retardation of tumor growth and enhancement of animal lifespan.
Figure 7: Enhanced survival of orthotopic 4T1 tumor bearing mice with FA-TLR7 treatment.

(A) 4T1 Orthotopic tumors were established as described in Fig. 5 and treated with 10 nmol/mouse daily with FA-TLR7 by IV injection (5 days on 2 days off) for 5 weeks total. Mice were monitored for tumors that reached 2 cm in any direction or open ulcerated tumors. (B) Histology sections from each tumor were analyzed at the time of harvest for CD8+ infiltration by counting 10 fields of the tumor section at 20x magnification. Total counts were quantified from each FA-TLR7a treated tumor and each control tumor.
Discussion
Studies reported above support several major conclusions regarding MDSC and TAM biology; namely that i) FRβ expression is predominantly restricted to MDSCs and TAMs in the TME for both human and murine tumors, ii) FRβ expression identifies the functionally suppressive subpopulations of MDSCs and TAMs, and iii) targeting TAMs and MDSCs in the TME with potent immune modulating compounds is safe, reprograms TAM and MDSC phenotypes, and induces antitumor activity.
Identification of FRβ as a marker for immunosuppressive MDSCs and TAMs is important to tumor biology because it provides a tool for isolating, modifying, and characterizing this functionally significant subset of myeloid cells. Because the customary markers for MDSCs and TAMs have typically been the markers used for characterization of general myeloid populations, identification of a cell as a suppressive MDSC or TAM has historically required an additional test to confirm functionality (8). While FRβ has been previously documented as a marker for activated macrophages (9), its significance as a marker of immunosuppressive myeloid cells in the TME was never established. The data herein support the idea that FRβ can be used to both isolate MDSCs and TAMs for study in vitro and modify their behavior for analysis of their interactions with other tumor cells in vivo.
One of the most accepted markers for M2 macrophages is Arginase 1, an enzyme that metabolizes the arginine required for NO production (37). Not surprisingly, upregulation of Arg1 in M2 macrophages has led to the dogma that M2-like macrophages do not synthesize NO (38). However, as shown in Fig. 3, our data demonstrate that M2-like M-MDSCs and TAMs rely on NO production to perform their immunosuppressive function. Indeed, Massi et al. have demonstrated that macrophages in melanoma tissues are induced to generate NO rather than Arg1 (39). Nagaraj et al. (27) have further shown that this NO inactivates T cells via nitrosylation of the T cell receptor, and Bingisser and colleagues (40) have added that macrophage-derived NO also suppresses T cell activation via reversible disruption of Jak3/STAT5 signaling. Riquelme et al. (41) have supported this concept by demonstrating that prolonged survival of tissue allografts requires direct immunosuppression of T cells by macrophage-generated NO. Collectively these data establish that TAMs and MDSCs in the tumor microenvironment can employ NO as a means to regulate T cell activity.
A second major conclusion of this paper is that folate receptor targeting can be exploited to deliver folate-conjugated drugs specifically to FRβ+ tumor-associated myeloid cells without delivery of compound to cell types that will not be directly impacted. There are mixed data on the efficacy of delivery of TLR7 agonists to epithelial cells with the majority of TLR7/8/9 expression restricted to the immune compartment and stroma of a tumor (31,42). Not only was FRβ shown to be highly upregulated on tumor site M-MDSCs and TAMs, but the activities of these cells were also found to be controllable through FRβ-mediated internalization of immunomodulatory folate-drug conjugates, while the tumor lines used in these studies were found to be insensitive to the folate conjugates. Although PI3K inhibitors were already reported to enhance checkpoint therapy through suppression of MDSCs (43), we have shown here that a TLR7 agonist can constitute an even more potent suppressor of M-MDSC and TAM functions. Thus, despite a known role for PI3Ki in the elimination of MDSCs (44), our study suggests that TLR7a-mediated reprogramming of these myeloid cells may be the preferred option (Fig. 6B). Together with data from Lu et al., 2017 (44), our study makes a clear case for design of methods to control MDSCs at the tumor site and suggests that exploration of additional approaches to regulating MDSC properties may lead to more universal strategies for treatment of solid tumors.
It is important to note that TLR7 agonists are not new additions to the immunotherapy toolbox. Thus, TLR7 agonists currently hold FDA approval as topical treatments for cutaneous lymphomas, basal cell carcinomas, actinic keratoses, and other skin lesions (45). Unfortunately, due to their potent ability to activate the immune system, TLR7 agonists have invariably proven to be too toxic for systemic administration. Even topical absorption of imiquimod through the skin is sufficient to generate flu-like symptoms including fever, fatigue and headaches (46). Our data on systemic administration of free/nontargeted TLR7 agonist confirmed the drug to be highly toxic as previously reported by others (47). Also, studies that have explored systemic delivery of TLR7a, such as, Nishii et al. have done so using much smaller doses (5 nmol), less frequent administrations and less potent agonists than we have employed in this study (48). Therefore, the ability to target highly effective but toxic compounds like TLR7 agonists solely to cells whose reprogramming would improve tumor outcomes could enable novel strategies for training the immune system to suppress tumor growth. Drugs that would otherwise have severe side effects can now be repurposed for use through folate receptor-mediated targeting.
The question of whether it would be more effective to destroy or reprogram a tumor’s MDSCs and TAMs was also addressed in this study. Thus, when MDSCs and TAMs were depleted from tumors by folate-targeted photodynamic therapy, a rebound in the MDSC and TAM populations was observed within 72 hours, and this rebound was accompanied by a recovery in the growth rate of the tumor. These data unfortunately suggest that the ability of a tumor to recruit new myeloid populations may outpace the ability of a therapy to eradicate them. Repolarization of the myeloid cells, however, was found to generate a more lasting change in TME myeloid cell phenotype that was not rapidly overcome by recruitment of new myeloid cell populations. Moreover, repolarization induced a significant increase in CD8 T cell infiltration into the tumor whereas depletion with photodynamic therapy had no significant effect on CD8 T cell infiltration into the tumor. Multiple laboratories have studied blocking or depleting MDSCs to enhance adoptive T cell therapies, but repolarization of existing myeloid populations to enhance immunotherapies has remained largely unexplored until now (49).
Remarkably, despite a correlative link between FRβ expression and M-MDSC and TAM function, it remains unclear if FRβ is required for immunosuppression or if the receptor simply segregates with the suppressive population. In endothelial cells, folate and tetrahydrobiopterin are required for the production of NO (50), but this requirement has not been demonstrated in MDSCs or TAMs. We showed that FRβ marks the sole immunosuppressive subpopulations of M-MDSCs and TAMs under hypoxic conditions and that their immunosuppression is dependent on NO production. These observations fit with the initial Corzo et al. observation on hypoxia and NO production (19). While we did not see an impact on T cell suppression when PD-L1 was blocked during a suppression assay under antigen specific conditions, we did see an impact of anti-PD-L1 when assaying suppression of anti-CD3/anti-CD28 activated T cells. These data suggest the activation of different immunosuppression mechanisms depending on antigen specific vs non-specific conditions. Nagaraj et al. demonstrated that MDSCs inhibit T cells in a primarily antigen specific manner (27). However, not all studies utilize the same suppression assay conditions and a mix of antigen-specific vs. non-specific assays are used. Together, these data suggest that careful attention to suppression assay conditions is necessary due to the potential for different suppressive mechanisms under different immunological conditions.
In conclusion, we have demonstrated that FRβ constitutes a highly specific marker of immunosuppressive MDSCs and TAMs that can be exploited to image, isolate, and manipulate the functional properties of MDSCs/TAMs in the TME. Because MDSCs and TAMs are not mutating like cancer cells and since MDSCs and TAMs contribute prominently to the properties of nearly all solid tumors, use of folate-targeted drugs to reprogram tumor MDSCs and TAMs could enable new approaches to controlling the immune environment in many cancer types.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Significance.
FRβ serves as both a means to identify and target MDSC and TAM within the tumor, allowing for delivery of immunomodulatory compounds to tumor myeloid cells in a variety of cancers.
Acknowledgements
Funding:
Endocyte 40001138 P. Low, Purdue University Center for Cancer Research T. Ratliff, RO1 DK084454 T. Ratliff
Conception and Design: G. Cresswell, B. Wang, T. L. Ratliff, and P. Low
Development and Methodology: G. Cresswell, B. Wang, T. L. Ratliff, P. Low, E. Kischuk, and R. A. Alfar, M. Broman
Acquisition of Data: G. Cresswell, B. Wang, E. Kischuk, R. A. Alfar, R. Vickman, M. Broman, E. Eruslanov
Analysis and Interpretation of Data: G. Cresswell, B. Wang, T. L. Ratliff, P. Low, R. A. Alfar, E. Kischuk, and M. Broman, B. D. Elzey, and S. Crist
Writing of Manuscript: G. Cresswell, B. Wang, T. L. Ratliff, and P. Low
Study Supervision: P. Low and T. L. Ratliff
Other (reagents and tissue collection): D. S. Dimitrov and S. Kularatne, C. Sundaram, S. Singhal
We would like to thank the Purdue University Flow Cytometry and Cell Separation Facility, NIH grant P30 CA023168 and the NIH Shared Instrumentation Grant S10DO20029 for carrying out live cell sorting and coordinating use of instrumentation. Dr. L. Tiffany Lyle and Victor A. Bernal-Crespo of the Purdue University Histology Research Laboratory for processing, sectioning, and staining of all immunohistochemistry samples. Veronika Slivova and Alexandra Gol-Chambers of the Indiana University Health Biorepository for collection of human tissue specimens from the IU Health Methodist Hospital at Indianapolis, IN. On Target Laboratories for providing OTL38 compound. Endocyte for providing folate-conjugated drug compounds, FA-TLR7a and FA-PI3Ki.
Footnotes
Competing Interests: Dr. Low received funding from Endocyte under supervision of Purdue University.
References
- 1.Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol 2016;37:208–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ozkan B, Lim H, Park SG. Immunomodulatory Function of Myeloid-Derived Suppressor Cells during B Cell-Mediated Immune Responses. Int J Mol Sci 2018;19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tartour E, Pere H, Maillere B, Terme M, Merillon N, Taieb J, et al. Angiogenesis and immunity: a bidirectional link potentially relevant for the monitoring of antiangiogenic therapy and the development of novel therapeutic combination with immunotherapy. Cancer and Metastasis Reviews 2011;30:83–95 [DOI] [PubMed] [Google Scholar]
- 4.Deng J, Liu Y, Lee H, Herrmann A, Zhang W, Zhang C, et al. S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell 2012;21:642–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature 2016;539:443–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alberto Carretero-González DC, Sepúlveda Juan M., Lora David, Ghanem Ismael, López-Martin José A., Zugazagoitia Jon, de Velasco Luis Paz-Ares Guillermo. Analysis of response rate with ANTI PD1/PD-L1 monoclonal antibodies in advanced solid tumors: a meta-analysis of radnomized clinical trials. Oncotarget 2018;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest 2015;125:3365–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 2016;7:12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puig-Kroger A, Sierra-Filardi E, Dominguez-Soto A, Samaniego R, Corcuera MT, Gomez-Aguado F, et al. Folate Receptor beta Is Expressed by Tumor-Associated Macrophages and Constitutes a Marker for M2 Anti-inflammatory/Regulatory Macrophages. Cancer Research 2009;69:9395–403 [DOI] [PubMed] [Google Scholar]
- 10.Xia W, Hilgenbrink AR, Matteson EL, Lockwood MB, Cheng JX, Low PS. A functional folate receptor is induced during macrophage activation and can be used to target drugs to activated macrophages. Blood 2008;113:438–46 [DOI] [PubMed] [Google Scholar]
- 11.Vu-Quang H, Vinding MS, Jakobsen M, Song P, Dagnaes-Hansen F, Nielsen NC, et al. Imaging Rheumatoid Arthritis in Mice Using Combined Near Infrared and (19)F Magnetic Resonance Modalities. Sci Rep 2019;9:14314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elo P, Li XG, Liljenback H, Helin S, Teuho J, Koskensalo K, et al. Folate receptor-targeted positron emission tomography of experimental autoimmune encephalomyelitis in rats. J Neuroinflammation 2019;16:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu Y, Wang B, Shen J, Low SA, Putt KS, Niessen HWM, et al. Depletion of activated macrophages with a folate receptor-beta-specific antibody improves symptoms in mouse models of rheumatoid arthritis. Arthritis Res Ther 2019;21:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haverkamp JM, Crist SA, Elzey BD, Cimen C, Ratliff TL. In vivo suppressive function of myeloid-derived suppressor cells is limited to the inflammatory site. European Journal of Immunology 2011;41:749–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quatromoni JG, Singhal S, Bhojnagarwala P, Hancock WW, Albelda SM, Eruslanov E. An optimized disaggregation method for human lung tumors that preserves the phenotype and function of the immune cells. J Leukoc Biol 2015;97:201–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahalingam SM, Kularatne SA, Myers CH, Gagare P, Norshi M, Liu X, et al. Evaluation of Novel Tumor-Targeted Near-Infrared Probe for Fluorescence-Guided Surgery of Cancer. J Med Chem 2018;61:9637–46 [DOI] [PubMed] [Google Scholar]
- 17.Low PS, Wang Bingbing, Leamon Christopher Paul, Lu Yingjuan J. ; Method of Treating Cancer by Targeting Myeloid-Derived Suppressor Cells 2017. [Google Scholar]
- 18.Jiayin Shen KSP, Daniel W. Visscher Linda Murphy, Cynthia Cohen, Sunil Singhal, George Sandusky, Yang Feng, Dimitrov Dimiter S., and Low Phillip S.. Assessment of folate receptor-beta expression in human neoplastic tissues. Oncotarget 2015;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sunil Singhal JS, Annunziata Michael J., Rao Abhishek S., Bhojnagarwala Pratik S., O’Brien Shaun, Moon Edmund K., Cantu Edward, Danet-Desnoyers Gwenn, Ra Hyun-Jeong, Litzky Leslie, Akimova Tatiana, Beier Ulf H., Hancock Wayne W., Albelda Steven M., Eruslanov Evgeniy B.. Human tumor-associated monocytes/macrophages and their regulation of T cell responses in early-stage lung cancer. Science Translational Medicine 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng Y, Shen J, Streaker ED, Lockwood M, Zhu Z, Low PS, et al. A folate receptor beta-specific human monoclonal antibody recognizes activated macrophage of rheumatoid patients and mediates antibody-dependent cell-mediated cytotoxicity. Arthritis Res Ther 2011;13:R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rademakers SE, Lok J, van der Kogel AJ, Bussink J, Kaanders JH. Metabolic markers in relation to hypoxia; staining patterns and colocalization of pimonidazole, HIF-1alpha, CAIX, LDH-5, GLUT-1, MCT1 and MCT4. BMC Cancer 2011;11:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med 2010;207:2439–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jankovic B, Aquino-Parsons C, Raleigh JA, Stanbridge EJ, Durand RE, Banath JP, et al. Comparison between pimonidazole binding, oxygen electrode measurements, and expression of endogenous hypoxia markers in cancer of the uterine cervix. Cytometry B Clin Cytom 2006;70:45–55 [DOI] [PubMed] [Google Scholar]
- 24.Gottlieb CE, Mills AM, Cross JV, Ring KL. Tumor-associated macrophage expression of PD-L1 in implants of high grade serous ovarian carcinoma: A comparison of matched primary and metastatic tumors. Gynecol Oncol 2017;144:607–12 [DOI] [PubMed] [Google Scholar]
- 25.Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med 2014;211:781–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marcin Kowanetz XW, Lee John, Tan Martha, Hagenbeek Thijs, Qu Xueping, Yu Lanlan, Ross Jed,, TC Nina Korsisaari, Bou-Reslan Hani, Kallop Dara, Weimer Robby, Ludlam Mary J. C., Kaminker Joshua S., Modrusan Zora, van Bruggen Nicholas, Peale Franklin V., Carano Richard, Meng Y. Gloria, and Ferrara1 Napoleone. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci U S A 2010;107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagaraj S, Schrum AG, Cho HI, Celis E, Gabrilovich DI. Mechanism of T Cell Tolerance Induced by Myeloid-Derived Suppressor Cells. The Journal of Immunology 2010;184:3106–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang M, McKay D, Pollard JW, Lewis CE. Diverse Functions of Macrophages in Different Tumor Microenvironments. Cancer Res 2018;78:5492–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.RMJ D.D. Rees, Palmer R. Schulz, Hodson HF & Moncada S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br J Pharmacol 1990;101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho SS, Jeon J, Buch L, Nag S, Nasrallah M, Low PS, et al. Intraoperative near-infrared imaging with receptor-specific versus passive delivery of fluorescent agents in pituitary adenomas. J Neurosurg 2018;131:1974–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mullins SR, Vasilakos JP, Deschler K, Grigsby I, Gillis P, John J, et al. Intratumoral immunotherapy with TLR7/8 agonist MEDI9197 modulates the tumor microenvironment leading to enhanced activity when combined with other immunotherapies. J Immunother Cancer 2019;7:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Michaelis KA, Norgard MA, Zhu X, Levasseur PR, Sivagnanam S, Liudahl SM, et al. The TLR7/8 agonist R848 remodels tumor and host responses to promote survival in pancreatic cancer. Nat Commun 2019;10:4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chafe SC, Lou Y, Sceneay J, Vallejo M, Hamilton MJ, McDonald PC, et al. Carbonic anhydrase IX promotes myeloid-derived suppressor cell mobilization and establishment of a metastatic niche by stimulating G-CSF production. Cancer Res 2015;75:996–1008 [DOI] [PubMed] [Google Scholar]
- 34.Zhang W, Zhu XD, Sun HC, Xiong YQ, Zhuang PY, Xu HX, et al. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin Cancer Res 2010;16:3420–30 [DOI] [PubMed] [Google Scholar]
- 35.DuPre SA, Redelman D, Hunter KW Jr. The mouse mammary carcinoma 4T1: characterization of the cellular landscape of primary tumours and metastatic tumour foci. Int J Exp Pathol 2007;88:351–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.SPA Christopher B. Rodell, Cuccarese Michael F., Garris Christopher S., Li Ran, Ahmed Maaz S., Kohler Rainer H., Pittet Mikael J., and Weissleder Ralph. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumor-associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng 2018;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murray PJ. Macrophage Polarization. Annu Rev Physiol 2017;79:541–66 [DOI] [PubMed] [Google Scholar]
- 38.Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado Jde D, Popovich PG, Partida-Sanchez S, et al. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS One 2015;10:e0145342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Massi D, Marconi C, Franchi A, Bianchini F, Paglierani M, Ketabchi S, et al. Arginine metabolism in tumor-associated macrophages in cutaneous malignant melanoma: evidence from human and experimental tumors. Hum Pathol 2007;38:1516–25 [DOI] [PubMed] [Google Scholar]
- 40.PAT Roland M. Bingisser, Holt Patrick G. and Kees Ursula R.. Macrophage-Derived Nitric Oxide Regulates T-Cell Activation via Reversible Disruption of the Jak3/STAT5 Signaling Pathway. The Journal of Immunology 1998 [PubMed] [Google Scholar]
- 41.Riquelme P, Tomiuk S, Kammler A, Fandrich F, Schlitt HJ, Geissler EK, et al. IFN-gamma-induced iNOS expression in mouse regulatory macrophages prolongs allograft survival in fully immunocompetent recipients. Mol Ther 2013;21:409–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petes C, Odoardi N, Gee K. The Toll for Trafficking: Toll-Like Receptor 7 Delivery to the Endosome. Front Immunol 2017;8:1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davis RJ, Moore EC, Clavijo PE, Friedman J, Cash HA, Chen Z, et al. Anti-PD-L1 efficacy can be enhanced by inhibition of myeloid derived suppressor cells with a selective inhibitor of PI3Kdelta/gamma. Cancer Res 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu X, Horner JW, Paul E, Shang X, Troncoso P, Deng P, et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature 2017;543:728–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rook AH, Gelfand JM, Wysocka M, Troxel AB, Benoit B, Surber C, et al. Topical resiquimod can induce disease regression and enhance T-cell effector functions in cutaneous T-cell lymphoma. Blood 2015;126:1452–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nerurkar L, McColl A, Graham G, Cavanagh J. The Systemic Response to Topical Aldara Treatment is Mediated Through Direct TLR7 Stimulation as Imiquimod Enters the Circulation. Sci Rep 2017;7:16570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dowling DJ. Recent Advances in the Discovery and Delivery of TLR7/8 Agonists as Vaccine Adjuvants. Immunohorizons 2018;2:185–97 [DOI] [PubMed] [Google Scholar]
- 48.HTaMA Naoto Nishii, Kashima Yuta Kondo, Ohno Tatsukuni, Nagai Shigenori, Harada Lixin Li, Xia Yulong, Lau Walter, Hiroyuki Yoshihisa. Systemic administration of a TLR7 agonist attenuates regulatory T cells by dendritic cell modification and overcomes resistance to PD-L1 blockade therapy. Oncotarget 2018;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Che J, Song R, Chen B, Dong X. Targeting CXCR1/2: The medicinal potential as cancer immunotherapy agents, antagonists research highlights and challenges ahead. Eur J Med Chem 2019:111853 [DOI] [PubMed] [Google Scholar]
- 50.Stanhewicz AE, Kenney WL. Role of folic acid in nitric oxide bioavailability and vascular endothelial function. Nutr Rev 2017;75:61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.