

Abstract
Cancer stem cells (CSCs) play a critical role in metastasis, relapse, and therapy resistance in colorectal cancer. While characterization of the normal lineage of cell development in the intestine has led to the identification of many genes involved in the induction and maintenance of pluripotency, recent studies suggest significant heterogeneity in CSC populations. Moreover, while many canonical colorectal cancer CSC marker genes have been identified, the ability to use these classical markers to annotate stemness at the single-cell level is limited. In this study, we performed single-cell RNA sequencing on a cohort of 6 primary colon, 9 liver metastatic tumors, and 11 normal (non-tumor) controls to identify colorectal CSCs at the single-cell level. Finding poor alignment of the 11 genes most used to identify colorectal CSC, we instead extracted a single-cell stemness signature (SCS_sig) that robustly identified ‘gold-standard’ colorectal CSCs that expressed all marker genes. Using this SCS_sig to quantify stemness, we found that while normal epithelial cells show a bimodal distribution, indicating distinct stem and differentiated states, in tumor epithelial cells stemness is a continuum, suggesting greater plasticity in these cells. The SCS_sig score was quite variable between different tumors, reflective of the known transcriptomic heterogeneity of CRC. Notably, patients with higher SCS_sig scores had significantly shorter disease-free survival time after curative intent surgical resection, suggesting stemness is associated with relapse.
Keywords: Colorectal cancer, Cancer stem cells, Plasticity, scRNA-seq
Introduction
The prognosis for patients with colorectal cancer (CRC) remains poor, particularly for those with advanced disease (1) with only incremental improvement over the last two decades(2). Recent evidence suggests that cancer stem cells (CSCs), a subpopulation of tumor cells that possess the ability of unlimited self-renewal as well as ability to differentiate into non-stem cell types, may play a critical role in the development, progression and poor prognosis of CRC.
The concept of CSCs was first introduced with the recognition that malignant cell populations are derived from a small subgroup of cancer cells that are distinguished from the bulk of tumor cells by the ability to self-renewal and regenerate the malignant cell population indefinitely (3). Particularly in aggressive and therapeutically resistant cancers, a subset of CSCs can survive and promote cancer relapse following initially effective treatment due to their ability to establish higher invasiveness and less treatment sensitivity. This concept emphasizes the need to develop innovative treatment strategies targeting CSCs to achieve cures in these cancers (4). However, defining what is a CSCs and identifying these cells has been challenging to date. Growing evidence suggests that CSCs within individual tumors represent multiple pools of phenotypically and functionally heterogeneous cell populations, each of which can have unique biological characteristics (5). Additionally, the plasticity of individual CSCs is more widespread than previously thought; CSC can transition back-and-forth between stem and differentiated states in response to therapeutic insults or other stimuli within the tumor microenvironment (TME) (6–8), which could help explain the heterogeneity observed in tumors. Understanding the impact of CSC plasticity and other properties on disease progression and therapy resistance is critical to the development of more effective therapeutic strategies (4).
Many canonical CRC CSC marker genes have been identified, including LGR5, ASCL2, EPHB2, PROM1, AXIN2, LEFTY1, RNF43, CD44 and SLC12A2 (3,9–13). However, CSC identification by cell sorting according to individual cell surface markers has limitations, and there is the potential to miss a large population of CSCs (14). Single-cell RNA sequencing (scRNA-seq) is a powerful technique for identifying and characterizing CSCs by profiling the transcriptome of individual cells; however, scRNA-seq has not-to-date been used to identify CRC stemness. Here we performed scRNA-seq on primary CRC tumors, CRC liver metastases, and normal tissue to identify, characterize, and evaluate the impact of cancer cell stemness throughout the tumor ecosystem.
Materials and Methods
Patient and sample collection
All patients with diagnosis of colorectal cancer undergoing surgical resection at The University of Texas MD Anderson Cancer center between August 8, 2019 and November 30, 2021, there was no selection based on molecular or prior treatment criteria. A total of 22 samples were collected, however 4 samples failed quality control metrics. A total of 26 specimens from 18 patients with primary or liver metastatic CRC were included in this study (Supplementary table 1). Tumor and nonmalignant tissue samples were surgically removed and kept short-term in sterile cold DMEM medium for transport. Human research was conducted in accordance with the Helsinki Declaration ethical guidelines. Human tumor specimens were collected at The University of Texas MD Anderson Cancer Center under Institutional Review Board approved protocol LAB10–0982 (PI: Dr. Scott Kopetz) after written informed consent was obtained from each participant.
scRNA-seq library preparation and sequencing
After rinsing with cold PBS, fresh specimens were minced to approximately 1 mm3 pieces, transferred to a 50-ml conical tube with 30 ml dissociation solution and incubated for 30 min on a rotor at 37°C. The dissociated cells were subsequently passed through a 70 μm Cell-Strainer (BD) centrifuged at 400 g for 10 min; the supernatant was removed. If red blood cells (RBCs) were present in the pellet, the pelleted cells were suspended in 20 ml of 1× MACS RBC lysis buffer and incubated on ice for 10 min to lyse the RBCs. To make dissociation solution, collagenase A (Sigma, 11088793001) was dissolved in 75% (vol/vol) DMEM F12/HEPES medium (Gibco, 113300) and 25% (vol/vol) BSA fraction V (Gibco, 15260037) to prepare a concentration of 1 mg ml−1. The isolated cell was examined by countess and microscope, if mostly dead cells seen, the sample was excluded from library preparation.
Chromium single-cell sequencing technology from 10x Genomics was used to perform single-cell capture, barcoding and library preparation by following the 10X Genomics Single-Cell Chromium 3’ (PN-120237) protocol using V3 chemistry reagents (10X Genomics). The HS dsDNA Qubit Kit was used to determine the concentrations of both the cDNA and the libraries. The HS DNA Bioanalyzer was used for quality-tracking purposes and size determination for cDNA and lower-concentrated libraries. Sample libraries were normalized to 7.5 nM and equal volumes were added to each library for pooling. The concentration of the library pool was determined using the Library Quantification qPCR Kit (KAPA Biosystems) before sequencing. The barcoded libraries were sequenced at 100 cycles on an S2 flow cell on the Novoseq 6000 system (Illumina). Sequence reads were converted to FASTQ files and UMI read counts using the CellRanger software (10X Genomics).
scRNA-seq data preprocessing and quality control (QC)
The analysis started with a gene count matrix that had genes in rows and cell IDs in columns. Genes detected in fewer than three cells and cells detected with fewer than 500 reads, fewer than 200 genes and more than 50% of transcripts derived from mitochondrial genes were filtered out and excluded from subsequent analysis. Following the initial clustering, we removed likely cell doublets from all clusters as previously described (15). Briefly, potential doublets were identified by the following methods: (1) cells in distinct clusters with similar expression features and an aberrantly high gene count, and (2) cells of a cluster expressing markers from distinct lineages. A similar percentage of potential doublets (approximately 5%) was removed from each sample. A summary of the samples after QC is listed in Supplementary table 2.
Variable gene selection, dimensionality reduction and clustering
After poor quality cells were filtered out and doublets were removed, 111,224 cells (83%) were retained for downstream analyses. From these filtered cells, the gene count matrix was normalized to the total UMI counts per cell and transformed to the natural log scale. To identify highly variable genes, the FindVariableFeatures method in the Seurat V3 package (16) was used. The numbers of significant principal components (PCs) were determined by an elbow plot, which was generated with the ElbowPlot function in Seurat. The first fifteen PCs and top 2000 highly variable genes were used for unsupervised clustering with a resolution set to 0.6. For visualization, the dimensionality was further reduced using the Uniform Manifold Approximation and Projection (UMAP) method with the Seurat RunUMAP function. The cell types were annotated by comparing the canonical marker genes and the differentially expressed genes for each cluster.
Defining single-cell gene signature scores
Cells were scored to a gene signature as previously described (17), using the AddModuleScore function in Seurat, with which analyzed features are binned based on averaged expression, and the control features are randomly-selected from each bin. Given a pre-defined set of genes (Gj) reflecting an expression signature of a specific cell type or biological function, we generated for each cell i, a score, SCj(i), quantifying the degree to which sample i expressed Gj. SCj(i) was calculated by subtracting the average relative expression (Er) of a control gene set (Gj cont) from the average relative expression of Gj: SCj (i) = average[Er(Gj,i)] − average[Er(Gj cont,i)]. The control gene set (Gj cont) was defined by first binning all analyzed genes into 24 bins of equal size based on their aggregate expression levels. Next, we randomly select 100 genes from the same bin for each gene in Gj, such that its average expression was analogous to averaging over 100 randomly-selected gene sets of the same size as Gj. Each patient’s tumor gene signature score was represented by the median of all gene signature scores from their tumor tissue.
Enrichment analysis
Gene set enrichment analysis (GSEA) (18) was based on Hallmark 50 gene sets to identify the CSC transcriptome differences between each patient. The genes were ranked by log2 fold change, using the FindAllMarkers analysis in the Seurat package. The ranked gene lists were subjected to GSEA using the GSEA software run using the GSEA-Preranked method.
Statistical analysis
All statistical analyses were performed using the two-tailed Student’s t-test to assess the statistical significance between two groups in R statistical software (version 4.0.0) or GraphPad Prism 8. The p-value of ≤ 0.05 was considered statistically significant in the hypothesis testing.
Data availability
The raw single-cell RNA sequencing and processed data generated from this study (MDA cohort) are available on Gene Expression Omnibus under accession number GSE231559. Other datasets referenced in this study are available from the GEO database under the accession codes GSE132465 and GSE178341.
Results
Single-cell transcriptome map of CRC
We performed droplet-based encapsulation scRNA-seq on six primary CRC specimens, nine liver metastatic CRC specimens, three matched normal colon specimens, and eight normal liver specimens. After removing cells that did not pass quality control (QC), 111,224 cells remained for further analysis. A median of 1389 genes were detected per cell. The mixture samples from each cluster and cell origin site/class indicated the minimal bench effect of scRNA-seq data (Fig. 1A and Supplementary Fig. 1A). A two-stage clustering strategy was employed in which cells were first annotated into four major populations and further partitioned into minor populations (subtypes).
Figure 1: A single-cell transcriptome map of primary and liver metastatic CRC.
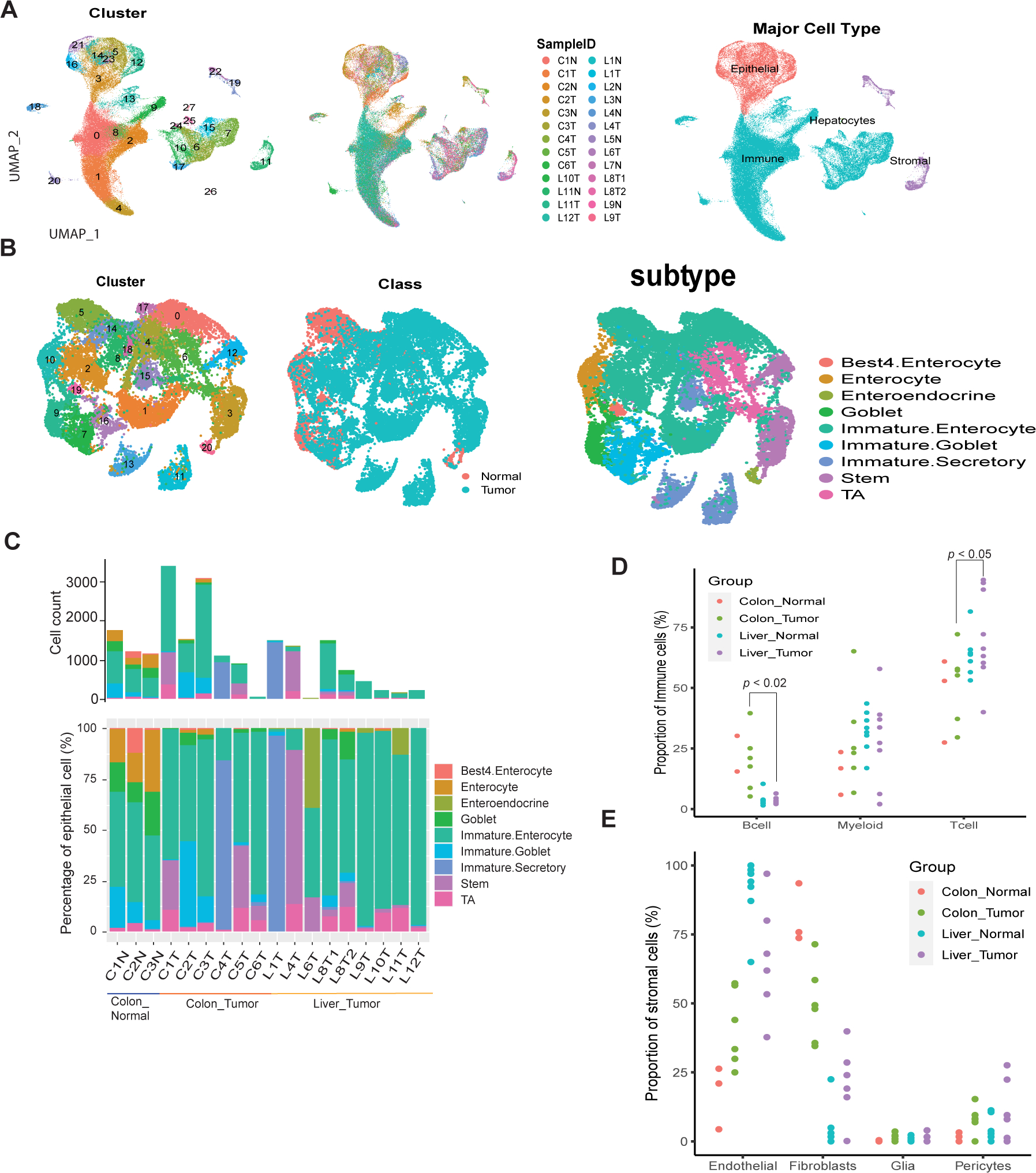
A single-cell transcriptome map of primary and liver-met CRC. A, UMAP plots displaying 111,224 cells from colon normal, colon primary CRC, liver normal and liver metastatic CRC tissues. Cells were colored by cluster (left), sample ID (middle) and major cell type (right), respectively. B, UMAP plot of colon and liver epithelial cells, color-coded by cluster (left), sample class (normal/tumor, middle) and epithelial subtype (right). C, Number and relative proportions of epithelial subtype in colon and liver metastatic CRC samples. D, Percentage of immune subtypes grouped by the sample type of site and class. E, Percentage of stromal subtypes grouped by the sample type of site and class, samples with total stromal cells < 20 were not presented.
Canonical marker genes were used to identify each cell cluster. Four major cell types, including epithelial cells (EPCAM, KRT8, KRT18), immune cells (PTPRC, CD68, CD163, CD3D, CD3E, CD79A, CD19), stromal cells (COL1A1, COL3A1, COL6A1, VWF, CDH5), and hepatocytes (CYP2E1, ASGR1), were annotated (Fig. 1A). Unsurprisingly, while barely detected in normal liver specimens, nearly all colon specimens had a substantial proportion of epithelial cells (Supplementary Fig. 1B). The low percentage of epithelial cells in some of the liver-metastatic tissue is likely due to little tumor remaining after neoadjuvant chemotherapy, which may be low in patients who received chemotherapy before surgery within 3 months. This is because the chemotherapy may have killed the tumor cells, as confirmed by a professional pathologist who observed extremely low tumor cell viability in samples L6T and L9T (Supplementary Table 1). For the partitioned epithelial, immune, and stromal cells, we further annotated the subtypes of each compartment based on known marker genes and found that most of the subtypes were recovered.
In the epithelial cell compartment (Fig. 1B), many of the tumor cell clusters did not express canonical epithelial subtype marker genes (Fig. 1B and Supplementary Fig. 1C). Combining our observations in these clusters with published epithelial subtype gene signatures (12) (Supplementary Fig. 1D), we annotated these tumor cells to the normal epithelial subtype with confidence (Fig. 1B, right). Liver metastatic CRC specimens showed less diversity in the number of transcriptional subtypes present in each sample. For example, L4T was predominated by stem cells and L1T was predominated by immature secretory type cells (Fig. 1C). A simpler or more uniform cell type in liver metastatic tumors may arise from either monoclonal seeding or clonal selection in the metastatic microenvironment (19). Notably, there were more immature/undifferentiated cells in tumor specimens compared to normal specimens, indicating that cancer represents a less differentiated state relative to normal tissue but with significant heterogeneity at a cell-to-cell level.
Hepatocytes were identified only in liver specimens, though counts were low likely due to the known fragility of these cells with respect to the dissociation process (Supplementary Fig. 1B). For immune cells, we assigned T cell, B cell and myeloid components (Supplementary Fig. 1E), the proportion of B cells was significantly reduced in liver metastatic CRC compared to the primary tumor, whereas the proportion of T cells was observed to be increased (Fig. 1D). In the stromal cell compartment (Supplementary Fig. 1F), liver metastatic tumors were predominantly endothelial cells, while primary tumors were predominantly fibroblasts (Fig. 1E). Interestingly, we found the fibroblast proportion was significantly decreased in liver metastatic tumor compared to primary CRC (Fig. 1E). This result is consistent with recent findings that cancer-associated fibroblasts were significantly more abundant in primary CRC than in liver metastatic tumor (20). Studies using scRNA-seq to investigate the TME and its role in disease pathogenesis and treatment response are becoming increasingly common. Lambrechts et al. found that fibroblasts and myofibroblasts in the TME exhibited a distinct phenotype compared to those in normal lung tissue, and this phenotype was associated with poor prognosis. They also identified potential therapeutic targets for modulating the TME (21). The stem-like malignant progenitors have phenotypic plasticity to adapt to specific microenvironments and overcome metastasis barriers (22).
Single-cell stemness signature for CRC
Single-cell RNA sequencing revealed heterogeneity of known CSC marker genes expression in epithelial cells, indicating that the stemness expression profiles cannot be fully captured by a few marker genes (Fig. 2A). To address these challenges, we used CRC scRNA-seq data to identify a novel stemness signature (SCS_sig) to annotate CSCs. We first annotated a ‘gold-standard’ set of CSCs that expressed nine canonical colon CSC marker genes (LGR5, ASCL2, EPHB2, PROM1, AXIN2, LEFTY1, RNF43, CD44 and SLC12A2) (3,9–13) (Fig. 2B); 346 gold-standard CSCs were identified (1.7% of epithelial cells) (Fig. 2C). Myc targets, Wnt/β-catentin pathways (Fig. 2D), and previously published stemness gene sets (Supplementary Fig. 2A), were enriched in these gold-standard stem cells compared to other tumor cells, suggesting that these CSCs were robustly identified.
Figure 2: Identification of a single-cell stemness signature.
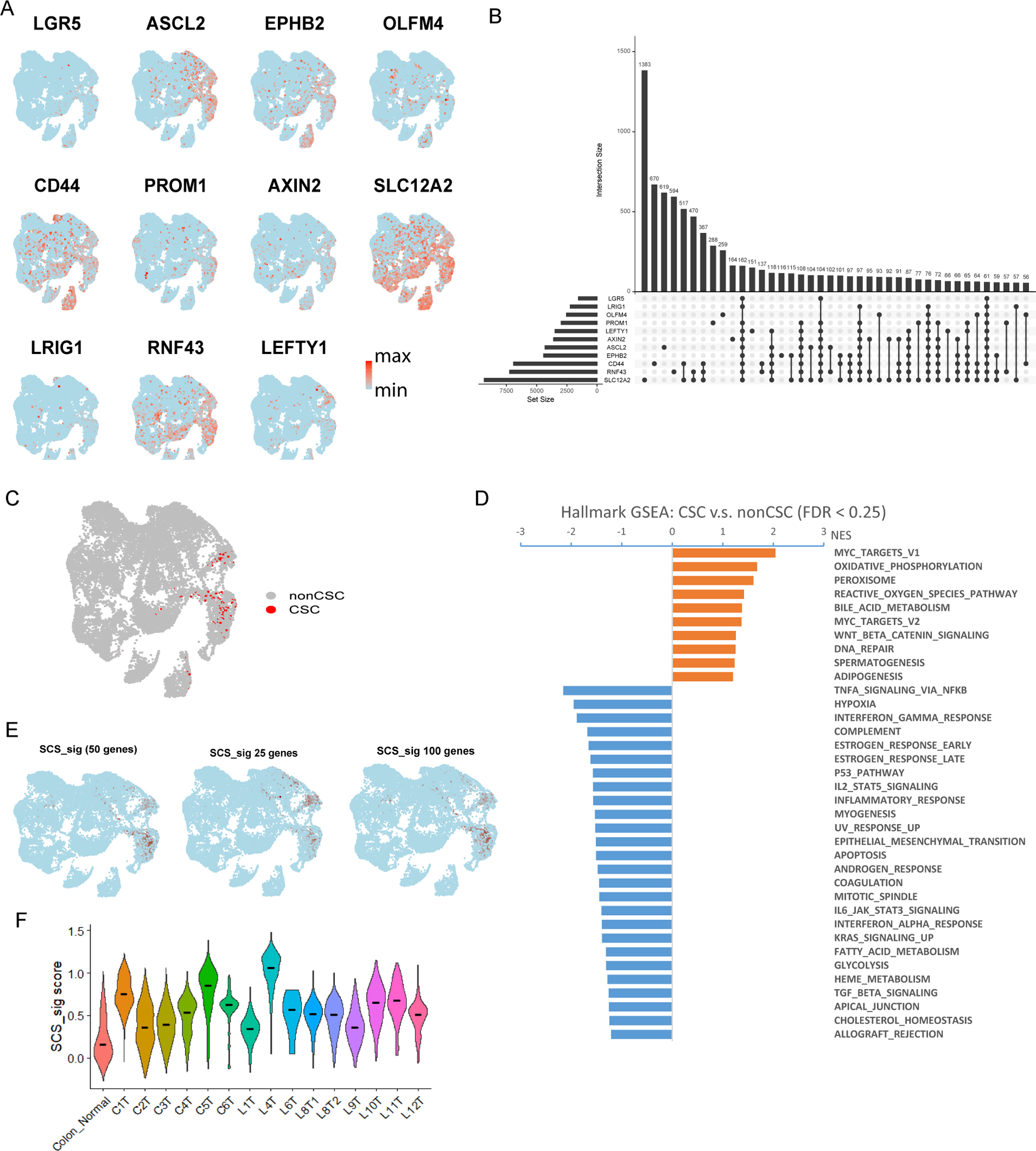
A, UMAP plots showing expression of canonical intestinal CSC marker genes. Color bar represents scaled expression. B, Intersection epithelial cell number that CSC gene is non-zero. LGR5, ASCL2 and EPHB2 are marked as primary CSC genes. C, UMAP showing the ‘gold-standard’ CSC expressing most of the canonical intestinal CSC marker genes (LGR5, ASCL2, EPHB2, PROM1, AXIN2, LEFTY1, RNF43, CD44 and SLC12A2). D, Gene set enrichment analysis (GSEA) results showing the hallmark gene sets related to ‘gold-standard’ CSC compared with other epithelial cells. Gene sets with FDR < 0.25 are shown. E, Feature plot showing the 10% epithelial cells (colored by dark red) with the highest SCS_sig score of 50, 25 and 100 genes. F, Violin plot of the SCS_sig score by tumor samples and merged normal colon samples. The line in each violin represents the median value.
To characterize and capture the unique transcriptomic features of stemness from these gold standard cells, we selected the 50 most significantly up-regulated genes (relative to other tumor cells) as a single-cell stemness signature (SCS_sig) (Supplementary table 3); this signature was positively correlated with the previously published 500-gene intestinal stem cell signature (23). We then determined the SCS_sig score at the single-cell level using AddModuleScore (17). In epithelial cells, the highest SCS_sig scores were well aligned with gold-standard CSCs (Fig. 2E, Supplementary Fig. 2B). Evaluation of the correlation between CSC marker genes and SCS_sig expression in epithelial cells indicated that the SCS_sig was statistically significantly associated with all CSC genes (Spearman p < 0.0001), most correlated with ASCL2 expression (Supplementary Fig. 2C). In selecting the optimal number of genes for the gene set, we also evaluated the signature scores using 25, 50, and 100 genes. We found that all three gene sets were significantly correlated with each other (Supplementary Fig. 2D), and the high signature score cells were largely overlapping for 25, 50, and 100 gene sets in the scRNASeq profile (Fig. 2E), indicating that a 50-gene set provided a robust approach for identifying cancer stem cells in colorectal cancer.
Furthermore, comparing individual tumors, while the stemness of each tumor was higher relative to normal colon controls there was considerable variability in stemness among different tumors from different patients (Fig. 2F). To evaluate the stemness of tumor cells from different patients, we used unsupervised clustering to identify the cells with the most stemness; cells within the highest 10% of SCS_sig scores of each sample were defined as having high stemness (Supplementary Fig. 2E). When the UMAP plot was annotated by sample, some tumor cells formed distinct clusters, while other clusters included a mix of primary, metastatic and normal cells (Supplementary Fig. 2E). The four individual clusters (1, 2, 4, 5) with higher SCS_sig scores were separated from each other as well as from the group of clusters (0, 3) with lower SCS_sig scores (Supplementary Fig. 2E). These results suggest that SCS_sig could serve as an indicator of tumor heterogeneity, as UMAP has good preservation of large-scale distances (24).
Single-cell stemness signature characterization and distribution
To examine if the SCS_sig can measure single-cell stemness, we calculated the differentiation states by CytoTRACE, an unsupervised framework for predicting relative differentiation states from single-cell transcriptomes (17). We found both SCS_sig and CytoTRACE scores were significantly higher in both gold-standard CSC compared to non-CSC cells and tumor cells compared with normal cells (Fig. 3A and Fig. 3B). Indeed, the SCS_sig and CytoTRACE scores were highly correlated at the single-cell level (R=0.75, p<0.0001; Fig. 3C). Our comparative analysis between the SCS signature and the CytoTRACE-derived top 50 gene lists revealed a substantial overlap, with 30 out of the 50 CytoTRACE genes coinciding with the SCS signature genes, reinforcing the validity of our SCS signature in representing stem cell characteristics (Supplementary Fig. 3F). Furthermore, we calculated the SCS_sig in epithelial cells from the Samsung Medical Center cohort (32 patient) (25) and Broad Institute cohort (62 patients) (26) and found SCS_sig score was higher in tumor compared with normal cells (Fig. 3D), and positively correlated with CytoTRACE score (Supplementary Fig. 3A). There was no significant difference of SCS_sig and CytoTRACE scores between primary and liver metastatic CRC (Supplementary Fig. 3B), suggesting that liver resident tumor cells did not acquire more stemness than primary tumor cells. In our analysis of 4,312 normal epithelial cells, we identified only one cell meeting the criteria for a gold-standard cancer stem cell (CSC), highlighting their scarcity in normal colon tissue. Comparative studies of primary colon tumors and liver metastases revealed significant inter-tumor variability in CSC numbers, but no discernible difference between the primary and metastatic sites (Supplementary Fig. 3G). The distribution of SCS_sig scores in tumor cells followed a normal and continuous pattern, which implies that the level of stemness in tumor cells is highly adaptable. In contrast, the SCS_sig score distribution in normal cells was bimodal in both our dataset and the Broad cohort data (26) (Fig. 3E).
Figure 3: SCS_sig characterization.
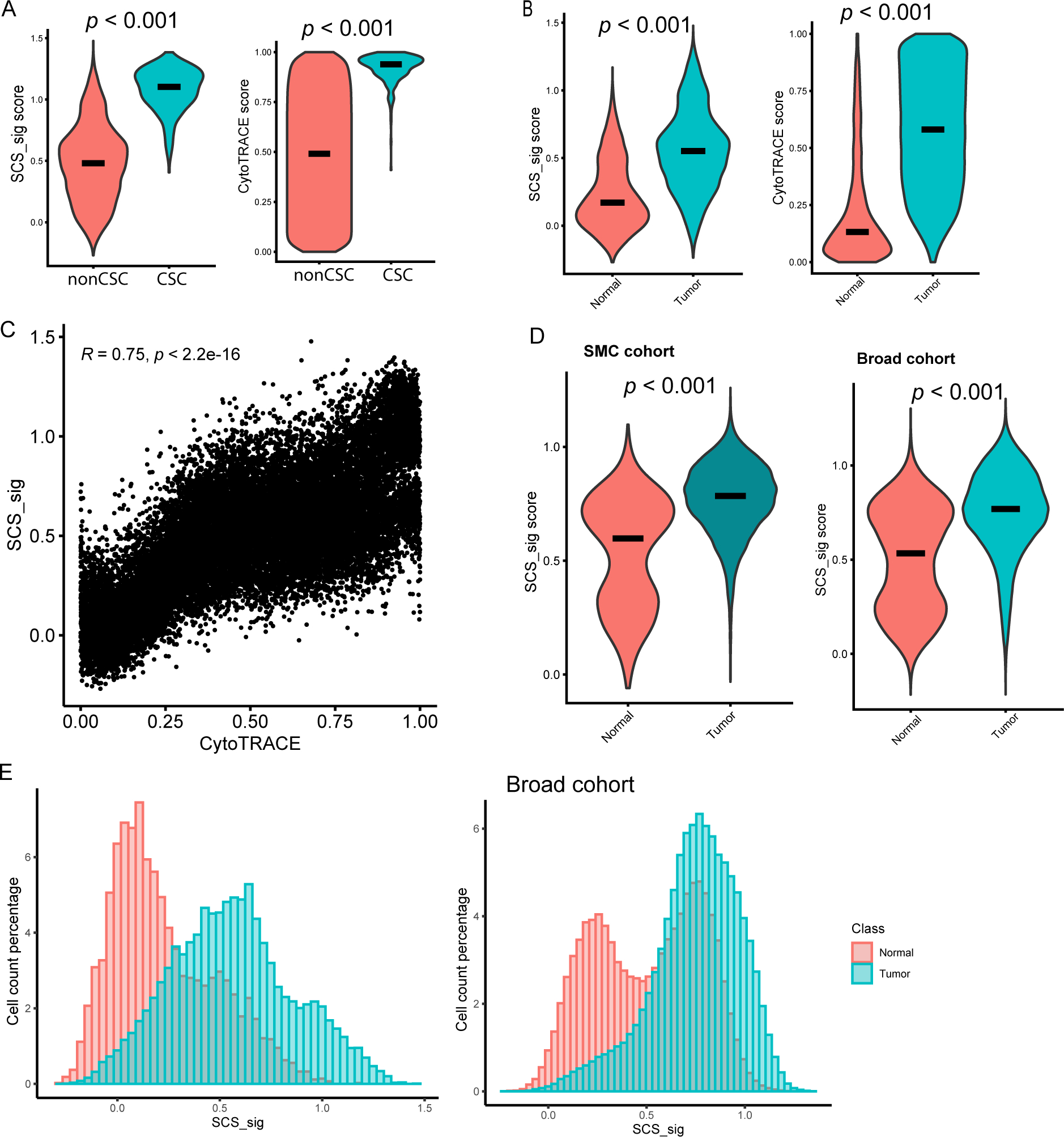
A and B, SCS_sig score (left) and CytoTRACE score (right) in non-CSC/CSC (A) and in normal/tumor cells (B). C, Spearman correlation coefficient of SCS_sig score and CytoTRACE score in epithelial cells. D, SCS_sig score in the Samsung Medical Center (SMC) cohort (left) and Broad cohort (right) epithelial cells by normal/tumor. E, Histogram showing the distribution of SCS_sig score in normal and tumor cells (MD Anderson data, left; Broad cohort data, right).
Association between single-cell stemness signature, tumor features and clinical outcomes
We investigated the potential relationship between SCS_sig score, patient treatment history, and survival outcomes (Fig. 4A). We found that patients with higher SCS_sig score tended to experience relapse shortly after surgery. For instance, patient L4, who had the tumor with highest SCS_sig score, relapsed within two months after surgery, while patients C3, C4, L1, and L9 with the four lowest SCS_sig scores have not yet relapsed (Fig. 4A, B). Splitting the cohort into half, the patients with higher SCS_sig scores had markerly shorter DFS relative to those with lower SCS_sig, average DFS time of 5.0 months vs. 19.2 months (Fig. 4B). Survival analysis by Kaplan-Meier plot and log-rank test revealed statistical significance (p=0.025) between high SCS_sig group and low group (Fig. 4C). Relapse after surgical resection is a major challenge in cancer treatment, and these data fit with emerging evidence in other tumor types that suggests that cancer cell stemness is a critical contributor(27).
Figure 4: Associate of SCS_sig with clinical outcomes and tumor features.
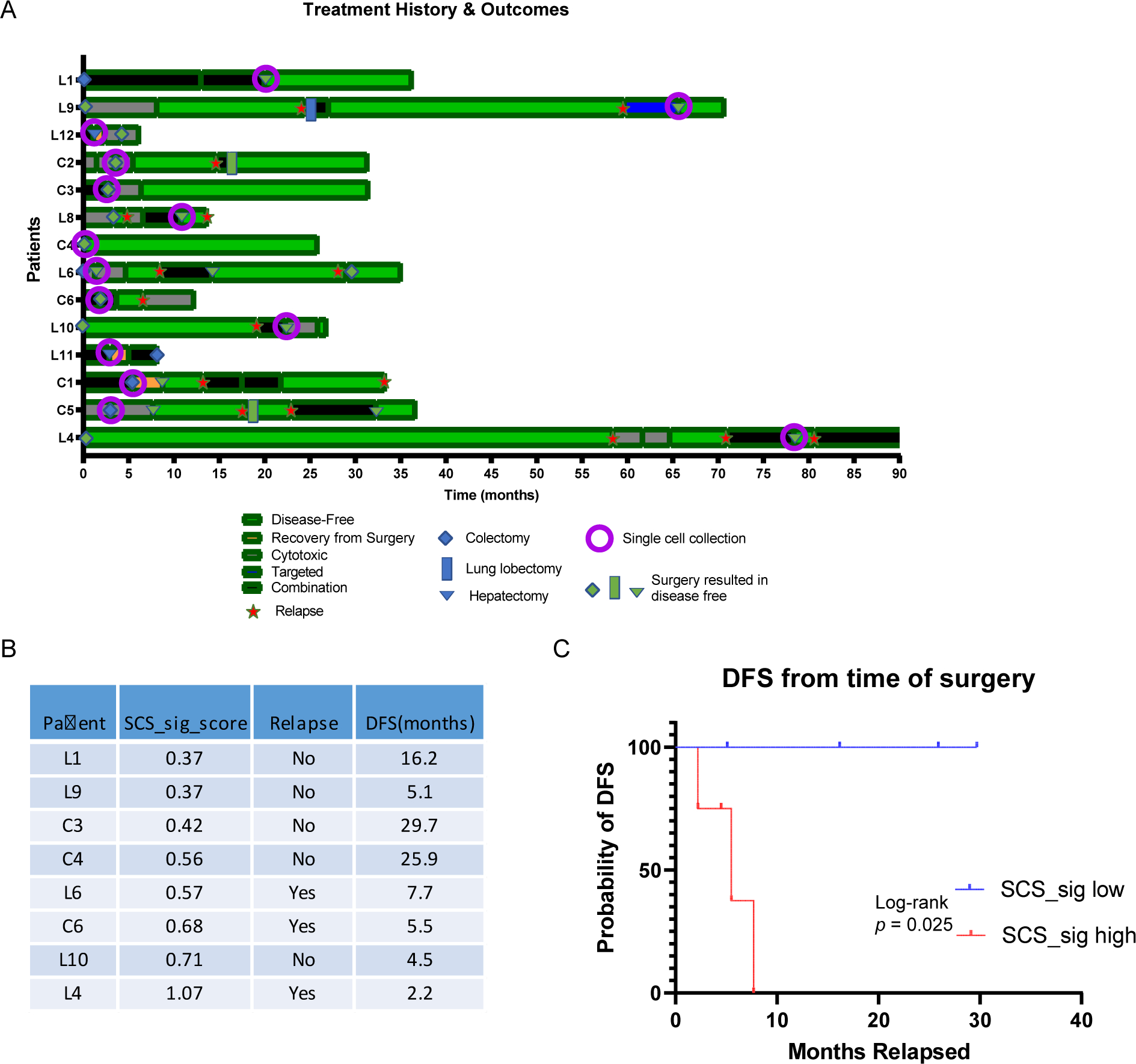
A, Swimmer plot showing the patients’ treatment history and outcomes, patients were ordered by SCS_sig score. B, SCS_sig score and disease-free survival (DFS) from time of single cell collection surgery. Each patient’s SCS_sig score is represented by the median SCS_sig score of tumor cells from that patient. C, Kaplan-Meier plot showing the DFS curves for patients grouped by median of SCS_sig score.
Discussion
Prior research has suggested identifying Cancer Stem Cells (CSCs) in single-cell RNA sequencing profiles based on the expression of single-gene or few-gene markers ((3), (9–13)). However, this approach resulted in higher heterogeneity among the CSCs that were identified in scRNA-seq profiles of CRC (Fig. 2A). We also observed that the canonical CRC CSC marker genes, some of which overlap with markers of normal intestinal stem cells, present complexities in their expression patterns, making it challenging to differentiate between normal and cancer stem cells using individual markers. Additionally, our analysis of specific markers like ALDH1 and BMI1 demonstrated a dispersed expression across the epithelial cell cluster (Supplementary Fig. 3C), emphasizing the intricacy and potential pitfalls of relying solely on individual markers for stem cell identification in colorectal cancer. In this study, we identified a novel gene set with high expression levels in CSCs. This gene set, which we refer to as the single-cell stemness signature (SCS_sig), is postulated to delineate the stemness characteristic of epithelial cells. The 50-gene set encompasses a wide range of biological functions, including well-established cancer-related genes, such as AREG and ASCL2, as well as unexpected genes involved in oxidative stress like PRDX4 and PRDX5, and ribosomal protein genes (eg, RPS14, RPS15A, RPL10, etc.)(28–32). Additional bioinformatic analyses of ribosomal protein-associated networks may also shed light on mechanisms maintaining stemness. Interestingly, markers of epithelial differentiation like KRT20 are notably absent. The lack of mature intestinal markers supports the stem-like nature of the identified cell subset(33). This comprehensive and balanced composition not only validates the robustness of our gene set but also opens new avenues for research, enhancing its reliability and scope for further validation and applications in colorectal cancer research.
Our analysis revealed that tumor cells with the top 10% signature score from four known ISC gene sets (ISC_Munoz, ISC_Merlos, Stemness_Biton, and Stemness_Li) did not congregate into a distinct cluster (Supplementary Fig. 3D), suggesting that the SCS_sig gene set encapsulates the similarity of stem cells more comprehensively than other ISC gene sets(14,23,34,35). We also found that the single cells with the top 10% signature score of these four ISC gene sets differ significantly in terms of their overlap with the top 10% using SCS_sig (Supplementary Fig. 3E). This, combined with low correlation between SCS_sig and ISC_Munoz (Supplementary Fig. 2B), implies that the new SCS_sig gene set may lead to different results on what would be considered stem cells in the tumor compared to other published ISC gene sets, providing a more comprehensive and nuanced understanding of stemness in tumor cells.
Our study emphasizes the importance of considering cell states over cell types of tumor cells. Utilizing this SCS_sig gene set, we quantified the stemness attribute of individual epithelial cells in both normal and tumor specimens. Interestingly, the stemness scores of malignant epithelial cells presented a unimodal normal distribution, in contrast to the bimodal distributions observed in epithelial cells of normal colonic tissue. These data suggest that stemness in normal, non-tumor cells is a regulated, binary on/off state. In contrast, in tumor cells, there is greater plasticity and stemness exists on a continuum. This observation is consistent with the continuous and reversible state theory of tumor cell stemness plasticity, which explains the role of CSCs in tumor growth, recurrence (6), metastasis (22), and treatment resistance (27). This theory posits that CSCs can transition back and forth between stem and non-stem cell states, allowing them to adapt to changing environmental conditions for a survival advantage (36). Thus, CSCs are thought to be a key factor in the aggressiveness and resistance of tumors, as they are able to persist and initiate new tumors even after conventional treatments such as chemotherapy and radiation. Such a perspective offers a more comprehensive understanding of tumor heterogeneity, potentially reshaping therapeutic strategies that target the intricate and adaptable nature of cancer stem cells.
We also propose that SCS_sig may be utilized as an indicator of inter-tumor heterogeneity. By assigning the top 10% of SCS_sig scoring cells in each tumor sample as ‘high stemness’, we were able to create a distinct grouping of cells exhibiting marked stemness traits. Tumor cells’ clusters exhibiting higher SCS_sig scores demonstrated clear separation from each other and also from the group of clusters that displayed lower SCS_sig scores. This differentiation between clusters based on their SCS_sig scores underlines the potential of the SCS_sig as a tool for dissecting tumor heterogeneity. Indeed, we observed a patient-specific bias in the distribution of cells with high SCS_sig scores (>1). We found that using a high SCS_sig score as a threshold would identify single cells from only three samples, and single cells from other samples were under-represented.This insight is critical, as understanding tumor heterogeneity is a key aspect of developing personalized therapeutic strategies and predicting treatment responses (37–39). Our study also revealed that patients exhibiting a high SCS_sig generally have a less favorable Disease-Free Survival (DFS) post-surgery of patients with CRC, highlighting its prognostic value as the tumor cell recurrence following surgical removal represents a significant obstacle in cancer therapy. However, our study acknowledges a notable limitation concerning the limited data on MSI-H status, available for only one patient, and the small sample size, which might not fully represent the diverse clinical scenarios encountered in larger populations. Future studies will aim to enhance the dataset to explore the potential correlations between stemness markers and significant prognostic factors in colorectal cancer more comprehensively. In this study, we emphasize the potential of our computational groundwork to pave the way for future studies, where detailed experimental validations such as lineage tracing and in vitro or in vivo assays can be integrated to confirm the functional properties and behaviors of identified CSC populations, thereby fostering a more nuanced understanding of colorectal cancer dynamics. Moreover, further investigations and validations are required to assess the functional implications of high and low SCS_sig scores in different tumor cells and exploring how these scores might be influenced by the tumor microenvironment or patient-specific factors. The potential application of this novel approach in a clinical setting could greatly aid the management and treatment of complex and heterogeneous colorectal cancers.
In summary, our study provides a novel approach for annotating stemness using CRC scRNA-seq data that can help improve our understanding of the heterogeneity and plasticity of these cells. Further studies are needed to validate the SCS_sig in larger patient cohorts and to explore its potential as a prognostic or therapeutic target in CRC.
Supplementary Material
Implications.
This study reveals significant heterogeneity of expression of genes commonly used to identify colorectal CSCs, and identifies a novel stemness signature to identify these cells from scRNAseq data.
Acknowledgments
We would like to thank to the Dr. John Paul Shen lab members for their valuable discussion and feedback during the development of this work; Sendurai Mani, PhD, (Professor, Pathology and Laboratory Medicine, Brown University) for the conceptualization of cancer stem cell; the tumor dissociation and single cell isolation in this study was supported by the NIH/NCI under award number P30 CA016672 and used the Oncology Research and Immuno-monitoring Core (ORION); the CPRIT Single Cell Genomics Core (RP180684, MD Anderson Cancer Center) for support with scRNA-seq experiments; Jennifer K. Peterson, PhD, (scientific editor, Gastrointestinal Medical Oncology, MD Anderson Cancer Center) for the invaluable scientific editing and insightful feedback that greatly improved the quality of our manuscript; Oscar Villarreal was supported by the CPRIT Training Program (RP210028).
Financial support
This work was supported by the National Cancer Institute (L30 CA171000 and K22 CA234406 to J.P.S., and The Cancer Center Support Grant P30 CA016672 to J.P.S), the Cancer Prevention & Research Institute of Texas (RR180035 to J.P.S.; J.P.S. is a CPRIT Scholar in Cancer Research), and the Col. Daniel Connelly Memorial Fund. This study was also supported by the Colorectal Cancer Moonshot Program and SPORE program (P50CA221707) of The UT MD Anderson Cancer Center.
Footnotes
Conflict of interest disclosure statement
S.K. stock and other ownership interests: MolecularMatch, Navire; consulting or advisory role: Roche, Genentech, EMD Serono, Merck, Karyopharm Therapeutics, Amal Therapeutics, Navire Pharma, Symphogen, Holy Stone, Biocartis, Amgen, Novartis, Eli Lilly, Boehringer Ingelheim; research funding: Amgen (Inst), Sanofi (Inst), Biocartis (Inst), Guardant Health (Inst), Array BioPharma (Inst), Genentech (Inst), EMD Serono (Inst), MedImmune (Inst), Novartis (Inst). J.P.S. consulting or advisory role: Engine Biosciences; research funding: Celsius Therapeutics (Inst), BostonGene (Inst). All other authors have declared no conflicts of interest.
References
- 1.Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin. 2020;70(3):145–64. [DOI] [PubMed] [Google Scholar]
- 2.Zeineddine FA, Zeineddine MA, Yousef A, Gu Y, Chowdhury S, Dasari A, et al. Survival improvement for patients with metastatic colorectal cancer over twenty years. NPJ Precis Oncol. 2023. Feb 13;7(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017. Oct 6;23(10):1124–34. [DOI] [PubMed] [Google Scholar]
- 4.Saygin C, Matei D, Majeti R, Reizes O, Lathia JD. Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem Cell. 2019. Jan 3;24(1):25–40. [DOI] [PubMed] [Google Scholar]
- 5.Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010. Jan 8;140(1):62–73. [DOI] [PubMed] [Google Scholar]
- 6.Shimokawa M, Ohta Y, Nishikori S, Matano M, Takano A, Fujii M, et al. Visualization and targeting of LGR5+ human colon cancer stem cells. Nature. 2017. May;545(7653):187–92. [DOI] [PubMed] [Google Scholar]
- 7.Lambert AW, Weinberg RA. Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat Rev Cancer. 2021. May;21(5):325–38. [DOI] [PubMed] [Google Scholar]
- 8.Bayik D, Lathia JD. Cancer stem cell-immune cell crosstalk in tumour progression. Nat Rev Cancer. 2021. Aug;21(8):526–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007. Jan 4;445(7123):106–10. [DOI] [PubMed] [Google Scholar]
- 10.van der Flier LG, van Gijn ME, Hatzis P, Kujala P, Haegebarth A, Stange DE, et al. Transcription factor achaete scute-like 2 controls intestinal stem cell fate. Cell. 2009. Mar 6;136(5):903–12. [DOI] [PubMed] [Google Scholar]
- 11.Koo BK, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M, et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature. 2012. Aug 30;488(7413):665–9. [DOI] [PubMed] [Google Scholar]
- 12.Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, et al. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell. 2019. Jul 25;178(3):714–730.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong VWY, Stange DE, Page ME, Buczacki S, Wabik A, Itami S, et al. Lrig1 controls intestinal stem-cell homeostasis by negative regulation of ErbB signalling. Nat Cell Biol. 2012. Apr;14(4):401–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Merlos-Suárez A, Barriga FM, Jung P, Iglesias M, Céspedes MV, Rossell D, et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell. 2011. May 6;8(5):511–24. [DOI] [PubMed] [Google Scholar]
- 15.Wang R, Dang M, Harada K, Han G, Wang F, Pool Pizzi M, et al. Single-cell dissection of intratumoral heterogeneity and lineage diversity in metastatic gastric adenocarcinoma. Nat Med. 2021. Jan;27(1):141–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018. May;36(5):411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016. Apr 8;352(6282):189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005. Oct 25;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dang HX, Krasnick BA, White BS, Grossman JG, Strand MS, Zhang J, et al. The clonal evolution of metastatic colorectal cancer. Sci Adv. 2020. Jun;6(24):eaay9691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Che LH, Liu JW, Huo JP, Luo R, Xu RM, He C, et al. A single-cell atlas of liver metastases of colorectal cancer reveals reprogramming of the tumor microenvironment in response to preoperative chemotherapy. Cell Discov. 2021. Sep 7;7(1):80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. 2018. Aug;24(8):1277–89. [DOI] [PubMed] [Google Scholar]
- 22.Fumagalli A, Oost KC, Kester L, Morgner J, Bornes L, Bruens L, et al. Plasticity of Lgr5-Negative Cancer Cells Drives Metastasis in Colorectal Cancer. Cell Stem Cell. 2020. Apr 2;26(4):569–578.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muñoz J, Stange DE, Schepers AG, van de Wetering M, Koo BK, Itzkovitz S, et al. The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent “+4” cell markers. EMBO J. 2012. Jun 12;31(14):3079–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. 2019. Jan;37(1):38–44. [DOI] [PubMed] [Google Scholar]
- 25.Lee HO, Hong Y, Etlioglu HE, Cho YB, Pomella V, Van den Bosch B, et al. Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat Genet. 2020. Jun 1;52(6):594–603. [DOI] [PubMed] [Google Scholar]
- 26.Pelka K, Hofree M, Chen JH, Sarkizova S, Pirl JD, Jorgji V, et al. Spatially organized multicellular immune hubs in human colorectal cancer. Cell. 2021. Sep 2;184(18):4734–4752.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holmberg R, Robinson M, Gilbert SF, Lujano-Olazaba O, Waters JA, Kogan E, et al. TWEAK-Fn14-RelB Signaling Cascade Promotes Stem Cell-like Features that Contribute to Post-Chemotherapy Ovarian Cancer Relapse. Mol Cancer Res MCR. 2023. Feb 1;21(2):170–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joanito I, Wirapati P, Zhao N, Nawaz Z, Yeo G, Lee F, et al. Single-cell and bulk transcriptome sequencing identifies two epithelial tumor cell states and refines the consensus molecular classification of colorectal cancer. Nat Genet. 2022. Jul;54(7):963–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Basu S, Gavert N, Brabletz T, Ben-Ze’ev A. The intestinal stem cell regulating gene ASCL2 is required for L1-mediated colon cancer progression. Cancer Lett. 2018. Jun 28;424:9–18. [DOI] [PubMed] [Google Scholar]
- 30.Jia W, Chen P, Cheng Y. PRDX4 and Its Roles in Various Cancers. Technol Cancer Res Treat. 2019. Jan 1;18:1533033819864313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agborbesong E, Zhou JX, Li LX, Harris PC, Calvet JP, Li X. Prdx5 regulates DNA damage response through autophagy-dependent Sirt2-p53 axis. Hum Mol Genet. 2023. Jan 27;32(4):567–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kang J, Brajanovski N, Chan KT, Xuan J, Pearson RB, Sanij E. Ribosomal proteins and human diseases: molecular mechanisms and targeted therapy. Signal Transduct Target Ther. 2021. Aug 30;6(1):323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JA, Seo MK, Yoo SY, Cho NY, Kwak Y, Lee K, et al. Comprehensive clinicopathologic, molecular, and immunologic characterization of colorectal carcinomas with loss of three intestinal markers, CDX2, SATB2, and KRT20. Virchows Arch Int J Pathol. 2022. Mar;480(3):543–55. [DOI] [PubMed] [Google Scholar]
- 34.Biton M, Haber AL, Rogel N, Burgin G, Beyaz S, Schnell A, et al. T Helper Cell Cytokines Modulate Intestinal Stem Cell Renewal and Differentiation. Cell. 2018. Nov 15;175(5):1307–1320.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh JJL, et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet. 2017. May;49(5):708–18. [DOI] [PubMed] [Google Scholar]
- 36.Vasquez EG, Nasreddin N, Valbuena GN, Mulholland EJ, Belnoue-Davis HL, Eggington HR, et al. Dynamic and adaptive cancer stem cell population admixture in colorectal neoplasia. Cell Stem Cell. 2022. Aug 4;29(8):1213–1228.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.El Baba R, Pasquereau S, Haidar Ahmad S, Diab-Assaf M, Herbein G. Oncogenic and Stemness Signatures of the High-Risk HCMV Strains in Breast Cancer Progression. Cancers. 2022. Sep 1;14(17):4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramamoorthi G, Kodumudi K, Gallen C, Zachariah NN, Basu A, Albert G, et al. Disseminated cancer cells in breast cancer: Mechanism of dissemination and dormancy and emerging insights on therapeutic opportunities. Semin Cancer Biol. 2022. Jan;78:78–89. [DOI] [PubMed] [Google Scholar]
- 39.Serrano-Oviedo L, Nuncia-Cantarero M, Morcillo-Garcia S, Nieto-Jimenez C, Burgos M, Corrales-Sanchez V, et al. Identification of a stemness-related gene panel associated with BET inhibition in triple negative breast cancer. Cell Oncol Dordr. 2020. Jun;43(3):431–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw single-cell RNA sequencing and processed data generated from this study (MDA cohort) are available on Gene Expression Omnibus under accession number GSE231559. Other datasets referenced in this study are available from the GEO database under the accession codes GSE132465 and GSE178341.