

Abstract
The histone lysine demethylases KDM4A-C are involved in physiologic processes including stem cell identity and self-renewal during development, DNA-damage repair, and cell cycle progression. KDM4A-C are overexpressed and associated with malignant cell behavior in multiple human cancers and are therefore potential therapeutic targets. Given the role of KDM4A-C in development and cancer, we aimed to test the potent, selective KDM4A-C inhibitor QC6352 on oncogenic cells of renal embryonic lineage. The anaplastic Wilms tumor cell line WiT49 and the tumor-forming human embryonic kidney cell line HEK293 demonstrated low nanomolar QC6352 sensitivity. The cytostatic response to QC6352 in WiT49 and HEK293 cells was marked by induction of DNA damage, a DNA repair-associated protein checkpoint response, S-phase cell cycle arrest, profound reduction of ribosomal protein gene and rRNA transcription, and blockade of newly synthesized proteins. QC6352 caused reduction of KDM4A-C levels by a proteasome-associated mechanism. The cellular phenotype caused by QC6352 treatment of reduced migration, proliferation, tumor spheroid growth, DNA damage, and S-phase cell cycle arrest was most closely mirrored by knockdown of KDM4A as determined by siRNA knockdown of KDM4A-C. QC6352 sensitivity correlated with high basal levels of ribosomal gene transcription in over 900 human cancer cell lines. Targeting KDM4A may be of future therapeutic interest in oncogenic cells of embryonic renal lineage or cells with high basal expression of ribosomal protein genes.
Introduction
Histone methylation regulates gene transcription, DNA repair, stem cell self-renewal, and cell differentiation during embryonic development by altering the accessibility of DNA at a given genetic locus.(1) Histone methylation is dynamically regulated by histone lysine methyltransferases and histone lysine demethylases (KDMs). The KDM4 family are histone lysine demethylases that specifically catalyze removal of methyl groups from di- and tri-methylated histone 3 lysine 9 (H3K9) and 36 (H3K36).(2) Trimethylated H3K9 (H3K9me3) is a mark of transcriptionally inactive, condensed heterochromatin and therefore regarded as a transcriptionally repressive histone mark.(3) The role of H3K36 methylation is more complex, as trimethylated H3K36 (H3K36me3) is found in actively transcribed, open euchromatin(4), but H3K36me3 may also act in concert with H3K9me3 or gene body DNA methylation to prevent initiation of aberrant transcription.(5,6)
The complete KDM4 gene family consists of KDM4A-F. KDM4A-C contain both N-terminal demethylase (Jumonji-C) and C-terminal “reader” (PHD/Tudor) protein domains, allowing recognition and demethylation of di- and tri-methylated lysine residues H3K9 and H3K36.(2) In contrast, KDM4D lacks “reader” domains and has expression limited to the testes, and KDM4E-F are non-coding pseudogenes.(2) KDM4A-C are expressed in many normal human tissues and are involved in physiologic processes including stem cell self-renewal(7,8), cell differentiation(9,10), DNA-damage repair(11–16), cell cycle(17,18), and cell metabolism(19,20). KDM4A-C expression is dysregulated and associated with disease pathogenesis in human cancers including breast, prostate, colorectal, acute myeloid leukemia, neuroblastoma and PAX3-FOXO1-driven alveolar rhabdomyosarcoma(21–26). KDM4A was shown to facilitate transcription of ribosomal genes in the nucleolus and found to regulate protein synthesis in the cytoplasm.(27,28)
QC6352 is a potent and selective KDM4 inhibitor developed using structure-function based design and shown to inhibit the catalytic, demethylase domain of KDM4A-D.(29) In breast cancer patient-derived xenograft models, QC6352 was shown to reduce tumor growth and deplete tumor-initiating, stem-like cell populations.(30) Pharmacologic inhibition of KDM4 using QC6352 was shown to have anticancer activity in PAX3-FOXO1 alveolar rhabdomyosarcoma cells and patient-derived xenografts primarily mediated through KDM4B inhibition.(26)
Wilms tumor (WT) is the most common childhood kidney cancer and arises from abnormal development and persistence of renal progenitor cells during kidney development.(31–33) Given the relevance of KDM4 to stem-cell renewal and cellular differentiation, and KDM4 dysregulation in other human cancers including the pediatric cancers neuroblastoma and rhabdomyosarcoma, we sought to investigate the role of KDM4 inhibition using QC6352 in embryonic kidney-associated cell lines including WT cells and the tumor-forming cell line HEK293.
Materials and Methods
Cell culture
The anaplastic WT WiT49 cell line (RRID: CVCL_0583) was obtained from Dr. Herman Yeger and cultured in DMEM (Corning, 10–017-CV) with 15% fetal bovine serum (FBS) supplemented with 600μL human insulin solution (Sigma I9278) and 3μL 2-mercaptoethanol 50mM (Gibco 31350–010). The human embryonic kidney cell line, HEK293 (RRID: CVCL_0045), was obtained from American Type Culture Collection (ATCC, CRL-1573) and cultured in MEM (Corning, 10–010-CV) with 10% FBS. The anaplastic WT cell line 17.94 (RRID: CVCL_D707) was obtained from the European Collection of Authenticated Cell Cultures and cultured in DMEM (Gibco, 11965–092) with 20% FBS.(34) The favorable histology WT COGW408 cell line was obtained from the Children’s Oncology Group/Alex’s Lemonade Stand Childhood Cancer Repository and cultured in IMDM (Gibco, 12440–053) with 10% FBS supplemented with 1X ITS solution (Corning 25–800-CR). The favorable histology WT cell line PDM182 (HCM-BROD-0051-C64; ATCCPDM-182) was obtained from ATCC and cultured in Conditional Medium (Propagenix, 256–100). Short tandem repeat (STR) profiling and screening using the LookOut Mycoplasma Detection kit (Sigma, MP0035) and JumpStart Taq DNA Polymerase (Sigma, D9307) were performed monthly.
Compounds and siRNA
The following chemicals and siRNAs were used: QC6352 (MedChemExpress, HY-104048), polyethylene glycol vehicle (Sigma, 202371–500G), Doxorubicin (Sigma, D1515), control non-targeting siRNA (Dharmacon, 001810–01-05), KDM4A siRNA (Invitrogen, s18636), KDM4B siRNA (Invitrogen, s22867) and KDM4C siRNA (Invitrogen, s225930).
Crystal violet staining
Three thousand cells were plated in 6-well culture plates; QC6352 was added the next day. The cells were allowed to grow for 2 weeks. Cells were washed with phosphate buffered saline without calcium or magnesium (PBS, Gibco) and fixed with 4% paraformaldehyde in PBS (PFA, J19943-K2, Thermo Scientific) for 15–20 minutes. Cells were washed with PBS and stained with 0.3% crystal violet (Sigma, HT90132) for 1 hour.
PrestoBlue assay
Cells (3,000 per well) were plated in 96-well plates and incubated at 37°C for 24 hours. Cells were treated with QC6352 at different final concentrations (10,000, 1,000, 500, 250, 50, 25, 12.5, 5, 2.5, 0.5 and 0 nM) for 5 days. Next, 10 μl of Prestoblue reagent (Invitrogen, A13262) were added to each well and plates were incubated at 37°C for 30 – 60 minutes. Fluorescence (560 nm-excitation and 590-nm emission) was measured using a BioTeck SYNERGY H1 microplate reader. GraphPad Prism was used to generate curves and to calculate IC50 values.
Cell cycle analysis
One hundred thousand cells were plated in 6-well plates and treated with QC6352 (25 nM) for 72 hours. Cells were trypsinized and centrifuged at 3000 rpm for 3 minutes and washed with ice cold-PBS. Cells were washed with staining media (PBS, 5% FBS) and resuspended in propidium iodide (PI) (0.05 mg/ml PI, 0.1 % (weight/volume) sodium citrate, 0.1 % (volume/volume) Triton X-100) at 1×10e6 cells/ml. Cells were vortexed and treated with 10 μl RNAse (0.2 mg/ml) for 30 minutes, filtered and analyzed by flow cytometry using an LSR Fortessa (BD Biosciences) instrument. Data were analyzed using ModFit modeling software.
Annexin V
After QC6352 treatment, 3×105 cells were centrifuged and washed with staining media (PBS, 5% FBS) and resuspended in 100 μl 1X Annexin Binding Buffer (BD, 556454) with 1:20 Annexin V APC (Tonbo, 20–6409-T100) and 1:10 DAPI solution. Cells were incubated at room temperature for 15 minutes in the dark. After incubation, 150 μl of 1X Annexin Binding Buffer was added to cells. Cells were analyzed by flow cytometry using BD FACSDiva (RRID:SCR_001456).
Western blot
Antibodies used are listed in Supplementary Table 1. Cells were washed with ice-cold PBS and lysed using 2x sample loading buffer (1 M Tris HCL pH 6.8, 10% SDS, 20% glycerol, 5% β-mercaptoethanol and 0.001% bromophenol blue). On ice, cell lysates were sonicated with 5–10s bursts at 30% amplitude using a VIBRA cell sonicator (Sonics and Materials Inc.). Lysates were heated at 95°C for 10 minutes. 15–20 μl of lysates were separated by 4–12% gradient SDS polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane using iBlot 2 dry transfer system (Life Technologies). Membranes were blocked in 5% skim milk for 1 hour at room temperature with mild shaking. Membranes were incubated with primary antibody overnight at 4°C. Membranes were washed 3 times with TBST. Membranes were incubated with secondary antibody (Invitrogen, A24537, anti-rabbit or A24518, anti-mouse, 1:4,000) for 1 hour at room temperature, followed by 3 TBST washes. Membranes were incubated with SuperSignal West Pico Plus Chemiluminescent Substrate for 1–2 minutes and imaged using a LI-COR Odyssey system.
Comet assay
The single cell electrophoresis Comet assay measured DNA damage (Abcam, ab238544) per the manufacturer’s protocol. Results were visualized under a fluorescent microscope (EVOS M5000) at 20X magnification. Tail length, tail moment, and tail DNA percentage were analyzed using OpenComet software.(35)
Scratch wound migration assay
Cells were seeded in a 96-well ImageLock plate at a density of 30,000–40,000 per well and incubated at 37°C for 24 hours. Linear scratches were created with the IncuCyte Wound Maker (Essen). Cells were washed twice with media. Cells were treated with QC6352 or KDM4 siRNAs. Plates were scanned using the IncuCyte S3 system every 6 hours for 5 days (10x objective).
Proliferation assay
Cells (3,000 – 4,000) were plated in 96-well ImageLock plates and placed in the IncuCyte system. Plates were scanned every 6 hours. The next day, media were replaced with 100 μl QC6352 or vehicle control in fresh media and scanning was continued for 5 – 7 days (10x objective).
Spheroid growth
Cells (5,000 in 80 μl) were plated in ultra-low-attachment 96-well plates and centrifuged at 150g for 10 minutes. Plates were then placed in the IncuCyte system and spheroid growth was monitored every six hours using the IncuCyte Spheroid module. On day 3, spheroids were treated with 20 μl of fresh media containing QC6352 and monitored every 6 hours for 2 weeks.
Immunofluorescence
Cells were fixed in 4% PFA in PBS for 5–10 minutes. Subsequently, cells were permeabilized with 0.3 % Triton X-100 for 15–20 minutes. After incubation with 5% normal goat serum for 1 hour at room temperature, cells were probed for KDM4A-C, fibrillarin, RNA194 (RNA polymerase I), or H3K9me3 overnight (Supplementary Table 1) at 4°C followed by incubation with secondary antibodies (1:400; Invitrogen, A31572, Alexa Fluor 555 anti- Rabbit (Red) or Alexa Fluor 488 anti- Mouse (Green)) for 1 hour at room temperature. Slides were mounted with prolong antifade with DAPI, and images were captured using a Zeiss LSM 780 microscope.
Immunoprecipitation
After washing cells with ice-cold PBS, 400 μl of ice-cold lysis buffer (Pierce IP lysis buffer, 87787; 25 mM Tris-HCL pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40 and 5% glycerol) were added to cells and incubated on ice for 5 to 10 minutes with periodic mixing. The cell extracts were cleared by centrifugation 13,000g for 10 minutes at 4°C. An equal amount of protein (400 μg) from the control and treatment groups was incubated overnight at 4°C with indicated antibody (Supplementary Table 1) followed by incubation with beads (Dynabeads Protein G, 10003D, Invitrogen) for 3 hours at 4°C with gentle shaking. Beads were separated using a column-based magnet and immunocomplexes were released by heating (75°C for 10 minutes) in 50 μl of 2X lysis buffer and studied by Western blotting.
Small interfering RNA transfection
When cells were 50–70% confluent, transfection mixture was prepared using siRNA (10nM) and Lipofectamine RNAiMAX (7μl, Invitrogen, cat 13778–075) in Opti-MEM and incubated for 15–20 minutes at room temperature. The mixture was added to cells and incubated for 48–72 hours at 37°C.
RNA-Seq
Total RNA was isolated using the RNeasy Plus Mini Kit (Qiagen, 74134). RNA-seq library preparation, sequencing, mapping, and generation of gene level reads were previously described(36). Raw RNA-seq FastQ files were input into the Workflow for the Analysis of RNA-seq Differential Expression (WARDEN) program using the St. Jude Cloud-based analysis tool (37). Using WARDEN, log2 counts per million (log2CPM) values were used to quantify gene expression according to mapped RNA-seq reads. WARDEN was used to generate input files for gene set enrichment analysis (GSEA)(38). GSEA software v4.0.3 (UC San Diego and Broad Institute; RRID: SCR_005724) analyzed differential expression of Gene Ontology (GO) lists.
PRISM Multiplexed Cell Line Profiling Analysis
PRISM Multiplexed Cell Line Profiling (Broad Institute) was performed as previously described.(39,40) Over 930 Cancer Cell Line Encyclopedia (CCLE; https://sites.broadinstitute.org/ccle/)-indexed cell lines were stably transfected with a unique DNA barcode. Cell lines were assigned to assay-ready pools according to similar doubling times. Pools of cells were treated for 5 days with QC6352 (13.7 nM, 41.2 nM, 123 nM, 370.4 nM, 1.11μM, 3.33 μM, 10 μM), cells were lysed, and mRNA was isolated. Transcribed barcode sequences were amplified by PCR and detected by a Luminex FlexMap 3D scanner. The QC6352 cell line sensitivity signature was derived from the post-treatment quantity of each barcode and used to fit dose-response curves for each cell line. The resulting log2 area under curve was compared to baseline genome-wide RNA expression (https://depmap.org) to correlate gene expression and QC6352 sensitivity. A lineage regressed out analysis in which cellular lineage was considered a confounder was used. The top 100 genes with expression associated with QC6352 sensitivity in this analysis were used for gene set analysis in the Enrichr database using Jensen COMPARTMENT, GO Cellular Components, and GO Biological Process sets.(41–43)
In vivo efficacy of QC6352
Animal experiments were performed under a St Jude Children’s Research Hospital Animal Care and Use Committee-approved protocol (#641). Male and female NSG mice (strain NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ; RRID:IMSR_JAX:005557), 6–7 weeks old were subcutaneously injected with HEK293 cells (3×106; 50% Matrigel). When tumors reached 100–200 mm3, control and treatment groups were established. QC6352 (25 mg/kg) or vehicle control were administered via oral gavage for 3 weeks (2 times a day for 5 consecutive days and two days off). Murine body weight and tumor volume were measured twice weekly. After euthanasia, tumors were collected and immediately snap-frozen in liquid nitrogen.
Electron microscopy
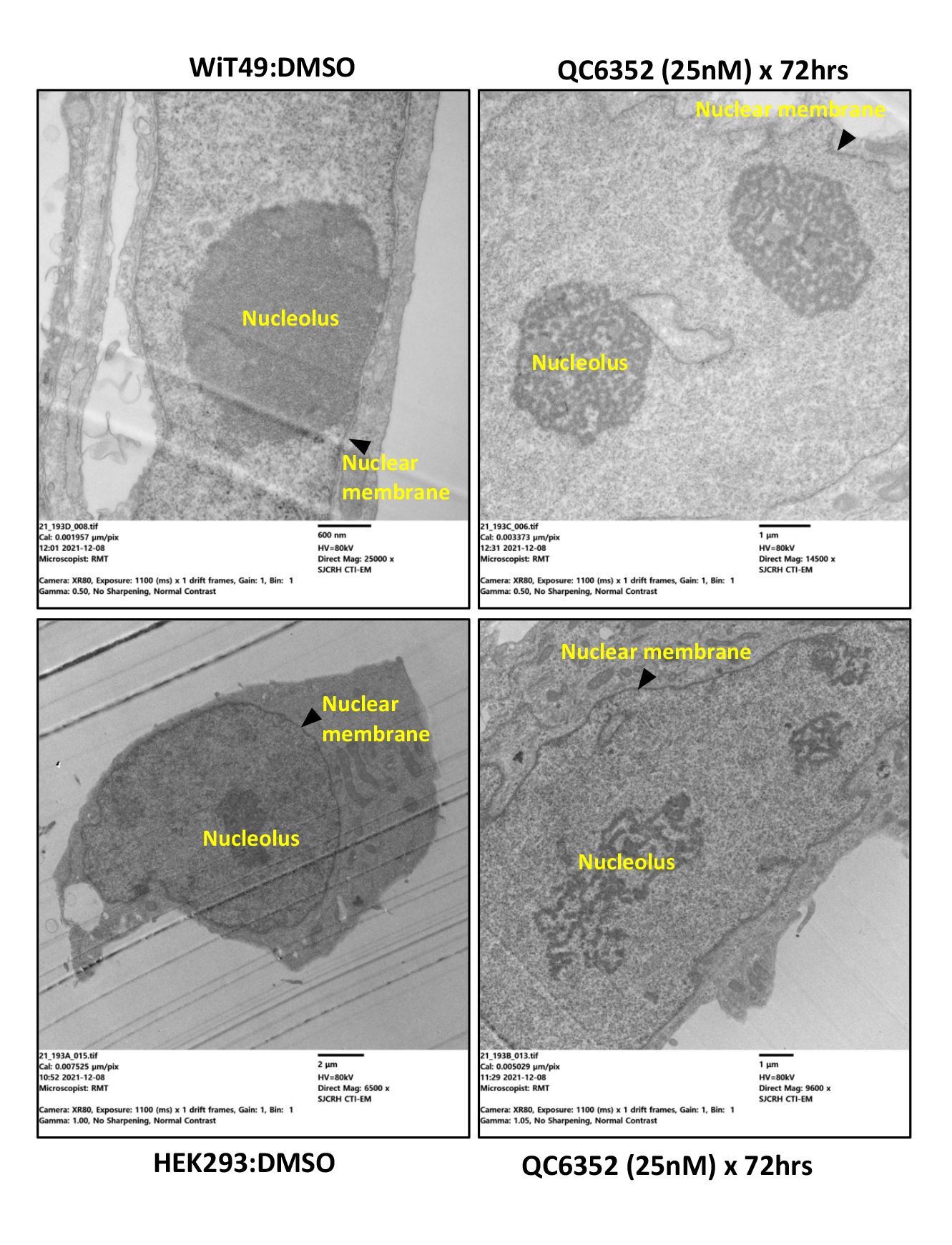
After treatment with QC6352 (25 nM) for 72 hours, cells were washed with PBS followed by fixation in 0.1M cacodylate buffer containing 2.5% glutaraldehyde and 2% paraformaldehyde. Samples were post fixed in reduced osmium tetroxide and contrasted with aqueous uranyl acetate. Dehydration was by an ascending series of ethanol to 100% followed by 100% propylene oxide. Samples were infiltrated with EmBed-812 and polymerized at 60°C. Embedded samples were sectioned at 70nm on a Leica ultramicrotome and examined in a ThermoFisher Scientific TF20 transmission electron microscope at 80kV. Digital micrographs were captured with an Advanced Microscopy Techniques imaging system.
qRT-PCR for rRNA transcription, ChIP-PCR for KDM4A occupancy at rDNA promoter, and Click-IT assay for new protein synthesis,
Methods are outlined in the Supplementary Methods section and primer sequences are provided in Supplementary Table 2.
Statistics
In vitro experiments were performed using a minimum of three technical replicates. Data were tested for a normal distribution using the Kolmogorov-Smirnov test. Normally distributed data were compared using the two-tailed paired t-test. Non-normally distributed data were compared using the Kruskal-Wallis test. Data are displayed as mean with standard deviation or standard error of the mean as indicated. Statistical comparisons were made with GraphPad Prism software (RRID:SCR_002798). Throughout the manuscript and supplementary figures, the following indicators correspond to the listed level of statistical significance: non-significant (ns), p>0.05, * p≤0.05, ** p≤0.01, ***P≤0.001, ****P≤0.0001.
Data Availability Statement
The datasets generated during the current study are available from the corresponding authors on reasonable request. The WiT49 and HEK293 QC6352 RNA-seq data are available on the Gene Expression Omnibus Database (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE226112.
Results
KDM4A expression and Wilms tumor
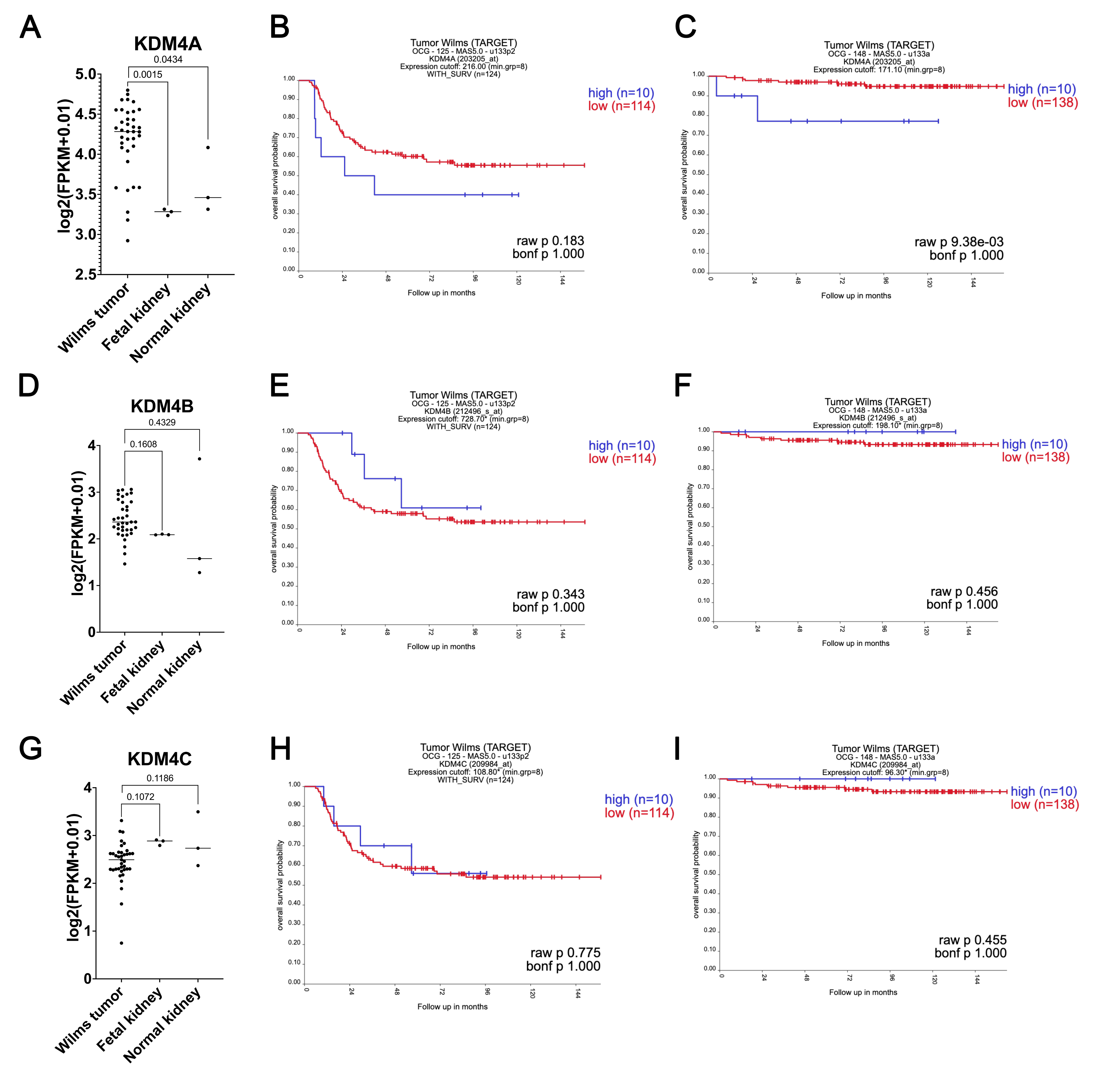
Publicly available RNA-seq data were queried to compare KDM4A, B, and C expression among WT, fetal kidney, and normal kidney samples. KDM4A (but not KDM4B or C) was found to exhibit significantly higher expression in WT compared to fetal kidney (p=0.0015) and normal kidney (p=0.0434) samples (Supplementary Figure 1).(36) The NCI-TARGET gene expression microarray data were queried using the KaplanScanner tool of the R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl) to determine potential association between high versus low tumor expression of KDM4A, B, and C and overall survival probability. Among validation cohort samples (n=148; predominantly favorable histology WT) high expression of KDM4A (but not KDM4B or C) was associated with inferior overall survival (p<0.0001; Supplementary Figure 1). Among discovery cohort samples (n=124; exclusively anaplastic WT or favorable histology WT with disease relapse) there was a nonsignificant association between high KDM4A expression and inferior overall survival (p=0.183); however, this trend was not observed for KDM4B or C (Supplementary Figure 1).
QC6352 sensitivity among embryonic renal tumor cells
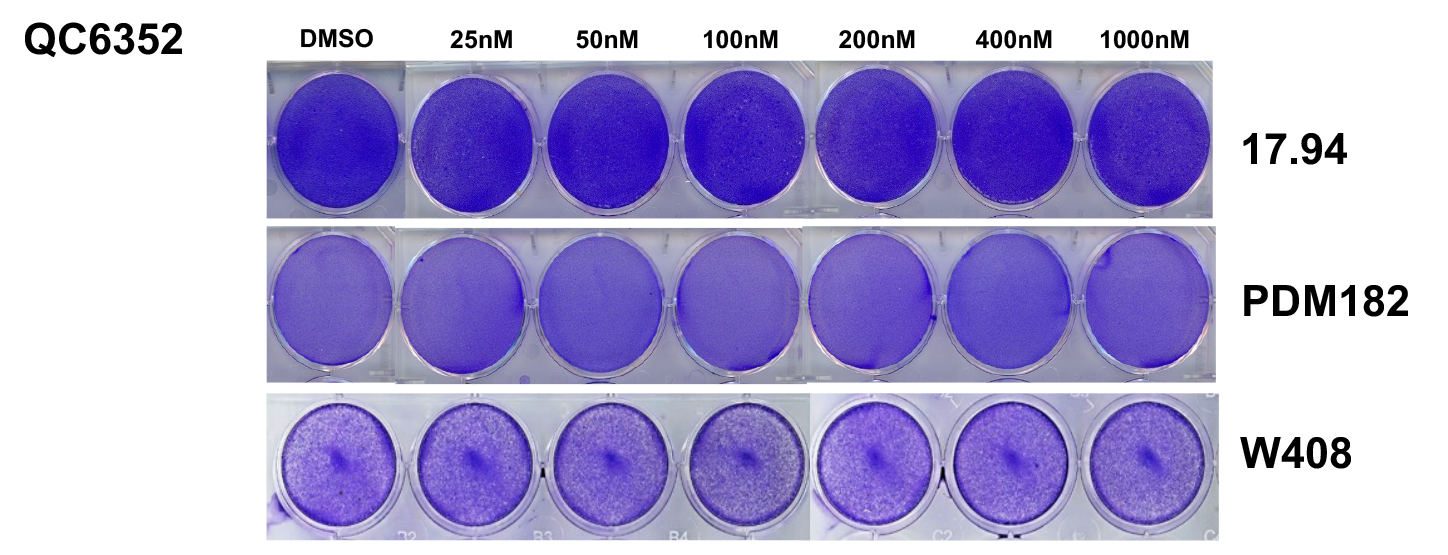
QC6352 sensitivity of anaplastic WT cell lines WiT49 and 17.94, favorable histology WT cell lines COG-W-408 and PDM182, and the tumor-forming human embryonic kidney cell line HEK293 was assessed using the PrestoBlue assay. WiT49 (IC50 36.55 nM) and HEK293 (IC50 4.24 nM) were sensitive to QC6352 at low nanomolar concentrations, while remaining lines were not sensitive (IC50s > 10,000 nM; Figure 1A). Of note, the QC6352 LC50 in WiT49 and HEK293 was >10,000 nM, indicative of a cytostatic, but not cytotoxic, response. The dose-dependent sensitivities of WiT49 and HEK293 to QC6352 (and insensitivity of other cell lines) were confirmed using crystal violet assays (Figure 1B; Supplementary Figure 2). QC6352 treatment caused reduced proliferation and migration in WiT49 and HEK293 assessed using phase-contrast live-cell microscopy (Figure 1C–D).
Fig.1: QC6352 sensitivity among oncogenic cells of renal embryonic lineage.
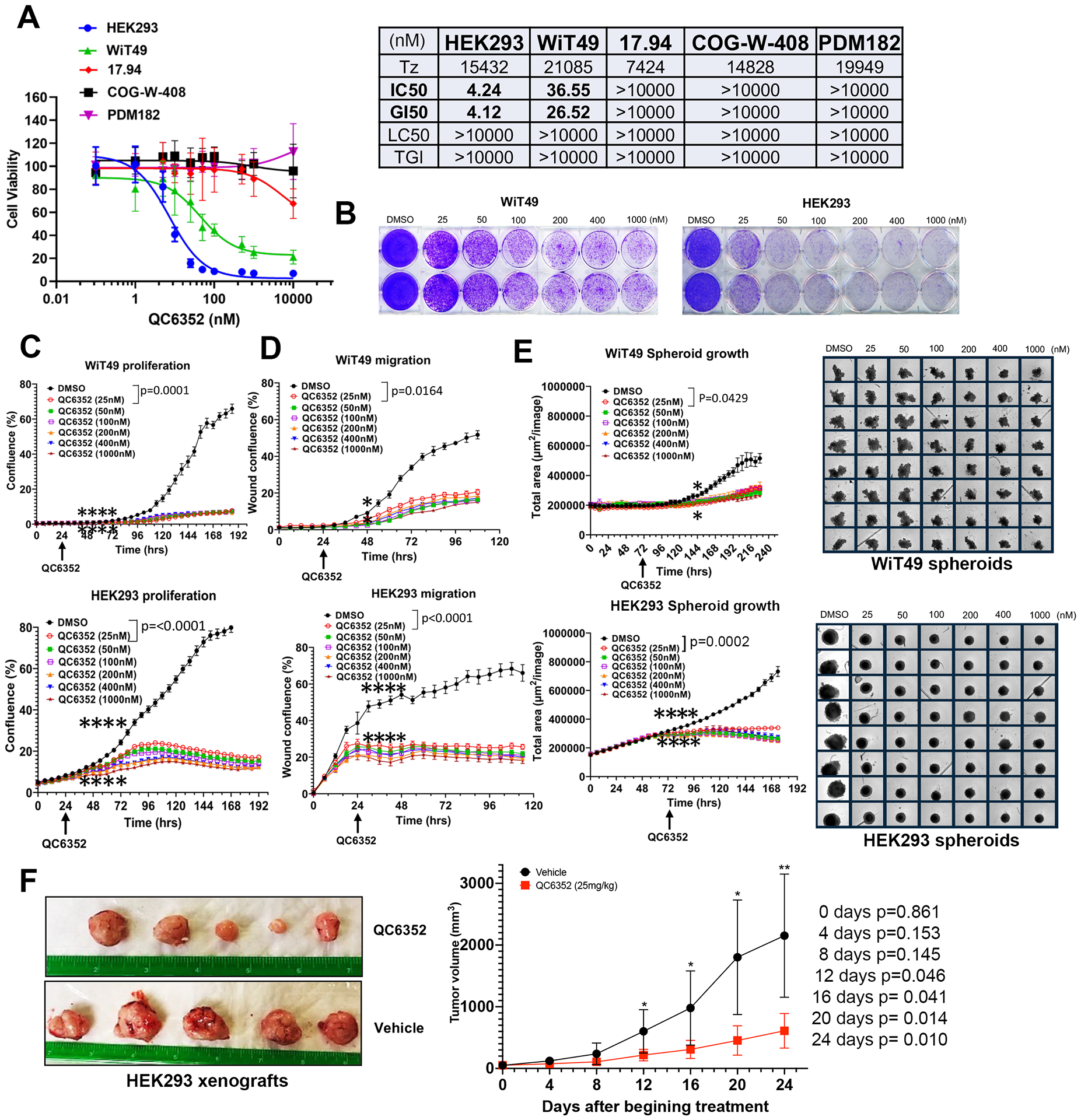
(A) PrestoBlue assay dose response curves display the sensitivity (IC50) of Wilms tumor cell lines (WiT49, 17.94, COGW408, and PDM182) and the tumor-forming embryonic kidney cell line, HEK293 to the KDM4 inhibitor QC6352. (B) Crystal violet assay demonstrates a dose-dependent response to QC6352 in WiT49 and HEK293. (C) Live-cell microscopy proliferation assay demonstrates reduced proliferation in QC6352-treated WiT49 and HEK293. (D) Scratch wound cell migration assay performed using phase-contrast live-cell microscopy demonstrates decreased migration in QC6352-treated WiT49 and HEK293. (E) QC6352 significantly reduces spheroid growth in WiT49 and HEK293. For C-E, the rightmost asterix indicates the timepoint at which separation in curves is statistically significant (p<0.05; paired two-tailed t-test) and persists throughout remaining indicated duration. (F) Tumor photographs and volume curves from HEK293 subcutaneous xenografts in NSG mice demonstrate a volumetric response to QC6352 (25 mg/kg oral gavage for three weeks) compared to vehicle control (paired two tailed t-test values shown).
To more closely approximate cell-cell contacts characteristic of human tumors in vitro, the QC6352 sensitivity was tested in 3D culture conditions using spheroids formed from WiT49 and HEK293. Spheroid growth was significantly impaired by QC6352 treatment in WiT49 and HEK293 (Figure 1E). Subcutaneous HEK293 xenografts formed tumors in NSG mice, while WiT49 did not despite multiple attempts. Treatment of HEK293 tumors with 25 mg/kg QC6352 caused significant reduction in tumor growth (Figure 1F).
QC6352 causes cell cycle arrest and DNA damage
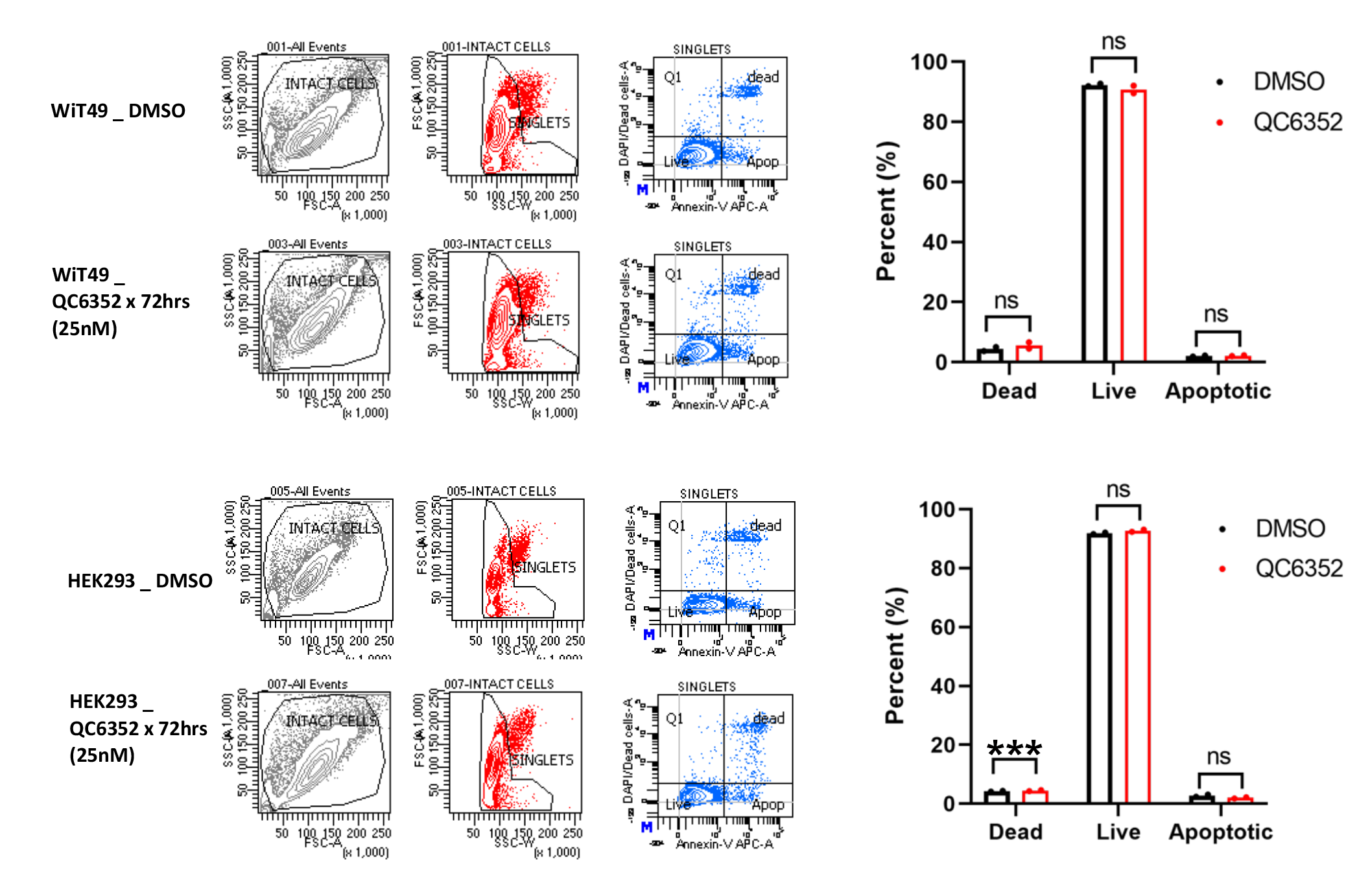
Treatment with 25nM QC6352 for 72 hours resulted in an increased proportion of WiT49 and HEK293 cells in the S-phase (paired t-test p=0.0097 WiT49; p=0.0175 HEK293) of the cell cycle and a decreased proportion of cells in the G0/G1 phase (p=0.0274 WiT49, p=0.0079 HEK293) as determined by propidium iodide staining and flow cytometry (Figure 2A). However, Annexin-V flow cytometry did not demonstrate a biologically significant increase in the proportion of dead (p=0.205 WiT49, p<0.0001 HEK293, but absolute difference less than 1%) or apoptotic (p=0.795 WiT49, p=0.125 HEK293) cells with QC6352 treatment (Supplementary Figure 3). These data demonstrate a cytostatic response to QC6352 marked by S-phase cell cycle arrest.
Fig.2: QC6352 causes cell cycle arrest and DNA damage in WiT49 and HEK293 cells.
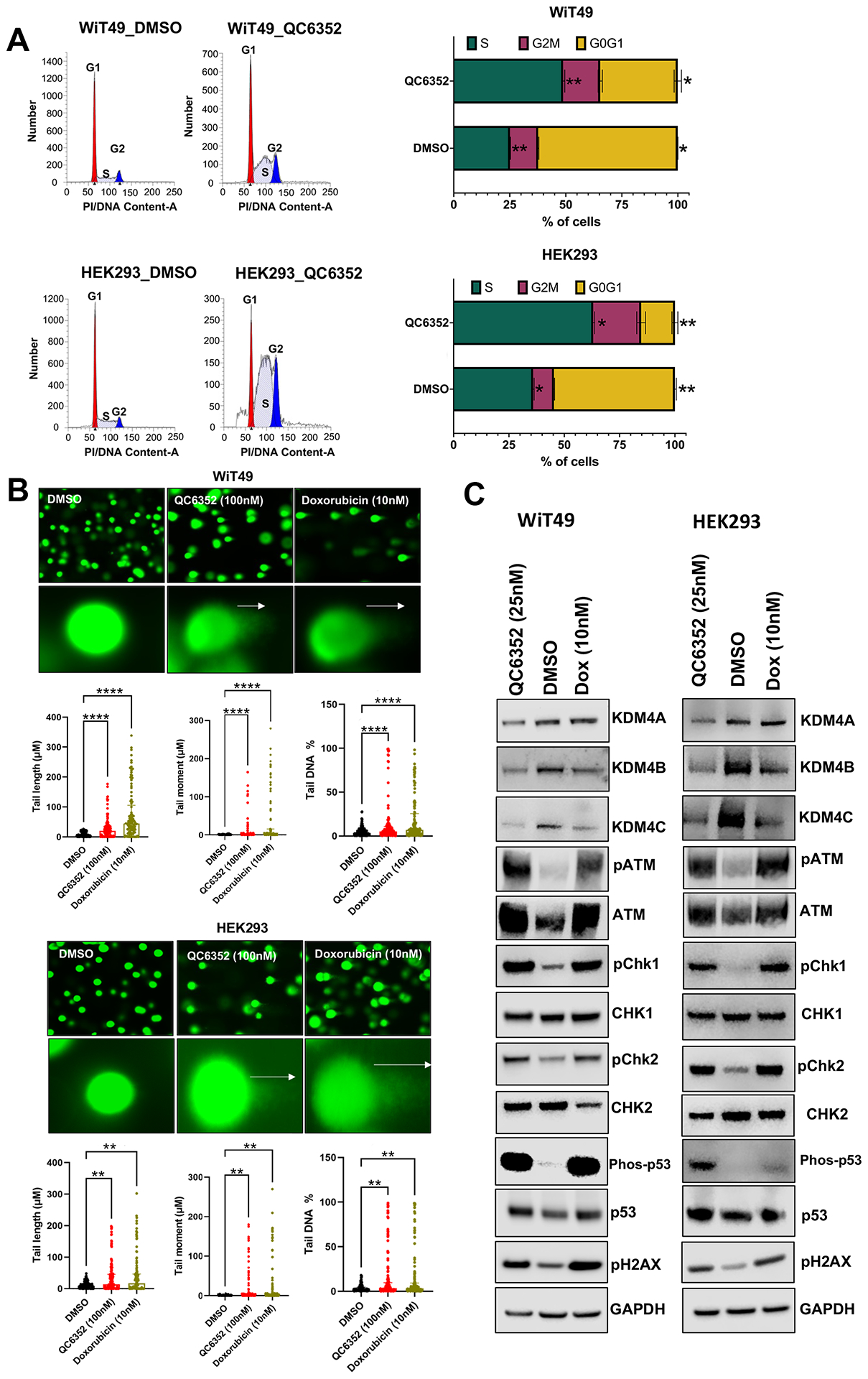
(A) Propidium iodide staining with flow cytometry demonstrates an increased proportion of QC6352-treated (25nM for 72 hours) cells in the S-phase (paired two-tailed t-test p=0.0097 WiT49; p=0.0175 HEK293) and reduced proportion in the G0G1 phase (p=0.0274 WiT49; p=0.0079 HEK293). (B) QC6352 (100nM for 72 hours) induces DNA damage detected by the Comet assay. White arrows show “comet tails” indicative of DNA damage. Images were taken at 20X magnification. Doxorubicin-treated cells (10nM for 72 hours) are included as positive control. Kruskal-Wallis p-values are represented. (C) Western blot analysis for DNA damage and activation of checkpoint response proteins. Cells were treated with QC6352 (25nM) for 72 hours. Doxorubicin-treated cells (10nM for 72 hours) are included as a positive control. GAPDH is used as a loading control.
Next, the reason for S-phase cell cycle arrest with QC6352 treatment was interrogated. The Comet assay demonstrated a pattern consistent with double strand DNA breaks in WiT49 and HEK293 (Figure 2B). To validate DNA damage and interrogate the consequences of this phenomenon, DNA damage checkpoint response protein activation was assessed by western blot. Doxorubicin, which is known to induce DNA double strand breaks by inhibiting Top II, was used as a positive control. Like doxorubicin, QC6352 treatment caused upregulation of pATM, pCHK1, pCHK2, phospho-p53, and pH2AX compared to vehicle control in both WiT49 and HEK293 (Figure 2C). These data indicate that QC6352 causes double strand DNA breaks that result in S-phase cell cycle arrest and trigger a DNA-damage checkpoint response in WiT49 and HEK293.
QC6352 causes impaired ribosome biogenesis
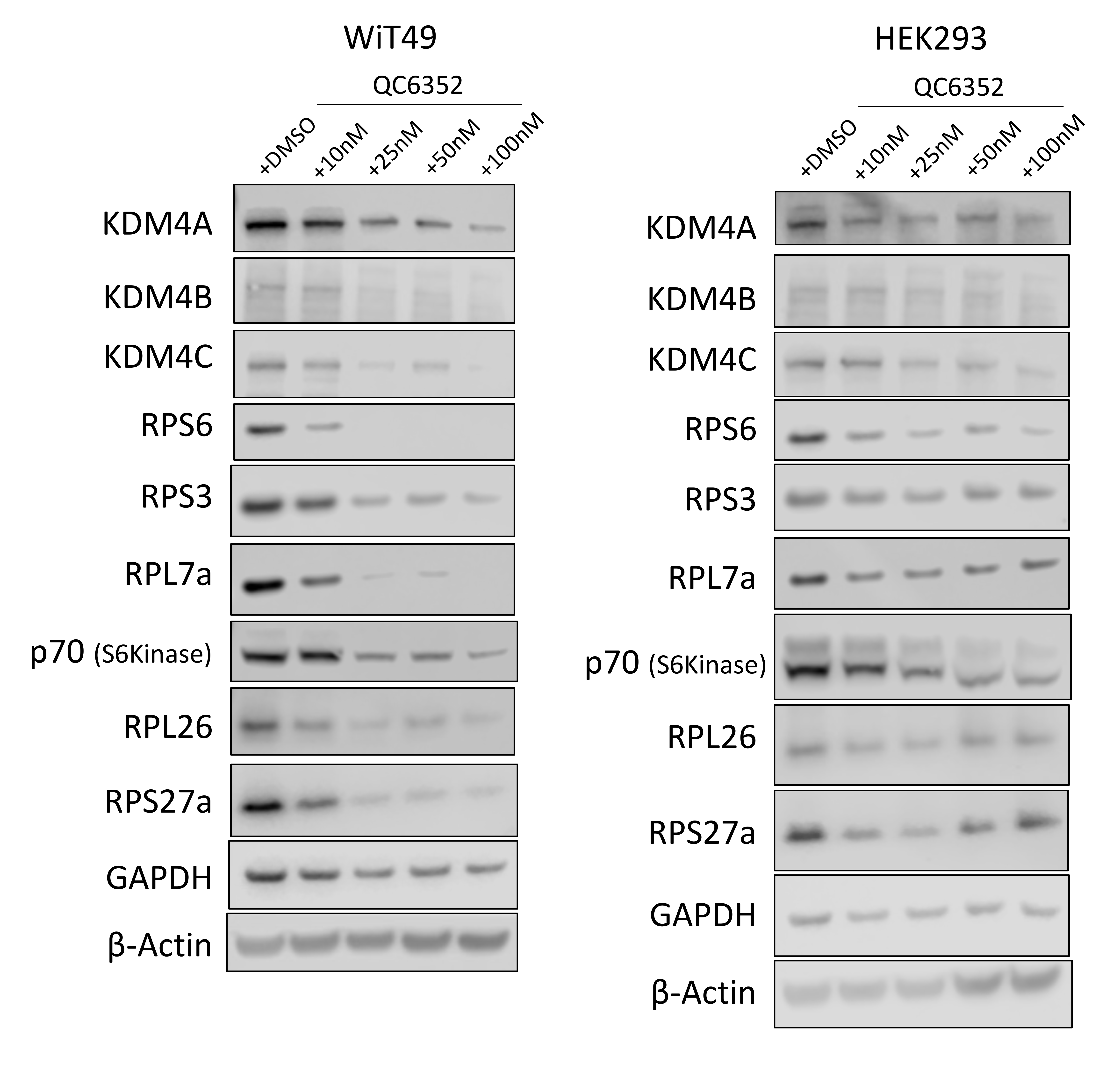
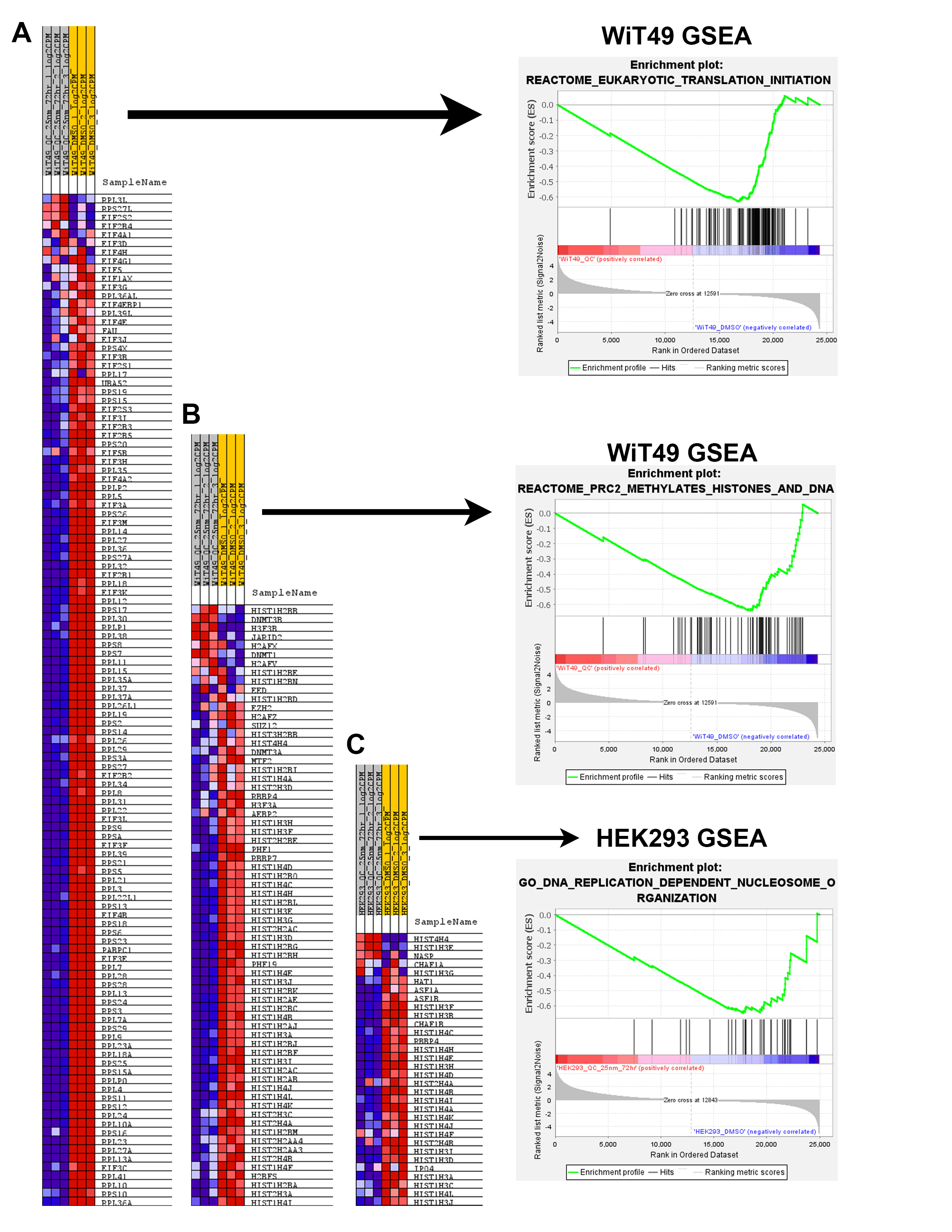
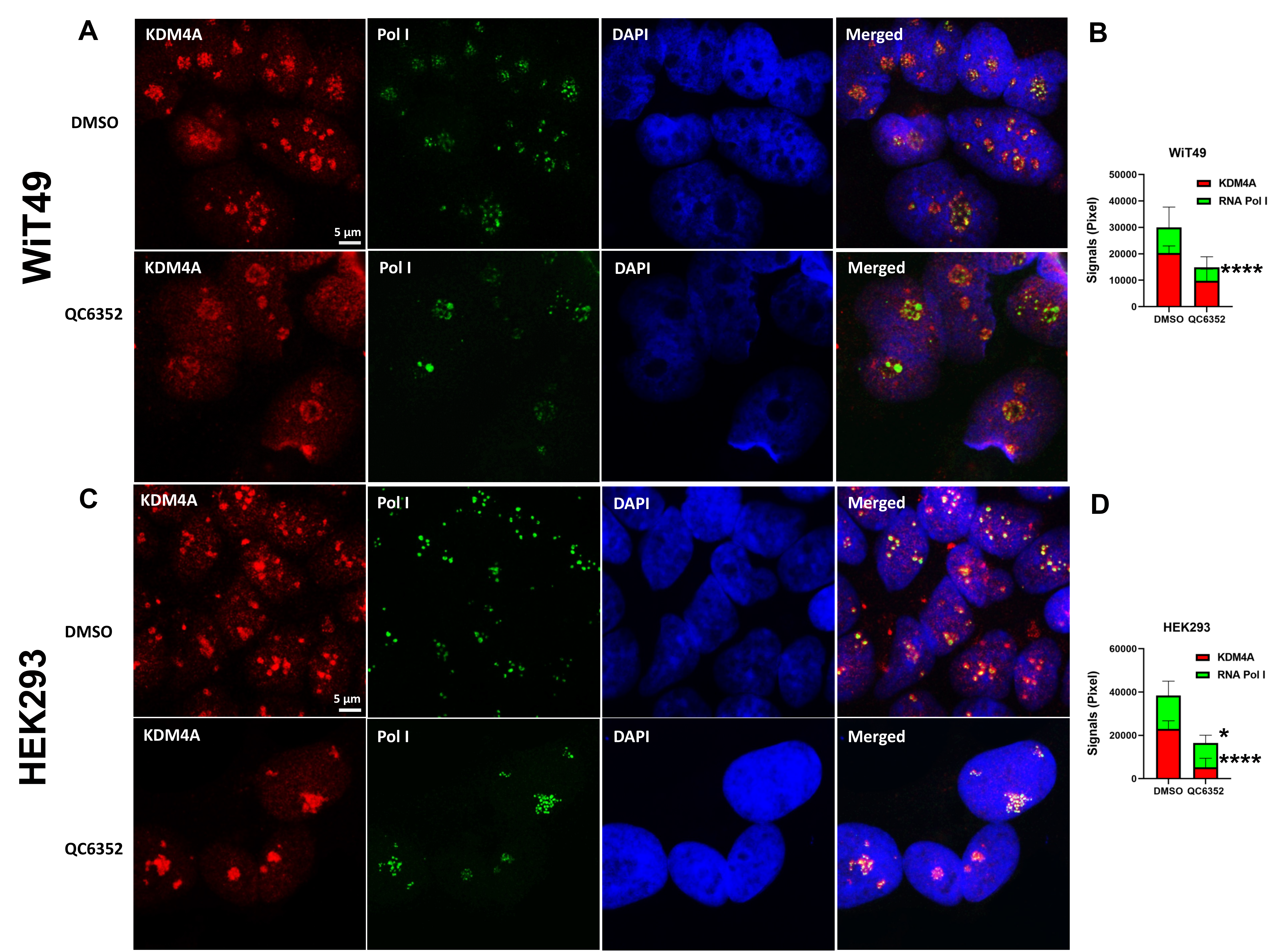
RNA-sequencing was performed on WiT49 and HEK293 cells that were treated with 25 nM QC6352 for 72 hours and compared to vehicle control. Gene set enrichment analysis (GSEA) demonstrated the KEGG_RIBOSOME pathway to be the second most downregulated pathway in WiT49 (Normalized Enrichment Score (NES)=−2.18, Familywise Error Rate (FWER) p-value<0.001) and the most significantly downregulated pathway in HEK293 (NES=−2.44; FWER p<0.001; Figure 3A–B; Supplementary Data File 1). Ribosomal RNA (rRNA) transcription was assessed by qRT-PCR of the human 47S-ITS1 transcript, which demonstrated a dose-dependent reduction in rRNA transcription with increasing doses of QC6352 treatment in WiT49 and HEK293 (Figure 3C). QC6352 treatment reduced KDM4A binding to the promoter and non-transcribed regions of rDNA as assessed by KDM4A ChIP-PCR (Figure 3D). We observed downregulation of the ribosomal proteins RPS6, RPS3, RPL7a, RPL26, and RPS27a and p70s6 kinase (phosphorylates RPS6 to induce protein synthesis in the ribosome) in QC6352-treated WiT49 and HEK293 cells by western blot (Figure 3E, Supplementary Figure 4). Furthermore, snap frozen HEK293 QC6352-treated tumors demonstrated a marked reduction in KDM4A, KDM4B, and KDM4C expression levels and reduction in ribosomal protein S6, RNA polymerase I, and N-MYC by western blot (Figure 3F). Because we observed reduction of rRNA and ribosomal protein gene transcription/expression in response to QC6352 treatment and the function of the ribosome is to synthesize proteins, we performed the Click-iT assay for metabolic labeling of newly synthesized proteins. We noted blockade of protein synthesis in response to QC6352 treatment in WiT49 and HEK293 cells (Figure 3G).
Fig.3: QC6352 causes impaired ribosome biogenesis in WiT49 and HEK293 cells.
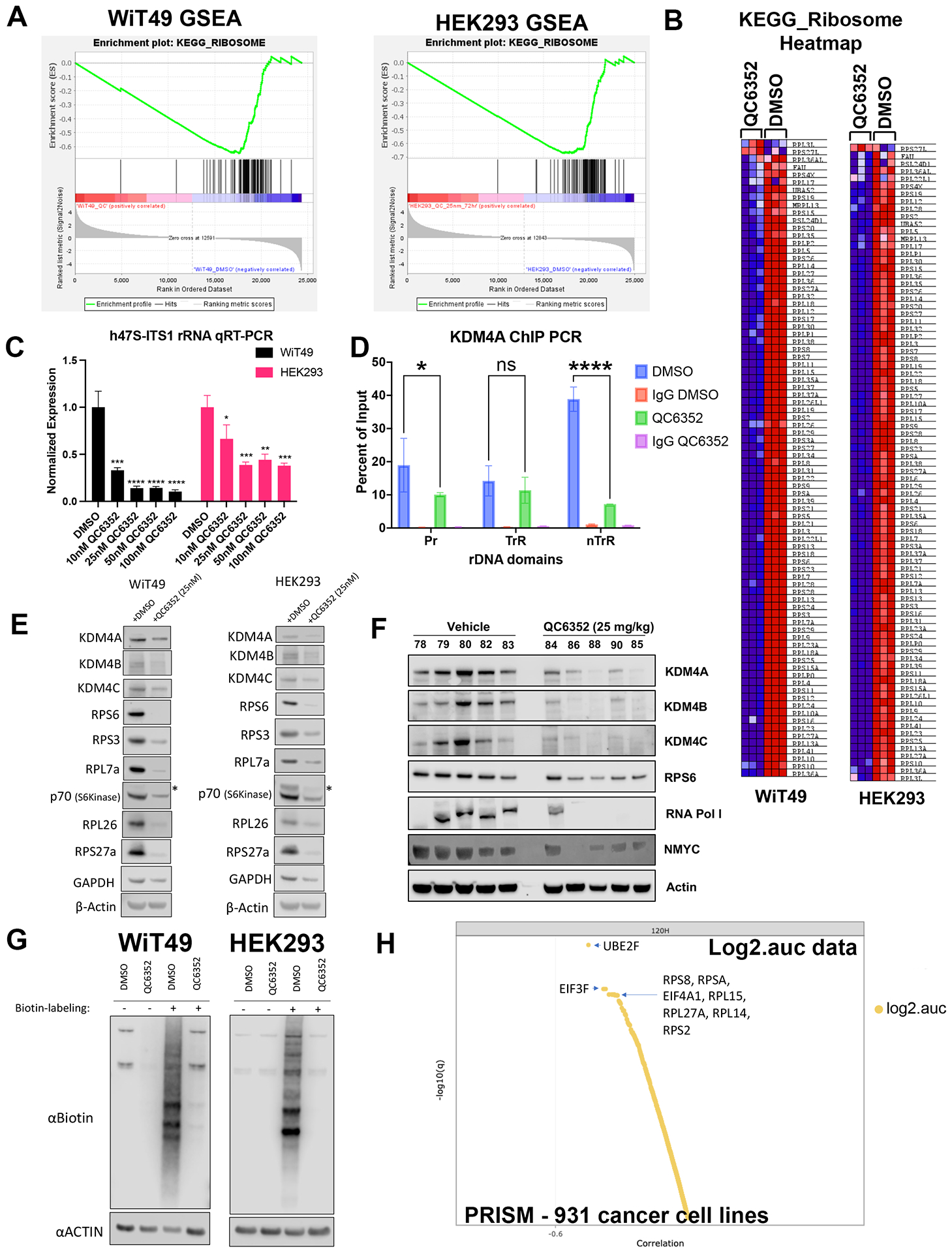
RNA-seq was performed on WiT49 and HEK293 cells treated with 25 nM QC6352 for 72 hours. (A) Gene set enrichment analysis demonstrated the KEGG_RIBOSOME pathway to be significantly downregulated in both WiT49 and HEK293 cells. (B) Heatmap display of the KEGG_RIBOSOME pathway gene set demonstrates marked reduction of ribosomal protein gene expression with QC6352 treatment in HEK293 and WiT49. (C) qRT-PCR for human 47S rRNA demonstrates a dose-dependent reduction of ribosomal RNA transcription with QC6352 treatment for 72 hours in WiT49 and HEK293 cells (t-test p values *<0.05, **<0.01, ***<0.001, ****<0.0001). (D)25nM QC6352 treatment for 72 hours resulted in decreased KDM4A occupancy at the human rDNA promoter region (Pr) and nontranscribed regions (nTrR), but not the transcribed regions (TrR) in WiT49 cells as assessed by KDM4A ChIP PCR. (E) Western blot shows downregulation of KDM4A, KDM4B, KDM4C, the ribosomal proteins RPS6, RPS3, RPL7a, RPL26, and RPS27a, and P70S6 kinase in response to QC6352 treatment. (F) HEK293 xenograft tumors harvested at the completion of QC6352 treatment (25 mg/kg oral gavage for three weeks) demonstrate a reduction in KDM4A, KDM4B, KDM4C, ribosomal protein S6, RNA polymerase I, and N-MYC by western blot compared to vehicle control. (G) Click-iT metabolic labeling of nascent protein synthesis demonstrated blockade of new protein synthesis in response to QC6352 treatment in WiT49 and HEK293 cells. (H) PRISM multiplexed cell line profiling RNA expression and compound sensitivity correlation analysis data from 931 cancer cell lines demonstrated that six of the top ten genes with basal expression that correlated to QC6352 sensitivity were ribosomal genes: RPS8, RPSA, RPL15, RPL27A, RPL14, and RPS and two of the top 10 genes were translational elongation factors: EIF3F and EIF4A1. The top “hit” was the ubiquitination-associated NEDD8-conjugating enzyme UBE2F.
Other notable downregulated pathways on RNA-seq associated with QC6352 treatment included the REACTOME_EUKARYOTIC_TRANSLATION_INTIATION pathway (WiT49 NES=−2.19; FWER p<0.001; HEK293 NES=−2.30; FWER p<0.001) which contains many ribosomal protein genes and translational initiation factors. Additionally, the REACTOME_PRC2_METHYLATES_HISTONES_AND_DNA pathway in WiT49 (NES=−2.10; FWER p=0.001) and the GO_DNA_REPLICATION_DEPENDENT_NUCLEOSOME_ORGANZATION in HEK293 (NES=−1.90; FDR q value 0.033; FWER p=0.38) were found to be among the top downregulated pathways with QC6352 treatment and both contain replicative canonical histone genes (Supplementary Figure 5, Supplementary Data File 1).
Transcription of ribosomal protein genes is associated with QC6352 sensitivity across human cancer cell lines
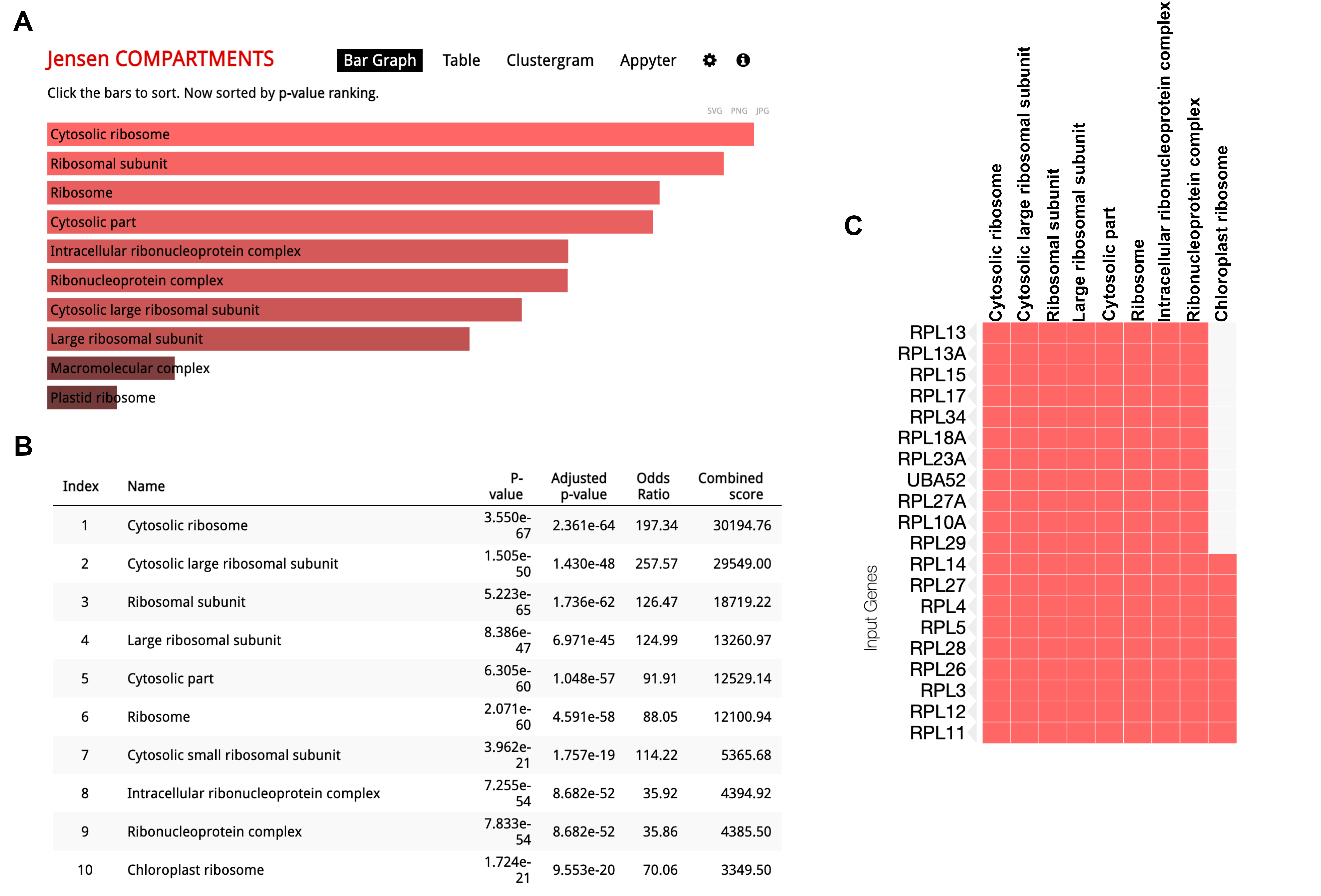
To determine QC6352 sensitivity beyond embryonic renal tumor cell lines, QC6352 was tested at 8 concentrations in 931 barcoded cancer cell lines using PRISM multiplexed cell line profiling. QC6352 sensitivity (log2 area under the dose-response curve) was correlated with genome-wide basal mRNA expression across the panel of cell lines, with lineage treated as a confounder. Six of the top ten genes with expression that correlated to QC6352 sensitivity were ribosomal protein genes: RPS8, RPSA, RPL15, RPL27A, RPL14, and RPS2 (Figure 3H). Two translational elongation factors EIF3F and EIF4A1 were also among the top ten genes with RNA transcript levels associated with QC6352 sensitivity. Gene set analysis was performed on the top 100 genes with expression that correlated with QC6352 sensitivity using the Enrichr database. Nine of the top 10 Jensen COMPARTMENT pathways analyzed using the Enrichr database were associated with the ribosome (Supplementary Figure 6). These data show that high basal expression of ribosomal protein genes is associated with increased QC6352 sensitivity across the spectrum of cancer cell lines, which reinforces the finding of QC6352 impairing ribosome biogenesis as a mechanism of action in oncogenic cells of renal embryonic lineage.
KDM4A and the nucleolus
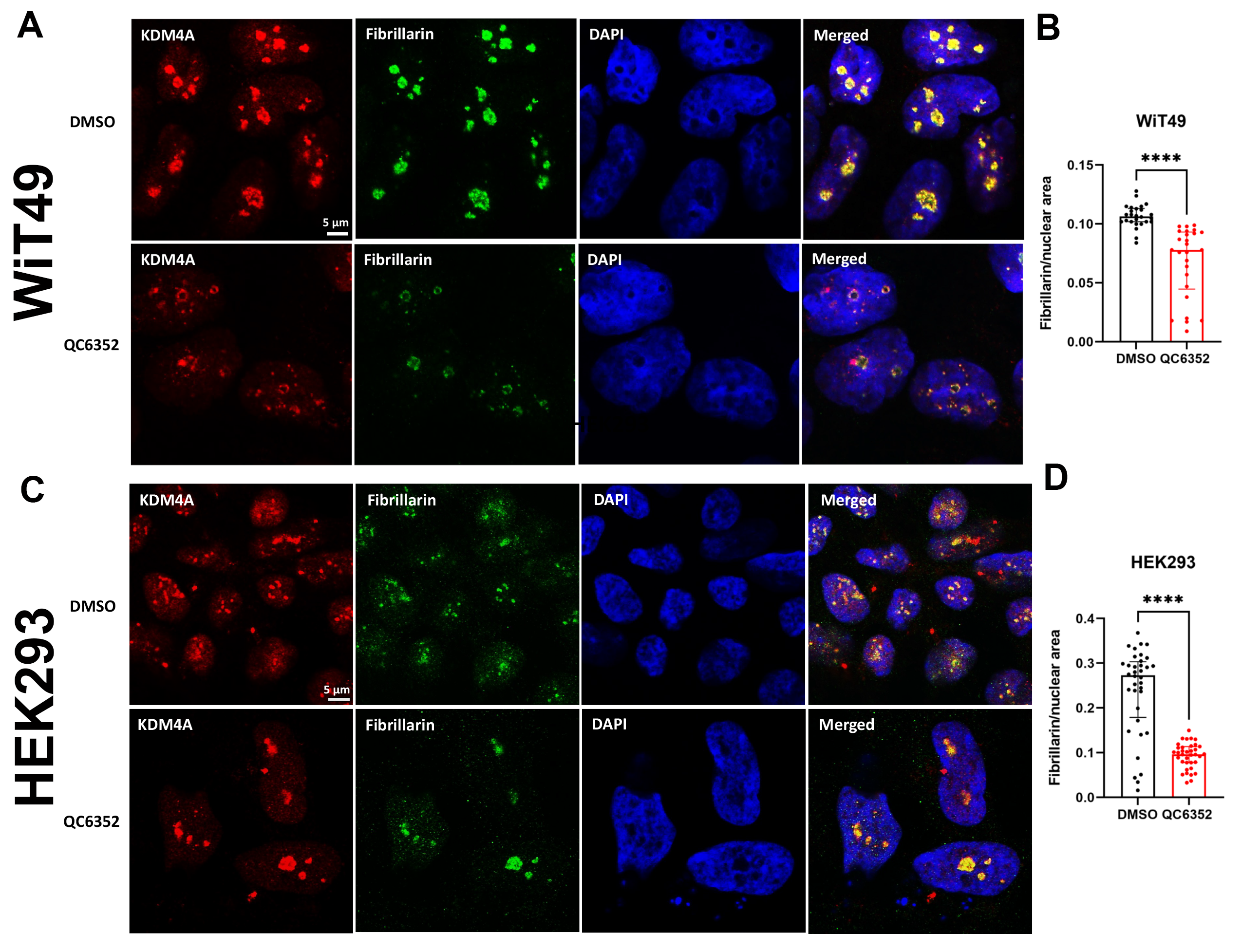
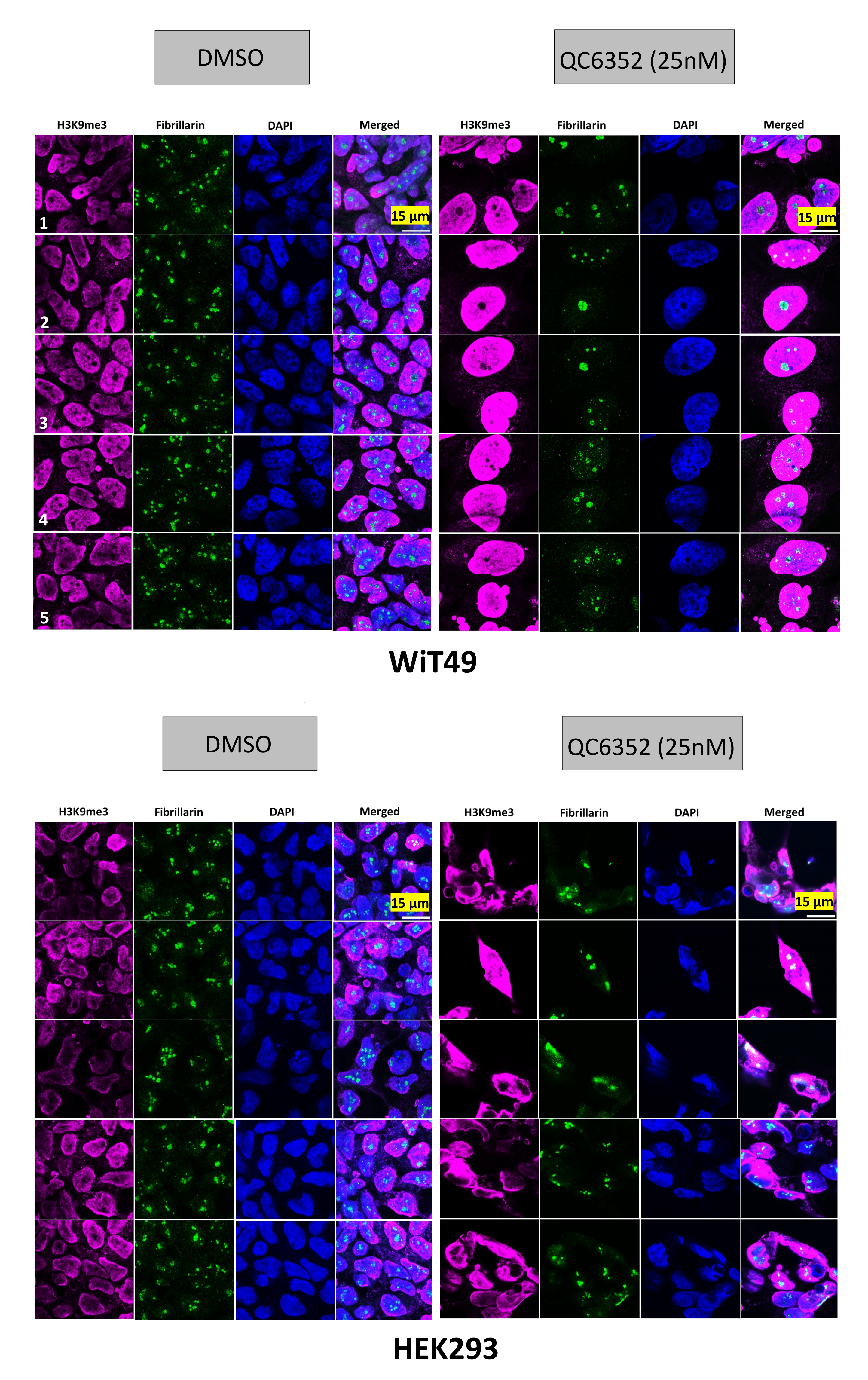
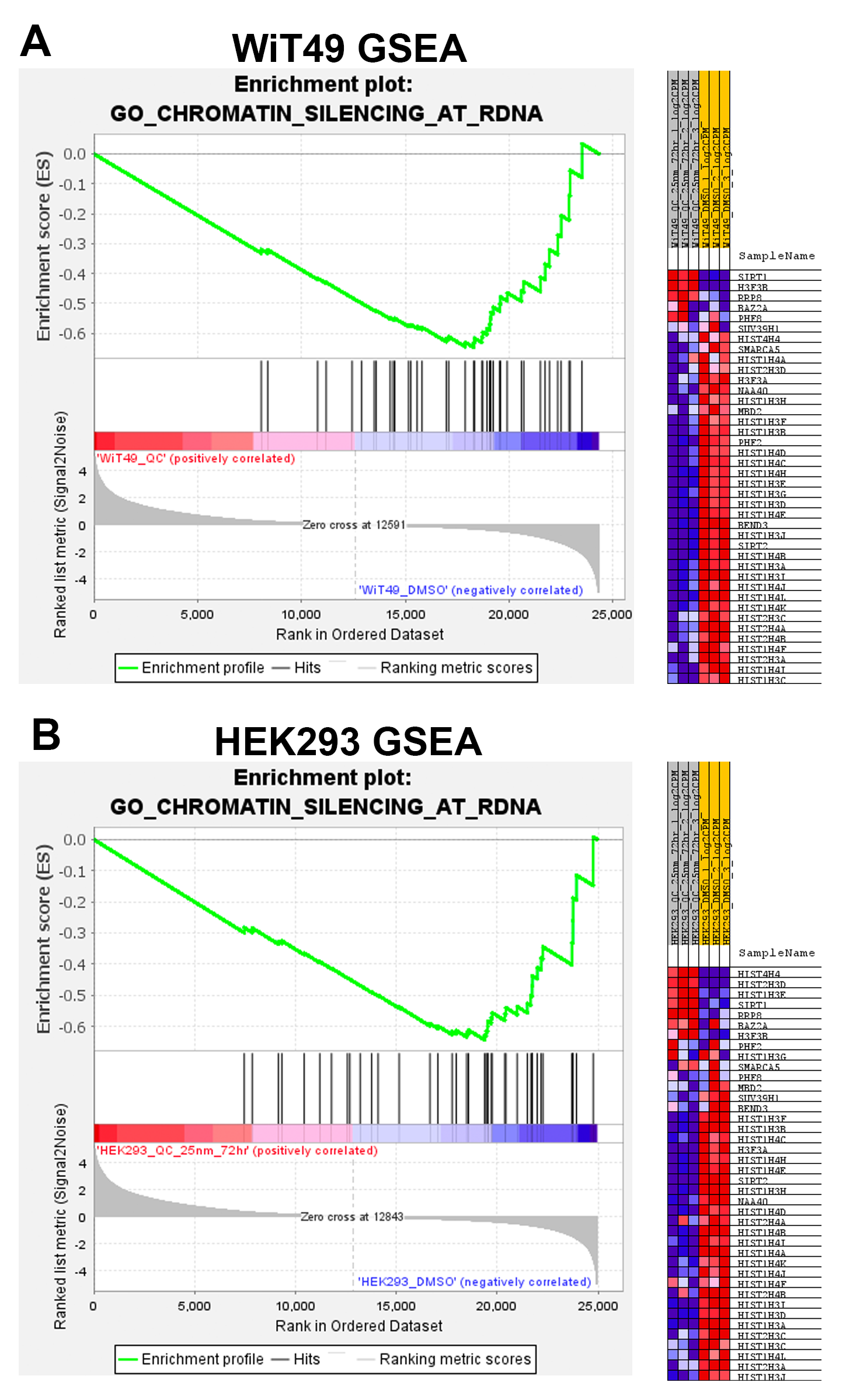
A link between KDM4A and transcription of ribosomal genes (rRNA synthesis in the nucleolus) was previously demonstrated in U2OS osteosarcoma cells.(28) We demonstrated that KDM4A (but not KDM4B or KDM4C) and the nucleolar marker fibrillarin co-localize to the nucleolus in WiT49 and HEK293 (Figure 4). QC6352 treatment caused reduction of the nucleolar to nuclear area ratio (Supplementary Figure 7), reduction of KDM4A and RNA polymerase I expression (Supplementary Figure 8), and increased nuclear detection of H3K9me3 (Supplementary Figure 9) in WiT49 and HEK293. QC6352 treatment resulted in alteration of nucleolar structure with reduction of dense fibrillar centers that mark active nucleoli as assessed by scanning electron microscopy (Supplementary Figure 10). Furthermore, GSEA in WiT49 and HEK239 demonstrated GO_CHROMATIN_SILENCING_AT_RDNA to be among the top 20 altered pathways with QC6352 treatment (Supplementary Figure 11). These experiments demonstrate that KDM4A (but not KDM4B or C) localizes to the nucleolus and inhibition of KDM4 by QC6352 reduces active nucleoli, resulting in impaired ribosome biogenesis.
Fig.4A: KDM4A, but not KDM4B or C is located in the nucleolus in WiT49 and HEK293.
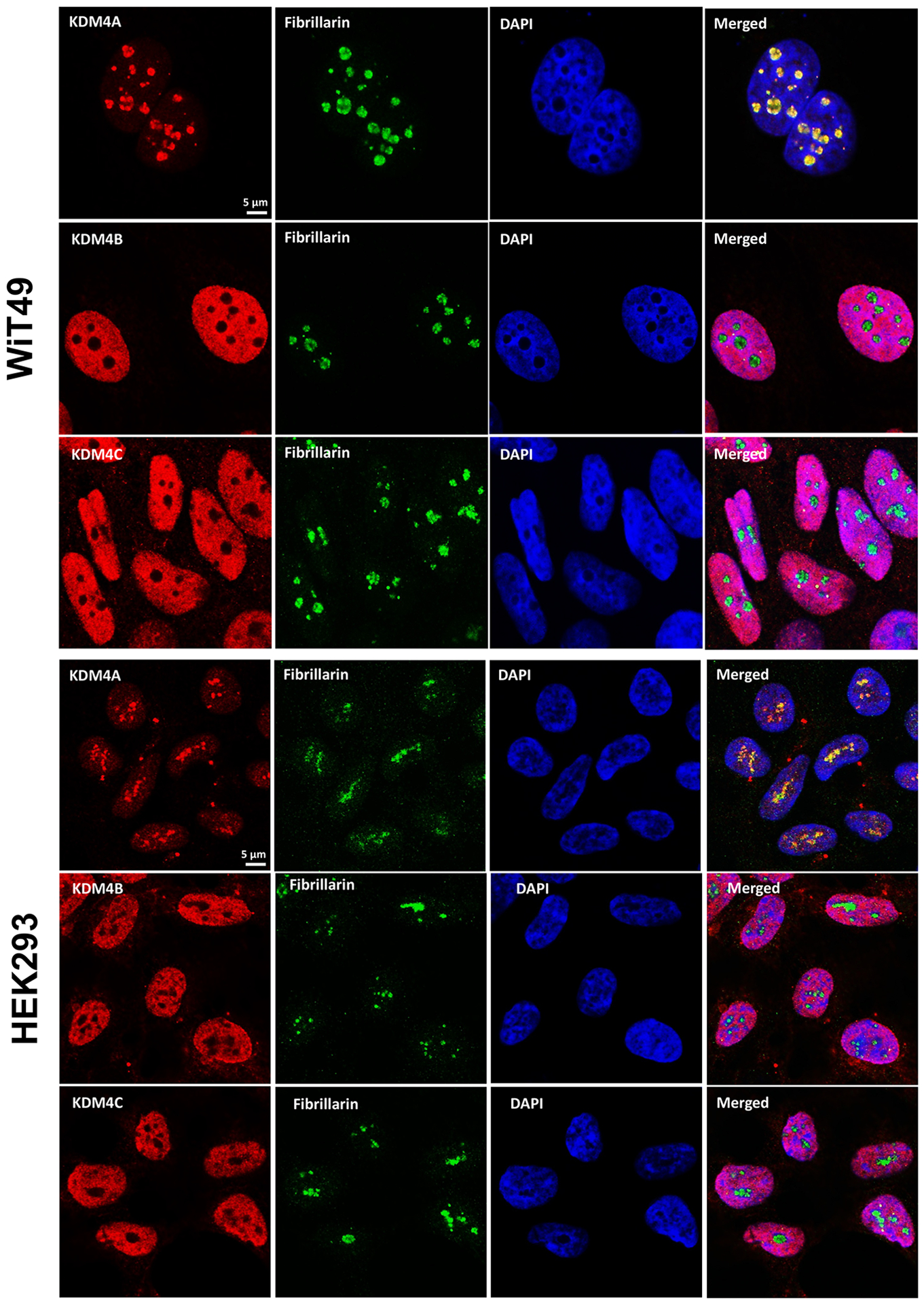
KDM4A (red) and the nucleolar marker fibrillarin (green) co-localize in the nucleoli [also represented by areas of reduced DAPI staining (blue) in the nucleus due to incompact chromatin in nucleoli] in WiT49 and HEK293 cells. KDM4B and KDM4C localize to the nucleus, but not the nucleolus in WiT49 and HEK293 cells.
The QC6352 cellular phenotype is associated with KDM4A inhibition
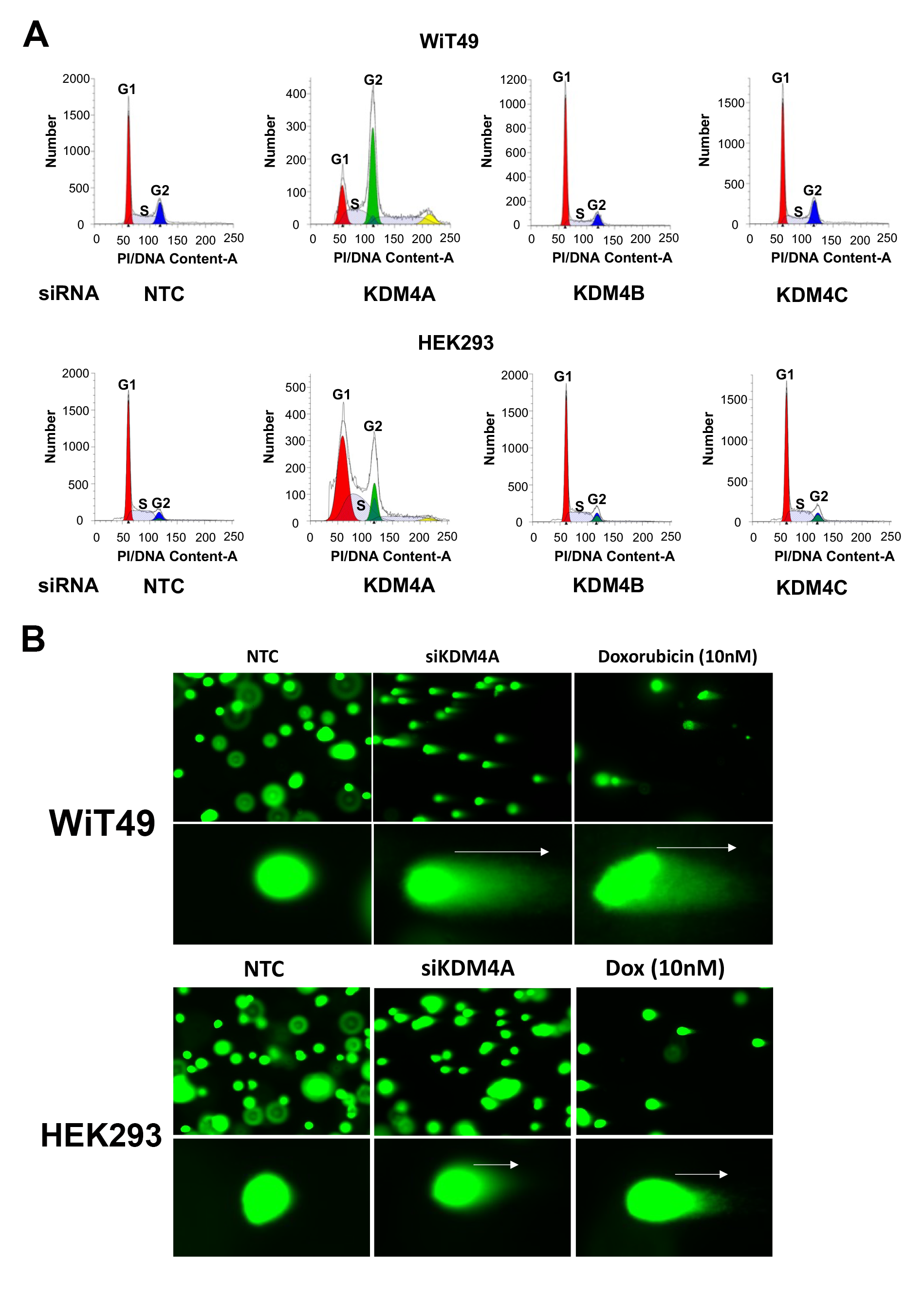
Because of the link between KDM4A and ribosomal gene transcription in the nucleolus, we hypothesized that the QC6352 cellular phenotype was most associated with KDM4A inhibition. To test this hypothesis, siRNAs against KDM4A, KDM4B, KDM4C, and non-targeting control siRNA were used to knockdown KDM4A-C in WiT49 and HEK293 (Figure 5A). SiRNA for KDM4A caused reduced proliferation by crystal violet assay in WiT49 and HEK293 (Figure 5B). A less prominent reduction was observed for siKDM4C in WiT49 cells compared to non-targeting control. KDM4A knockdown caused reduced proliferation and migration compared to KDM4B, KDM4C, and NTC siRNA in both WiT49 and HEK293 determined by phase-contrast live-cell microscopy (Figure 5C–D). SiKDM4A reduced spheroid growth in WiT49 and HEK293 compared to non-targeting control (Figure 5E). siKDM4C also reduced spheroid growth in WiT49 (Figure 5E). SiKDM4A most closely replicated the QC6352 cell cycle effects by increasing the proportion of S-phase cells in both WiT49 and HEK293 (Supplementary Figure 12). SiKDM4A caused DNA damage as determined by the COMET assay (Figure 5F, Supplementary Figure 12) and increased detection of pH2AX by western blot (Figure 5G).
Fig.5: The QC6352 treatment phenotype is mirrored by knockdown of KDM4A.
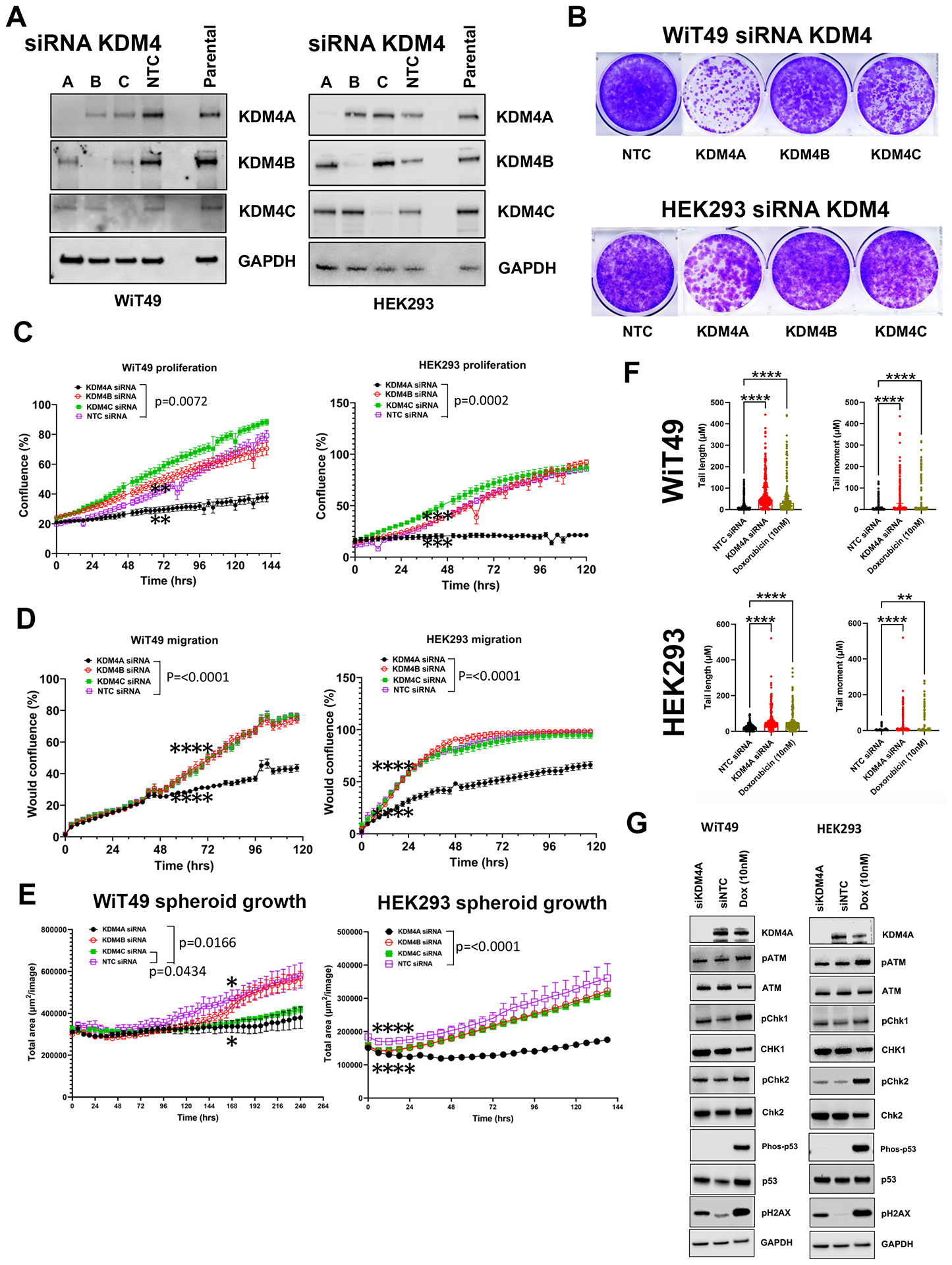
(A) Western blot confirmed siRNA-mediated knockdown of KDM4A, KDM4B, KDM4C and non-targeting control (NTC) in WiT49 and HEK293 (10nM siRNAs). (B) Crystal Violet assay demonstrated that KDM4A knockdown caused reduced proliferation in WiT49 and HEK293. KDM4C knockdown also caused reduced proliferation in WiT49. KDM4A knockdown caused reduced proliferation (C) and migration (D) compared to KDM4B, KDM4C, and NTC siRNA in both WiT49 and HEK293 by live-cell microscopy. (E) Spheroid growth was reduced by KDM4A knockdown in WiT49 and HEK293. (F) SiKDM4A induces DNA damage detected by the Comet assay. Doxorubicin-treated cells (10nM for 72 hours) are included as positive control. Kruskal-Wallis p-values are shown. (G) Western blot corroborates DNA damage caused by siKDM4A marked by increased detection of pH2AX in WiT49 and HEK293. A DNA damage checkpoint protein response does not appear to be induced by siKDM4A. For C-E, the rightmost asterix indicates timepoint at which separation in curves is statistically significant (paired two-tailed t-test p<0.05) and persists throughout remaining indicated duration.
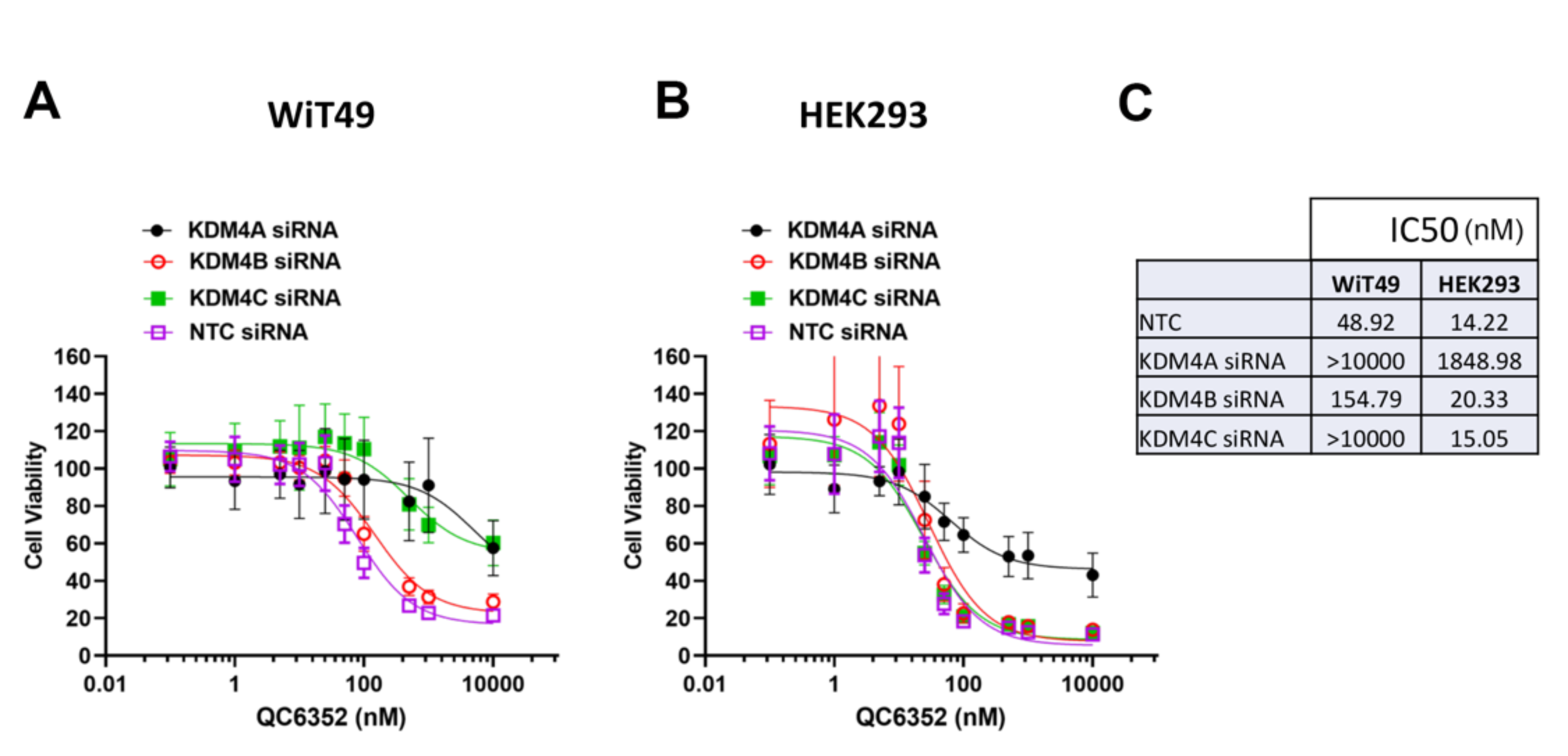
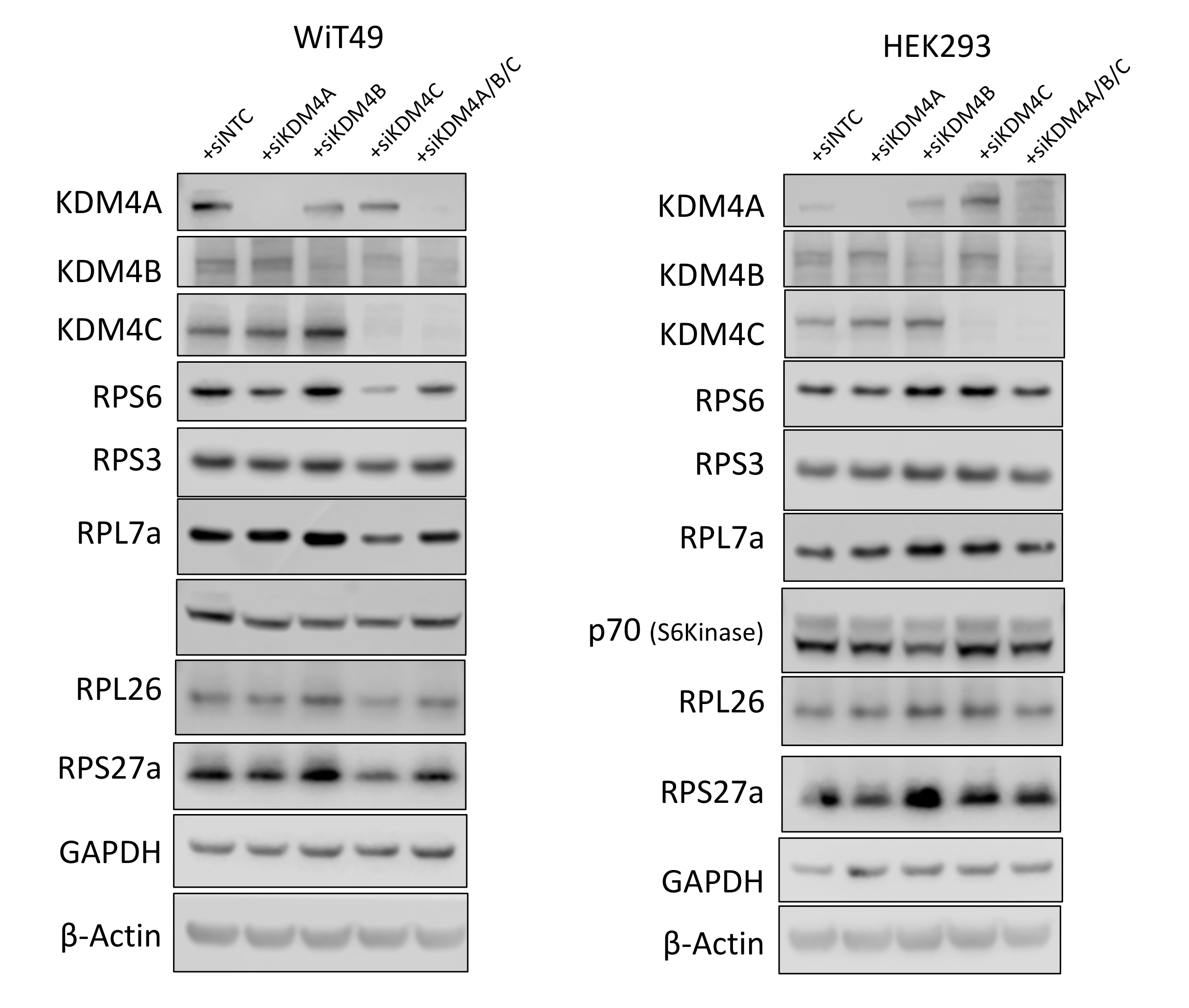
Furthermore, siRNA-mediated KDM4A knockdown decreased QC6352 sensitivity in WiT49 and HEK293 (Supplementary Figure 13). Like the spheroid data, knockdown of KDM4C also decreased QC6352 sensitivity in WiT49, but not HEK293 (Supplementary Figure 13). These data suggest that KDM4A is the KDM4 family member most responsible for the QC6352 cellular phenotype. However, RNA-seq with siRNA to KDM4A, B, or C and KDM4A-C did not demonstrate a transcriptomic signature consistent with impaired ribosome biogenesis (Supplementary Data File 2–3). SiRNA to KDM4 A, B, or C and KDM4A-C did not reduce ribosomal protein expression by Western blot (Supplementary Figure 14).
QC6352 causes decreased KDM4A-C protein levels mediated by ubiquitination
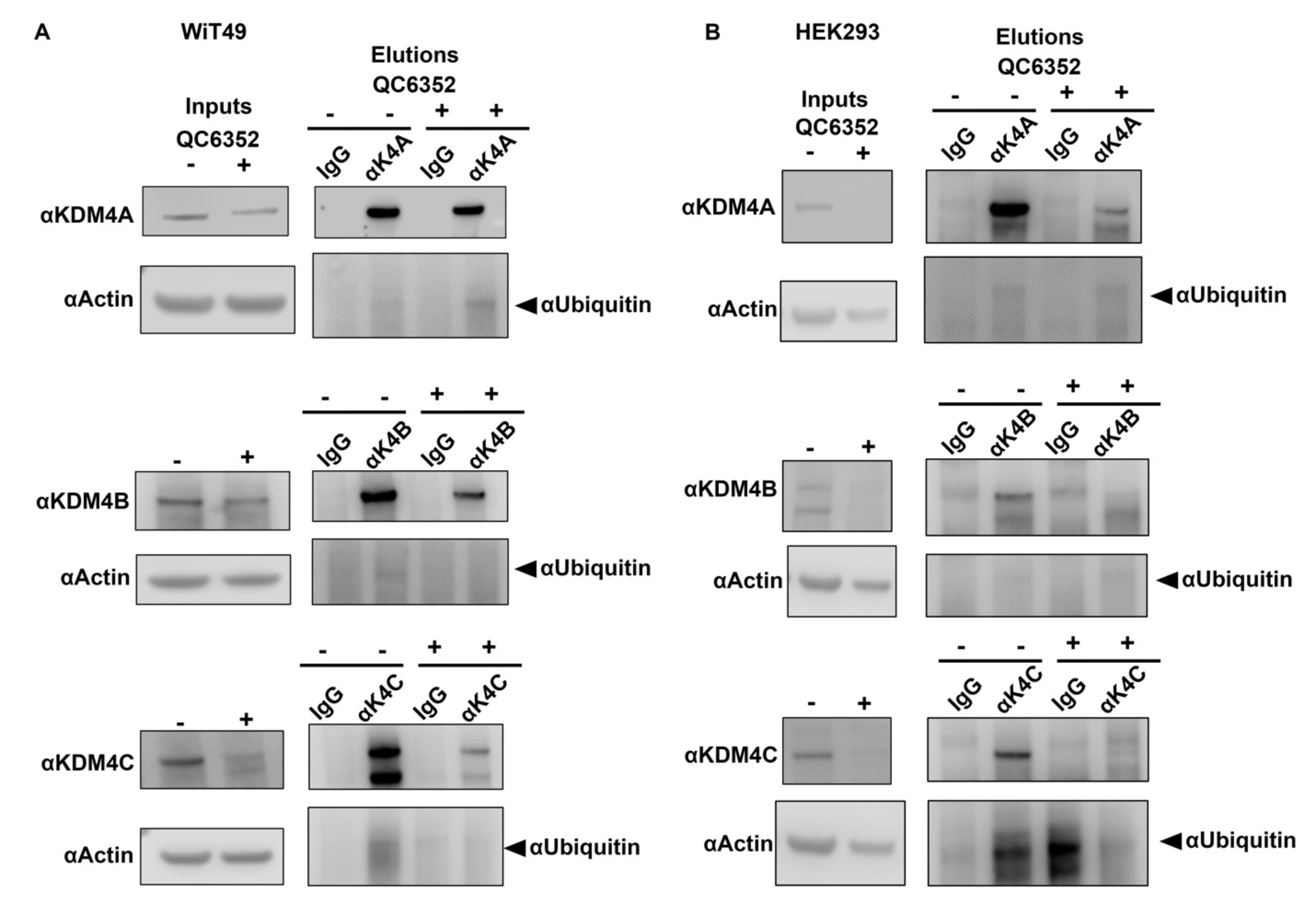
Treatment of WiT49 and HEK293 with 25 nM QC6352 caused decreased KDM4A-C protein levels observable beginning at six hours and continuing for at least 72 hours (Figure 6A). QC6352 treatment was associated with the expected increase in H3K9me3 and H3K36me3. However, treatment with QC6352 did not cause decreased expression of KDM4A-C mRNA (Supplementary Figure 15). Co-treatment with 25nM QC6352 and 500nM MG132 (proteasome inhibitor) rescued KDM4A-C levels in WiT49 and HEK293, indicating that the decreased protein levels are likely caused by a proteasome-mediated mechanism (Figure 6B). Immunoprecipitation of ubiquitin confirmed a direct protein-protein interaction between KDM4A-C and ubiquitin (Figure 6C) that is enhanced by QC6352 treatment and reduced by MG132 co-treatment in WiT49 (Figure 6D). Reciprocal immunoprecipitation of KDM4A, B, and C with subsequent western blot of ubiquitin supported a direct interaction between KDM4A and C and ubiquitin (Supplementary Figure 16). In addition to the previously demonstrated catalytic inhibition of KDM4A-C histone demethylase function, the current study demonstrates that QC6352 causes a ubiquitin-mediated decrease in KDM4A-C. Of note, a reduction in KDM4A-C was not observed in QC6352 insensitive cell lines (Supplementary Figure 17).
Fig.6: QC6352 causes decreased KDM4A-C protein levels caused by ubiquitination.
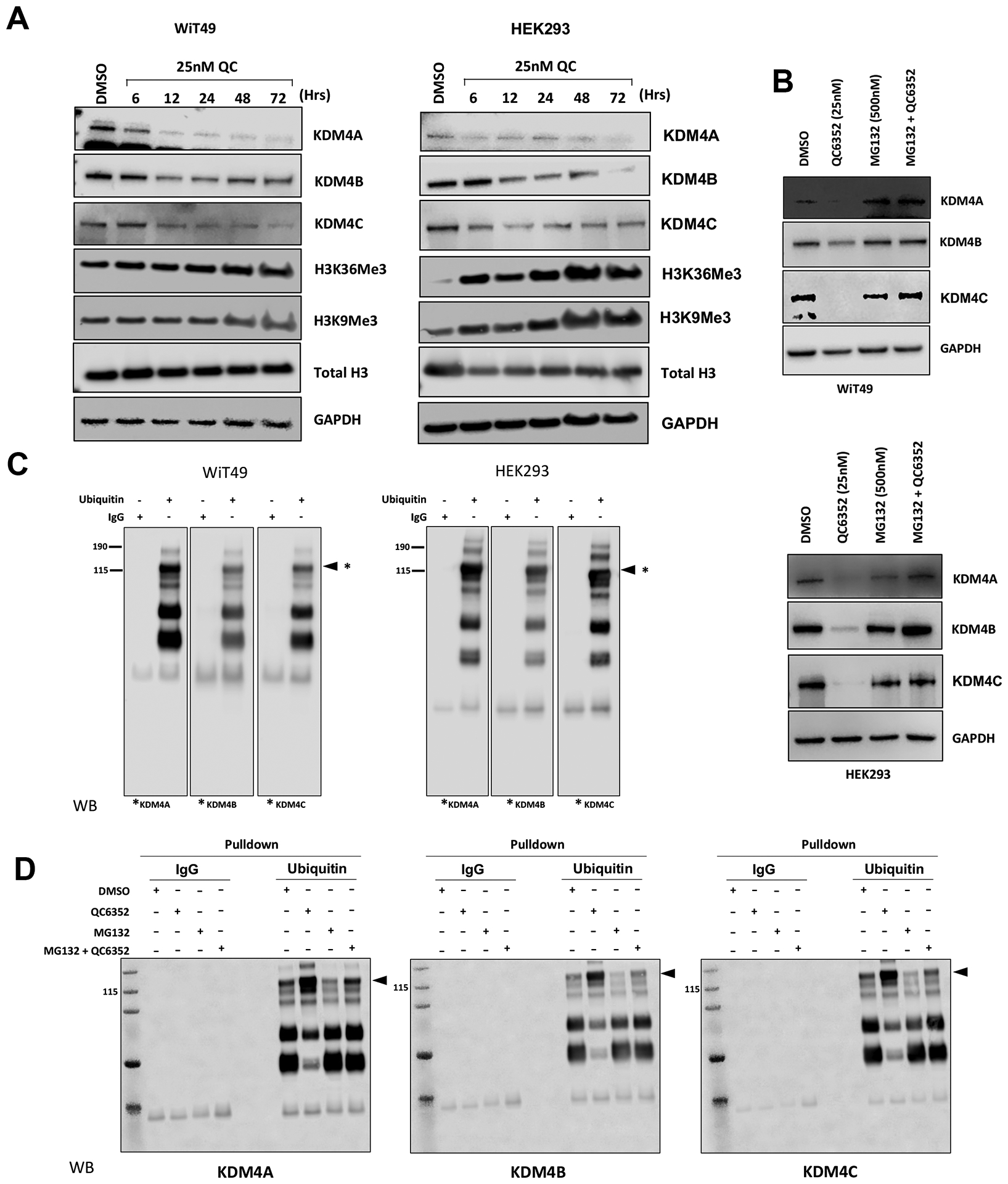
(A) QC6352 treatment of WiT49 and HEK293 causes reduction of KDM4A, B, and C on the protein level starting at six hours and persisting for at least 72 hours. This is accompanied by an increase in H3K36me3 and H3K9me3 (KDM4A-C targets). (B) The reduction in KDM4A, KDM4B, and KDM4C associated with QC6352 is abrogated by co-treatment with the proteasome inhibitor MG132. (C) Immunoprecipitation of ubiquitin followed by western blot for KDM4A, B, and C demonstrated a direct protein-protein interaction between KDM4A-C and ubiquitin in both untreated WiT49 and HEK293 cells. Equal amounts of protein from group were immunoprecipitated with ubiquitin antibody and the immunocomplexes were analyzed by western blot for KDM4A, KDM4B and KDM4C. Asterices indicate the relevant band and associated molecular weight for KDM4A, B, and C. (D) Immunoprecipitation of ubiquitin followed by western blot for KDM4A, B, and C demonstrated a direct protein-protein interaction between KDM4A, B, and C and ubiquitin that is increased in the presence of QC6352 treatment, and decreased by co-treatment with QC6352 and MG132 (proteasome inhibitor).
Discussion
This study demonstrates that treatment with the potent, selective KDM4 inhibitor QC6352 is cytostatic in some oncogenic cells of embryonic renal lineage including the anaplastic WT cell line WiT49 and the tumor-forming human embryonic kidney cell line HEK293. The QC6352 mechanism includes the known KDM4 catalytic domain inhibition and a newly characterized reduction of KDM4 protein levels associated with ubiquitination. QC6352 treatment was also associated with induction of DNA-damage, DNA-repair associated protein checkpoint response, S-phase cell cycle arrest, and profound reduction of ribosomal protein gene transcription, rRNA transcription, and ribosome biogenesis. KDM4A siRNA knockdown caused reduced QC6352 sensitivity, supporting its role as a target of the compound. These cellular pathways converge on the process of impaired ribosome biogenesis. To corroborate and expand this QC6352 response signature observed in embryonic kidney-associated oncogenic cells, high basal expression of ribosomal genes correlated with QC6352 sensitivity across a broad spectrum of more than 900 human cancer cell lines.
In this study, QC6352 treatment caused DNA damage and S-phase cell cycle arrest in WiT49 and HEK293 cells. The in vitro phenotype associated with these phenomena (decreased cell proliferation, migration, spheroid growth, S-phase arrest, DNA damage) was most closely phenocopied by siRNA knockdown of KDM4A rather than KDM4B or C. In addition, knockdown of KDM4A decreased the QC6352 sensitivity, reinforcing its role as a QC6352 target. KDM4A has been previously implicated in the repair of DNA double strand breaks.(12) Furthermore, previous studies have shown that KDM4A protein levels are regulated in a cell-cycle dependent manner and that KDM4A overexpression increased chromatin accessibility for DNA replication, resulting in S-phase progression. In contrast, KDM4A depletion caused slowed DNA replication and increased ATR/p53-dependent apoptosis.(17) In addition, Van Rechem et. al. demonstrated that KDM4A controlled the cell-cycle expression of replicative canonical histone genes.(44) In support of this finding, our current results demonstrated significant reduction of histone gene transcription in addition to the predominant signature of downregulated ribosomal gene transcription in response to QC6352. In the context of our current results and the prior literature, the observed DNA damage and S-phase cell cycle arrest in WiT49 and HEK293 cells are most likely mediated through KDM4A inhibition in response to QC6352 treatment.
The predominant effect of QC6352 treatment was impaired ribosome biogenesis. Ribosome biogenesis originates in the nucleolus, where RNA polymerase I transcribes rRNA genes and RNA polymerase II transcribes ribosomal protein genes. Our data demonstrate that KDM4A localizes to the nucleolus, and that KDM4 inhibition by QC6352 causes impaired ribosome biogenesis and function marked by decreased ribosomal protein gene transcription, decreased rRNA transcription, and blockade of protein synthesis.
These findings were corroborated using PRISM RNA expression and compound sensitivity correlation analysis. In summary, sensitivity to the KDM4 inhibitor QC6352 was associated with increased basal expression of ribosomal genes in over 900 human cancer cell lines. KDM4A has been previously demonstrated to co-localize and associate with the RNA-polymerase I complex in the nucleolus and regulate transcription of rRNA in response to growth factor availability.(28)
QC6352 was developed as an inhibitor of the catalytic, demethylase domain of KDM4A-D.(29) In addition to this foundational mechanism, our current analysis also demonstrates that QC6352 treatment causes reduction in KDM4A-C by ubiquitination. Ipenberg et. al. demonstrated that heat shock protein 90 (Hsp90) regulated stability of KDM4B and that pharmacological inhibition of Hsp90 with geldanamycin caused proteasome-mediated degradation of KDM4B associated with ubiquitination on lysines 337 and 562.(45) However, KDM4C levels were not affected by this mechanism, suggesting that the turnover of KDM4 family members may be regulated by distinct mechanisms. Tan et. al. demonstrated that FBXO22 targets KDM4A for ubiquitin-mediated proteasomal degradation.(46) Van Rechem et. al. also demonstrated that KDM4A levels are regulated by the proteasome.(47) Our data show a direct interaction between ubiquitin and KDM4A-C and demonstrate that QC6352 treatment may recruit KDM4 family ubiquitination mechanisms to lower KDM4A-C levels. The specific mechanisms of QC6352-induced KDM4A ubiquitination are a future direction of this work.
This study has limitations. First, QC6352 sensitivity was tested in WT and embryonic kidney cell lines. A cytostatic phenotype was observed in WiT49 and HEK293. HEK293 are a transformed, immortalized tumor-forming cell line derived from human embryonic kidney tissue but have been linked to a possible adrenal/neuronal lineage using comparative transcriptomic studies.(48,49) To mitigate this limitation, we demonstrated a link between QC6352 sensitivity and high basal ribosomal gene expression in many human cancer cell lines. Second, the testing of an in vivo phenotype was limited to only HEK293 because we could not establish subcutaneous xenografts from WiT49 despite exhaustive efforts. We attempted to overcome this limitation by performing assays of 3D spheroid growth, which more closely resembles the cell-cell interactions and oxygen dynamics of human tumors when compared to conventional 2D culture conditions. Finally, while the cellular phenotype of QC6352 treatment was mirrored by siRNA-mediated knockdown of KDM4A, RNA-seq analysis of siRNA-mediated knockdown of KDM4A-C (individually and in combination) did not reveal a signature consistent with impaired ribosome biogenesis. This discrepancy between chemical inhibition and siRNA-mediated knockdown of KDM4 could be due to additional histone demethylase targets of QC6352 or compensatory activity of other histone demethylases in the presence of KDM4 knockdown.
In conclusion, in sensitive cells of renal embryonic lineage, QC6352 promotes reduction of KDM4A levels through a proteasome-associated mechanism, resulting in a cytostatic response marked by DNA damage, DNA-damage checkpoint response initiation, S-phase cell cycle arrest, and marked reduction of ribosome biogenesis. Therefore, KDM4A inhibition may be a future target of therapeutic interest in cancer cells associated with an embryonic renal lineage or with high basal expression of ribosomal protein genes.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This research was funded in part by the American Lebanese Associated Charities (ALSAC)/St. Jude Children’s Research Hospital and by the National Institutes of Health (NIH)/National Cancer Institute (NCI) Grant 5K08CA25556903 (AJM), American Cancer Society-Research Scholar grant 130421-RSG-17071-01-TBG (JY), National Cancer Institute 1R01CA22973901 (JY) and R01CA266600 (JY). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Scanning electron microscopy Images were acquired at the Cell & Tissue Imaging Center which is supported by SJCRH and NCI P30 CA021765.
Abbreviations list:
- KDM
histone lysine demethylase
- H3K9
histone 3 lysine 9
- H3K36
histone 3 lysine 36
- H3K9me3
trimethylated H3K9
- H3K36me3
trimethylated H3K36
- WT
Wilms tumor
- FBS
fetal bovine serum
- ATCC
American Type Culture Collection
- MEM
minimum essential media
- DMEM
Dulbecco’s modified Eagle’s medium
- IMDM
Iscove’s modified Dulbecco’s medium
- ITS solution
insulin-transferrin-selenium solution
- STR
short tandem repeat
- siRNA
small interfering RNA
- PBS
phosphate buffered saline
- PFA
paraformaldehyde
- nm
nanometer
- nM
nanomolar
- IC50
half-maximal inhibitory concentration
- PI
propidium iodide
- APC
allophycocyanin
- DAPI
4’,6-diamidino-2-phenylindole
- μl
microliter
- SDS
sodium dodecyl-sulfate
- RNA194
RNA polymerase I
- IP
immunoprecipitation
- Opti-MEM
reduced serum minimal essential medium
- WARDEN
Workflow for the Analysis of RNA-seq Differential Expression
- log2CPM
log2 counts per million
- GSEA
Gene Set Enrichment Analysis
- GO
Gene Ontology
- PRISM
Profiling Relative Inhibition Simultaneously in Mixtures
- NSG
Nod scid gamma
- NES
normalized enrichment score
- rRNA
ribosomal RNA
- rDNA
ribosomal DNA
- FWER
familywise error rate
- qRT-PCR
quantitative real-time polymerase chain reaction
- ChIP-PCR
chromatin immunoprecipitation followed by polymerase chain reaction
Footnotes
Competing Interests Statement: The authors have no competing interests to declare.
References
- 1.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 2012;13(5):343–57 doi 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu Q, Young B, Wang Y, Davidoff AM, Rankovic Z, Yang J. Recent Advances with KDM4 Inhibitors and Potential Applications. J Med Chem 2022;65(14):9564–79 doi 10.1021/acs.jmedchem.2c00680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicetto D, Zaret KS. Role of H3K9me3 heterochromatin in cell identity establishment and maintenance. Curr Opin Genet Dev 2019;55:1–10 doi 10.1016/j.gde.2019.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res 2011;21(3):381–95 doi 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chantalat S, Depaux A, Héry P, Barral S, Thuret JY, Dimitrov S, et al. Histone H3 trimethylation at lysine 36 is associated with constitutive and facultative heterochromatin. Genome Res 2011;21(9):1426–37 doi 10.1101/gr.118091.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teissandier A, Bourc’his D. Gene body DNA methylation conspires with H3K36me3 to preclude aberrant transcription. EMBO J 2017;36(11):1471–3 doi 10.15252/embj.201796812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pedersen MT, Kooistra SM, Radzisheuskaya A, Laugesen A, Johansen JV, Hayward DG, et al. Continual removal of H3K9 promoter methylation by Jmjd2 demethylases is vital for ESC self-renewal and early development. EMBO J 2016;35(14):1550–64 doi 10.15252/embj.201593317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Das PP, Shao Z, Beyaz S, Apostolou E, Pinello L, De Los Angeles A, et al. Distinct and combinatorial functions of Jmjd2b/Kdm4b and Jmjd2c/Kdm4c in mouse embryonic stem cell identity. Mol Cell 2014;53(1):32–48 doi 10.1016/j.molcel.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verrier L, Escaffit F, Chailleux C, Trouche D, Vandromme M. A new isoform of the histone demethylase JMJD2A/KDM4A is required for skeletal muscle differentiation. PLoS Genet 2011;7(6):e1001390 doi 10.1371/journal.pgen.1001390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strobl-Mazzulla PH, Sauka-Spengler T, Bronner-Fraser M. Histone demethylase JmjD2A regulates neural crest specification. Dev Cell 2010;19(3):460–8 doi 10.1016/j.devcel.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pfister SX, Ahrabi S, Zalmas LP, Sarkar S, Aymard F, Bachrati CZ, et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep 2014;7(6):2006–18 doi 10.1016/j.celrep.2014.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janssen A, Colmenares SU, Lee T, Karpen GH. Timely double-strand break repair and pathway choice in pericentromeric heterochromatin depend on the histone demethylase dKDM4A. Genes Dev 2019;33(1–2):103–15 doi 10.1101/gad.317537.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mallette FA, Mattiroli F, Cui G, Young LC, Hendzel MJ, Mer G, et al. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J 2012;31(8):1865–78 doi 10.1038/emboj.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sulkowski PL, Oeck S, Dow J, Economos NG, Mirfakhraie L, Liu Y, et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 2020;582(7813):586–91 doi 10.1038/s41586-020-2363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsieh HJ, Zhang W, Lin SH, Yang WH, Wang JZ, Shen J, et al. Systems biology approach reveals a link between mTORC1 and G2/M DNA damage checkpoint recovery. Nat Commun 2018;9(1):3982 doi 10.1038/s41467-018-05639-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young LC, McDonald DW, Hendzel MJ. Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following γ-irradiation. J Biol Chem 2013;288(29):21376–88 doi 10.1074/jbc.M113.491514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Black JC, Allen A, Van Rechem C, Forbes E, Longworth M, Tschöp K, et al. Conserved antagonism between JMJD2A/KDM4A and HP1γ during cell cycle progression. Mol Cell 2010;40(5):736–48 doi 10.1016/j.molcel.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 18.Kupershmit I, Khoury-Haddad H, Awwad SW, Guttmann-Raviv N, Ayoub N. KDM4C (GASC1) lysine demethylase is associated with mitotic chromatin and regulates chromosome segregation during mitosis. Nucleic Acids Res 2014;42(10):6168–82 doi 10.1093/nar/gku253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao E, Ding J, Xia Y, Liu M, Ye B, Choi JH, et al. KDM4C and ATF4 Cooperate in Transcriptional Control of Amino Acid Metabolism. Cell Rep 2016;14(3):506–19 doi 10.1016/j.celrep.2015.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng Y, Yuan Q, Vergnes L, Rong X, Youn JY, Li J, et al. KDM4B protects against obesity and metabolic dysfunction. Proc Natl Acad Sci U S A 2018;115(24):E5566–E75 doi 10.1073/pnas.1721814115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patani N, Jiang WG, Newbold RF, Mokbel K. Histone-modifier gene expression profiles are associated with pathological and clinical outcomes in human breast cancer. Anticancer Res 2011;31(12):4115–25. [PubMed] [Google Scholar]
- 22.Wissmann M, Yin N, Müller JM, Greschik H, Fodor BD, Jenuwein T, et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol 2007;9(3):347–53 doi 10.1038/ncb1546. [DOI] [PubMed] [Google Scholar]
- 23.Li H, Yang X, Wang G, Li X, Tao D, Hu J, et al. KDM4B plays an important role in mitochondrial apoptosis by upregulating HAX1 expression in colorectal cancer. Oncotarget 2016;7(36):57866–77 doi 10.18632/oncotarget.11077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boila LD, Chatterjee SS, Banerjee D, Sengupta A. KDM6 and KDM4 histone lysine demethylases emerge as molecular therapeutic targets in human acute myeloid leukemia. Exp Hematol 2018;58:44–51.e7 doi 10.1016/j.exphem.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 25.Yang J, AlTahan AM, Hu D, Wang Y, Cheng PH, Morton CL, et al. The role of histone demethylase KDM4B in Myc signaling in neuroblastoma. J Natl Cancer Inst 2015;107(6):djv080 doi 10.1093/jnci/djv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh S, Abu-Zaid A, Jin H, Fang J, Wu Q, Wang T, et al. Targeting KDM4 for treating PAX3-FOXO1-driven alveolar rhabdomyosarcoma. Sci Transl Med 2022;14(653):eabq2096 doi 10.1126/scitranslmed.abq2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Rechem C, Black JC, Boukhali M, Aryee MJ, Gräslund S, Haas W, et al. Lysine demethylase KDM4A associates with translation machinery and regulates protein synthesis. Cancer Discov 2015;5(3):255–63 doi 10.1158/2159-8290.CD-14-1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salifou K, Ray S, Verrier L, Aguirrebengoa M, Trouche D, Panov KI, et al. The histone demethylase JMJD2A/KDM4A links ribosomal RNA transcription to nutrients and growth factors availability. Nat Commun 2016;7:10174 doi 10.1038/ncomms10174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen YK, Bonaldi T, Cuomo A, Del Rosario JR, Hosfield DJ, Kanouni T, et al. Design of KDM4 Inhibitors with Antiproliferative Effects in Cancer Models. ACS Med Chem Lett 2017;8(8):869–74 doi 10.1021/acsmedchemlett.7b00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Metzger E, Stepputtis SS, Strietz J, Preca BT, Urban S, Willmann D, et al. KDM4 Inhibition Targets Breast Cancer Stem-like Cells. Cancer Res 2017;77(21):5900–12 doi 10.1158/0008-5472.CAN-17-1754. [DOI] [PubMed] [Google Scholar]
- 31.Hohenstein P, Pritchard-Jones K, Charlton J. The yin and yang of kidney development and Wilms’ tumors. Genes Dev 2015;29(5):467–82 doi 10.1101/gad.256396.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davidoff AM. Wilms tumor. Adv Pediatr 2012;59(1):247–67 doi 10.1016/j.yapd.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coorens THH, Treger TD, Al-Saadi R, Moore L, Tran MGB, Mitchell TJ, et al. Embryonal precursors of Wilms tumor. Science 2019;366(6470):1247–51 doi 10.1126/science.aax1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown KW, Charles A, Dallosso A, White G, Charlet J, Standen GR, et al. Characterization of 17.94, a novel anaplastic Wilms’ tumor cell line. Cancer Genet 2012;205(6):319–26 doi 10.1016/j.cancergen.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 35.Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, Clement MV. Corrigendum to OpenComet: An automated tool for comet assay image analysis [Redox Biol. Volume 2, 2014, Pages 457–465]. Redox Biol 2021;40:101876 doi 10.1016/j.redox.2021.101876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy AJ, Chen X, Pinto EM, Williams JS, Clay MR, Pounds SB, et al. Forty-five patient-derived xenografts capture the clinical and biological heterogeneity of Wilms tumor. Nat Commun 2019;10(1):5806 doi 10.1038/s41467-019-13646-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McLeod C, Gout AM, Zhou X, Thrasher A, Rahbarinia D, Brady SW, et al. St. Jude Cloud: A Pediatric Cancer Genomic Data-Sharing Ecosystem. Cancer Discov 2021;11(5):1082–99 doi 10.1158/2159-8290.CD-20-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu C, Mannan AM, Yvone GM, Ross KN, Zhang YL, Marton MA, et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat Biotechnol 2016;34(4):419–23 doi 10.1038/nbt.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corsello SM, Nagari RT, Spangler RD, Rossen J, Kocak M, Bryan JG, et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat Cancer 2020;1(2):235–48 doi 10.1038/s43018-019-0018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 2013;14:128 doi 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016;44(W1):W90–7 doi 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, et al. Gene Set Knowledge Discovery with Enrichr. Curr Protoc 2021;1(3):e90 doi 10.1002/cpz1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Rechem C, Ji F, Mishra S, Chakraborty D, Murphy SE, Dillingham ME, et al. The lysine demethylase KDM4A controls the cell-cycle expression of replicative canonical histone genes. Biochim Biophys Acta Gene Regul Mech 2020;1863(10):194624 doi 10.1016/j.bbagrm.2020.194624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ipenberg I, Guttmann-Raviv N, Khoury HP, Kupershmit I, Ayoub N. Heat shock protein 90 (Hsp90) selectively regulates the stability of KDM4B/JMJD2B histone demethylase. J Biol Chem 2013;288(21):14681–7 doi 10.1074/jbc.C113.462770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tan MK, Lim HJ, Harper JW. SCF(FBXO22) regulates histone H3 lysine 9 and 36 methylation levels by targeting histone demethylase KDM4A for ubiquitin-mediated proteasomal degradation. Mol Cell Biol 2011;31(18):3687–99 doi 10.1128/MCB.05746-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Rechem C, Black JC, Abbas T, Allen A, Rinehart CA, Yuan GC, et al. The SKP1-Cul1-F-box and leucine-rich repeat protein 4 (SCF-FbxL4) ubiquitin ligase regulates lysine demethylase 4A (KDM4A)/Jumonji domain-containing 2A (JMJD2A) protein. J Biol Chem 2011;286(35):30462–70 doi 10.1074/jbc.M111.273508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stepanenko AA, Dmitrenko VV. HEK293 in cell biology and cancer research: phenotype, karyotype, tumorigenicity, and stress-induced genome-phenotype evolution. Gene 2015;569(2):182–90 doi 10.1016/j.gene.2015.05.065. [DOI] [PubMed] [Google Scholar]
- 49.Shaw G, Morse S, Ararat M, Graham FL. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J 2002;16(8):869–71 doi 10.1096/fj.01-0995fje. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during the current study are available from the corresponding authors on reasonable request. The WiT49 and HEK293 QC6352 RNA-seq data are available on the Gene Expression Omnibus Database (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE226112.