

Abstract
We aimed to characterize the genetic basis of disease in a family with multiple autoimmune manifestations, including systemic lupus erythematosus (SLE), immune thrombocytopenia, and autoimmune thyroiditis. Whole exome sequencing (WES) was conducted to identify candidate variants, which were analyzed by flow cytometry, immunoprecipitation, and luciferase reporter assay in transfected 293T cells. Gene expression in peripheral blood mononuclear cells (PBMC) was profiled by bulk RNA sequencing and plasma cytokines were measured by proximity extension assay.
We studied two siblings with early-onset SLE and immune thrombocytopenia. WES identified two maternally inherited in cis variants (p. Pro50Leu and p.Ala76Gly) in Suppressor of cytokine signaling 1 (SOCS1), flanking the kinase inhibitory domain that interacts with Janus kinases (JAK). Both variants were predicted to be benign by most in silico algorithms and neither alone affected the ability of SOCS1 to inhibit JAK-STAT1 signaling by functional studies. When both variants were expressed in cis, the mutant SOCS1 protein displayed decreased binding to JAK1 and reduced capacity to inhibit type I interferon (IFN-I) signaling by ~20–30% compared to the wildtype protein. PBMC from the probands and their mother showed increased expression of interferon-inducible genes compared to healthy controls, supporting defective regulation of IFN-I signaling. Cells from all three subjects displayed heightened sensitivity to IFN-I stimulation, while response to IFN-γ, IL-4, and IL-6 was comparable to healthy controls.
Our work illustrates the critical fine-tuning of IFN-I signaling by SOCS1 to prevent autoimmunity. We show that a combination of genetic variants that are individually benign may have deleterious consequences.
Keywords: SOCS1, in cis variants, interferon, autoimmunity, autoimmune disease
1. Introduction
Autoimmune diseases are caused by a breach of tolerance to self-antigens. Systemic lupus erythematosus (SLE) is a prototypical autoimmune disease characterized by aberrant activation of T lymphocytes and B lymphocytes leading to the development of autoantibodies. Complement activation and chronic inflammation cause further damage to organs, including the skin and kidneys.[1]
While hereditary factors are important for the development of autoimmunity, the genetic risk in most cases is determined by a combination of susceptibility alleles.[2] Monogenic causes of autoimmunity are rare, but may explain cases with early disease onset and strong familial links.[3] The convergence of polygenic and monogenic effects in driving autoimmunity is demonstrated by the relationship between type I interferon (IFN-I) dysregulation and SLE.
Enhanced production of IFN-I in patients with SLE was first observed two decades ago.[4] The pathogenic role of these anti-viral cytokines is well demonstrated by animal models and highlighted by the recent approval of anifrolumab (monoclonal antibody against IFN-I receptor).[5] Polymorphisms in genes involved in IFN-I signaling have been identified as susceptibility alleles for SLE. Monogenic lupus is associated with gain-of-function variants in mediators of IFN-I signaling or loss-of-function variants in regulators of the pathway.[3]
In this study, we describe two in cis variants in Suppressor of Cytokine Signaling 1 (SOCS1) in a family with multiorgan manifestations of autoimmunity. We show that in cis expression of variants that are individually benign can disturb the interaction between SOCS1 and Janus kinase 1 (JAK1), leading to aberrant activation of IFN-I signaling.
2. Material and Methods
2.1. Study approval
This study was approved by the Institutional Review Board at Boston Children’s Hospital. Informed consent and assent were provided by participants and legal guardians.
2.2. Clinical presentation
Proband 1 (P1) was a 13-year-old female who presented with vomiting, abdominal pain, periorbital edema, and dark-colored urine. Her initial labs were notable for pancytopenia, hypocomplementemia, proteinuria, and positive testing for antinuclear antibodies and anti-dsDNA antibodies (Table 1). She had acute renal failure and her kidney biopsy revealed Class IV lupus glomerulonephritis and microthrombotic disease (Figure 1A). Her initial therapy included pulse-dose glucocorticoids, intravenous immunoglobulin, and plasmapheresis. Her early disease course was complicated by posterior reversible encephalopathy syndrome and peritonitis. P1 subsequently received cyclophosphamide and rituximab, then transitioned to mycophenolate mofetil and hydroxychloroquine with glucocorticoid taper. Six years after her initial presentation, P1 attained medication-free remission with normal cell counts and complement levels.
Table 1:
Demographics and laboratory data
| Parameter | Proband 1 | Proband 2 | Reference range |
|---|---|---|---|
|
| |||
| Age on set (y) | 13 | 12 | - |
| Routine parameters | |||
| Hemoglobin (g/dL) | 7.0 | 13.7 | 11.3 – 13.4 |
| WBC (103 cells/μL) | 4.5 | 11.32 | 5.52 – 9.29 |
| Platelet (103 cells/μL) | 60 | 5 | 189 – 342 |
| Creatinine (mg/dL) | 3.47 | 0.65 | 0.30 – 1.00 |
| BUN (mg/dL) | 70 | 12 | 5 – 17 |
| Urine protein (mg/dL) | 437.8 | 11.0 | 0 – 12.0 |
| Rheumatologic/Immunologic parameters | |||
| Anti-nuclear Ab | 1:320 | 1:640 | <1:40 |
| Anti-dsDNA Ab | Positive | Positive | Negative |
| Anti-Ro60 | Negative | Positive | Negative |
| Anti-β2-glycoprotein IgG Ab | Negative | Positive | Negative |
| Anti-Cardiolipin IgG Ab | Negative | Positive | Negative |
| Anti-Cardiolipin IgM Ab | Negative | Positive | Negative |
| ESR (mm/hr) | 26 | 8 | 0 – 30 |
| CRP (mg/dL) | 0.10 | 0.23 | 0 – 0.50 |
| IgA (mg/dL) | < 4 | 109 | 70 – 312 |
| IgG (mg/dL) | 594 | 1,163 | 639 – 1,344 |
| IgM (mg/dL) | 18 | 70 | 40 – 240 |
| C3 (mg/dL) | < 40 | 143 | 83 – 177 |
| C4 (mg/dL) | 7 | 25 | 14 – 42 |
Ab: antibody; BUN: blood urea nitrogen; ESR: erythrocyte sedimentation rate; CRP: C-reactive protein; C3: Complement C3; C4: Complement C4
Figure 1. Identification of in cis SOCS1 variants in a family with multiple manifestations of autoimmunity.
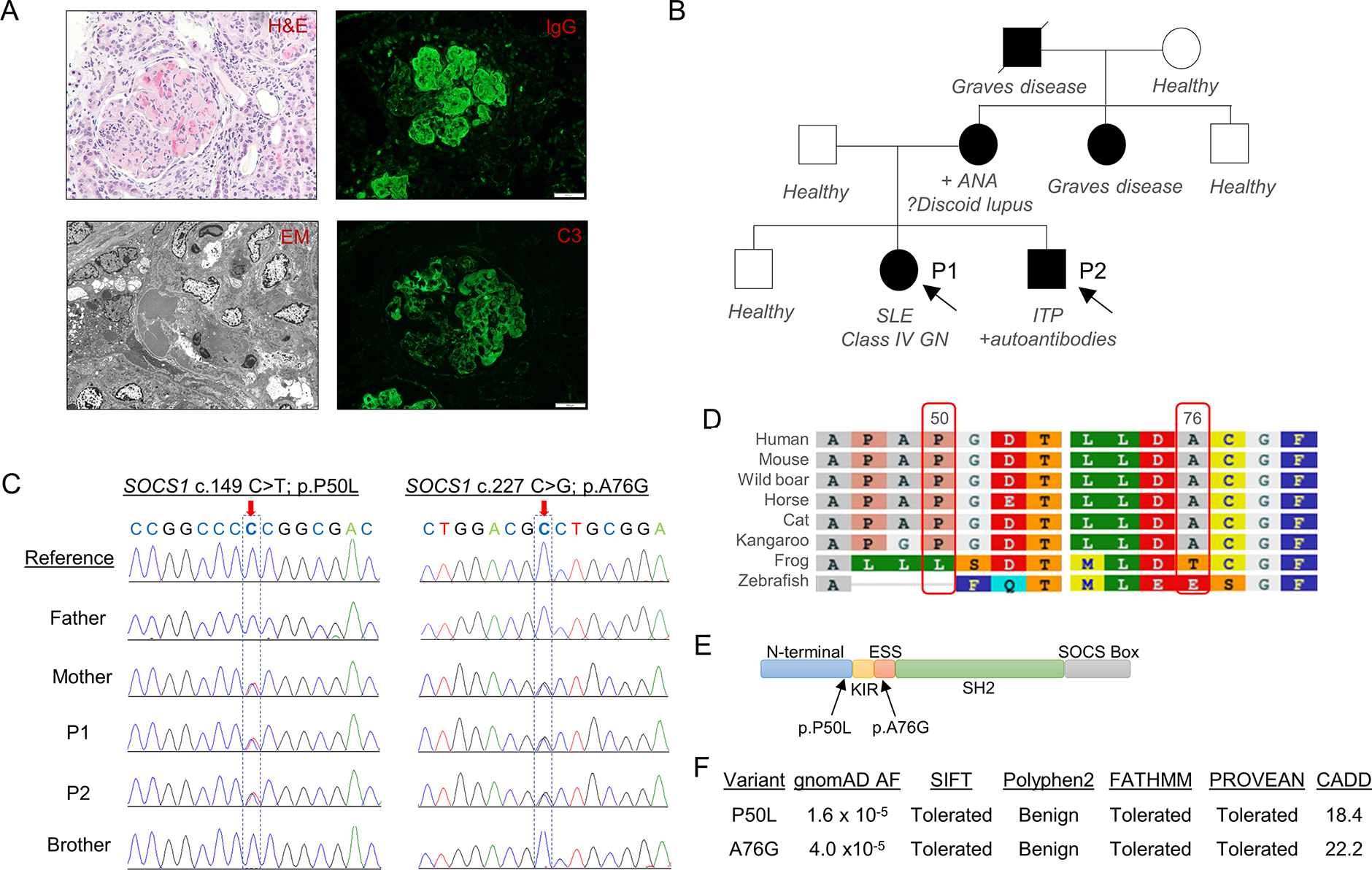
A) Evidence of lupus glomerulonephritis in Proband 1 as shown by glomerular hypercellularity and presence of multiple thrombi (Hematoxylin and eosin staining; top left), prominent electron dense subendothelial and mesangial deposits (electron microscopy, bottom left), and deposition of immunoglobulins and complements (immunofluorescence; right panels; white scale bar = 100 μM). B) Pedigrees of the Probands’ family with multiple autoimmune diseases. C) Chromatogram from Sanger sequencing analysis confirming in cis SOCS1 variants c.149 C>T (p.P50L) and c.227 C>G (p.A76G) in P1, P2, and their mother. D) Alignment of SOCS1 amino acid sequence among different species. E) Location of candidate variants relative to the functional domains of SOCS1. F) Allelic frequency (AF) of candidate variants in gnomAD and in silico prediction of pathogenicity using SIFT, Polyphen2, FATHMM, PROVEAN, and CADD.
Proband 2 (P2; younger sibling of P1) was a 10-year-old male who presented with petechiae, epistaxis, and a platelet count of 5 × 103/μL (normal, 180–320 × 103 cells/μL) about 4 months after P1’s initial hospitalization. P2 was diagnosed with immune thrombocytopenic purpura (ITP) and rheumatology evaluation additionally revealed positive ANA, anti-dsDNA, anti-cardiolipins, and anti-Ro60 antibodies (Table 1). His thrombocytopenia improved with prednisone treatment, but relapsed after tapering. P2 was subsequently treated with intravenous immunoglobulins and maintained on hydroxychloroquine without recurrence of petechiae or epistaxis. His platelet count has remained stable between 50–80 × 103/μL.
The probands’ family history was notable for mother with whole body skin rash at age 15 (diagnosed with possible erythema annulare centrifugum vs. discoid lupus), anemia and positive ANA (at age 25), and hypertension secondary to fibromuscular dysplasia (at age 48). The father and an elder sibling of the probands are healthy. Maternal grandfather was diagnosed with Graves’ disease in his 40’s and died from bile duct cancer at age 81. The probands’ maternal aunt was diagnosed with Graves’ disease at age 12. A pedigree of the family is displayed in Figure 1B. The mother’s ancestry is from South Korea while the father’s family is of English origin.
2.3. Whole exome sequencing (WES)
WES was performed by GeneDx on DNA isolated by buccal swab as previously described.[6] Sequencing utilized IDT xGen probes. Average coverage was ~100x and with more than 95% of targets covered at >20x. WES datasets were processed through a variant calling pipeline managed by WuXi NextCODE. Variants were filtered and prioritized using Emegene software. Candidate variants were confirmed via Sanger sequencing.
2.4. Functional analyses
Detailed methods for ectopic expression of SOCS1, flow cytometry, luciferase reporter assay, western blotting, immunoprecipitation, RNA sequencing, quantitative PCR, and proximity extension assay are provided in Supplemental Methods. Primer sequences for quantitative PCR and site-directed mutagenesis are provided in Supplemental Table 1.
2.5. Statistical analysis
For quantitative variables, differences between 2 groups were analyzed by Mann-Whitney U test. All tests were 2-sided, and P less than 0.05 was considered statistically significant. Statistical analyses were performed using Prism 9.0 software (GraphPad Software, San Diego, Calif).
2.6. Data Availability
All primary data are available from the corresponding author upon request.
3. Results
3.1. Identification of in cis SOCS1 variants
We aimed to identify the genetic basis of disease in a family with multiple manifestations of autoimmunity, including SLE with class IV glomerulonephritis, ITP, and Graves’ disease (Figure 1A, B and Table 1). WES results from P1 and P2 revealed two heterozygous missense variants in SOCS1 (c.149C>T; p.Pro50Leu and c.227C>G; p.Ala76Gly) inherited in cis from their mother. The results were confirmed by Sanger sequencing and the variants were absent in the father and healthy sibling (Figure 1C). The extended family members were not available for genetic testing. Other rare variants shared by the mother and affected siblings are listed in Supplemental Table 2. To our knowledge, these variants have not been linked to polygenic or monogenic autoimmune diseases.
SOCS1 was considered a candidate gene because it functions as an endogenous regulator of type I (IFN-α/β) and type II IFN (IFN-γ) signaling by inhibiting the activation of JAKs. We and others have shown that haploinsufficiency of SOCS1, typically associated with monoallelic loss-of-function variants (i.e., frameshift variants, nonsense variants, or complete deletion), is associated with pleiotropic manifestations of autoimmunity.[7–9]
Both SOCS1 variants found in the probands are rare in the Genome Aggregation Database (gnomAD), with an allelic frequency of 1/63,488 (1.6 × 10−5) for SOCS1P50L and 6/153,814 (3.9 × 10−5) for SOCS1A76G. Interestingly, all individuals with these variants in gnomAD are East Asians and variant co-occurrence analysis suggested that one subject possessed both variants in cis. Pertinent to the mother’s Korean genetic background, interrogation of the Korean Genome Project database revealed an allelic frequency of ~0.1% for both variants in the Korean population, although data on variant co-occurrence was not available.
The two affected amino acids are conserved among mammalian species and flank the kinase inhibitor region (KIR; amino acid residues 55–66) of SOCS1 that interacts with the kinase domain of JAKs to prevent JAK phosphorylation (Figure 1D,E). In silico algorithms including SIFT, PolyPhen2, PROVEAN and FATHMM predicted that both P50L and A76G are benign substitutions (Figure 1F). On the other hand, analysis by combined annotation dependent depletion (CADD) suggested that the variants may be deleterious (using a conventional cutoff score of 15).
3.2. Functional analysis of SOCS1 variants
To evaluate the impact of the identified variants on SOCS1 function, we transfected HEK293T cells with plasmids encoding FLAG-tagged wildtype or mutant SOCS1 (p. P50L, p. A76G, and both variants in cis). Western blotting of whole cell lysates confirmed comparable expression of all SOCS1 variants with the expected molecular weight (Figure 2A). Consistent with the known function of SOCS1, IFN-α2-induced STAT1 phosphorylation in transfected cells was suppressed by ectopic expression of wildtype SOCS1, as assessed by phospho-flow cytometry (Figure 2B,C and Supplemental Figure 1A) and by western blot (Figure 2D,E). Neither P50L nor A76G variant alone affected the inhibitory function of SOCS1, but mutant SOCS1 with both substitutions displayed a modest reduction of its inhibitory capacity. This difference was not observed for IFN-α2-induced STAT3 phosphorylation (Supplemental Figure 1B,C). Although SOCS1 is known to modulate the signaling of both IFN-I and IFN-γ, the P50L and A76G variants (individually or in cis) did not affect the inhibitory activity of SOCS1 on IFN-γ-induced STAT1 phosphorylation (Figure 2F–H).
Figure 2. Functional analysis of SOCS1 variants.
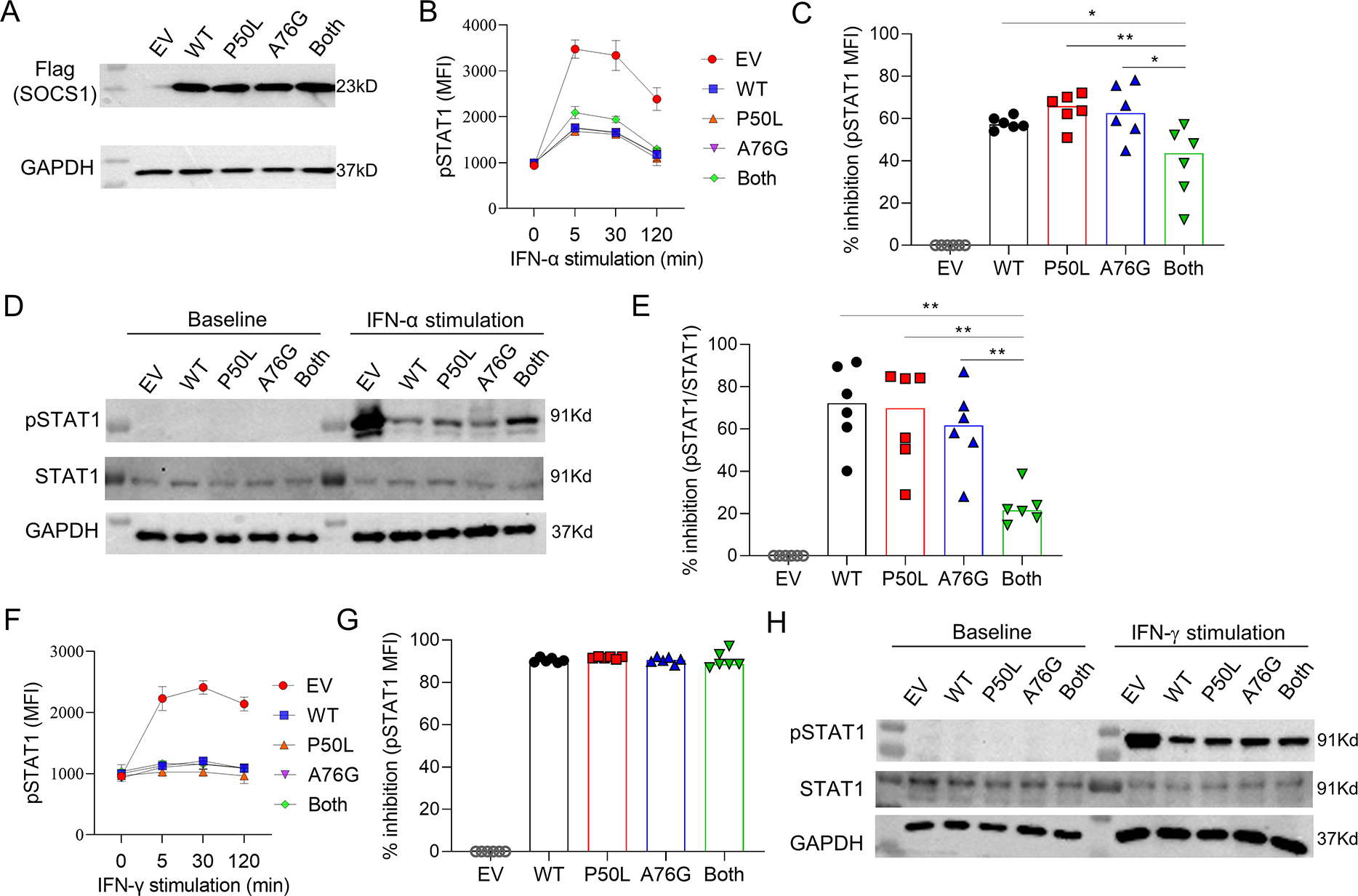
A) Wildtype and mutant SOCS1 expression in transfected HEK293T cells. B) Intracellular phospho-STAT1 staining, as measured by mean fluorescence intensity (MFI), in transfected HEK293T cells treated with PBS or IFN-α2 (100 ng/mL) analyzed at the indicated timepoints and C) quantification at 5 minutes after stimulation. D) Representative immunoblot and E) quantification of STAT1 phosphorylation in transfected HEK293T cells treated with PBS or IFN-α2 (100 ng/mL) for 5 minutes. F) Intracellular phospho-STAT1 staining in transfected HEK293T cells treated with PBS or IFN-γ (100 ng/mL) analyzed at the indicated timepoints and G) quantification at 30 minutes after stimulation. H) Immunoblot of STAT1 phosphorylation in transfected HEK293T cells treated with PBS or IFN-γ (100 ng/mL) for 30 minutes. Panels C and E illustrate the percentage inhibition (relative to EV) from 6 independent experiments. EV, empty vector; MFI, mean fluorescence intensity; GAPDH, glyceraldehyde 3-phosphate dehydrogenase (loading control). * p < 0.05 (Mann Whitney U test for panels C and E).
Next, we assessed the effect of wildtype and mutant SOCS1 on the expression of interferon inducible genes (ISGs) in transfected cells treated with IFN-α2. Wildtype SOCS1 suppressed the induced expression of IFI44, IFIT1, IFTM3, LY6E, MX1, MX2 and CXCL10, as measured by quantitative PCR. While mutant SOCS1 with both P50L and A76G variants seemed less potent in suppressing the expression of ISGs compared to the wildtype protein, the disruption was mild and statistical significance was shown only for IFI44 and IFIT1 (Figure 3A).
Figure 3. The impact of SOCS1 on the expression of interferon-stimulated genes (ISG).
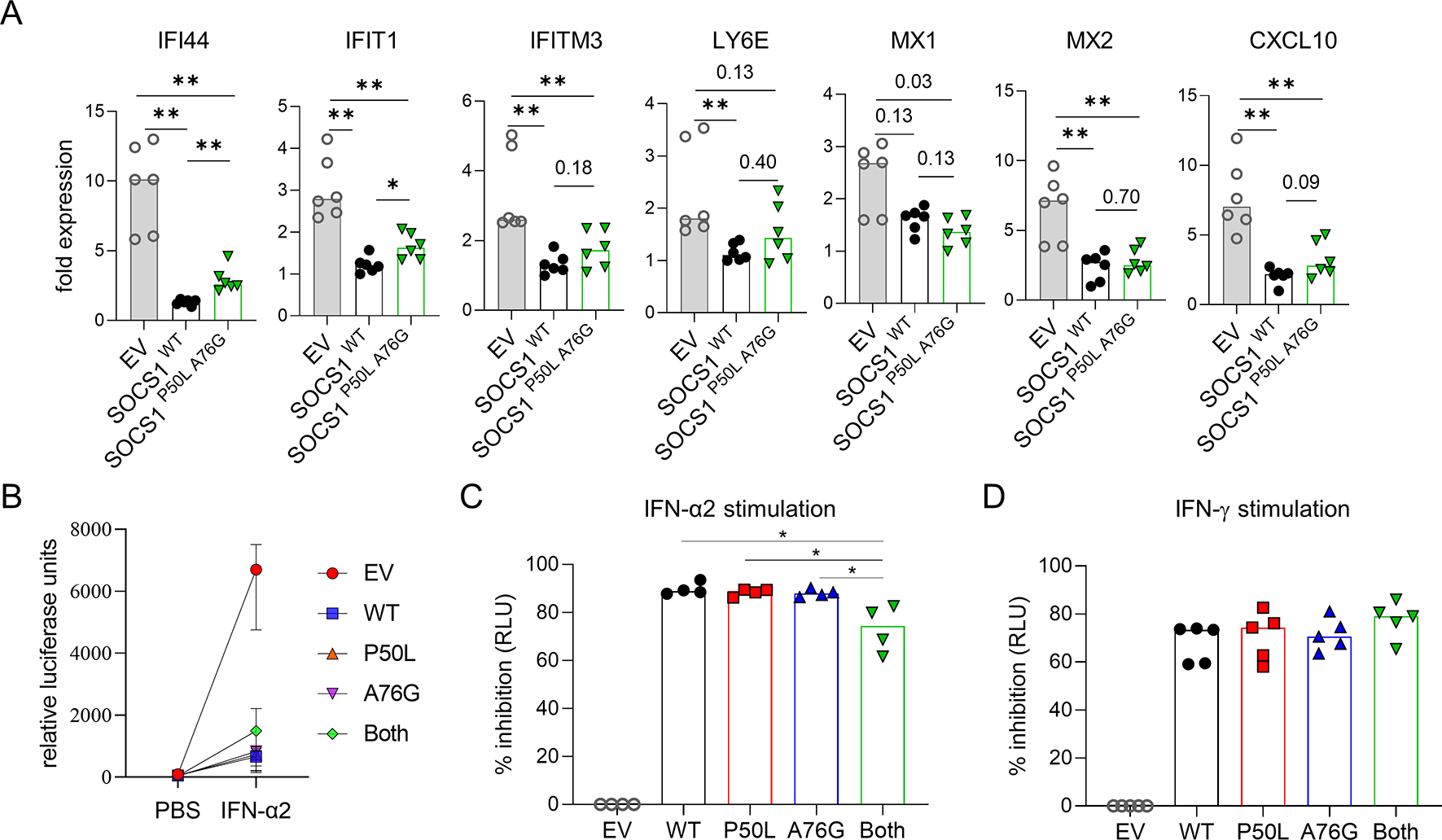
A) Quantitative PCR of select ISGs in HEK293T cells transfected with wildtype or mutant SOCS1 plasmids and stimulated with IFN-α2 (100 ng/mL) for 8 hours. B) Luciferase activity in ISRE-luc 293T cells transfected with wildtype or mutant SOCS1 plasmids and stimulated with IFN-α2 (100 ng/mL) for 24 hours. C) Percentage inhibition of luciferase activity from IFN-α2 (100 ng/mL) or IFN-γ (100 ng/mL) stimulation (pooled from 4–5 independent experiments). * p < 0.05 (Mann Whitney U test).
Using another method to evaluate IFN-I-induced gene expression, we analyzed HEK293T IFN-I-luciferase reporter cells that produce luciferase upon IFN-I stimulation. Overexpression of wildtype SOCS1 inhibited IFN-α2-induced luciferase production by 90% (Figure 3B,C). In line with the data on STAT1 phosphorylation, SOCS1P50L and SOCS1A76G individually displayed normal inhibitory capacity while mutant SOCS1 with both variants was ~20% less potent in suppressing IFN-α2-induced luciferase production. Parallel experiments using IFN-γ-luciferase reporter cells confirmed that SOCS1P50L A76G did not impact IFN-γ signaling (Figure 3D). Given the proximity of both P50L and A76G variants to the KIR domain, we investigated whether these variants affect the interaction between SOCS1 and JAKs. We performed immunoprecipitation using cell lysates from transfected HEK293T cells to pull down FLAG-tagged wildtype or mutant SOCS1. JAK1 binding to SOCS1 was quantified by western blotting. The ratio of JAK1 and SOCS1-FLAG expression was comparable in the input cell lysates (Figure 4A; left panel). Immunoprecipitation of SOCS1 mutant with P50L and A76G variants in cis revealed reduced binding of JAK1, while neither variant alone affected JAK1 binding (Figure 4A, right panel). Similar effects were shown when the transfected cells were stimulated with IFN-α2 (Figure 4B). These data collectively demonstrate that SOCS1P50L and SOCS1A76G are benign missense variants individually, but in cis expression of both variants leads to impaired inhibition of IFN-I associated with diminished JAK1 binding.
Figure 4. SOCS1 mutants and interaction with JAK1.
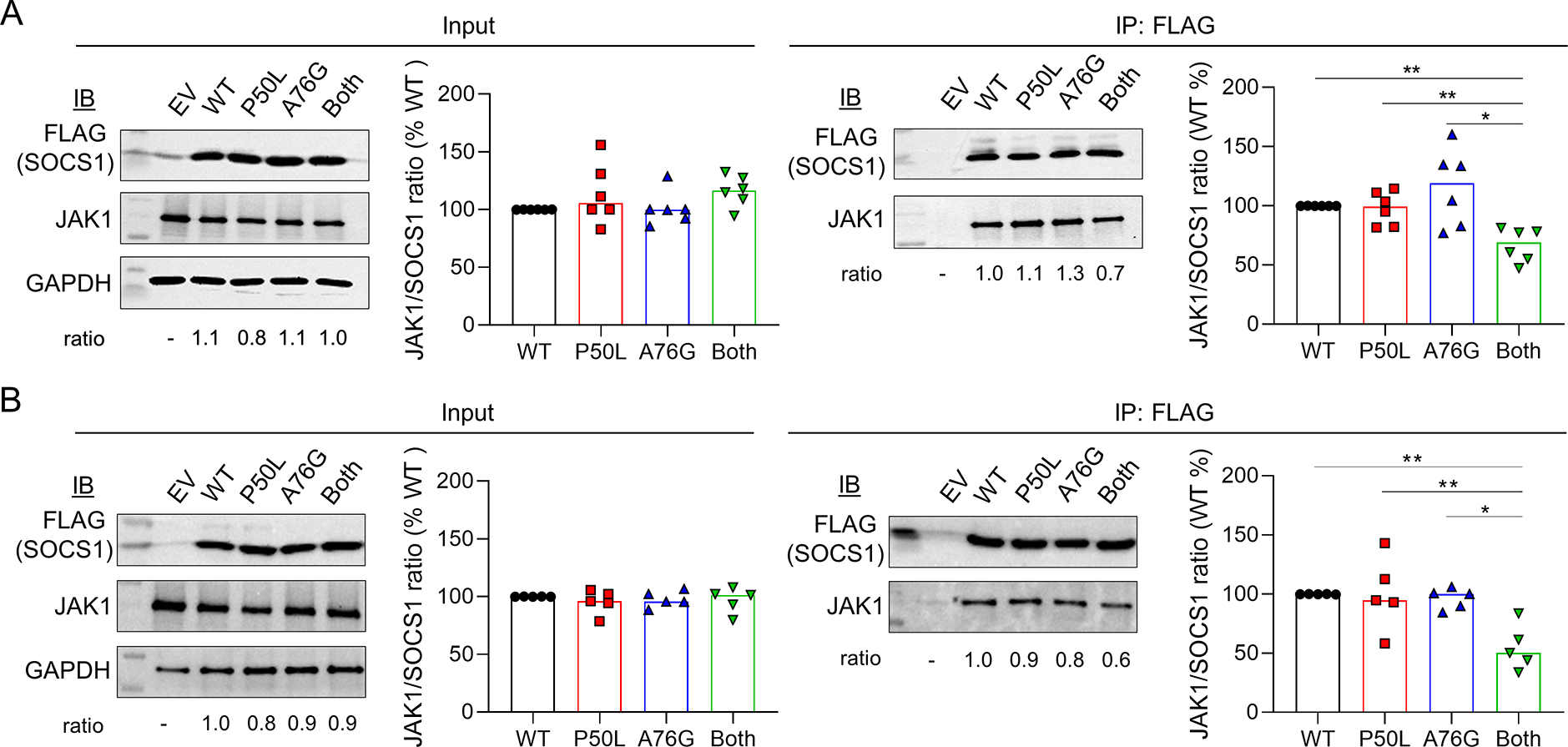
A) Immunoblot and densitometry analysis of JAK1/SOCS1 in transfected HEK293T cells before or after immunoprecipitation using anti-FLAG beads. The ratio of JAK1/SOCS1 expression is displayed. B) Immunoblotting and immunoprecipitation for the analysis of JAK1/SOCS1 interaction in IFN-α2-treated cells. Bands were quantified by densitometry analysis using Image J Software. Bar graphs illustrate the median of 6 independent experiments in panel A and 5 independent experiments in panel B. p < 0.05 (Mann Whitney U test).
2.3. IFN-I dysregulation in individuals with SOCS1P50L A76G
To investigate if heightened IFN-I signaling occurs in individuals with the SOCS1P50L A76G variant, we performed bulk RNA sequencing analysis of PBMC. The SOCS1P50L A76G group included P1, P2, and their mother, all of whom were asymptomatic and without recent illness at the time of sample collection. A substantial proportion of the most upregulated genes in the SOCS1P50L A76G group were ISGs according to the Interferome database (19/68 genes; Figure 5A).[10] Enrichment of ISGs was also confirmed by gene set enrichment analysis using the Hallmark interferon-alpha response gene set (Figure 5B). Differential expression of leading-edge genes is displayed in Figure 5C. Quantitative PCR confirmed the group differences in several ISGs (Figure 5D). Consistent with the hypomorphic nature of the SOCS1P50L A76G variant, the enhancement of ISG expression in all three family members was less prominent compared to a patient with SOCS1 haploinsufficiency due to a complete monoallelic deletion.[11]
Figure 5. In cis SOCS1P50L A76G variant is associated with enhanced IFN-I signaling.
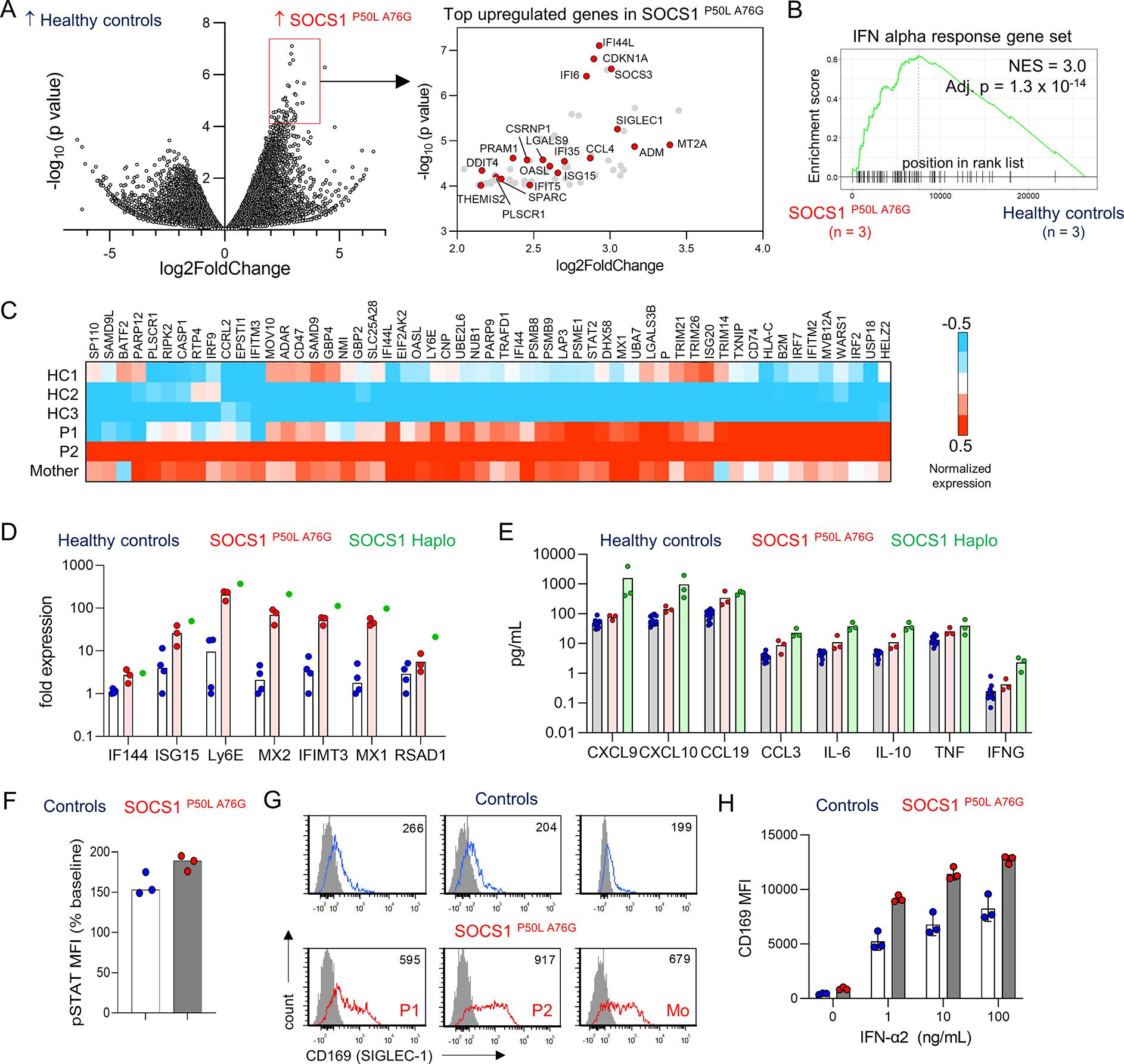
A) Volcano plot showing the differentially expressed genes in 3 healthy controls vs 3 individuals with SOCS1P50L A76G. Among the top 68 upregulated genes in the SOCS1P50L A76G group, 18 were ISGs (red circles; right panel); B) GSEA plot for IFN-alpha response genes in healthy controls vs individuals with SOCS1P50L A76G. C) Heatmap display of leading-edge interferon alpha response genes from GSEA; D) Quantitative PCR of ISGs using cDNA from PBMCs (n = 3 per group for healthy controls and SOCS1P50L A76G; n = 1 for SOCS1 haploinsufficiency). E) Quantification of cytokine levels in the plasma of healthy controls, individuals with SOCS1P50L A76G and previous cases of SOCS1-haplosufficiency by proximity extension assay (n = 3 per group). F) Flow cytometry quantification of IFN-I-induced STAT1 phosphorylation in CD14+ monocytes from healthy controls and individuals with SOCS1P50L A76G (n = 3 per group). G) Flow cytometry plot of surface CD169 expression on CD14+ monocytes at baseline (n = 3 per group). H) MFI of CD169 expression on monocytes 24 hours after stimulation with IFN-α2 (0 – 100 ng/mL; n = 3 per group).
Analysis of plasma cytokines and chemokines by proximity extension assay showed elevation of the interferon-induced chemokine CXCL10 in individuals with SOCS1P50L A76G compared to healthy controls. Mild increases in IL-6, TNF, CCL19, and IL-10 were also noted, illustrating that the immunodysregulation in individuals with SOCS1P50L A76G variant was not limited to the IFN-I axis. Supporting the differential response to IFN-I and IFN-γ, neither the probands or the mother displayed increased levels of IFN-γ nor the IFN-γ-inducible chemokine CXCL9. In contrast to the mild findings associated with SOCS1P50L A76G, higher levels of these cytokines and chemokines were observed in the plasma of 3 patients with SOCS1 haploinsufficiency due to loss-of-function variants (Figure 5E).[8, 11] Lastly, we tested whether cells from individuals with SOCS1P50L A76G are hyperresponsive to IFN-I signaling. We found a trend for increased IFN-α2-induced STAT1 phosphorylation in CD14+ monocytes from the probands and the mother compared to healthy controls (Figure 5F). Accordingly, greater surface expression of the ISG CD169 (SIGLEC-1) at baseline in individuals with SOCS1P50L A76G was drastically augmented by IFN-α2 stimulation (Figure 5G,H). In contrast, response to IFN-γ as measured by IFN-γ-induced expression of CD64 and CD274 on monocytes was comparable between the groups (Supplemental Figure 2A,B). Moreover, we did not detect differences in IL-6-induced STAT3 phosphorylation or IL-4-induced STAT6 phosphorylation in monocytes from individuals with SOCS1P50L A76G compared to healthy controls (Supplemental Figure 2C,D). Taken together, these studies demonstrate that SOCS1P50L A76G is associated with increased IFN-I signaling at baseline and hypersensitivity to IFN-I stimulation.
4. Discussion
Using whole exome sequencing, we describe two SOCS1 variants, P50L and A76G in cis configuration, in a family with early-onset autoimmune manifestations. Both variants were predicted to be benign by in silico algorithms and ectopic expression studies confirmed that neither variant alone affected the inhibitory function of SOCS1. Uniquely, in cis expression of both variants partially disrupted the binding of SOCS1 to JAK1 and conferred enhanced response to IFN-I stimulation. Supportive studies using primary cells from individuals with SOCS1P50L A76G showed increased baseline expression of ISGs and hypersensitivity to IFN-I stimulation.
By illustrating that a combination of “benign” variants can elicit deleterious consequences, our study demonstrates that interactions of minor structural modifications within the same gene may contribute to the development of monogenic diseases. Functional studies are needed to clarify the impact on protein function in these scenarios as most in silico prediction algorithms are not designed to analyze in cis variants.
SOCS1 interacts with JAK via the KIR and SH2 domains and prevents the phosphorylation of STAT. SOCS1 has the highest affinity for the kinase domain of JAK1 and JAK2; it has lower affinity for TYK2 and does not interact with JAK3.[12] The regulatory function of SOCS1 is critical as complete deficiency of SOCS1 in mice causes neonatal lethality due to excessive IFN-γ signaling and T cell-driven inflammation [13, 14]. Interestingly, heterozygous SOCS1 deficiency is associated with the development of a lupus-like disease in female mice.[15]
Human SOCS1 haploinsufficiency was first described in 2020 and most cases are caused by monoallelic LOF variants (e.g., large deletions, indels with frameshift and nonsense variants).[7–9, 11] The clinical manifestations in the probands including SLE, lupus nephritis, and ITP, are within the heterogenous clinical spectrum of SOCS1 haploinsufficiency. The degree of immunodysregulation as assessed by gene expression signature and plasma cytokine / chemokine levels was milder in individuals with SOCS1P50L A76G compared to those with LOF variants. Nevertheless, these data highlight the fine-tuning of cytokine signaling by SOCS1 as functional disruption of one allele by ~20–30% may be sufficient to drive aberrant IFN-I signaling. These findings raise the possibility that differential expression of SOCS1 among individuals, even in the absence of deleterious variants, may modulate the risk for autoimmunity. While SOCS1 expression at the transcript level was comparable between healthy controls and patients with SLE in several publicly available datasets (Supplemental Figure 3),[16–19] whether a difference exists at the protein level warrants further assessment. Additional genetic and/or environmental factors may also play a role in disease penetrance as P1 and P2 were asymptomatic until adolescence and both presented to medical attention within a span of 4 months. In line with the role of IFN-I in the development of autoimmune disease, PBMC from both probands and the mother in our study displayed the IFN-I signature seen in patients with SLE. While the use of JAK inhibitors has been described in patients with SOCS1 haploinsufficiency,[9, 11] the probands have responded well to standard therapy thus far. Dysregulation of IFN-I signaling is not the only connection between SOCS1 and autoimmunity. SOCS1 interacts with several tyrosine kinases (JAK1, JAK2, and TYK2) that mediate the signaling of multiple cytokines and growth factors. Curiously, hyperresponsiveness of the SOCS1P50L A76G variant appears to be specific to IFN-I stimulation as phosphorylation of STAT1, STAT3, and STAT6 induced by IFN-γ, IL-6, and IL-4, respectively, was comparable to wildtype SOCS1. Future studies are required to understand the differential requirement of SOCS1 in the regulation of these cytokines.
A limitation of this study is the timing of biospecimen collection. Analyses of the probands were performed during clinical remission and the degree of immunodysregulation would likely be more pronounced in the setting of active disease. While we suspect that the SOCS1P50L A76G variant is likely linked to the finding of Graves disease in the probands’ maternal grandfather and aunt, DNA was not available to confirm this association.
5. Conclusion
In summary, our work illustrates the precise regulation of IFN-I signaling by SOCS1 as mild disruption of SOCS1 function in one allele is associated with heterogenous autoimmune manifestations. The unique finding of in cis SOCS1 variants in this study illustrates that a combination of variants that are individually benign may have deleterious consequences.
Supplementary Material
Highlights.
In cis SOCS1 variants P50L and A76G are associated with early-onset autoimmunity
SOCS1P50L A76G, but neither variant alone, impairs the binding of SOCS1 with JAK1
SOCS1P50L A76G confers hypersensitivity to IFN-I but not IFN-γ, IL-4, or IL-6
Combined effects of benign variants may have deleterious consequences
Acknowledgement
We thank the patients and families for their participation. PYL is supported by the National Institute of Health / National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) K08-AR074562 and the Rheumatology Research Foundation K Supplement Award.
Abbreviations
- IFN-I
Type I interferon
- IFN-II
Type II interferon
- ISG
IFN-stimulated gene
- ITP
Immune thrombocytopenia
- JAK
Janus kinase
- SOCS
Suppressor of cytokine signaling
- SLE
Systemic lupus erythematosus
- STAT
Signal transducer and activator of transcription
- WES
Whole exome sequencing
Footnotes
Conflict of interest statement:
The authors declare the following conflicts of interest related to this work: PYL is a consultant for Fresh Track Therapeutics and Exo Therapeutics and receives royalties from UpToDate. The other authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Tsokos GC. Systemic lupus erythematosus. N Engl J Med, 2011;365:2110–21. [DOI] [PubMed] [Google Scholar]
- [2].Crow MK. Collaboration, genetic associations, and lupus erythematosus. N Engl J Med, 2008;358:956–61. [DOI] [PubMed] [Google Scholar]
- [3].Lo MS. Insights Gained From the Study of Pediatric Systemic Lupus Erythematosus. Front Immunol, 2018;9:1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity, 2006;25:383–92. [DOI] [PubMed] [Google Scholar]
- [5].Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N Engl J Med, 2020;382:211–21. [DOI] [PubMed] [Google Scholar]
- [6].Rockowitz S, LeCompte N, Carmack M, Quitadamo A, Wang L, Park M et al. Children’s rare disease cohorts: an integrative research and clinical genomics initiative. NPJ Genom Med, 2020;5:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Thaventhiran JED, Lango Allen H, Burren OS, Rae W, Greene D, Staples E et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature, 2020;583:90–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lee PY, Platt CD, Weeks S, Grace RF, Maher G, Gauthier K et al. Immune dysregulation and multisystem inflammatory syndrome in children (MIS-C) in individuals with haploinsufficiency of SOCS1. J Allergy Clin Immunol, 2020;146:1194–200.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hadjadj J, Castro CN, Tusseau M, Stolzenberg MC, Mazerolles F, Aladjidi N et al. Early-onset autoimmunity associated with SOCS1 haploinsufficiency. Nat Commun, 2020;11:5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H et al. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res, 2013;41:D1040–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Michniacki TF, Walkovich K, DeMeyer L, Saad N, Hannibal M, Basiaga ML et al. SOCS1 Haploinsufficiency Presenting as Severe Enthesitis, Bone Marrow Hypocellularity, and Refractory Thrombocytopenia in a Pediatric Patient with Subsequent Response to JAK Inhibition. J Clin Immunol, 2022;42:1766–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liau NPD, Laktyushin A, Lucet IS, Murphy JM, Yao S, Whitlock E et al. The molecular basis of JAK/STAT inhibition by SOCS1. Nat Commun, 2018;9:1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS et al. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell, 1999;98:597–608. [DOI] [PubMed] [Google Scholar]
- [14].Marine JC, Topham DJ, McKay C, Wang D, Parganas E, Stravopodis D et al. SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell, 1999;98:609–16. [DOI] [PubMed] [Google Scholar]
- [15].Fujimoto M, Tsutsui H, Xinshou O, Tokumoto M, Watanabe D, Shima Y et al. Inadequate induction of suppressor of cytokine signaling-1 causes systemic autoimmune diseases. Int Immunol, 2004;16:303–14. [DOI] [PubMed] [Google Scholar]
- [16].Jiang SH, Athanasopoulos V, Ellyard JI, Chuah A, Cappello J, Cook A et al. Functional rare and low frequency variants in BLK and BANK1 contribute to human lupus. Nat Commun, 2019;10:2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lee HM, Mima T, Sugino H, Aoki C, Adachi Y, Yoshio-Hoshino N et al. Interactions among type I and type II interferon, tumor necrosis factor, and beta-estradiol in the regulation of immune response-related gene expressions in systemic lupus erythematosus. Arthritis research & therapy, 2009;11:R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lee HM, Sugino H, Aoki C, Nishimoto N. Underexpression of mitochondrial-DNA encoded ATP synthesis-related genes and DNA repair genes in systemic lupus erythematosus. Arthritis research & therapy, 2011;13:R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Toro-Domínguez D, Martorell-Marugán J, Goldman D, Petri M, Carmona-Sáez P, Alarcón-Riquelme ME. Stratification of Systemic Lupus Erythematosus Patients Into Three Groups of Disease Activity Progression According to Longitudinal Gene Expression. Arthritis & rheumatology (Hoboken, NJ), 2018;70:2025–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All primary data are available from the corresponding author upon request.