

Abstract
Duchenne muscular dystrophy (DMD) is the most prevalent herediatry disease in men, characterized by dystrophin deficiency, progressive muscle wasting, cardiac insufficiency, and premature mortality, with no effective therapeutic options. Here, we investigated whether adenine base editing can correct pathological nonsense point mutations leading to premature stop codons in the dystrophin gene. We identified 27 causative nonsense mutations in our DMD patient cohort. Treatment with adenine base editor (ABE) could restore dystrophin expression by direct A-to-G editing of pathological nonsense mutations in cardiomyocytes generated from DMD patient-derived induced pluripotent stem cells. We also generated two humanized mouse models of DMD expressing mutation-bearing exons 23 or 30 of human dystrophin gene. Intramuscular administration of ABE, driven by ubiquitous or muscle-specific promoters could correct these nonsense mutations in vivo, albeit with higher efficiency in exon 30, restoring dystrophin expression in skeletal fibers of humanized DMD mice. Moreover, a single systemic delivery of ABE with human single guide RNA (sgRNA) could induce body-wide dystrophin expression and improve muscle function in rotarod tests of humanized DMD mice. These findings demonstrate that ABE with human sgRNAs can confer therapeutic alleviation of DMD in mice, providing a basis for development of adenine base editing therapies in monogenic diseases.
Keywords: MT: RNA/DNA Editing, adenine base editing, DMD, nonsense mutation, humanized mouse model
Graphical abstract
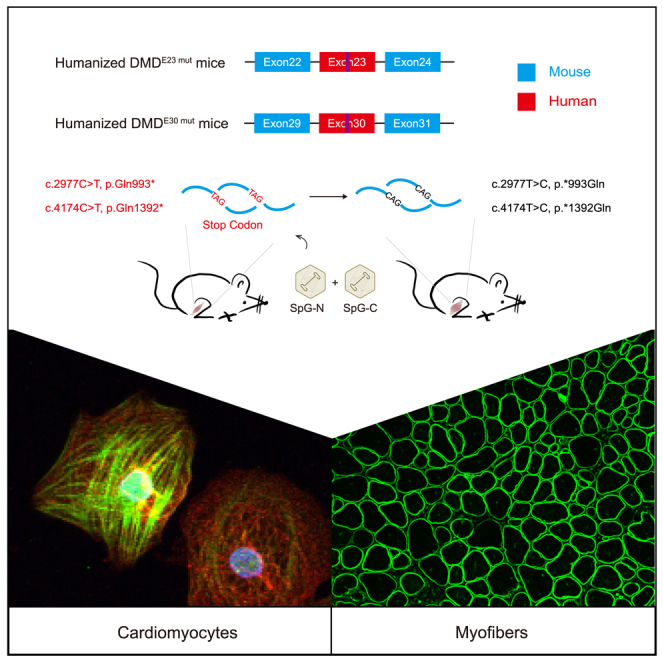
Li and colleagues demonstrated the efficacy of the SpG-ABE system in correcting several human nonsense mutations and restoring dystrophin expression in both patient iPSC-derived cardiomyocyte cells and humanized mouse models of DMD, indicating the potential of adenine base editing for the intervention of diseases caused by nonsense mutations.
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked lethal disease caused by genetic mutations in the dystrophin gene and has become the most common neuromuscular disorder in childhood whose global prevalence reaches up to 1 in every 3,500 male births.1,2 The dystrophin serves as a key scaffold protein to stabilize muscle fiber membrane and guide signaling molecule organization, thereby ensuring muscular system homeostasis and function.3,4 Consequently, the deficiency of functional dystrophin brings about disruptions in the sarcolemmal integrity and muscle cell regeneration, leading to progressive muscle atrophy and weakness. Therefore, DMD patients gradually experience ambulation loss, pulmonary insufficiency, heart failure, and early death in their twenties.5,6 DMD still cannot be completely cured. Recent Food and Drug Administration (FDA)-approved mini-dystrophin, in principle, only changes DMD into the milder phenotype of Becker muscular dystrophy and does not cure it. Among thousands of DMD-causing mutations, the nonsense point mutations are estimated to account for approximately 10% of all DMD cases.7,8 Both non-homologous end-joining (NHEJ) and homology-directed repair strategies based on the CRISPR-Cas system have been harnessed to treat pathological point mutations and partially restore dystrophin expression in the mdx or mdx4Cv mice model of DMD bearing a nonsense point mutation in mouse exon 23 or 53, respectively.9,10,11,12,13,14,15,16 However, the aforementioned strategies rely on random and low-efficiency repair events after DNA double-strand break (DSB) induced by wild-type (WT) Cas9 nuclease leading to limited therapeutic efficacy with large and unneglectable risk for the introduction of errant genome modifications such as large DNA insertion/deletion (indel), inversion, and chromosome rearrangements.17,18,19,20 Hence, there is an urgent need to explore safe and efficient gene editing approaches to rectify pathological nonsense mutations affecting DMD patients without genomic DSB induction.
Recently, adenine base editing became an attractive strategy for the treatment of genetic muscular diseases such as DMD and spinal muscular atrophy due to its high accuracy and safety profile.21,22,23,24 The adenine base editor (ABE) is made up of Cas9 nickase and engineered adenosine deaminase,25,26,27 which recognizes the target site in a single guide RNA (sgRNA)-dependent manner and directly catalyzes A-to-G substitution without exogenous template and DNA cleavage. Thus, the base editing capability of ABE avoids the route dependent on DSB generation and the NHEJ pathway,26 making the ABE a safe tool for gene therapies. Notably, the G⋅C to A⋅T conversion has been found as the pathological source of ∼174 DMD point mutations,22 suitable for base correction via ABE. Previously, the fusion of ABE7.10 or miniABE(GG) variant with Streptococcus pyogenes Cas9 nickase (nSpCas9) has been taken to treat two different DMD mouse models carrying point mutations in mouse exons 4 and 20 respectively.21,22 However, sequence divergence between mouse and human dystrophin genes impeded mouse-specific sgRNA from use in patients, calling for generating genetically humanized mice for human-specific sgRNA test. Furthermore, adenine base editing efficacy for various point mutations distributed among 79 different exons in the dystrophin gene might be affected by distinct sequence context,28,29 and remains to be investigated.
In the present study, we demonstrated the efficacy of the SpG-ABE system with a PAM-relaxed SpCas9 variant30,31 in correcting several human nonsense mutations identified from the DMD cohort and restoring dystrophin expression in both patient iPSC-derived cardiomyocyte cells and humanized mouse models of DMD. Notably, SpG-ABE treatment also improve muscular function of DMD mice, indicating the potential of adenine base editing for the intervention of other diseases caused by nonsense mutations.
Results
Identification and in vitro adenine base editing of nonsense mutations from DMD patients’ cohorts
To identify DMD-related nonsense mutations as potential targets for adenine base editing, we performed whole-exome sequencing in human peripheral leukocytes obtained from DMD patients admitted to our hospital. These patients presented with clinically dystrophic symptoms such as abnormal gait and muscular hypertrophy. Histopathological analysis of bicep muscle samples confirmed that dystrophin protein was absent in these patients (Figure 1A), suggesting a pathological cause of the dystrophic phenotype. Subsequent whole-exome sequencing and bioinformatics analysis identified 12 candidate nonsense point mutations in 27 patients, including c.4174C>T, p.Gln1392∗ in exon 30 found in our previous work32 (Table S1). These nonsense mutations were randomly distributed in exons of the dystrophin gene and could potentially induce premature translation termination of dystrophin transcripts. Among these nonsense mutations, 52% (n = 14) generated a TGA termination codon, while 30% (n = 8) and 18% (n = 5) resulted in TAG and TAA termination codons, respectively (Figure 1B).
Figure 1.

Adenine base editing correct pathological nonsense mutations in vitro
(A) Immunohistochemistry of dystrophin protein in the left bicep muscles of normal donor and DMD patient. Dystrophin is present in brown. Scale bar, 50 μm. (B) The percentages of premature termination codons arising from pathological nonsense mutations. (C) Schematic for the fluorescent reporter assay to determine the optimal sgRNAs. The reporter construct comprises the mCherry, human exonic mutant sequence, and the EGFP. The correction of nonsense mutation in the exonic mutant sequence allows EGFP expression. (D) Representative flow cytometry plots of EGFP expression in HEK293T cells treated with reporter plasmid alone or its combination with ABE construct. (E) Quantification of the percentages of EGFP-positive cells after transfection of ABE and sgRNAs targeting nonsense mutation in human exons 23, 24, 30, and 44 (n = 3). Triangles are biological replicates and quantification is shown as mean ± SD.
To test the editing possibility of adenines in these termination codons by the ABE, we screened a panel of 33 sgRNA pairs spanning 12 different nonsense mutation sites in the dystrophin gene (see Table S2) that were individually inserted into an EGFP reporter plasmid to block expression of the fluorescent protein (Figure 1C). We co-transfected each nonsense mutation-reporter with a base editing plasmid expressing SpG-ABE with more broad PAM recognition30,31 than WT SpCas9 and the respective sgRNA in HEK293T cells, then quantified the efficiency of EGFP fluorescence restored by each sgRNA-guided ABE relative to that of an upstream mCherry reporter lacking a premature termination codon. As expected, the expression of ABE with a nonspecific sgRNA failed to restore detectable EGFP signal (Figures 1D and S1A), indicating the stringency of out reporter system. In contrast, we found that several sgRNAs could direct the correction of nonsense mutations to activate robust EGFP expression (Figures 1D and S1A), with several sgRNAs displaying particularly high efficiency in rescuing EGFP expression blocked by nonsense mutations in human dystrophin exons 23, 24, 30, 37, 44, or 55 (Figures 1E and S1B). Other sgRNAs we screened for nonsense point mutations in exons 6, 25, 35, 41, 54, or 59 could only guide marginal editing (Figure S1B). For the most efficient sgRNAs, fluorescence restoration efficiency was as high as 80% (Figures 1D, 1E, and S1B). These results indicated that an SpG-ABE DNA editing system could mediate the correction of several nonsense mutations in the human dystrophin gene.
SpG-ABE can restore dystrophin expression in human DMD cardiomyocytes by correcting nonsense mutations
Patient-derived induced pluripotent stem cells (iPSCs) are an important in vitro model for testing the efficacy of SpG-ABE in correcting DMD mutations and restoring dystrophin expression. To investigate whether SpG-ABE could restore dystrophin expression in human DMD cells, we generated iPSCs from a DMD patient with the c.4174C>T mutation (Figures 2A and S2A–S2D). The iPSC was corrected by the SpG-ABE system, and the monoclonal lines with the corrected mutation site were collected. Then the selected clones were differentiated into cardiomyocytes and assessed for dystrophin expression. We nucleofected DMD iPSCs harboring the c.4174C>T mutation with SpG-ABE and sgRNA-1∼5 to induce base editing. Deep sequencing revealed that SpG-ABE and hEx30 sgRNA-4 could provide average on-target editing rates of 58.5% ± 0.707% without detectable bystander editing (Figures 2B and 2C). By contrast, SpG-ABE guided by hEx30 sgRNA-2, sgRNA-3, or sgRNA-5 induced around 20%–30% on-target editing efficiency with 5%–20% bystander editing events (Figure S2E). Single corrected iPSC colonies were selected for expansion and differentiated into cardiomyocytes. Immunofluorescence staining for dystrophin showed high expression in these corrected cardiomyocytes, with levels comparable to that in normal cardiomyocytes derived from H9 cells (Figure 2D). Both genome and cDNA sequencing confirmed correction of c.4174C>T mutation in cardiomyocytes derived from ABE-edited DMD iPSCs (Figures S2F and S2G). Moreover, no significant genomic modifications above background were detected at any of the top nine predicted off-target sites (Figure S2H), supporting that the SpG-ABE system exhibited high specificity in editing the c.4174C>T nonsense mutation in dystrophin gene.
Figure 2.

Adenine base editing rescues dystrophin expression in human DMD cardiomyocytes
(A) Illustration of the application of adenine base editing in human DMD iPSC model. Human DMD iPSCs from patient fibroblasts were edited by the SpG-ABE components and subsequently differentiated into cardiomyocytes for downstream analyses. (B) Schematic for the binding position of hEx30 sgRNA-4 in the exon 30 mutant. The PAM and sgRNA are shown in red and blue respectively. The adenines in the editing window are numbered from the PAM. Genomic sequencing chromatogram of the exon 30 mutant in human iPSCs is present at the bottom. (C) The percentages of genomic editing events in human DMD iPSCs with ABE and hEx30 sgRNA-4 treatment (n = 3). On-target adenine (A15) is shown in blue. (D) Immunofluorescence analysis of dystrophin and cTnI levels in the normal, DMD, and corrected cardiomyocytes. Dystrophin and cTnI are shown in red and green, respectively. Nuclei are marked by DAPI stain in blue. Scale bar, 50 μm. Quantitative data are presented as mean ± SEM.
For in vivo administration, the SpG-ABE variant was split into two separate fragments using the trans-splicing inteins from Nostoc punctiforme (Npu),33 and each fragment was individually packaged in a single AAV vector (Figure S2I). A dual adeno-associated virus 9 (AAV9) system was chosen for delivery of SpG-ABE components due to the preferential tropism of AAV9 for skeletal and cardiac muscles. For ABEv1 construct, we adapted the framework established by Levy et al.,34 replacing nCas9 (D10A) with SpG. ABEv1 carrying ubiquitous promoter CBh was used to test the mutation editing feasibility in patient-derived iPSCs after splitting ABE with the intein system, whereas ABEv2 carrying muscle-specific promoter Spc5.12 was designed mainly for muscle-specific expression of ABE in vivo to avoid ABE expression in non-muscle cells. Previous research has suggested that higher sgRNA copy numbers can enhance efficiency.35,36 Therefore, we added additional sgRNA expression units in ABEv2 design. Nucleofection of the ABEv1 andABEv2 in human DMD iPSCs resulted in 75.7% ± 3.8% and 30.3% ± 13.0% on-target editing frequency, respectively, at the target adenine, A15 (Figure S2J), suggesting that both split-SpG-ABE systems could correct the c.4174C>T mutation, with the ubiquitous CBh promoter conferring higher efficiency in undifferentiated patient iPSCs. These results collectively illustrated that genome editing with SpG-ABE could restore dystrophin expression by correcting nonsense mutations in human DMD cells.
Local delivery of SpG-ABE can restore dystrophin expression in humanized DMD mice
Based on our in vitro results, we next investigated whether SpG-ABE could directly correct nonsense mutations in vivo using humanized DMD model mice generated by introducing exon 30 of human dystrophin gene harboring the c.4174C>T nonsense mutation into the murine dystrophin homolog (DMDE30mut mice).32 For in vivo evaluation of the split-SpG-ABE systems, we intramuscularly administered 5 × 1011 viral genomes (vg) for each AAV9 vector into the right tibialis anterior (TA) muscle of 3-week-old humanized dystrophic mice with left TA saline-treated as control (Figures 3A and 3B). At 4 weeks after local AAV injection, the skeletal muscles from SpG-ABE or saline-treated legs were collected for analysis. In the humanized DMDE30mut mice given a single administration of SpG-ABE with hEx30 sgRNA-4, we observed obvious A-to-G transition at A15, but not in mice treated with saline (Figure 3C). The average efficiency of on-target genome editing was 9.7% ± 0.9% and 12.7% ± 1.9% in the ABEv1 and ABEv2 treatment groups, respectively (Figure 3C). Dmd cDNA sequencing revealed A-to-G editing rates of 45.7% ± 1.2% for ABEv1 and 68.3% ± 10.9% for ABEv2 in transcripts (Figure 3D). The discrepancy of A-to-G editing rate at the genome and cDNA levels was potentially due to the high abundance of dystrophin transcripts in skeletal muscles. Consequently, the TA muscles receiving ABEv1 infusion exhibited dystrophin expression restoration in 61.0% ± 4.1% of skeletal fibers, and ABEv2 treatment resulted in a restoration of 72.4% ± 5.4%. (Figures 3E, 3G, and S3). Western blot analysis revealed a restoration of dystrophin protein levels to 22.2% ± 1.269% and 59.3% ± 10.1% of WT control levels in DMDE30mut mice treated with ABEv1 and ABEv2, respectively (Figures 3F and 3H). In contrast to in vitro results showing that ABEv1 outperformed ABEv2 in patient iPSCs, the DMDE30mut mice treated with ABEv2 exhibited almost 2-fold greater restoration of dystrophin than those treated with ABEv1 (Figures 3F and 3H).
Figure 3.

Local ABE administration restores dystrophin expression in adult humanized DMDE30mut mice
(A) Schematic for the c.4174C>T nonsense mutation in human exon 30 sequence of the humanized DMDE30mut mice. (B) Overview of the intramuscular application of split-ABE system components in 8-week-old DMDE30mut mice. The TA muscles in mouse right legs were infused with AAV-expressing N- and C-terminal ABEs at the dose of 5 × 1011 vg/leg/AAV, while the saline was injected into left legs as negative controls. All mice were dissected for analyses at 3 weeks post-injection. (C) The percentages of genomic edits in mouse TA muscles with ABE or saline treatment (n = 3). (D) Deep-sequencing of the percentage of editing events in the transcripts of mouse TA muscles with ABE or saline treatment (n = 3). (E) Immunofluorescence analysis of dystrophin and spectrin expressions in the TA muscles of age-matched WT mice and DMDE30mut mice with ABE or saline treatment. Dystrophin is shown in green. Scale bar, 200 μm. (F) Western blot analysis of dystrophin and vinculin proteins in mouse TA muscles with ABE or saline treatment. The proteins from age-matched WT mice were used to standardize dystrophin expression levels. (G) Quantification of dystrophin+ myofibers in the cross-sections of mouse TA muscles as in (E). (H) Quantification of dystrophin expression level in the TA muscles from treated DMDE30mut mice shown as in (F). Quantitative data are calculated after normalization to the vinculin expression. Quantification is shown as mean ± SEM (n = 3). Each triangle represents an individual mouse. p value was evaluated with Student t test. NS, not significant; ∗p < 0.05.
We next tested whether the split SpG-ABE system could also correct other nonsense mutations identified from our DMD cohort. For this purpose, we generated another humanized DMD mouse model carrying a c.2977C>T, p.Gln993∗ mutation in exon 23 of human dystrophin, designated as DMDE23mut mice (Figure S4A). Similar to local AAV treatment for DMDE30mut mice, AAV9 vectors carrying ABEv1 and ABEv2 with sgRNAs targeting the A13 site in exon 23 were injected into the skeletal muscle of humanized DMDE23mut mice. Three weeks post intramuscular AAV delivery in adult DMDE23mut mice, we observed ∼1.5% and ∼1% on-target genomic modifications at A13 for ABEv1 and ABEv2, respectively (Figures S4B–S4D), which also generated ∼0.5% and ∼1% of bystander editing events at A18 (Figure S4D). Average A-to-G substitution rates at A13 in dystrophin transcripts were 13% or 17%, while bystander editing rates at A18 were 3% and 14% for ABEv1 and ABEv2, respectively (Figure S4E). Immunofluorescence staining indicated that dystrophin was present in 26.4% ± 1.5% of myofibers in the TA muscles treated with ABEv1, and in 53.6% ± 5.8% of those treated with ABEv2 (Figures S4F, S4G, and S5). Western blotting revealed that dystrophin protein was restored to 18.2% ± 5.7% and 55.9% ± 2.8% of that in WT mice by ABEv1 and ABEv2, respectively (Figures S4H and S4I). Overall, these data suggested that SpG-ABE with specific sgRNAs for pathological nonsense mutation in human dystrophin gene could robustly rescue the expression of dystrophin in a humanized DMD mouse model.
Systemic delivery of SpG-ABE improved muscle function via dystrophin restoration in humanized DMD mice
In light of the high efficiency of dystrophin restoration by SpG-ABE and hEx30 sgRNA-4 in TA muscles (Figures 3G and 3H), we then evaluated the effects of systemic SpG-ABE administration in neonatal DMDE30mut mice. At 4 weeks after intraperitoneal AAV delivery of ABEv1 or ABEv2, we harvested heart (HE), diaphragm (DI), and TA muscle tissues of AAV- or saline-infused mice (Figure 4A). Deep-seq analysis revealed that systemic ABEv1 treatment resulted in A-to-G conversion rates of 37.2% ± 1.4%, 13.2% ± 4.8%, and 7.7% ± 1.9% in heart, diaphragm, and TA tissues, respectively (Figure 4B). By contrast, ABEv2 treatment generated 20.3% ± 4.3%, 4.7% ± 1.9%, and 12.0% ± 2.6% editing efficiency at A15 in heart, diaphragm, and TA tissues, respectively in DMDE30mut mice (Figure 4B). Notably, the genomic editing frequency in heart was markedly higher than that in diaphragm and TA muscles (Figure 4B). RT-PCR and cDNA sequencing indicated that ABEv1 could induce 89.1% ± 1.7%, 23.1% ± 12.3%, and 19.3% ± 3.2% base substitution at A15 in endogenous dystrophin transcripts in heart, diaphragm, and TA muscle, and ABEv2 could induce 84.3% ± 1.8%, 9.1% ± 5.5% and 24.7% ± 3.3%, respectively (Figure 4C). Consistent with genome analysis (Figure 4B), ∼90% of A15 sites were rectified in cardiac transcripts by both ABEv1 and ABEv2, around 4-fold higher than that in DI and TA tissue (Figure 4C).
Figure 4.

Systemic ABE delivery improves muscle function by the restoration of broad dystrophin in humanized DMDE30mut mice
(A) Overview for systemic administration of the split-ABE system components in neonatal DMDE30mut mice. The postnatal day 3 (P3) mice were intraperitoneally infused with AAV9-expressing split-ABE components at the total dose of 2 × 1011 vg, with the saline treatment as negative control. Four weeks later, all mice were dissected for downstream analyses. (B) The percentages of genomic edits in the HE, DI, and TA of DMDE30mut mice with ABE or saline treatment (n = 3). (C) Deep-sequencing analysis of modification events in the transcripts of HE, DI, and TA from DMDE30mut mice with ABE or saline treatment (n = 3). (D) Immunohistochemistry of dystrophin protein expression in the HE, DI, and TA of age-matched WT mice and DMDE30mut mice with AAV or saline treatment. Dystrophin is shown in green. Scale bar, 200 μm. (E) Quantification of dystrophin+ myofibers in the cross-sections of mouse TA muscles as in (D). (F) Western blot analysis of dystrophin protein expression in the heart, DI, and TA from WT mice and DMDE30mut mice with ABE or saline treatment. The vinculin was used as the loading control. (G) Rotarod running test of age-matched WT mice and DMDE30mut mice with ABE or saline treatment (n = 9). (H) Rodent treadmill running test of age-matched WT mice and DMDE30mut mice with ABE or saline treatment (n = 9). Quantitative data are shown as mean ± SEM. p value was evaluated with Student t test. NS, not significant; ∗∗p < 0.01.
Immunofluorescence staining for dystrophin further revealed that systemic ABEv1 and ABEv2 delivery could restore protein expression in 95.9% ± 0.5% or 94.0% ± 0.9% myofibers of heart samples and 44.3% ± 1.0% or 44.6% ± 11.9% of skeletal myofibers in the TA, whereas dystrophin was detected in 71.6% ± 21.3% or 26.3% ± 19.6% of DI myofibers in the ABEv1 and ABEv2 groups, respectively (Figures 4D, 4E, and S6). The comparatively lower efficiency of dystrophin restoration observed in skeletal muscle as opposed to the heart was likely attributable to the inefficient AAV transduction in skeletal muscle. Western blot analysis further validated the efficient restoration of full-length dystrophin protein in heart and TA muscle following systemic administration of ABEv1 and ABEv2, while only the ABEv1 treatment could restore robust dystrophin expression in the DI (Figure 4F), aligning well with the immunofluorescent staining assays (Figures 4D, 4E, and S6). Furthermore, we found the muscle fibrosis and blood creatine kinase activity were alleviated by ABE treatment as indicated by H&E histology staining and CK analysis results (Figure S7).
To examine muscle function after systemic SpG-ABE treatment, we performed rotarod running and rodent treadmill running test for the DMDE30mut mice receiving saline or SpG-ABE treatment. The DMDE30mut mice with only saline infusion showed a significant reduction in running activity compared with that of age-matched WT mice (Figures 4G and 4H), but both ABEv1- and ABEv2-treated DMDE30mut mice showed a dramatic improvement in motor performance, respectively (Figures 4G and 4H), further supporting the body-wide restoration of dystrophin and suggesting that systemic ABE treatment could alleviate muscle dysfunction due to nonsense mutation in dystrophin. Next, cardiac function was evaluated by echocardiography for untreated and treated DMD mice at 6 weeks after AAV administration. Consistent with previous study that DMD mutant mice carried no cardiac defects due to the compensation of utrophin expression for dystrophin loss in the heart,19 we detected no difference of several electrocardiogram indices among treated and untreated DMD mice (Figure S8).
These findings together indicated that systemic ABE application could broadly restore dystrophin levels and partially improve muscle function in humanized dystrophic mice. Additionally, the CBh promoter system does not exhibit a significant disparity when compared with our Spc5.12 promoter system.
Discussion
Millions of people worldwide suffer from DMD, but current guideline recommended therapeutic strategy remains limited to corticosteroid treatments that only ameliorate secondary symptoms, such as inflammation or impaired angiogenesis.37,38,39 The efficacy of the four FDA-approved exon-skipping agents and a gene therapy supplement still has potential for further enhancement. Previous studies have shown that SpCas9 with a single sgRNA targeting 5′-TGG-3′ PAM sequence has been found to trigger exon 51 skipping and restore the correct reading frame at a high frequency to restore body-wide dystrophin expression in muscle tissue of DMD mice and dog models with exon 50 deletion.40,41 However, the same sgRNA also reportedly targets a 5′-TAG-3′ PAM in human dystrophin exon 51 but with low efficiency,42 suggesting that genetically humanized animal models would be helpful for in vivo evaluation of patient-specific mutations. To address this issue, we generated humanized DMD model mice by introducing short DNA fragments from human dystrophin carrying the DMD patient mutations. In the present study, we identified 12 point mutations that could lead to premature translation termination and demonstrated that SpG-ABE administration could restore dystrophin in two different humanized DMD mouse lines carrying nonsense mutations in human exons 23 or 30.
In addition to exons 23 and 30, nonsense mutations in exons 24, 37, 44, and 55 were also efficiently rectified by the SpG-ABE system in EGFP reporter assays in vitro. Since no relevant DMD animal models yet exist for these mutations, future studies may examine editing outcomes of these sites by the SpG-ABE system using cardiomyocyte or skeletal myocyte derivatives of human DMD iPSCs. Despite its relatively high efficiency in correcting nonsense mutation in the abovementioned exons, the SpG-ABE system did not efficiently edit nonsense mutations in exons 6, 25, 35, 41, 54, or 59. Previously, the nSpCas9-ABEmax system was shown to induce skipping of exon 50 and rectify exon 51 deletion mutation in DMD ΔEx51 mice by mutating the splice donor site of exon 50.43 Further studies are necessary to determine whether adenine base editing is capable of the induction of skipping the mutant exons carrying pathological nonsense mutations when direct correction of nonsense mutations was low efficient.
Local delivery of SpG-ABE with the most efficient sgRNA identified by in vitro screening into skeletal muscles of DMDE30mut mice resulted in 10%–15% on-target genome editing, almost 4-fold higher than that of nSpCas9-ABE7.10 in DMDE23mut mice.21 This effect is likely due to the higher activity of SpG-ABE compared with the nSpCas9-ABE7.10 variant. Notably, intramuscular delivery of ABEv2 components in adult DMD mice conferred apparently stronger therapeutic alleviation of DMD in mice compared with ABEv1, based on the greater number of dystrophin-positive myofibers and higher dystrophin protein expression in the ABEv2-treated muscles. This effect may be attributable to the 3:1 ratio of sgRNA:ABE in the ABEv2 system. We opted to use the 3:1 sgRNA:ABE ratio based on the additional space provided by the smaller SPc5-12 promoter in the AAV vector since previous studies have shown that sgRNA-to-Cas9 ratio represents a major limiting factor for in vivo efficiency of SpCas9-driven genomic modifications and dystrophin restoration.10,35 Through further modification, it may be possible to increase this ratio to 6:1, potentially boosting the extent of dystrophin rescue in vivo. Furthermore, despite the comparable efficiency between hEx30 sgRNA-4 and hEx23 sgRNA-1 in restoring EGFP expression in vitro, local injection of SpG-ABE with hEx23 sgRNA-1 resulted in a genome editing frequency of only ∼1% in adult DMDE23mut mice, ∼9-fold lower than that of SpG-ABE with hEx30 sgRNA-4 in DMDE30mut mice. It is plausible that this difference may arise from relatively poor in vivo access to human exon 23 by the SpG-ABE variant. It is possible that further directed evolution of SpG-ABE and sgRNA optimization may improve the in vivo editing efficiency of this site.
The restoration of as little as 4% of functional dystrophin is sufficient to provide therapeutic benefits in dystrophic mouse models.44,45,46 In this study, we found that full-length dystrophin protein was restored to 20%–80% of the normal level after 3-week administration of SpG-ABE and human sgRNA in adult dystrophic mice. Consistently, one-time intraperitoneal delivery of SpG-ABE and hEx30 sgRNA-4 was shown to repair 95% of functional dystrophin in heart, and 45% of DMDE30mut mice, so the muscle strength and running activity of ABE-treated dystrophic mice were enhanced dramatically in comparison with the saline-treated mice, suggesting the functional improvement of ABE-treated skeletal muscles.
Currently, numerous therapeutic strategies are undergoing clinical trials, but AAV gene therapy stands out as the most promising therapeutic approach for DMD. Micro-dystrophin and gene editing using AAV have demonstrated wide effectiveness in preclinical models and relative safety in early clinical trials. Consequently, the current focus of research is directed toward successfully translating this advanced therapy medicinal product from the laboratory to clinical practice. This involves addressing issues related to dose levels, repeat treatment, immune modulation, and pharmaceutical-scale manufacture and supply. Among the several remaining issues in AAV gene therapy, potency and efficacy are key areas of concern. The systemic nature of DMD, which necessitates body-wide dystrophin restoration, may require a high AAV dosage for effective treatment. Therefore, the utilization of high-dose AAV in gene therapy can be enhanced through the following aspects. A more potent gene expression cassette would mean in theory that less virus will be needed to achieve therapeutic effect. One way of enhancing gene expression is via addition of gene regulatory elements such as novel promoters and introns into the gene expression cassette. Additionally, an AAV serotype that is muscle-tropic and preferentially transfers genes into muscle cells would be ideal, especially because currently, a high dosage to achieve body-wide transduction is needed to treat DMD. Delivering therapeutic constructs using muscle-specific AAVs will improve the clinical outcome of the treatments.
Since ABE-driven nonsense mutation correction occurs at the genomic level, it is plausible that the extent of dystrophin restoration and muscle function improvement would be higher after a longer treatment duration owing to the survival advantage of corrected muscle cells. A major concern of ABEs in the therapeutic application is their bystander mutation and off-target editing. Here, the bystander editing activity of SpG-ABE was only detected at the A18 site in humanized DMDE23mut mice, rather than in the DMDE30mut mice. Nevertheless, the above bystander edits at A18 led to a synonymous mutation without changing the amino acid sequence, so the restored dystrophin protein by the SpG-ABE remained to be functional. In this scenario, applying the SpG-ABE system is believed to not trigger detrimental effects in the DMDE23mut mice and the relevant DMD patients. Notably, ABEs are known to generate inappreciable off-target DNA edits and is much safer than cytosine base editors and other genome-editing tools.47,48,49 Supporting this notion, we did not observe any notable off-target events at the genomic sites predicted by the computer algorithm. The off-target sites contained at least two or three mismatches to the target sequence of the sgRNA, which might prevent the binding of nSpCas9 to these predicated genomic loci. Though ABEs are described to possess off-target RNA editing activity,49,50 we did not detect byproduct events around the off-target sites of cellular transcripts, further evidencing the safety of the nSpCas9-ABE system in therapeutic application. Since the dCas12Max-ABE fused between TadA8e and nuclease-inactive xCas12i is recently shown to present high on-target editing activity but induce negligible off-target edits,27 future investigations are necessary to explore whether the dCas12Max-ABE system can efficiently and safely correct diverse DMD mutations in humanized animal models.
Materials and methods
Study approval
All applicable institutional and/or national guidelines for the care and use of animals were followed. The study was approved by the ethics committee of First Affiliated Hospital of Fujian Medical University. The informed consent was obtained from all individual participants. All experimental procedures on the mice were approved by the Institutional Animal Care and Use Committee (IACUC) of HuidaGene Therapeutics. The humanized dystrophic mice were generated as previously described.32 All mice were housed with a constant temperature (24°C–26°C) and humidity (40%–60%) with a 12-h light-dark cycle, and fed with the standard food.
Plasmid construction
The SpG-ABEmax plasmid (#140002, Addgene) was a gift from David Liu Lab and reconstructed to form pU6-BpiI-EF1α-ABEmax-SpG-CBh-BFP vector. The reporter vector contains CMV-driven mCherry cassette, ATG-removed EGFP, and human exon mutant sequence identified in the patients with DMD. The sgRNAs targeting pathological nonsense mutations were designed using the CCTOP tool (https://cctop.cos.uni-heidelberg.de:8043/) and then synthesized as DNA oligos. The sgRNA oligos were annealed and cloned into the NG-ABEmax vector to form the CRISPR-targeting plasmids (Table S2). The split-SpCas9 variants in Cbh_v5 AAV-ABE N terminus (#137177, Addgene) and Cbh_v5 AAV-ABE C terminus (#137178, Addgene) plasmids were replaced by the SpG at the same split point to form V1-ABE system. The V2-ABE system was further constructed by replacing CBh with the SPc5-12 promoter and inserting one additional U6-sgRNA into N- and C-terminal plasmids, respectively.
Cell culture and transfection
The HEK293T cells from American Type Culture Collection (ATCC) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (#11965092, Gibco) supplemented with 10% fetal bovine serum (FBS) (#04-001-1ACS, Biological Industries) and 1% penicillin/streptomycin (#15140122, Thermo Fisher Scientific). For sgRNA screening, HEK293T cells were seeded on 12-well culture plates (#3513, Corning) at the same amount. ABE-expressing plasmids and the reporter plasmid were then co-transfected using the polyethylenimine (PEI) reagent (#101000029, Polyplus). Two days later, the percentages of EGFP-positive cells in the mCherry-positive cell population were detected with a Beckman CytoFlex flow-cytometer.
Establishment and maintenance of human DMD iPSCs
The fibroblasts were derived from a male patient carrying a c.4174C>T mutation in exon 30 of the DMD gene. This donor had no other disorders including infectious diseases and cancers. The written informed consent for cell donation was obtained from this donor and his family members. The fibroblasts were maintained in DMEM with 10% FBS and 1% penicillin/streptomycin and passaged with Trypsin-EDTA (#25200056, Gibco). Human DMD fibroblasts were reprogrammed into DMD iPSCs using the CytoTune-iPS Sendai Reprogramming Kit (#A16517, Thermo Fisher Scientific). Human DMD iPSCs were plated in cell-culture dishes coated with matrigel (#354277, Corning) and grown in the ncTarget medium (#RP01020, Nuwacell Biotechnologies) at 37°C, 5% CO2. The iPSCs were passaged at 80% confluence using hPSC dissociation buffer (#RP01007, Nuwacell Biotechnologies).
Human iPSC nucleofection and sorting
One hour before nucleofection, human DMD iPSCs were treated with 10 μM ROCK inhibitor Y-27632 (#10005583, Cayman). The iPSCs were dissociated into single cells with Accutase (#7920, STEMCELL Technologies). After 3 × 106 cells were mixed with 6 μg plasmids in nucleofection buffer (#RP01005, Nuwacell Biotechnologies), the nucleofection process was performed with Lonza 2B Nucleofector, employing program B016. Forty-eight hours later, fluorescence-positive cells were sorted out by BD FACSAria III Sorter. For sgRNA screening, 5 × 103 cells were collected, and their lysis was amplified with different primer sets (Table S2). For obtaining single iPSC clone, the cells were immediately seeded on a matrigel-coated 100-mm culture dish (#430167, Corning) and maintained in the ncTarget medium with 10 μM Y-27632. After 7 days, a single cell colony was picked and transferred to the 12-well culture plate. After being subjected to genome sequencing, the desired cells were expanded in the ncTarget medium.
Cardiomyocyte differentiation of human iPSCs
Human DMD iPSCs with or without ABE treatment were digested into single cells and seeded on the matrigel-coated 12-well culture plate at a density of 5 × 105 cells/well. When confluence was over 95%, cardiomyocyte differentiation was induced using the STEMdiff Cardiomyocyte Differentiation Kit (#05010, STEMCELL Technologies). The differentiation medium was changed in a successive sequence.
Immunofluorescence staining of human iPSC-derived cardiomyocytes
The cardiomyocytes were fixed in 4% paraformaldehyde (#P0099, Beyotime) for 10 min and soaked in blocking buffer (#P0260, Beyotime) for 20 min. Following three washes, the cardiomyocytes were stained with primary antibodies against dystrophin (#D8168, Sigma) and cTnI (#21652-1-AP, Thermo Fisher Scientific) at 4°C overnight. The cells were washed in PBS and then probed with secondary antibodies for 2 h at room temperature (RT). The secondary antibodies are listed: Alexa Fluor 488 AffiniPure donkey anti-rabbit IgG (#711-545-152, Jackson ImmunoResearch Labs) and Alexa Fluor 647 AffiniPure donkey anti-mouse IgG (#715-605-151, Jackson ImmunoResearch Labs). After DAPI staining, the images were captured under an immunofluorescence microscopy (Nikon C2).
AAV administration
The AAV9 particles were produced by PackGene Biotech (Guangzhou, China). In brief, the pHelper, pRepCap, and GOI plasmids at the ratio 2:1:1 were co-transfected into the cells when the confluency was between 70% and 90%. After 72 h, the iodixanol density gradient centrifugation was used to purify AAV9 particles. For intramuscular injection, 3-week-old DMDE30mut and DMDE23mut mice were anesthetized, and their TA muscles were injected with AAV9 preparations at the dose of 5 × 1011 vg/leg/AAV or with the same volume saline solution. For intraperitoneal infusion, the P3 mice were administrated with AAV9 vectors at the dose of 1 × 1011 vg per virus or with the saline solution. Four weeks later, mouse HE, DI, and TA muscles were isolated and then cut into small pieces for the experiments described below.
Genomic DNA extraction and deep sequencing
Human cells or mouse tissues were digested in the lysis buffer containing proteinase K, and their genomic DNA was extracted following the manufacturer’s instructions. The DNA was amplified with Phanta max super-fidelity DNA polymerase (#P505-d1, Vazyme) and specific primer sets before Sanger or deep sequencing. For the construction of deep-sequencing products, Illumina flow cell binding sequences and barcodes were added to the 5′ and 3′ ends of primer sequences. The DNA products were purified with Gel extraction kit (Omega) and analyzed by 150-base pair paired-end reads on an Illumina NovaSeq 6000 platform (Genewiz Co. Ltd.). The deep-sequencing data were first de-multiplexed by Cutadapt (v.2.8) based on sample barcodes. The de-multiplexed reads were then processed by CRISPResso2 for the quantification of editing efficiency, including indels, A-to-G or C-to-T conversions at each target site.
RNA isolation, cDNA synthesis, and sequencing
Total mRNAs were isolated from mouse tissues or human iPSC-derived cardiomyocytes by TRIzol reagent (#15596-018, Ambion) according to the manufacturer’s instructions. The mRNAs were reverse transcribed into cDNAs with HiScript II One Step RT-PCR Kit (#P611-01, Vazyme). The cDNAs were amplified using Phanta max super-fidelity DNA polymerase and performed with Sanger and deep sequencing to analyze A-to-G conversation efficiency.
Western blotting
Mouse tissues were crushed into powder and homogenized in RIPA lysis buffer (#P0013B, Beyotime) containing protease inhibitor cocktail. Protein concentrations were determined with BCA Protein Assay Kit (#23225, Thermo Fisher Scientific) and then adjusted to an identical concentration. Ten micrograms of total proteins were loaded into each lane of the 3%–8% tris-acetate gels (#EA03752BOX, Invitrogen) and electrophoresed for 1 h. The proteins were transferred onto the PVDF membranes under a wet condition. Later, the membranes were blocked in 5% non-fat milk/TBST buffer and then incubated with primary antibodies overnight at 4°C. Primary antibodies are listed as follows: dystrophin (#D8168, Sigma) and vinculin (#13901S, Cell Signaling Technology). Following three-time washes, the membranes were probed with HRP-conjugated secondary antibodies at RT for 1 h. After the incubation with Chemiluminescent substrates (#WP20005, Invitrogen), the membranes were viewed by the Image Lab Software 5.2.
Immunohistochemical analyses
For immunofluorescence staining, mouse tissues were embedded in the O.C.T. compound and then snap-frozen in liquid nitrogen-cooled isopentane. They were cut into 10-μm sections with a microtome and transferred onto the slides. The sections were fixed in 4% paraformaldehyde for 2 h and permeabilized with 0.4% Triton X-/PBS for 30 min at RT. After being blocked in 10% goat serum/PBS, the sections were probed with primary antibody against dystrophin (#ab15277, Abcam) at 4°C overnight. Following three washes, the sections were stained with secondary antibodies and DAPI for 3 h at RT. After the wash in PBS, the coverslips were sealed with permanent synthetic mounting media. All pictures were observed and captured under an inverted Olympus FV3000 microscope.
Rotarod and rodent treadmill test
The mice were trained daily for a week prior to the experiment. Three mice were put simultaneously on the rotarod (Ugo Basile Inc.) with an accelerating speed from 4 to 40 rpm over 30 s. When the mice fell off and onto the lever, the test was stopped and the time was recorded. Each mouse was tested five times and the average value was used for further comparison. The mice were trained daily for a week prior to the experiment. Three mice were put simultaneously on the rodent treadmill (Shanghai TOW Intelligent Technology Co., Ltd, AT-5MR) with an accelerating speed from 0 to 15 m/s over 30 s and the running time was recorded before first falling.
Echocardiographic analysis
Echocardiographic analysis in WT and DMDE30mut mice was assessed at 6 weeks post-AAV administration using transthoracic echocardiography (Vevo 3100, Visual Sonics) with a 25-MHz transducer, following the method outlined in reference51. In brief, mice were anesthetized with isoflurane, placed on a platform with ECG leads attached, and their fur was removed. Pre-warmed ultrasound gel was applied to the chest area, and the ultrasound probe was positioned to ensure clear visualization of the left ventricle and atria in B-Mode, with the outflow tract appearing horizontal on the screen. Measurements such as left ventricular posterior wall and left ventricular internal dimension were taken in M-mode, averaging values over three heart cycles. Echocardiographic data collection and analysis were performed without knowledge of the mice’s genotypes or treatments.
Statistical analysis
Quantitative data were derived from at least three independent experiments. The statistical significance of group differences was calculated by the unpaired Student’s t test between two groups among multiple groups. A p value of less than 0.05 was considered to be significant.
Data and code availability
The data will be available from the corresponding author upon reasonable request.
Acknowledgments
We thank Ni Xie for drawing the cute mice. We thank Guofang Chen for helpful discussions. This work was supported by grants from National Natural Science Foundation of China (82202052 to M.J., 81870902 to N.W. and 31925016, 82021001 to H.Y.); STI2030-Major Projects(2021ZD0200900 to H.Y.); Lingang Laboratory (LG202106-01-02 to H.Y.); Strategic Priority Research Program of Chinese Academy of Sciences (XDB32060000 to H.Y.); Basic Frontier Scientific Research Program of Chinese Academy of Sciences From 0 to 1 original innovation project (ZDBS-LY-SM001 to H.Y.); Major Scientific Research Project for Middle-age and Youth of Fujian Provincial Health Commission grant (2021ZQNZD003 to W.-J.C.); Major Scientific Research Project of Fujian Provincial Health Commission grant (2022ZD01002 to W.-J.C.) and Joint Funds for the Innovation of Science and Technology of Fujian Province (2018Y9082 to N.W.). C.X. was funded by Lingang Laboratory Fund and Shanghai City Committee of Science and Technology Project (22QA1412300).
Author contributions
H.Y., G.L., and M.J. jointly conceived the project and designed experiments. H.Y., W.C., N.W., and C.X. supervised the project. G.L., M.J., and J.L. designed vectors, performed in vivo virus injection, tissue dissection, histological immunostaining, and muscle function experiments. G.L. and Y.W. conducted confocal imaging. Z.W. and L.C. derived the DMD fibroblasts. M.J. and H.L. performed the iPSC reprograming, cardiomyocyte differentiation, and immunofluorescence staining. C.X. and H.L. assisted with construction of plasmids. G.L. and M.J. performed in vivo virus injection, tissue dissection, histological immunostaining, and muscle function experiments. J.L. and D.Y. assisted with tissue dissection, immunostaining, and animal breeding. G.L., M.J., Z.L., H.L., D.Y., and C.X. analyzed the data and organized figures. W.C., H.L., and C.X. wrote the manuscript with data contributed by all authors who participated in the project.
Declaration of interests
H.Y. is a founder of HuidaGene Therapeutics.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2024.102165.
Contributor Information
Chunlong Xu, Email: xucl@lglab.ac.cn.
Hui Yang, Email: huiyang@huidagene.com.
Wan-Jin Chen, Email: wanjinchen75@fjmu.edu.cn.
Guoling Li, Email: guolingli@huidagene.com.
Supplemental information
References
- 1.Hoffman E.P., Brown R.H., Jr., Kunkel L.M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 2.Chemello F., Bassel-Duby R., Olson E.N. Correction of muscular dystrophies by CRISPR gene editing. J. Clin. Invest. 2020;130:2766–2776. doi: 10.1172/JCI136873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Constantin B. Dystrophin complex functions as a scaffold for signalling proteins. Biochim. Biophys. Acta. 2014;1838:635–642. doi: 10.1016/j.bbamem.2013.08.023. [DOI] [PubMed] [Google Scholar]
- 4.Blake D.J., Weir A., Newey S.E., Davies K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- 5.Mercuri E., Muntoni F. Muscular dystrophies. Lancet. 2013;381:845–860. doi: 10.1016/S0140-6736(12)61897-2. [DOI] [PubMed] [Google Scholar]
- 6.Chen G., Wei T., Yang H., Li G., Li H. CRISPR-Based Therapeutic Gene Editing for Duchenne Muscular Dystrophy: Advances, Challenges and Perspectives. Cells. 2022;11:2964. doi: 10.3390/cells11192964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tuffery S., Bareil C., Demaille J., Claustres M. Four novel dystrophin point mutations: detection by protein truncation test and transcript analysis in lymphocytes from Duchenne muscular dystrophy patients. Eur. J. Hum. Genet. 1996;4:143–152. doi: 10.1159/000472188. [DOI] [PubMed] [Google Scholar]
- 8.Bladen C.L., Salgado D., Monges S., Foncuberta M.E., Kekou K., Kosma K., Dawkins H., Lamont L., Roy A.J., Chamova T., et al. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum. Mutat. 2015;36:395–402. doi: 10.1002/humu.22758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long C., Amoasii L., Mireault A.A., McAnally J.R., Li H., Sanchez-Ortiz E., Bhattacharyya S., Shelton J.M., Bassel-Duby R., Olson E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science (New York, N.Y.) 2016;351:400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hakim C.H., Wasala N.B., Nelson C.E., Wasala L.P., Yue Y., Louderman J.A., Lessa T.B., Dai A., Zhang K., Jenkins G.J., et al. AAV CRISPR editing rescues cardiac and muscle function for 18 months in dystrophic mice. JCI Insight. 2018;3 doi: 10.1172/jci.insight.124297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelson C.E., Hakim C.H., Ousterout D.G., Thakore P.I., Moreb E.A., Castellanos Rivera R.M., Madhavan S., Pan X., Ran F.A., Yan W.X., et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science (New York, N.Y.) 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu L., Lau Y.S., Gao Y., Li H., Han R. Life-Long AAV-Mediated CRISPR Genome Editing in Dystrophic Heart Improves Cardiomyopathy without Causing Serious Lesions in mdx Mice. Mol. Ther. 2019;27:1407–1414. doi: 10.1016/j.ymthe.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bengtsson N.E., Hall J.K., Odom G.L., Phelps M.P., Andrus C.R., Hawkins R.D., Hauschka S.D., Chamberlain J.R., Chamberlain J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017;8 doi: 10.1016/j.braindev.2017.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bengtsson N.E., Tasfaout H., Hauschka S.D., Chamberlain J.S. Dystrophin Gene-Editing Stability Is Dependent on Dystrophin Levels in Skeletal but Not Cardiac Muscles. Mol. Ther. 2021;29:1070–1085. doi: 10.1536/ihj.20-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Long C., McAnally J.R., Shelton J.M., Mireault A.A., Bassel-Duby R., Olson E.N. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science (New York, N.Y.) 2014;345:1184–1188. doi: 10.1126/science.1254445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y., Long C., Li H., McAnally J.R., Baskin K.K., Shelton J.M., Bassel-Duby R., Olson E.N. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv. 2017;3 doi: 10.1126/sciadv.1602814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelson C.E., Wu Y., Gemberling M.P., Oliver M.L., Waller M.A., Bohning J.D., Robinson-Hamm J.N., Bulaklak K., Castellanos Rivera R.M., Collier J.H., et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019;25:427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adikusuma F., Piltz S., Corbett M.A., Turvey M., McColl S.R., Helbig K.J., Beard M.R., Hughes J., Pomerantz R.T., Thomas P.Q. Large deletions induced by Cas9 cleavage. Nature. 2018;560:E8–E9. doi: 10.1038/s41586-018-0380-z. [DOI] [PubMed] [Google Scholar]
- 20.Shin H.Y., Wang C., Lee H.K., Yoo K.H., Zeng X., Kuhns T., Yang C.M., Mohr T., Liu C., Hennighausen L. CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat. Commun. 2017;8 doi: 10.1038/ncomms15464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryu S.M., Koo T., Kim K., Lim K., Baek G., Kim S.T., Kim H.S., Kim D.E., Lee H., Chung E., Kim J.S. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol. 2018;36:536–539. doi: 10.1038/nbt.4148. [DOI] [PubMed] [Google Scholar]
- 22.Xu L., Zhang C., Li H., Wang P., Gao Y., Mokadam N.A., Ma J., Arnold W.D., Han R. Efficient precise in vivo base editing in adult dystrophic mice. Nat. Commun. 2021;12:3719. doi: 10.1038/s41467-021-23996-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin X., Chen H., Lu Y.Q., Hong S., Hu X., Gao Y., Lai L.L., Li J.J., Wang Z., Ying W., et al. Base editing-mediated splicing correction therapy for spinal muscular atrophy. Cell Res. 2020;30:548–550. doi: 10.1038/s41422-020-0304-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Šikrová D., Cadar V.A., Ariyurek Y., Laros J.F.J., Balog J., van der Maarel S.M. Adenine base editing of the DUX4 polyadenylation signal for targeted genetic therapy in facioscapulohumeral muscular dystrophy. Mol. Ther. Nucleic Acids. 2021;25:342–354. doi: 10.1016/j.omtn.2021.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of AT to GC in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porto E.M., Komor A.C., Slaymaker I.M., Yeo G.W. Base editing: advances and therapeutic opportunities. Nat. Rev. Drug Discov. 2020;19:839–859. doi: 10.1038/s41573-020-0084-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang H., Kong X., Xue M., Hu J., Wang Z., Wei Y., Wang H., Zhou J., Zhang W., Xu M., et al. An Engineered xCas12i with High Activity, High Specificity and Broad PAM Range. Protein Cell. 2022;14:538–543. doi: 10.1093/procel/pwac052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muntoni F., Torelli S., Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2:731–740. doi: 10.1016/s1474-4422(03)00585-4. [DOI] [PubMed] [Google Scholar]
- 29.Wasala N.B., Chen S.J., Duan D. Duchenne muscular dystrophy animal models for high-throughput drug discovery and precision medicine. Expet Opin. Drug Discov. 2020;15:443–456. doi: 10.1080/17460441.2020.1718100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishimasu H., Shi X., Ishiguro S., Gao L., Hirano S., Okazaki S., Noda T., Abudayyeh O.O., Gootenberg J.S., Mori H., et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science (New York, N.Y.) 2018;361:1259–1262. doi: 10.1126/science.aas9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walton R.T., Christie K.A., Whittaker M.N., Kleinstiver B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science (New York, N.Y.) 2020;368:290–296. doi: 10.1126/science.aba8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li G., Jin M., Li Z., Xiao Q., Lin J., Yang D., Liu Y., Wang X., Xie L., Ying W., et al. Mini-dCas13X-mediated RNA editing restores dystrophin expression in a humanized mouse model of Duchenne muscular dystrophy. J. Clin. Invest. 2023;133 doi: 10.1172/jci162809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Truong D.J.J., Kühner K., Kühn R., Werfel S., Engelhardt S., Wurst W., Ortiz O. Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic Acids Res. 2015;43:6450–6458. doi: 10.1093/nar/gkv601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levy J.M., Yeh W.H., Pendse N., Davis J.R., Hennessey E., Butcher R., Kobl-an L.W., Comander J., Liu Q., Liu D.R. Cytosine and adenine base editing ofthe brain, liver, retina, heart and skeletal muscle of mice via adenoassociated viruses. Nat. Biomed. Eng. 2020;4:97–110. doi: 10.1038/s41551-019-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Min Y.L., Li H., Rodriguez-Caycedo C., Mireault A.A., Huang J., Shelton J.M., McAnally J.R., Amoasii L., Mammen P.P.A., Bassel-Duby R., Olson E.N. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 2019;5:eaav4324. doi: 10.1126/sciadv.aav4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y., Li H., Min Y.L., Sanchez-Ortiz E., Huang J., Mireault A.A., Shelton J.M., Kim J., Mammen P.P.A., Bassel-Duby R., Olson E.N. Enhanced CRISPR-Cas9 correction of Duchenne muscular dystrophy in mice by a self-complementary AAV delivery system. Sci. Adv. 2020;6:eaay6812. doi: 10.1126/sciadv.aay6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang T., Kong X. Recent advances of glucocorticoids in the treatment of Duchenne muscular dystrophy (Review) Exp. Ther. Med. 2021;21:447. doi: 10.3892/etm.2021.9875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Podkalicka P., Mucha O., Dulak J., Loboda A. Targeting angiogenesis in Duchenne muscular dystrophy. Cell. Mol. Life Sci. 2019;76:1507–1528. doi: 10.1007/s00018-019-03006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miyatake S., Shimizu-Motohashi Y., Takeda S., Aoki Y. Anti-inflammatory drugs for Duchenne muscular dystrophy: focus on skeletal muscle-releasing factors. Drug Des. Dev. Ther. 2016;10:2745–2758. doi: 10.2147/DDDT.S110163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amoasii L., Long C., Li H., Mireault A.A., Shelton J.M., Sanchez-Ortiz E., McAnally J.R., Bhattacharyya S., Schmidt F., Grimm D., et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aan8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amoasii L., Hildyard J.C.W., Li H., Sanchez-Ortiz E., Mireault A., Caballero D., Harron R., Stathopoulou T.R., Massey C., Shelton J.M., et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science (New York, N.Y.) 2018;362:86–91. doi: 10.1126/science.aau1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y., Li H., Nishiyama T., McAnally J.R., Sanchez-Ortiz E., Huang J., Mam-men P.P.A., Bassel-Duby R., Olson E.N. A humanized knockin mouse model of Duchenne muscular dystrophy and its correction by CRISPR-Cas9 therapeutic gene editing. Mol Ther. Nucl. Acids. 2022;29:525–537. doi: 10.1016/j.omtn.2022.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chemello F., Chai A.C., Li H., Rodriguez-Caycedo C., Sanchez-Ortiz E., Atmanli A., Mireault A.A., Liu N., Bassel-Duby R., Olson E.N. Precise correction of Duchenne muscular dystrophy exon deletion mutations by base and prime editing. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abg4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Putten M., Hulsker M., Young C., Nadarajah V.D., Heemskerk H., van der Weerd L., 't Hoen P.A.C., van Ommen G.J.B., Aartsma-Rus A.M. Low dystrophin levels increase survival and improve muscle pathology and function in dystrophin/utrophin double-knockout mice. FASEB J. 2013;27:2484–2495. doi: 10.1096/fj.12-224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li D., Yue Y., Duan D. Marginal level dystrophin expression improves clinical outcome in a strain of dystrophin/utrophin double knockout mice. PLoS One. 2010;5 doi: 10.1371/journal.pone.0015286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Putten M., Hulsker M., Nadarajah V.D., van Heiningen S.H., van Huizen E., van Iterson M., Admiraal P., Messemaker T., den Dunnen J.T., 't Hoen P.A.C., Aartsma-Rus A. The effects of low levels of dystrophin on mouse muscle function and pathology. PLoS One. 2012;7 doi: 10.1371/journal.pone.0031937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zuo E., Sun Y., Wei W., Yuan T., Ying W., Sun H., Yuan L., Steinmetz L.M., Li Y., Yang H. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science (New York, N.Y.) 2019;364:289–292. doi: 10.1126/science.aav9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jin S., Zong Y., Gao Q., Zhu Z., Wang Y., Qin P., Liang C., Wang D., Qiu J.L., Zhang F., Gao C. Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science (New York, N.Y.) 2019;364:292–295. doi: 10.1126/science.aaw7166. [DOI] [PubMed] [Google Scholar]
- 49.Gaudelli N.M., Lam D.K., Rees H.A., Solá-Esteves N.M., Barrera L.A., Born D.A., Edwards A., Gehrke J.M., Lee S.J., Liquori A.J., et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020;38:892–900. doi: 10.1038/s41587-020-0491-6. [DOI] [PubMed] [Google Scholar]
- 50.Grünewald J., Zhou R., Garcia S.P., Iyer S., Lareau C.A., Aryee M.J., Joung J.K. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. 2019;569:433–437. doi: 10.1038/s41586-019-1161-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pesce P., Cecchetto L., Brocco S., Bolognesi M., Sodhi K., Abraham N.G., Sac-erdoti D. Characterization of a murine model of cardiorenal syndrome type 1 by high-resolution Doppler sonography. J. Ultrasound. 2014;18:229–235. doi: 10.1007/s40477-014-0129-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data will be available from the corresponding author upon reasonable request.