

Abstract
Mitochondrial DNA (mtDNA) analysis is crucial for the diagnosis of mitochondrial disorders, forensic investigations, and basic research. Existing pipelines are complex, expensive, and require specialized personnel. In many cases, including the diagnosis of detrimental single nucleotide variants (SNVs), mtDNA analysis is still carried out using Sanger sequencing. Here, we developed a simple workflow and a publicly available webserver named Mitopore that allows the detection of mtDNA SNVs, indels, and haplogroups. To simplify mtDNA analysis, we tailored our workflow to process noisy long-read sequencing data for mtDNA analysis, focusing on sequence alignment and parameter optimization. We implemented Mitopore with eliBQ (eliminate bad quality reads), an innovative quality enhancement that permits the increase of per-base quality of over 20% for low-quality data. The whole Mitopore workflow and webserver were validated using patient-derived and induced pluripotent stem cells harboring mtDNA mutations. Mitopore streamlines mtDNA analysis as an easy-to-use fast, reliable, and cost-effective analysis method for both long- and short-read sequencing data. This significantly enhances the accessibility of mtDNA analysis and reduces the cost per sample, contributing to the progress of mtDNA-related research and diagnosis.
Keywords: mtDNA analysis, long-read sequencing, clinical mtDNA diagnosis, haplogroups, webserver, forensic mtDNA analysis, ONT, Mitopore, iPSCs quality control, mtDNA heteroplasmy
Graphical abstract
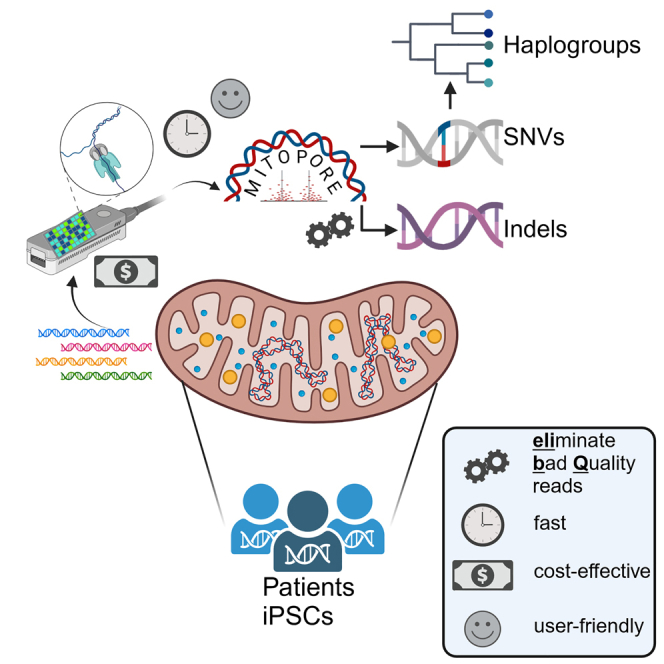
Rossi and colleagues describe a streamlined approach for the analysis of mtDNA using noisy long-read sequencing data. They introduce Mitopore, a user-friendly webserver for efficient, cost-effective mtDNA analysis of SNVs, indels, and haplogroups. The economic efficiency of Mitopore holds great promise to advance research and clinical mtDNA diagnosis.
Introduction
Mitochondria are greatly responsible for energy production in most eukaryotic organisms.1 They contain their own maternally inherited genome, collectively referred to as mtDNA.2,3 mtDNA encodes for 13 proteins, 22 tRNAs, and two rRNAs that ensure mitochondrial respiratory chain function.3 Mitochondrial DNA mutations—SNVs, indels, and large-scale deletions—are an important cause of inheritable mitochondrial diseases.4 In addition, somatic mtDNA mutations may accumulate upon environmental exposure, aging, and neurodegeneration, and may contribute to cancer development.5,6,7,8 Distinct inherited mtDNA SNV patterns define haplogroups, which are indicative of genetic ancestry.9 mtDNA analysis is thus also used for forensic analysis.10,11 In addition, mtDNA integrity needs to be guaranteed for the quality control of primary cells, including human induced pluripotent stem cells (iPSCs).12,13
Several mtDNA features need to be considered when performing sequencing data analysis. (1) Approximately 0.0087% of the nuclear genome has been reported to be mtDNA-derived nuclear mtDNA segments (NUMTs).14 This requires rigorous data processing to exclude genomic DNA contaminations. (2) Because mtDNA reference sequences are linear, accurate alignment of the ∼16.5 kbp circular mtDNA in the artificial breakage regions is difficult. (3) Mitochondria and mtDNA exist as multicopies.15 These up to thousands of mtDNA copies per cell can exhibit different SNVs, either in only a subset (heteroplasmy) or all (homoplasmy) copies.16,17 A high degree of sequencing and alignment accuracy are thus necessary to distinguish different variants.
mtDNA heteroplasmy can be disease associated or disease causative—for example, when crossing a certain threshold upon which the respiratory chain is negatively affected.18,19 mtDNA mutations are generally classified as follows: usually benign homoplasmic haplogroup-associated SNVs, organ-specific disease-associated mutations, or systemic disease-associated mutations.20,21,22 Accurate analysis is thus paramount to understand the relationship between mtDNA sequence and disease phenotypes.
Despite its importance, current mtDNA analysis approaches exhibit several limitations that preclude broad use. Many users rely on Sanger sequencing, which exhibits low sensitivity. Furthermore, it is impractical for higher throughput and multiplexed analyses due to the high per-sample costs, labor intensity, and need for meticulous primer design to account for NUMTs.
Next-generation sequencing (NGS) enables high throughput, but accessibility is limited by substantial upfront investments and the availability of software tools. Existing solutions are often user-unfriendly and require additional data postprocessing,23,24,25 requiring a certain level of understanding of the underlying concepts. Consequently, sophisticated bioinformatic analysis is still required, precluding routine analysis of mtDNA by many laboratories and clinicians.
To democratize mtDNA analysis, we developed Mitopore, a complete workflow and user-friendly webserver that uses cost- and time-effective noisy error-prone Oxford Nanopore Technologies (ONT) long-read sequencing data.
Results
Development of the Mitopore workflow to enrich and sequence mtDNA
To develop our pipeline, we defined the following pillars:
-
(1)
Accessibility: a pipeline used by a broad range of users needs to be cost-effective in terms of upfront investment and consumables.
-
(2)
Integrity analysis: integration of a mtDNA analysis workflow into quality control and diagnosis pipelines needs to cover the most important integrity aspects, including SNV/indel, and large-scale deletion detection.
-
(3)
Multiplexing: to enable simultaneous processing of multiple samples, a pipeline should support multiplexing.
To account for these pillars, we made use of ONT sequencing to develop our pipeline. ONT devices and low-depth Flongle flow cells are cost-effective and can be used without extensive training. ONT sequencing facilitates the identification of SNVs/indels26 and of larger deletions that may not be captured by short-read sequencing due to fragmentation and assembly challenges.27 Finally, multiplexing (e.g., of PCR amplicons) is enabled through barcoding by ligation and can be optimized to fit individual experimental needs, such as the number of necessary reads.28
We used a PCR-based enrichment step to prevent contamination by NUMTs and to enable low input amounts. The whole mtDNA was enriched using a set of nine overlapping primers (Table S1), as previously described.29 If one is interested in only a specific position, a reduced set of amplicons can be used to increase read depth per sample and multiplexing capacity. Followed by purification and library preparation, samples are loaded onto a Flongle flow cell and subjected to an 8- to 12-h sequencing run followed by analysis (Figure 1).
Figure 1.

The Mitopore workflow
The Mitopore workflow enables mtDNA analysis within 1 day. The workflow consists of 4 distinct steps: (1) cell lysis, DNA extraction, and mtDNA amplification by PCR; (2) PCR product purification and preparation of the sequencing library; (3) real-time adaptive sequencing using ONT long-read sequencing; and (4) upload to the Mitopore webserver and automated analysis. The final mutation analysis provides a comprehensive overview of identified mutations and sets them in the context of disease-causing known and potentially harmful unknown variants.
Importantly, ONT sequencing offers adaptive sequencing by providing real-time data output. This enables the optimization of run time to potentially further reduce time until results can be analyzed. Subsequently, FASTQ output files can directly be used for analysis without the need for further processing.
Estimation of read depth and sensitivity for mtDNA variant detection
Determining the minimum required number of reads per sample is essential to enable estimation of multiplexing capacity and to call a certain variant with high confidence. Focusing on ONT sequencing with Flongle flow cells, we aimed to determine this minimum number.
We adapted a mathematical model (see materials and methods) and calculated the number of reads per sample necessary to identify the heteroplasmy level of a variant X within 90–99% confidence intervals (Table S2). Based on our model, the required number of reads ranges from 18 to 43 (for a heteroplasmy level of 0.95) to 6,386–15,614 (for a heteroplasmy level of 0.05).
To test these estimations and benchmark our solution, we performed a NGS experiment on an Illumina MiSeq sequencing device. We used samples with known heteroplasmy levels under various PCR conditions to reduce the risk of PCR bias (see materials and methods). Depending on the conditions and the resulting number of reads (only samples with >1,000 reads were considered), the heteroplasmy levels were determined to be 0 (UMGi176-A cl. 1), 0.18–0.2 (UMGi176-A cl. 5), and 0.35–0.37 (UMGi176-A cl. 2; Table S3). We then performed ONT sequencing on the 0.18–0.2 clone (UMGi176-A cl. 5), downsampled the number of reads, and performed variant calling. By plotting the observed heteroplasmy levels against the number of reads, the observed heteroplasmy levels are within the desired measurement ± margin of error corridor predicted from our estimation (Figure 2A). The mutation frequency we detected with ONT sequencing was 0.183, which is in line with the NGS data. Based on our mathematical model, the necessary number of reads to detect a heteroplasmy level of 0.18 with 99% confidence is 2,456, which is in line with our data (Figure 2B). To determine the lower limit of detection, we mixed DNA of a sample homoplasmic for m4295G with a sample homoplasmic for m4295A at varying ratios and performed targeted mtDNA sequencing (Figure S1). Based on these results, we conclude that the lower limit of heteroplasmy detection is 0.05.
Figure 2.

Estimated number of reads to call a variant of interest with high confidence
(A) A sample with a known heteroplasmy level of 0.2 was downsampled to simulate datasets with fewer reads. These downsampled data were subjected to variant calling and the observed heteroplasmy for a given sample plotted against the number of reads at this position within the sample. The margin of error (here: 10% ± of the actual heteroplasmy) is marked by thin red lines on the y axis and the 90%, 95%, and 99% (from left to right) CIs by dashed blue lines on the x axis. Heteroplasmy for downsampled (5 times resampled using different seeds) samples derived from targeted ( i.e., 1 specific ∼2 kbp amplicon) mtDNA analysis. (B) CIs and margins of error are adjusted based on the actual detected mutation rate of 0.18 after data analysis and are based on the number of necessary reads estimation for a heteroplasmy of 0.18, demonstrating the accuracy of the reads estimation model.
Development of the Mitopore webserver
We recognized that merely describing the workflow is insufficient to promote widespread adoption. The main challenges are the need for bioinformatics expertise and the lack of user-friendly tools to process FASTQ data.
To tackle these problems, we developed a user-friendly software called Mitopore and implemented it as a webserver using Python, Java, and R (Figure S2). Our initial goal was to find the most suitable aligner for the analysis of ONT sequencing-derived mtDNA data. Based on own tests and reports from the literature,29,30 we decided to implement Minimap231 for reference sequence alignment because it outperforms other aligners in terms of speed and coverage.28,30
Next, we ensured that Mitopore accurately identifies and robustly classifies SNVs, indels, and haplogroups. To enable the execution of these analyses, we integrated existing pipelines processing NGS-derived data in Mitopore: Mutserve for SNV calling, Mutect2 for indel calling, and Haplogrep 3 for haplogroup calling.24,32,33 Mutserve outperforms other existing tools for mtDNA variant calling and is able to process hundreds of binary alignment map (BAM) files within a couple of minutes.34 Mutect2 is a part of the genome analysis toolkit (GATK) maintained by the Broad Institute35,36 and was recently updated by a mitochondria mode enabling SNV and indel calling.33 Since FASTQ quality filtering performed by the sequencer does not take individual base quality into account, we aimed to improve per-base read quality. Therefore, we developed an additional step to eliminate bad quality (eliBQ) reads. The eliBQ tool removes individual bases below a quality threshold of 6. Thus, reads are split into smaller pieces above a minimum length of 50 nt (Figure 3A). Based on these settings, quality scores are improved by ∼20% (Figure 3B) due to the removal of low-quality bases that are otherwise “protected” by the higher average quality across the whole read. This is a major step forward, because only by using this “short read-like”-based approach can FASTQ files from noisy ONT sequencing be subjected to indel calling by Mutect2. By using semisynthetic datasets based on original sequencing data, we demonstrate the applicability of eliBQ to enable indel calling with Mutect2 (Figure 3C). Taken together, these three tools are a solid foundation upon which to build a comprehensive analysis software. By implementing these pipelines, Mitopore offers an integrated solution for analyzing and identifying mtDNA indels and SNVs, eliminating the need for bioinformatics knowledge. Mitopore provides an intuitive interface and streamlined functionalities to facilitate the interpretation of mtDNA analysis results. Identified SNVs are annotated with the latest data from MITOMAP.37,38 Haplotype analysis helps to evaluate identified SNVs because they are, for example, less likely disease relevant when they are haplogroup defining. By developing Mitopore, we aim to enhance accessibility and standardization in mtDNA analysis for researchers across various expertise levels. The results are presented as an interactive Circular Genome Viewer (CGV) plot (Figure 4A) and contain information on read quality and length (Figure 4B) and on mapping accuracy to the reference genome (Figure 4C). In addition, identified variants are annotated based on the MITOMAP database38 and visualized in a disease variant plot (Figure 4D). This visualization enables the user to quickly assess the mtDNA integrity of an analyzed sample and identify suspicious variants that may warrant further investigation. All of the data are included as a comprehensive table containing information on identified variants and their heteroplasmy levels as well as haplogroup identification.
Figure 3.

Quality improvement by the eliBQ tool enables mtDNA indel calling
(A) The eliBQ principle is based on taking individual base quality into account. Low-quality bases are removed and reads are split accordingly into smaller pieces above a certain length threshold. Resulting short-read sequences are stored in the eliBQ FASTQ format, where each read is split into fragments and displays improved quality per base. (B) Quality score comparison between original FASTQ files and eliBQ postprocessed data. The eliBQ filtering increases per read Phred quality by removing low-quality bases below a Phred score of 6. Average Phred base quality (log2) is plotted against the proportion of reads assigned to this group. The graph demonstrates how eliBQ improves ONT-derived basecalling by postprocessing FASTQ files. (C) eliBQ enables indel calling of noisy long-read sequencing data by Mutect2 at 2 positions that were introduced artificially in an original sequencing dataset obtained in our laboratory.
Figure 4.

The Mitopore.de analysis output
(A) The circular genome viewer implementation enables users to dynamically and interactively scan through the mtDNA representation, including variant calling results from the analyzed samples. Seamless zooming facilitates the understanding of results and their categorization. The linear mode further increases details by providing base-level information of depth and detected variants. (B) Analysis of FASTQ input is performed and visualized as comprehensive quality plots providing information on read lengths and average read quality. This facilitates spotting sampling errors between analyzed samples. (C) The tabular representation of alignment parameters provides the most important alignment information after mapping to the reference sequence; in particular, the percentage of reads mapped to the mitochondrial genome indicates the sample purity and fidelity of the experiment. (D) The disease variant plot enables quick assessment of mtDNA integrity of analyzed samples with color-coding SNVs according to their status as likely benign known polymorphic (green) or potentially suspicious (red) variants. The x axis indicates the position in the linear mtDNA reference, and the y axis represents the level of heteroplasmy for a given position. Protein-coding sequences are indicated by the respective gene name and blue boxes. In the given example, no suspicious variants are present.
Haplogroup classification using Mitopore
To demonstrate the usability of Mitopore for iPSC quality control, we analyzed the mtDNA of nine iPSC lines. The median sequencing depth was 1,440–5,482 reads per position for the total and 1,438–5,443 reads per position for the protein-coding part of the mtDNA (Table 1).
Table 1.
Sample characteristics and detected SNVs of analyzed iPSC samples
| iPSC line | Median read depth per mtDNA position whole mtDNAa | Median read depth per mtDNA position protein-coding mtDNAa | SNVs detected above coverage filter (no described polymorphism) | Top haplogroup hit |
|---|---|---|---|---|
| 1 BIHi005 | 2,345 | 2,306 | 26 (1) | HV |
| 2 KOLF2.1J | 5,482 | 5,443 | 45 | K1a4a1a2a |
| 3 DU247b | 3,282 | 3,270 | 20 | H6bc |
| 4 DU225b | 3,700 | 3,481 | 17 | H6bc |
| 5 AS789b | 2,345 | 2,318 | 18 | V+@72 |
| 6 DU211rd | 2,012 | 1,900 | 13 | H5b2 |
| 7 IMR90 | 3,411 | 3,405 | 38 | W3a1 |
| 8 iPSC11 (no. 1) | 2,922 | 2,917 | 48 | T2c1d+152 |
| 9 iPSC11 (no. 2) | 1,440 | 1,438 | 38 | T2c1d+152 |
| 10 iPSC12 | 2,082 | 2,041 | 18 | H6bc |
A heteroplasmy level of at least 0.05 was set as the minimum to be considered. iPSC, induced pluripotent stem cell.
Depth at variant calling position.
Internal identifier (unpublished iPSC lines).
Same parental (iPSC12) cell line.
Internal identifier (alternative name: IUFi001).
Initially, haplogroup calling quality was low. Upon inspection, we found a high number of variants flagged with “STRAND_BIAS” and thus were excluded from the final variant call format (vcf) results file. Although our high-coverage long reads-based approach is not prone to strand bias, we nonetheless inspected the coverage from a representative BAM file containing a high number of strand bias-flagged SNVs. Even in this extreme sample, we found the coverage to be relatively uniform, with a maximum difference in favor of one strand over the other being 0.661 vs. 0.339 (Figure S3). To estimate the maximum effect of differing strand coverages, we calculated the effect size and assumed different heteroplasmy levels based on the reverse strand starting from 0.1 to 1 on both strands (Figure S4). We found that for a stable heteroplasmy level of 1 on the forward strand, the chance of underestimation for the observed maximum of 0.661 vs. 0.339 strand coverage was ∼0.3.
mtDNA haplogroups are usually defined by homoplasmic SNVs. Strand bias logically does not apply for final heteroplasmy levels of ≥0.9 at high read depths. Consequently, we ignored the strand bias flag for these cases based on the calculation of necessary reads for accurate population representation (Table S2). We then implemented the creation of a HaploGrep hsd file24 containing SNVs with heteroplasmy levels ≥0.75. By using this strategy, iPSC lines derived from a common parental cell line were identified to belong to the same haplogroup. This analysis helps to evaluate iPSC ancestry, and should be implemented in iPSC quality control.
To determine heteroplasmic variants, the strand bias filter needs to be active. This is crucial because the risk of under- or overestimating potential disease variants for a high-accuracy analysis where the identification of suspicious variants is paramount cannot be neglected. Therefore, we decided to generate the final results file based on the appropriate number of reads to call a heteroplasmy level at a high confidence interval (90%). From the SNVs detected in the analyzed iPSC lines, only one cell line exhibited a suspicious variant (Table 1; Figure S5). This demonstrates how Mitopore can serve as an integral and user-friendly part of iPSC quality control by allowing assessment of the mtDNA integrity in minimum time.
Detection of clinically relevant SNVs and large-scale deletions using Mitopore
To demonstrate the applicability and reliability of the Mitopore workflow for potential application in a clinical setting, we further analyzed fibroblast and iPSC lines derived from patients carrying known heteroplasmic or known homoplasmic SNVs.39,40,41,42 Remarkably, Mitopore was able to detect these SNVs with high accuracy, which was in the expected range and only slightly different (up to a 0.06 difference) from the original measurement with homoplasmic mutations (Table 2, homoplasmic samples). In cases of lower heteroplasmic levels of 0.2 or 0.4 determined by short-read amplicon sequencing (Table S3), the observed difference was in the same range of 0.04–0.05 (Table 2, heteroplasmic samples).
Table 2.
Sample characteristics and detected disease-associated SNVs for analyzed clinically relevant samples
| Sample | SNV | Knowna SNV heteroplasmy level | Mitopore-measured SNV heteroplasmy level | Difference measured/known |
|---|---|---|---|---|
| Homoplasmic samples (heteroplasmy >90%) | ||||
| DNA ATP6 iPS TDA2 | m.9185T>C | 1.0042 | 0.962 | 0.038 |
| DNA ATP6 8993-C11 | m.8993T>G | 0.97441 | 0.924 | 0.05 |
| DNA ATP6 8993-B12 | m.8993T>C | 0.98241 | 0.97 | 0.012 |
| DNA ATP6 Fib 8993-D | m.8993T>G | 0.999b,41 | 0.938 | 0.061 |
| DNA ATP6 Fib 8993-A | m.8993T>G | 0.975b,41 | 0.942 | 0.033 |
| DNA CTL iPS TFBJ | WT | 0 | 0 | 0 |
| DNA CTL NPC smXM001 | WT | 0 | 0 | 0 |
| Fibroblasts “WA” | m14487T>C | 0.99 | 0.937 | 0.053 |
| Fibroblasts “BL” | m10197G>A | 0.93 | 0.87 | 0.06 |
| Heteroplasmic samples | ||||
| UMGi176-A cl. 1a | WT | 0c | 0 | 0 |
| UMGi176-A cl. 5a | m.13513G>A | 0.18–0.2c | 0.204 | 0.004–0.024 |
| UMGi176-A cl. 2a | m.13513G>A | 0.35–0.39c | 0.403 | 0.013–0.053 |
| Fibroblasts “HE” | m10191T>C | 0.7 | 0.678 | 0.022 |
| Large-scale deletion sample | ||||
| Type | Long-range PCR | Mitopore | ||
| Fibroblasts “VH” | Large-scale deletion | 2 kbp band and 2 additional smaller fragments (diagnostic finding) | Large-scale deletion of ∼12 kbp | |
CTL, control; iPSC, induced pluripotent stem cell; NGS, next-generation sequencing; NPC, neural progenitor cells; WT, wild type.
Original heteroplasmy levels were measured with different techniques.
Same levels as the resulting iPSC lines described in the original publication.
Determined by amplicon short-read NGS (Table S3).
Analysis of mtDNA integrity further includes the detection of large-scale deletions associated with severe metabolic diseases.43 Long-read ONT sequencing enables the detection of large deletions and structural rearrangements.44 The use of nine overlapping amplicons and ONT sequencing is especially promising.45 To test our workflow in detecting large-scale deletions, we analyzed a sample of a patient who was originally diagnosed with a large deletion (∼14 kbp) associated with Pearson or Kearns-Sayre syndrome. Interestingly, using our mtDNA enrichment step, we detected 3 bands, each ∼2 kbp in size. In contrast to the long-range PCR, this corresponds to an ∼4.7 kbp stretch, suggesting a smaller deletion than anticipated. To enable automated detection of potential large deletions, we implemented a script that extracts read coverage from aligned BAM files. We then sequenced the sample demonstrating automated detection of potential large deletions (Figure S6). Conclusively, Mitopore enables detection not only of small indels but also large-scale mtDNA deletions.
Performance of Mitopore with Illumina and Pacbio data
Other NGS approaches have been shown to produce high-quality short (Illumina) and long (Pacbio) sequencing reads46,47 and are generally considered to display superior quality compared to ONT long-read sequencing. To benchmark the Mitopore workflow, we subjected mtDNA reads derived from whole-genome sequencing (WGS) data of the recently published iPSC reference line KOLF2.1J48 to our webserver and compared the results to the data from our workflow, including PCR enrichment and sequencing on a Flongle flow cell (Figure 5). The variant detection threshold was set to 0.05 because we determined this to be the lower detection limit for the Mitopore workflow (Figure S1). After applying the read-based coverage filter, the polynucleotide stretch filter, and the haplogroup filter for high level heteroplasmies (see materials and methods), the results from our Mitopore workflow were mostly in line with the data obtained from higher-quality sequencing reads and differed at only four positions. Only a few not-haplogroup-defining variants were erroneously called at low heteroplasmy levels. These resided mostly in poly C (m308–310, m5895, m6420, m8372), in poly A (m5747), and in front of a CA repeat stretch (m513). In three cases this inability of low-quality ONT sequencing to correctly resolve polynucleotide stretches resulted in the inability to detect haplogroup-defining variants (m73, m497, m12308). In one case, an additional variant was called, but manual inspection of the BAM file revealed the call to be erroneous (m8908). We thus recommend consulting the BAM file in such situations or to perform Sanger sequencing for clarification.
Figure 5.

Comparison of Mitopore with high-quality sequencing data
Heatmap showing the called variants above a threshold level of 0.05 for third-generation long-read WGS (Pacbio, ONT), next-generation short-read WGS (Illumina), and the Mitopore approach with or without the application of various filters. Positions marked with yellow are haplogroup-defining mutations. In addition, erroneously called variants for Illumina sequencing and the Mitopore approach are mainly in polynucleotide stretches or around the ambiguous mtDNA reference position m3107. ∗ m310 and m3107 are often blacklisted by mtDNA variant calling tools because variant calling in these regions is error prone. mutserver call, original call from mutserver without any of the filters applied; w/o cov filter, without the coverage filter applied; w/o haplo filter, without the haplogroup filter for high-level heteroplasmies applied; w/o poly filter, without the polynucleotide stretch filter applied. Red arrowheads mark the positions where application of the Mitopore filters improves the accuracy of the variant calling.
Discussion
The present study aims to address challenges in existing mtDNA analysis approaches by developing a unified, comprehensive mtDNA screening workflow and pipeline. The primary objective was to create an accessible, efficient workflow—from sample preparation to analysis—using cost-effective ONT sequencing. This process was designed to reliably identify mtDNA indels, SNVs, and large-scale deletions and to classify mtDNA haplogroups.
Recently, several pipelines have been described that use ONT sequencing data to identify SNVs using deep learning neural networks.49,50,51,52 Although they demonstrate the power of shallow ONT WGS, none of those approaches are optimized for mtDNA analysis. They require substantial computational power, including graphic processing units, and the computation of results entails long waiting times. In addition, these pipelines require implementation by bioinformaticians, precluding their broad use.
Keraite and colleagues recently published a less-resource-intensive approach for SNV detection using ONT sequencing, but it still requires bioinformatic expertise to analyze the data.53 They enriched mtDNA by digestion of genomic DNA and linearizing individual mtDNA molecules using CRISPR-Cas9. This provides a more targeted selection of the start and end of the resulting linear sequence compared to conventional linearization, but comes at the cost of reduced multiplexing capacity. We predict the data obtained from this method to be compatible with our software, but at the time of submitting this paper, they are not available for download.
In general populations, mtDNA indels (4%) are reported to be rare compared to SNVs (96%),33 but they can still have significant consequences for cellular function and contribute to mitochondrial disorders.54 Consequently, the identification of mtDNA indels of different sizes using ONT sequencing has not been widely reported to date. This challenge becomes particularly difficult when dealing with noisy reads that are susceptible to sequencing errors and systematic biases.55 To overcome this issue, we developed and effectively implemented the eliBQ tool, which considers individual rather than average base quality. This involves removal of low-quality bases, followed by the segmentation of reads into smaller fragments above a specific length threshold. By implementing eliBQ, both the accuracy and reliability of noisy reads are strongly improved, enabling small indel identification (Figure 4). However, larger deletions are easily detected by inspecting the coverage of whole-mtDNA sequencing samples. In addition, haplogroup analysis was implemented because it offers valuable information about genetic diversity, ancestry, and migration.
We rigorously tested and benchmarked the Mitopore workflow and software using a total of 23 cell lines, including >10 of each patient-derived and iPSC cell lines. Among the patient-derived cell lines, one was initially reported to exhibit a heteroplasmy level of 0.99. However, using Mitopore, we detected a heteroplasmy of 0.67. Upon further investigation, it was revealed that the actual heteroplasmy was initially indeed found to be between 0.6 and 0.7, highlighting the accuracy of Mitopore, which is of critical importance in a clinical setting.
The development of the Mitopore simplified pipeline and webserver is of great significance for the scientific community for several reasons. It overcomes the complexity and expertise required by existing pipelines, making mtDNA analysis more accessible to researchers and clinicians.
The existing pipelines for mtDNA analysis differ in several key aspects from the Mitopore workflow and webserver. Most of them are tailored to analyze data derived from high-quality short read-based NGS data and take BAM files as input.56,57,58 These approaches require significant computational power and bioinformatic expertise to process the data. In addition, they rely on expensive sequencing devices, making them impractical for the purpose described herein. We searched the literature for webserver-based solutions to analyze mtDNA. Nevertheless, most of them were offline, nonfunctional, or incapable of handling FASTQ or BAM file inputs (Figure S7). The only solution theoretically applicable to entry-level long-read sequencing data was mtDNA-Server 2,59 which is based on mutserver. However, it has not been optimized for low-quality ONT sequencing data. Consequently, there was low certainty in variant calls, including haplogroup-defining variants (Figure S8).
By providing a user-friendly interface and eliminating the need for additional bioinformatics tools, Mitopore streamlines the analysis process and facilitates the generation of reliable and comprehensive results in <24 h at reasonable cost (Table S4). The cost can be even further reduced by using flow cells with more pores in cases of higher sequencing throughput. Finally, the compatibility of Mitopore with both long- and short-read sequencing data enhances its versatility and applicability.
A noteworthy limitation of our study is the unavailability of biological samples with indels for assessing the accuracy of indel detection using eliBQ. In the future, we intend to include patient-derived samples carrying small mtDNA indels, which will further enhance our analysis pipeline.
Conclusions
Mitopore holds tremendous potential to accelerate mitochondrial research by offering a simplified and accessible mtDNA integrity analysis approach. Mitopore supports multiplexing (e.g., of 6–8 whole-mtDNA or 18–24 targeted analyses of individual amplicons with a Flongle flow cell), thereby reducing sequencing costs to ∼19€–22€ per sample compared to the widely used Sanger sequencing (Table S4). In addition, sample input is substantially lower—for example, even using the standard recommendations, 62.5 ng per sample are enough for whole-mtDNA sequencing using Mitopore compared to the ∼360 ng needed to sequence 16 amplicons of ∼1 kbp size by Sanger sequencing (Table S4). Mitopore aids in the identification of disease-causing mutations. By facilitating comprehensive and streamlined mtDNA analysis, Mitopore paves the way for further exploration of mitochondrial biology and its implications in various diseases.
Materials and methods
Cell culture
Human parental iPSCs were purchased from The Jackson Laboratory (Bar Harbor, ME, KOLF2.1J), WiCell (Madison, WI, IMR90), and Cell Applications (San Diego, CA, iPSC-11, iPSC-12), provided by the Stem Cell Unit of the University Medical Center Göttingen (UMGi176-A cl. 1, cl. 2, cl. 5; use covered by study no. 2020-967_4 approved on 10.01.2024 by the Ethic Commission of the Medical Faculty of the Heinrich Heine University Düsseldorf), or the Medical University Düsseldorf (use of patient-derived fibroblasts and iPSCs covered by ethic vote no. 4161 [ethical approval no. 5471]; study no. 2020-967_3 approved on 25.09.2022 of the Heinrich Heine University Düsseldorf), respectively. The reader should note that some of these lines were genetically modified in-house. iPSCs were cultured following a previously established protocol.60 In brief, iPSCs were grown in mTeSR plus complete medium on 6-well plates coated with growth factor-reduced Geltrex (Thermo Fisher Scientific, Waltham, MA). To extract the total DNA, cells were incubated with Proteinase K (Carl Roth, Karlsruhe, Germany, 0.2 mg/mL) for 15 min at 55°C, followed by heat inactivation for 10 min at 95°C. The DNA concentration was determined using a NanoDrop2000 spectrophotometer (Thermo Fisher Scientific) and adjusted to 1 ng/μL. A PCR amplification strategy (30 cycles to reduce risk of amplification bias) involving 9 overlapping amplicons29 was used (polymerase: Phanta Max Super-Fidelity DNA Polymerase, Vazyme [Nanjing, China]), and the resulting PCR products were pooled and purified. Afterward, DNA quality was assessed using a NanoDrop2000 spectrophotometer. Total DNA at 250 ng (∼200 fmol) per sample was subjected to library preparation.
Long-read sequencing and data analysis
Long-read sequencing analysis was conducted as previously described,26 using a MinION Mk1C sequencer and a Flongle adaptor (ONT, Oxford, UK). To prepare the sequencing library, the pooled PCR products were subjected to library preparation using the Sequencing by Ligation Kit (ONT, SQK-LSK109) in combination with the Native Barcoding kit (ONT, EXP-NBD104) according to the manufacturer’s instructions. The final library was quantified using a Qubit4 (Thermo Fisher Scientific), loaded at a total of 10–15 fmol onto a Flongle flow cell (chemistry R9.4.1) and subjected to an 8- to 12-h sequencing run. Notably, newer generations of Flongle flow cells and the ligation sequencing kit (chemistry R10.4.1) allow for less input, thus reducing the total library input to 5–10 fmol. FAST5 files were subjected to basecalling using the guppy basecaller software (version 6.4). The initial basecalling settings to determine optimal read quality included --min_qscore 7, --require_barcodes_both_ends, --enable_trim_barcodes, --barcode_kits “EXP-NBD104,” and used the guppy super-accuracy basecalling mode (-c dna_r9.4.1_450bps_sup.cfg). Alternatively, FASTQ files directly derived from the sequencing device were used. In this case, the minimum quality score is 9 by default and can be adjusted accordingly (see the section coding for further information).
Subsequently, the resulting FASTQ files were concatenated into single FASTQ files (Linux: cat/path/to/fastq/files/∗.fastq >/your/new/location/output.fastq; Windows: type \path\to\fastq\files\∗.fastq> \your\new\location\output.fastq; inexperienced users: please refer to our concatenation tool available at https://mitopore.de/static/concat.html) and aligned to the revised Cambridge Reference Sequence (rCRS, NC_012920) using Minimap2 (version 2.24).31 In general, for data derived from WGS experiments, mapping reads into the complete reference genome is advisable to reduce the risk of false alignment of NUMTs to mtDNA, which would negatively affect variant calling accuracy. However, our mtDNA enrichment approach (see the cell culture section above) eliminates the risk for NUMTs contamination, which justifies alignment to the much shorter rCRS for performance reasons. SNV calling was performed using Mutserve (version 2.0) with a heteroplasmy variant calling threshold level of 0.05.
Short-read sequencing and data analysis
To compare the fidelity of ONT sequencing with NGS (sequencing by synthesis), the MT-ND5 target sequence of a known heteroplasmic sample was enriched for either 10 or 15 cycles to reduce the risk of PCR bias. Afterward, 1 μL of this PCR product was used as a template to amplify a 241-bp target sequence encompassing m13513 and to add adaptor sequences for a second dual-barcoding PCR reaction. PCRs were performed using Phanta Max High Fidelity Polymerase (Vazyme) according to the manufacturer’s instructions for 15, 20, or 25 cycles. Subsequently, a second level barcoding PCR adding individual barcodes to each sample was performed using 1 μL of this PCR as a template with the same cycling conditions for 19 cycles. For the complete list of primer sequences, please refer to Table S1. PCR products were combined together, and then subjected to size separation using a 1% agarose gel. Subsequently, gel extraction (GeneJet, Thermo Fisher Scientific) was performed to purify the amplicons. Illumina (San Diego, CA) library preparation was carried out as previously described.60 The resulting low-diversity Illumina library was quantified by Qubit4 (Thermo Fisher Scientific), loaded at a concentration of 6 pM and balanced by adding 30% Phix on an Illumina MiSeq benchtop sequencer. The library was sequenced with the Nano V2 reagent kit (2 × 150) in a single-end sequencing configuration, with a total of 251 cycles. Data were obtained in FASTQ format and analyzed with Mitopore as described above and by using OutKnocker.61
Mathematical model and long-read parameter estimation
To estimate the number of reads necessary to call an SNV with a certain confidence, we used a formula based on population estimation. Briefly, we calculated the number of reads (n) necessary to call a phenotype with differing levels of confidence intervals (z∗), margin of error (M), and sensitivity of detection/error rate (s) for differing levels of estimated (here: target detection rates, for example, for known heteroplasmies) population proportions ():
Assuming different parameters, we suggest aiming for the number of reads listed in Table S2 to correctly represent the distribution of expected heteroplasmy levels when using our workflow. Assessment of the lower limit of detection was made by mixing DNA of samples with known homoplasmy at m4295 (G or A) at ratios of 1:1,000, 1:100, 1:50, 1:20, 1:10, 1:5, and 1:2, followed by ONT sequencing on a Flongle flow cell.
Downsampling and testing of the mathematical reads estimation
FASTQ files from a sample with a heteroplasmy of 0.18–0.2 as determined by NGS (sequencing by synthesis) were downsampled to varying numbers of reads using the sequence toolkit (seqtk, available from https://github.com/lh3/seqtk) with the sample command: seqtk sample input.fastq target_nr_of_reads > output.fastq. The command was resampled five times with different seeds to ensure reproducible sampling among reads. Afterward, FASTQ files were aligned to the rCRS using Minimap2 (version 2.24), subjected to SNV calling using Mutserve as described (sequencing data analysis), and visualized using R ggplot2 (version 3.4.2).62
Development of the Mitopore webserver
The Mitopore.de user interface is designed for effortless usage, requiring minimal adjustments of parameters. The user can upload one or multiple FASTQ or BAM files compressed to a single zip archive, select the reference sequence of interest (default: human rCRS), select the baseline reads threshold for the coverage visualization plot, choose FASTQ quality control and filtering, and choose variant calling algorithm (default: SNVs). Upon providing an e-mail address, users receive notifications when their results are available for download. The result reports are accessible for offline use and stored for 7 days. A CAPTCHA (Completely Automated Public Turing test to tell Computers and Humans Apart) serves as a safeguard against uncontrolled traffic and exploitation from multiple requests. Multiple requests are processed one after another; the task queue is thus speed defining. Since the computational time needed per request of, for example, four compressed FASTQ files is usually <10 min; fast delivery of results in <2 h can be expected for SNV detection. In cases of indel detection, waiting times can be longer, up to several hours. For large requests, we recommend downloading the command line tool (available at https://github.com/thachnguyen/mitopore_workflow), which can improve computational speed depending on the computer used. The maximum upload limit per submission is 1 Gb, ensuring that up to 10 complete human mitochondrial genomes can be analyzed simultaneously. An increasing number of samples can be analyzed in parallel for position-specific approaches. We have implemented multiple security measures. Connection with the server is secured with the https protocol, and original FASTQ files are deleted automatically from the server directly after processing. The user’s e-mail address is only stored temporarily for sending the results. The e-mail address and final results are kept on the server for 7 days. Thereafter, all of the data are removed automatically. No personal information is stored. Users should not use any personally identifiable information as sample names.
Coding
Mitopore.de is a web application developed in a full-stack of Python (version 3.10.6), R (version 4.2.1), Java (version openjdk 11.0.19), JSscript (version ES13) using the Django web framework (version 4.1.1).
To visualize the results, CGV.js (version 1.1.0) and R ggplot2 (version 3.4.2)62 were used. The primary algorithms of Mitopore have been seamlessly integrated into a webserver. In addition, Mitopore offers the flexibility to operate in a local mode through a command line interface or Docker container (https://hub.docker.com/repository/docker/thachdt4/mitopore_local). (Please visit https://github.com/thachnguyen/mitopore_workflow for all resources used in Mitopore.) The webserver functions as follows: initially, the output of the mtDNA sequencing, for example, from an ONT sequencer, in the form of FASTQ files, undergoes quality control based on R shortRead (version 1.56.1)63 and provides information on average base quality, read length, read number per sample, and GC content. In this step, the average read quality is given by the settings from the sequencing run. In general, a minimum quality threshold of 7 and at least the high accuracy basecalling model for most applications are recommended. If the mutations of interest are present at high frequencies (e.g., above 0.75), then quality score thresholds can be adjusted accordingly. An alternative quality improvement process based on eliBQ can be chosen optionally. eliBQ filters FASTQ sequences by taking individual base quality rather than average base quality per read into account (for details, see the section development of the mitopore webserver in the results). Note that eliBQ is indispensable for indel detection because Mutect2 does not process the provided data derived from Flongle flow cell-derived ONT sequencing. The eliBQ filtering is experimental because it lacks validation with patient-derived indel data. Indels are reported after additional stringent filtering post Mutect2 calling. Only indels at a read depth of >200 and a heteroplasmy between 0.2 and 0.8 are considered. The artificial breakage region of the rCRS (m16024-580) is excluded from indel calling due to error-prone ONT sequencing. Due to quality limitations and challenges in handling indels with ONT data, we filter out equal-sized base substitutions of >1 (e.g., AA>AC, GC>GG) and 1-nt deletions (e.g., AA>A, CT>C). This ensures the accuracy of our analysis. For SNV calling or indel calling from high-quality short-read sequencing data, the use of eliBQ is considered optional because its utilization does not enhance the calling quality enough to warrant the increased computational power needed. In addition, the eliBQ filter may not be necessary for indel calling on ONT sequencing-derived data in the future because high accuracy duplex basecalling yields drastically improved quality of sequencing data. The eliBQ code is available under https://github.com/thachnguyen/mitopore_workflow/blob/main/mitopore_local/apps.py in two functions: def convert_fastq and def select_subset.
Afterward, long reads from ONT sequencers are aligned to the rCRS or the mtDNA reference for Homo sapiens, Rattus norvegicus, Mus musculus, Danio rerio, Caenorhabditis elegans, or a custom reference using Minimap2 (version 2.24).31 Note that nonhuman sequences have not undergone extensive testing, but are anticipated to perform equally well depending on the quality of the sequencing experiment. For short reads (e.g., derived from Illumina sequencers), bwa64 is used as the alignment tool due to better performance. Alignment quality control of the resulting BAM files, including number of mapped sequences, number of reads, and duplicates, is performed based on samtools (version 1.7) flagstat.
Variants are called from aligned BAM files via Mutserve32 (default for SNV) or Mutect233 (for indels >1 nt; currently only positions in the coding region [tRNA, rRNA, mRNA] with the single major genotype additional to the reference are considered to be indels with high confidence when at a depth of ≥200 reads and a heteroplasmy level between 0.2 and 0.8), and identified SNVs are analyzed with Haplogrep 324 to provide haplogroup information. Notably, other variant calling tools designed for short-read sequencing (e.g., Strelka65) were unable to process noisy long-read sequencing data in our hands. The final output report is presented in html format and includes comprehensive mtDNA analysis. Due to the challenges of ONT sequencing in resolving polynucleotide stretches, those containing at least six identical nucleotides are excluded from the disease table and plot (poly filter). Called variants with insufficient read depth are filtered out according to Table S2 (at 90% confidence interval [CI]; cov filter) and the STRAND_BIAS filter is neglected for variants with a heteroplasmy level >0.75 when the coverage at the position of interest is high enough according to Table S2. These filters do not need to be applied for high-quality data as derived from short-read or high-quality long-read sequencing. For these methods, the webserver offers the respective Minimap2 alignment options. The vcfs resulting from SNV or indel calling are annotated with variant information from MITOMAP38 and visualized as a comprehensive dot plot using a custom R script. For dynamic visualization, an interactive plot is generated using CGV, version 1.1.0 implemented in Javascript (ES13).66 The CGV plot provides and highlights SNV information for every sample track, provides comprehensive reference sequence coverage information, and presents the according base changes all showcased to the reference sequence track. All of these software components are deployed within our Django/Apache web service. The server uses a moderate configuration with 6 CPU cores (Intel i5-8400) @ 2.80 GHz 64 bit (906EA), 32 GB DDR Memory and Ubuntu 22.04.2 LTS 64 bit. The webserver workflow is depicted as a flowchart in Figure S2.
Comparison of sequencing methods
Although the Mitopore workflow and webserver are primarily tailored to low-quality long-read ONT sequencing data, they also work with high-quality NGS sequencing data. To compare the performance of the Mitopore approach consisting of PCR enrichment of mtDNA followed by ONT sequencing on a Flongle flow cell with different sequencing technologies, we used publicly available Illumina short-read WGS data,48 ONT long-read WGS performed in our laboratory,67 and Pacbio long-read WGS performed by an external company (Bioscientia). Reads were aligned to the human genome (GRCh38), reads mapped to the mtDNA extracted, and unaligned FASTQ files generated, which were subjected to www.Mitopore.de using the appropriate alignment options for ONT, Illumina, and PacBio. Resulting vcf files were merged and variant, with a threshold of 0.05 considered for comparison. Comparison was carried out using R and visualized using ggplot2 (version 3.4.2)62 and ComplexHeatmap (version 2.14.0).68
Data and code availability
All of the sequencing data, including FASTQ files and variant calling results from iPSC lines, were deposited at Mendeley Data (Dobner, Jochen; Nguyen, Thach; Rossi, Andrea (2024), “Mitopore mtDNA integrity analysis data,” Mendeley Data, V2, https://doi.org/10.17632/6k5gmhwvd5.2).
Acknowledgments
The Institut für Umweltmedizinische Forschung (IUF) is funded by the federal and state governments of Germany, the Ministry of Culture and Science of North Rhine-Westphalia (MKW), and the Federal Ministry of Education and Research (BMBF). We are grateful for the support from the Deutsche Forschungsgemeinschaft (DFG) (RO5380/1-1 and PR1527/6-1), the Bundesministerium für Bildung und Forschung (BMBF) (01GM2002A), the Leibniz Competition (SAW) Cooperative Excellence project (K246/2019), and AFM-Téléthon (AR-25179). We thank Haribaskar Ramachandran, Kerstin Dobner, Stephanie Binder, and Patrick Chinnery for discussion and comments on the manuscript. We thank Kira Frye for excellent technical support. The graphical abstract and Figure 1 were generated using BioRender.
Author contributions
Conceptualization: A.R. Methodology and source code: J.D. and T.N. Investigation: J.D. and T.N. Visualization: J.D. and T.N. Resources: A.R., A.P., F.D., M.G.P.G., and L.C. Writing: A.R. and J.D., with the input of all of the authors. Supervision: A.R. Project administration and funding acquisition: A.R., A.P., and J.K..
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2024.101231.
Supplemental information
References
- 1.Chan D.C. Mitochondrial Fusion and Fission in Mammals. Annu. Rev. Cell Dev. Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- 2.Luo S., Valencia C.A., Zhang J., Lee N.-C., Slone J., Gui B., Wang X., Li Z., Dell S., Brown J., et al. Biparental Inheritance of Mitochondrial DNA in Humans. Proc. Natl. Acad. Sci. USA. 2018;115:13039–13044. doi: 10.1073/pnas.1810946115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor R.W., Turnbull D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gusic M., Prokisch H. Genetic basis of mitochondrial diseases. FEBS Lett. 2021;595:1132–1158. doi: 10.1002/1873-3468.14068. [DOI] [PubMed] [Google Scholar]
- 5.Meyer J.N., Leung M.C.K., Rooney J.P., Sendoel A., Hengartner M.O., Kisby G.E., Bess A.S. Mitochondria as a Target of Environmental Toxicants. Toxicol. Sci. 2013;134:1–17. doi: 10.1093/toxsci/kft102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shokolenko I.N., Wilson G.L., Alexeyev M.F. Aging: A mitochondrial DNA perspective, critical analysis and an update. World J. Exp. Med. 2014;4:46–57. doi: 10.5493/wjem.v4.i4.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin Y.-H., Lim S.-N., Chen C.-Y., Chi H.-C., Yeh C.-T., Lin W.-R. Functional Role of Mitochondrial DNA in Cancer Progression. Int. J. Mol. Sci. 2022;23:1659. doi: 10.3390/ijms23031659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berneburg M., Grether-Beck S., Kürten V., Ruzicka T., Briviba K., Sies H., Krutmann J. Singlet Oxygen Mediates the UVA-induced Generation of the Photoaging-associated Mitochondrial Common Deletion. J. Biol. Chem. 1999;274:15345–15349. doi: 10.1074/jbc.274.22.15345. [DOI] [PubMed] [Google Scholar]
- 9.Kenney M.C., Ferrington D.A., Udar N. Retina. Elsevier; 2013. Mitochondrial Genetics of Retinal Disease; pp. 635–641. [DOI] [Google Scholar]
- 10.Cavalli-Sforza L.L., Feldman M.W. The application of molecular genetic approaches to the study of human evolution. Nat. Genet. 2003;33:266–275. doi: 10.1038/ng1113. [DOI] [PubMed] [Google Scholar]
- 11.Syndercombe Court D. Mitochondrial DNA in forensic use. Emerg. Top. Life Sci. 2021;5:415–426. doi: 10.1042/ETLS20210204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tolle I., Tiranti V., Prigione A. Modeling mitochondrial DNA diseases: from base editing to pluripotent stem-cell-derived organoids. EMBO Rep. 2023;24 doi: 10.15252/embr.202255678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rossi A., Lickfett S., Martins S., Prigione A. Preprint at Cell Press; 2022. A Call for Consensus Guidelines on Monitoring the Integrity of Nuclear and Mitochondrial Genomes in Human Pluripotent Stem Cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hazkani-Covo E., Zeller R.M., Martin W. Molecular Poltergeists: Mitochondrial DNA Copies (numts) in Sequenced Nuclear Genomes. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1000834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taanman J.-W. The mitochondrial genome: structure, transcription, translation and replication. Biochim. Biophys. Acta. 1999;1410:103–123. doi: 10.1016/S0005-2728(98)00161-3. [DOI] [PubMed] [Google Scholar]
- 16.Gustafsson C.M., Falkenberg M., Larsson N.-G. Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem. 2016;85:133–160. doi: 10.1146/annurev-biochem-060815-014402. [DOI] [PubMed] [Google Scholar]
- 17.Stefano G.B., Kream R.M. Mitochondrial DNA heteroplasmy in human health and disease. Biomed. Rep. 2016;4:259–262. doi: 10.3892/br.2016.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wallace D.C., Chalkia D. Mitochondrial DNA Genetics and the Heteroplasmy Conundrum in Evolution and Disease. Cold Spring Harbor Perspect. Biol. 2013;5:a021220. doi: 10.1101/cshperspect.a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stewart J.B., Chinnery P.F. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat. Rev. Genet. 2015;16:530–542. doi: 10.1038/nrg3966. [DOI] [PubMed] [Google Scholar]
- 20.Chinnery P.F., Gomez-Duran A. Oldies but Goldies mtDNA Population Variants and Neurodegenerative Diseases. Front. Neurosci. 2018;12 doi: 10.3389/fnins.2018.00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kirches E. LHON: Mitochondrial Mutations and More. Curr. Genom. 2011;12:44–54. doi: 10.2174/138920211794520150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goto Y. Clinical features of melas and mitochondrial DNA mutations. Muscle Nerve. 1995;3:S107–S112. doi: 10.1002/mus.880181422. [DOI] [PubMed] [Google Scholar]
- 23.Calabrese C., Simone D., Diroma M.A., Santorsola M., Guttà C., Gasparre G., Picardi E., Pesole G., Attimonelli M. MToolBox: a highly automated pipeline for heteroplasmy annotation and prioritization analysis of human mitochondrial variants in high-throughput sequencing. Bioinformatics. 2014;30:3115–3117. doi: 10.1093/bioinformatics/btu483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schönherr S., Weissensteiner H., Kronenberg F., Forer L. Haplogrep 3 - an interactive haplogroup classification and analysis platform. Nucleic Acids Res. 2023;51:W263–W268. doi: 10.1093/nar/gkad284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhidkov I., Nagar T., Mishmar D., Rubin E. MitoBamAnnotator: A web-based tool for detecting and annotating heteroplasmy in human mitochondrial DNA sequences. Mitochondrion. 2011;11:924–928. doi: 10.1016/j.mito.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 26.Nguyen T., Ramachandran H., Martins S., Krutmann J., Rossi A. Identification of genome edited cells using CRISPRnano. Nucleic Acids Res. 2022;50:W199–W203. doi: 10.1093/nar/gkac440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amarasinghe S.L., Su S., Dong X., Zappia L., Ritchie M.E., Gouil Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020;21:30. doi: 10.1186/s13059-020-1935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whitford W., Hawkins V., Moodley K.S., Grant M.J., Lehnert K., Snell R.G., Jacobsen J.C. Proof of concept for multiplex amplicon sequencing for mutation identification using the MinION nanopore sequencer. Sci. Rep. 2022;12:8572. doi: 10.1038/s41598-022-12613-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramos A., Santos C., Alvarez L., Nogués R., Aluja M.P. Human mitochondrial DNA complete amplification and sequencing: A new validated primer set that prevents nuclear DNA sequences of mitochondrial origin co-amplification. Electrophoresis. 2009;30:1587–1593. doi: 10.1002/elps.200800601. [DOI] [PubMed] [Google Scholar]
- 30.Becht C., Schmidt J., Blessing F., Wenzel F. Comparative analysis of alignment tools for application on Nanopore sequencing data. Curr. Dir. Biomed. Eng. 2021;7:831–834. doi: 10.1515/cdbme-2021-2212. [DOI] [Google Scholar]
- 31.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weissensteiner H., Forer L., Fendt L., Kheirkhah A., Salas A., Kronenberg F., Schoenherr S. Contamination detection in sequencing studies using the mitochondrial phylogeny. Genome Res. 2021;31:309–316. doi: 10.1101/gr.256545.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laricchia K.M., Lake N.J., Watts N.A., Shand M., Haessly A., Gauthier L., Benjamin D., Banks E., Soto J., Garimella K., et al. Mitochondrial DNA variation across 56,434 individuals in gnomAD. Genome Res. 2022;32:569–582. doi: 10.1101/gr.276013.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ip E.K.K., Troup M., Xu C., Winlaw D.S., Dunwoodie S.L., Giannoulatou E. Benchmarking the Effectiveness and Accuracy of Multiple Mitochondrial DNA Variant Callers: Practical Implications for Clinical Application. Front. Genet. 2022;13 doi: 10.3389/fgene.2022.692257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van der Auwera G.A., Carneiro M.O., Hartl C., Poplin R., del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J., et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinformatics. 2013;43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lott M.T., Leipzig J.N., Derbeneva O., Xie H.M., Chalkia D., Sarmady M., Procaccio V., Wallace D.C. mtDNA Variation and Analysis Using Mitomap and Mitomaster. Curr. Protoc. Bioinformatics. 2013;44:1.23.1–1.23.26. doi: 10.1002/0471250953.bi0123s44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brandon M.C., Lott M.T., Nguyen K.C., Spolim S., Navathe S.B., Baldi P., Wallace D.C. MITOMAP: a human mitochondrial genome database--2004 update. Nucleic Acids Res. 2005;33:D611–D613. doi: 10.1093/nar/gki079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henke M.-T., Zink A., Diecke S., Prigione A., Schuelke M. Generation of two mother-child pairs of iPSCs from maternally inherited Leigh syndrome patients with m.8993 T > G and m.9176 T > G MT-ATP6 mutations. Stem Cell Res. 2023;67 doi: 10.1016/j.scr.2023.103030. [DOI] [PubMed] [Google Scholar]
- 40.Steiner T., Zink A., Henke M.-T., Cecchetto G., Buenning M., Rossi A., Schuelke M., Prigione A. RNA-based generation of iPSCs from a boy carrying the mutation m.9185 T>C in the mitochondrial gene MT-ATP6 and from his healthy mother. Stem Cell Res. 2022;64 doi: 10.1016/j.scr.2022.102920. [DOI] [PubMed] [Google Scholar]
- 41.Lorenz C., Zink A., Henke M.-T., Staege S., Mlody B., Bünning M., Wanker E., Diecke S., Schuelke M., Prigione A. Generation of four iPSC lines from four patients with Leigh syndrome carrying homoplasmic mutations m.8993T > G or m.8993T > C in the mitochondrial gene MT-ATP6. Stem Cell Res. 2022;61 doi: 10.1016/j.scr.2022.102742. [DOI] [PubMed] [Google Scholar]
- 42.Lorenz C., Lesimple P., Bukowiecki R., Zink A., Inak G., Mlody B., Singh M., Semtner M., Mah N., Auré K., et al. Human iPSC-Derived Neural Progenitors Are an Effective Drug Discovery Model for Neurological mtDNA Disorders. Cell Stem Cell. 2017;20:659–674.e9. doi: 10.1016/j.stem.2016.12.013. [DOI] [PubMed] [Google Scholar]
- 43.Goldstein A., Falk M.J. In: GeneReviews® [Internet] Adam M.P., Feldman J., Mirzaa G.M., Pagon R.A., Wallace S.E., Bean L.J.H., Gripp K.W., Amemiya A., editors. Seattle (WA); University of Washington, Seattle: 2003. Single Large-Scale Mitochondrial DNA Deletion Syndromes; pp. 1993–2024. [PubMed] [Google Scholar]
- 44.Frascarelli C., Zanetti N., Nasca A., Izzo R., Lamperti C., Lamantea E., Legati A., Ghezzi D. Nanopore long-read next-generation sequencing for detection of mitochondrial DNA large-scale deletions. Front. Genet. 2023;14 doi: 10.3389/fgene.2023.1089956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zascavage R.R., Hall C.L., Thorson K., Mahmoud M., Sedlazeck F.J., Planz J.V. Approaches to Whole Mitochondrial Genome Sequencing on the Oxford Nanopore MinION. Curr. Protoc. Hum. Genet. 2019;104:e94. doi: 10.1002/cphg.94. [DOI] [PubMed] [Google Scholar]
- 46.Stoler N., Nekrutenko A. Sequencing error profiles of Illumina sequencing instruments. NAR Genom. Bioinform. 2021;3 doi: 10.1093/nargab/lqab019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hon T., Mars K., Young G., Tsai Y.-C., Karalius J.W., Landolin J.M., Maurer N., Kudrna D., Hardigan M.A., Steiner C.C., et al. Highly accurate long-read HiFi sequencing data for five complex genomes. Sci. Data. 2020;7:399. doi: 10.1038/s41597-020-00743-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pantazis C.B., Yang A., Lara E., McDonough J.A., Blauwendraat C., Peng L., Oguro H., Kanaujiya J., Zou J., Sebesta D., et al. A reference human induced pluripotent stem cell line for large-scale collaborative studies. Cell Stem Cell. 2022;29:1685–1702.e22. doi: 10.1016/j.stem.2022.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goenka S.D., Gorzynski J.E., Shafin K., Fisk D.G., Pesout T., Jensen T.D., Monlong J., Chang P.-C., Baid G., Bernstein J.A., et al. Accelerated identification of disease-causing variants with ultra-rapid nanopore genome sequencing. Nat. Biotechnol. 2022;40:1035–1041. doi: 10.1038/s41587-022-01221-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Edge P., Bansal V. Longshot enables accurate variant calling in diploid genomes from single-molecule long read sequencing. Nat. Commun. 2019;10:4660. doi: 10.1038/s41467-019-12493-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shafin K., Pesout T., Chang P.-C., Nattestad M., Kolesnikov A., Goel S., Baid G., Kolmogorov M., Eizenga J.M., Miga K.H., et al. Haplotype-aware variant calling with PEPPER-Margin-DeepVariant enables high accuracy in nanopore long-reads. Nat. Methods. 2021;18:1322–1332. doi: 10.1038/s41592-021-01299-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang N., Xu M., Nie F., Ni P., Xiao C.-L., Luo F., Wang J. NanoSNP: a progressive and haplotype-aware SNP caller on low-coverage nanopore sequencing data. Bioinformatics. 2023;39 doi: 10.1093/bioinformatics/btac824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keraite I., Becker P., Canevazzi D., Frias-López C., Dabad M., Tonda-Hernandez R., Paramonov I., Ingham M.J., Brun-Heath I., Leno J., et al. A method for multiplexed full-length single-molecule sequencing of the human mitochondrial genome. Nat. Commun. 2022;13:5902. doi: 10.1038/s41467-022-33530-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alston C.L., Rocha M.C., Lax N.Z., Turnbull D.M., Taylor R.W. The genetics and pathology of mitochondrial disease. J. Pathol. 2017;241:236–250. doi: 10.1002/path.4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Delahaye C., Nicolas J. Sequencing DNA with nanopores: Troubles and biases. PLoS One. 2021;16 doi: 10.1371/journal.pone.0257521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Battle S.L., Puiu D., TOPMed mtDNA Working Group. Verlouw J., Broer L., Boerwinkle E., Taylor K.D., Rotter J.I., Rich S.S., Grove M.L., et al. A bioinformatics pipeline for estimating mitochondrial DNA copy number and heteroplasmy levels from whole genome sequencing data. NAR Genom. Bioinform. 2022;4 doi: 10.1093/nargab/lqac034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gupta R., Kanai M., Durham T.J., Tsuo K., McCoy J.G., Kotrys A.V., Zhou W., Chinnery P.F., Karczewski K.J., Calvo S.E., et al. Nuclear genetic control of mtDNA copy number and heteroplasmy in humans. Nature. 2023;620:839–848. doi: 10.1038/s41586-023-06426-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lareau C.A., Ludwig L.S., Muus C., Gohil S.H., Zhao T., Chiang Z., Pelka K., Verboon J.M., Luo W., Christian E., et al. Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling. Nat. Biotechnol. 2021;39:451–461. doi: 10.1038/s41587-020-0645-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weissensteiner H., Forer L., Fuchsberger C., Schöpf B., Kloss-Brandstätter A., Specht G., Kronenberg F., Schönherr S. mtDNA-Server: next-generation sequencing data analysis of human mitochondrial DNA in the cloud. Nucleic Acids Res. 2016;44:W64–W69. doi: 10.1093/nar/gkw247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramachandran H., Martins S., Kontarakis Z., Krutmann J., Rossi A. Fast but not furious: A streamlined selection method for genome-edited cells. Life Sci. Alliance. 2021;4 doi: 10.26508/lsa.202101051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmid-Burgk J.L., Schmidt T., Gaidt M.M., Pelka K., Latz E., Ebert T.S., Hornung V. OutKnocker: a web tool for rapid and simple genotyping of designer nuclease edited cell lines. Genome Res. 2014;24:1719–1723. doi: 10.1101/gr.176701.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wickham H. Springer-Verlag; New York: 2016. ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- 63.Morgan M., Anders S., Lawrence M., Aboyoun P., Pagès H., Gentleman R. ShortRead: a bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics. 2009;25:2607–2608. doi: 10.1093/bioinformatics/btp450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li H., Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saunders C.T., Wong W.S.W., Swamy S., Becq J., Murray L.J., Cheetham R.K. Strelka: accurate somatic small-variant calling from sequenced tumor–normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 66.Grant J.R., Stothard P. The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 2008;36:W181–W184. doi: 10.1093/nar/gkn179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dobner J., Nguyen T., Dunkel A., Prigione A., Krutmann J., Rossi A. Mitochondrial DNA integrity and metabolome profile are preserved in the human induced pluripotent stem cell reference line KOLF2. Stem Cell Rep. 2024;19:343–350. doi: 10.1016/j.stemcr.2024.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gu Z. Complex heatmap visualization. iMeta. 2022;e43 doi: 10.1002/imt2.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the sequencing data, including FASTQ files and variant calling results from iPSC lines, were deposited at Mendeley Data (Dobner, Jochen; Nguyen, Thach; Rossi, Andrea (2024), “Mitopore mtDNA integrity analysis data,” Mendeley Data, V2, https://doi.org/10.17632/6k5gmhwvd5.2).