

SUMMARY
Despite the successes of immunotherapy in cancer treatment over recent decades, less than <10%–20% cancer cases have demonstrated durable responses from immune checkpoint blockade. To enhance the efficacy of immunotherapies, combination therapies suppressing multiple immune evasion mechanisms are increasingly contemplated. To better understand immune cell surveillance and diverse immune evasion responses in tumor tissues, we comprehensively characterized the immune landscape of more than 1,000 tumors across ten different cancers using CPTAC pan-cancer proteogenomic data. We identified seven distinct immune subtypes based on integrative learning of cell type compositions and pathway activities. We then thoroughly categorized unique genomic, epigenetic, transcriptomic, and proteomic changes associated with each subtype. Further leveraging the deep phosphoproteomic data, we studied kinase activities in different immune subtypes, which revealed potential subtype-specific therapeutic targets. Insights from this work will facilitate the development of future immunotherapy strategies and enhance precision targeting with existing agents.
Graphical abstract
In brief
Immunotherapy holds strong promise for cancer treatment but at present benefits only a small proportion of cases. A pan-cancer analysis of the immune landscape in more than 1,000 tumors across ten cancer types reveals immune surveillance and immune evasion mechanisms as well as potential molecular target that could augment future immunotherapy and precision medicine strategies.
INTRODUCTION
A key component of cancer ecology is the tumor microenvironment (TME). Immune and stromal cells within a tumor can both promote and limit the malignant state in a context-dependent fashion.1,2 Understanding the cancer immune microenvironment can reveal how a patient’s immune system can be harnessed for anti-cancer therapies.
To explore the TME broadly, we examined 1,056 tumor samples from 10 cancers using the pan-cancer proteogenomic dataset from the NCI-supported Clinical Proteomic Tumor Analysis Consortium (CPTAC).3 This unique multi-omics dataset includes genomic, transcriptomic, epigenomic, and proteomic data. The latter comprises mass-spectrometry-based quantification of relative protein abundance and post-translational modifications. While multiple pan-cancer analyses focusing on cancer immunity using high-dimensional multi-omic data have been explored,4–6 this work advances the field by integrating novel insights gained from deep proteomic sample characterization.
Employing novel algorithms for dissecting the tumor microenvironment accounting for both RNA and protein expression,7 we illuminated the tumor molecular microenvironment, explored the pathways related to its function, and associated it with clinical outcomes. Despite many differences across the 10 cancers, our analysis revealed seven pan-cancer immune subtypes. This indicates a common molecular fingerprint of cancer patient immune response. The large sample size of the aggregated pan-cancer multi-omic data enabled us to detect a large collection of genomic, epigenetic, transcriptomic, and proteomic changes associated with each immune subtype. Coupling the deep phosphoproteomics with kinase enrichment analysis tools,8,9 we characterized kinase activities in different immune subtypes, revealing known and potentially novel targets. Furthermore, machine learning algorithms10,11 applied to digital pathology hematoxylin and eosin (H&E)-stained images demonstrated correlations between the immune subtypes and direct visualization of lymphocytic infiltrates.
Our work advances the understanding of the diverse immune activation and evasion strategies employed by tumors, casting light on potential immunotherapy strategies. Two dedicated user-friendly web server portals implemented to accompany this analysis12,13 will facilitate further exploration of the rich data resources generated in this and related work.
RESULTS
Multi-omic pan-cancer data
In recent proteogenomic studies by the NCI-supported CPTAC, 1,056 treatment-naive samples from patients representing 10 cancers were analyzed using proteogenomic approaches consisting of whole-genome sequencing (WGS), RNA-seq, quantitative proteomics, and phosphoproteomics.3,14–27 The sample distribution over different cancers is visualized in Figure 1A: breast cancer (BC, n = 113), clear cell renal cell carcinoma (CCRCC, n = 103), colon cancer (CO, n = 96), glioblastoma (GBM, n = 99), head and neck squamous carcinoma (HNSCC, n = 110), lung squamous carcinoma (LSCC, n = 108), lung adenocarcinoma (LUAD, n = 110), ovarian cancer (OV, n=82), pancreatic ductal adenocarcinoma (PDAC, n = 140), and uterine cancer (UCEC, n = 95).
Figure 1. Derivation of immune subtypes.

(A) Outline for the derivation of immune subtypes. First, multi-omic deconvolution was performed based on proteomics and RNA-seq to estimate cell type compositions in each tumor. In parallel, pathway scores of immune-related pathways derived based on proteomics were clustered to define 10 immune pathway modules. Finally, the estimated cell type fractions and the 10 immune module scores were integrated to cluster tumors into different immune subtypes.
(B) Heatmap showing, for each cancer, the average of tumor cell percentage, immune and stromal scores, and cell type fractions. Significant differential levels between cancers (Bonferroni’s adjusted p value < 1%) are highlighted with a (*) (STAR Methods).
(C) Association between cell type fractions and survival outcomes for each cancer. The heatmap displays p values (signed −log10 scale) from Cox-proportional hazard regression models. Associations significant at 10% FDR are displayed with black dots.
(D) Kaplan-Meier curves showing cancer-specific associations between fractions of CD8 T cells and patient survival outcomes. For each cancer, tumors with high and low fractions of CD8 T cells were derived using the 1st and the 3rd quartiles, respectively. p values from logrank test are reported.
(E) Heatmap showing, from top to bottom, (1) estimated cell type fractions by deconvolution analysis; (2 and 3) pathway scores of immune modules based on proteome and RNA, respectively; (4 and 5) protein and RNA expressions of cell type markers. The annotation track and the pie-plot on the top show the distribution of different tumors within immune subtypes.
(F) Bar plot showing the proportion of samples allocated to different immune subtypes within each cancer.
Cell type composition heterogeneity and its association with patient outcomes
The pan-cancer tumors exhibited substantial heterogeneity in tumor cell percentages, as well as immune and stromal cell percentages (STAR Methods). Specifically, CCRCC, LUAD, and PDAC emerged as the cancers with the highest immune infiltration, while CCRCC and PDAC also exhibited higher stromal component compared with other cancers. Conversely, UCEC showed the highest tumor cell percentages but the lowest immune and stromal composition (Figure 1B).
To gain insights into the infiltration pattern of different immune/stromal cell types in these tumors, we estimated cell type composition fractions in the tumor microenvironment (TME) using a recently developed deconvolution algorithm,7 which leverages matched bulk gene expression and proteomic profiles to perform tissue deconvolution (Table S1). The comparison of cell type fractions among different tumors revealed extensive cell type composition heterogeneity across different cancers (Figure 1B). CCRCC and LUAD featured notably higher CD8+ T cell infiltration, whereas GBM, CO, and UCEC exhibited lower CD8+ T cell fractions. B cells also showed higher infiltration in LUAD, while lower infiltration in GBM, BR, and UCEC. The low presence of CD8+ T cells and B cells in GBM aligns with the well-known phenomenon in brain tumors, in which T cells and B cells are outnumbered by microglia and macrophages.28 On the other hand, monocytes appeared to be more enriched in CCRCC, GBM, and OV compared with other cancers. Furthermore, CCRCC was the cancer with the highest enrichment of fibroblasts and endothelial cells (Figure 1B).
Percentages of different cell types were also found to be associated with progression-free survival (PFS) in different cancers, such as CCRCC, LUAD, PDAC, and UCEC (Figures 1C and 1D; STAR Methods). Specifically, increased CD8+ T cells resulted in superior PFS for CCRCC, LUAD, and PDAC patients, but worse PFS in UCEC. In CO, higher infiltration of different cell types resulted in worse overall survival (OS) (Figure 1C).
Immune subtypes spanning 10 cancers
In addition to cell type fractions, we also leveraged 427 immune-related signatures from the literature to characterize the TME of CPTAC tumors (STAR Methods; Table S1). Given the high correlation among these immune-related signatures, we first grouped them into 10 different immune modules based on their single-sample gene set enrichment scores derived from the pan-cancer proteomic data. Besides the myeloid and lymphocyte modules, we observed signature groups representing wound healing proliferation, interferon, and TGFB/stromal. We then derived module activity scores for each tumor sample based on proteomic data. We utilized them together with the cell type fractions to perform consensus clustering to detect immune subtypes with different TME (Figure 1A; STAR Methods).
We identified seven clusters: CD8+/IFNG+, eosinophils/endothelial, fibroblast/TGF-β, CCRCC/endothelial, brain/neuro, CD8−/IFNG+, and CD8−/IFNG− (Figures 1E and 1F). CD8+/IFNG+ contained tumors from all 10 cancers and was characterized by the enrichment of CD8+ T cells, the activation of interferon, and immune-related pathways such as T cell receptor signaling (Figures 1E, 1F, and 2B). Eosinophils/endothelial was enriched in PDAC, LUAD, and LSCC tumors and was characterized by the presence of eosinophilic cells (Figures 1E and 1F). Fibroblast/TGF-β was characterized by the upregulation of TGF-β, fibroblasts, and the activation of extracellular matrix-related pathways such as epithelial mesenchymal transition (EMT) and focal adhesion (Figure 2B; Table S2). It is well known that the activation of hypoxia together with TGF-β can affect the TME, stimulating the production of extracellular matrix components, a characteristic of EMT.29 Interestingly, upregulation of hypoxia was observed in fibroblast/TGF-β solely based on proteomics (Figure 2C; Table S2), suggesting the value of the integrative proteogenomic approach.
Figure 2. Associations of immune subtypes with treatment responses, pathway activities, and patient demographic variables.

.
(A) Kaplen-Meier curves displaying associations between CD8+/IFNG+ and PFS for samples in the immunotherapy (left) and chemotherapy (right) arms in the phase III OAK clinical trial.30 p values from logrank test are reported.
(B) Bubble plot showing summary statistics of association analyses between immune subtypes and biological pathways. Bubble sizes correspond to Benjamini-Hochberg adjusted p values (−log10 scale), while bubble colors correspond to the mean differences between the averaged pathway score for samples in one immune subtype and that of the other subtypes.
(C) Bubble plot showing pathway analysis results as in (B), but for pathways found activated solely based on proteomics.
(D) Pan-cancer association between immune subtypes and demographic variables. Error bars correspond to 95% confidence intervals of odds ratios.
(E) Boxplots for pathway scores of Epithelial Mesenchymal Transition (EMT) and Interferon Gamma Signaling (IFNG) pathways among HNSCC cancers stratified by smoking status. (*) indicates significant p values (< 0.05) based on the Wilcoxon signed rank test.
CD8−/IFNG+ represents an immune subtype characterized by low immune infiltration of CD8 T cells and B cells, but strong activation of interferon gamma signaling. In contrast, CD8−/IFNG− is characterized by the lowest fraction of all immune and stromal cell types. For both these two clusters, we observed upregulation of cell-cycle-related pathways such as DNA damage/repair, and MYC targets (Figure 2B; Table S2). Interestingly, allograft rejection was upregulated in CD8−/IFNG+ but downregulated in CD8−/IFNG−, aligning the changes observed in interferon gamma signaling between these two subtypes (Figure 2B). Notably, PPARA activates gene expression was found to be elevated in CD8−/IFNG− based on proteomic data alone (Figure 2C). It has been documented that the activation of PPAR-gamma can induce the suppression of immune responses and interferon gamma activity.31,32
The remaining two clusters, CCRCC/endothelial and brain/neuro, represent cancer-specific subtypes (Figure 1E). Despite both being characterized as ‘‘immune cold,’’ these subtypes exhibited notable differences compared with CD8−/IFNG−. The brain/neuro subtype displayed enrichment of neurons and upregulation of oxidative phosphorylation and pyruvate metabolism pathways (Figure 2C). This is consistent with previous findings that the less immunogenic GBM and pediatric brain tumors showed upregulation of metabolic pathways.22,24 CCRCC/Endothelial was the predominant immune subtype within CCRCC. Although similar to CD8−/IFNG− in terms of low T cell infiltration, this subtype exhibits significantly elevated levels of mast and endothelial cell infiltration, accompanied by an upregulation of the focal adhesion pathway (Figure 2B). The prevalence of the CCRCC/endothelial subtype in CCRCC was further confirmed in an independent cohort as illustrated in the subsequent validation section.
Investigating the association between immune subtypes and demographic variables (Table S2; STAR Methods), we found underrepresentation of females compared with males in CD8−/IFNG− (Figures 2D and S2C). East Asian were more enriched in CD8−/IFNG+ than European patients (Figures 2D and S2F). Furthermore, fibroblast/TGF-β was enriched for smokers (Figures 2D and S2D). Indeed, significantly different activity of related pathways, including EMT and IFNG, between never- and ever smokers was observed in HNSCC (Figure 2E).
Association between immune subtypes and treatment responses
In order to explore the association between immune subtypes and cancer treatment responses, we conducted an analysis using data from the phase III OAK clinical trial (NCT02008227), which involved 425 non-small cell lung cancer patients treated with immunotherapy (atezolizumab/MPDL3280A).30 We obtained RNA-seq data from pre-treatment tumor tissues for 344 patients.33 By utilizing an immune subtype predicting model trained on CPTAC pan-cancer RNA-seq data (STAR Methods), we identified 75 out of the 344 tumors belonging to CD8+/IFNG+. Strikingly, these patients showed significantly better PFS (Figure 2A, left). This association was not detected when considering an independent group of patients within the OAK trial (n = 355) who received chemotherapy (Docetaxel) (Figure 2A, right). These findings align with our expectations, supporting the notion of enhanced immunotherapeutic response in CD8+/IFNG+.
Validation of cell type fraction and immune subtypes
To validate both the immune composition estimates and the inferred immune subtypes, we analyzed a subset of tumors, for which FFPE blocks were available, using alternative experimental platforms, including immunohistochemistry (IHC), multiple reaction monitoring-mass spectrometry (MRM), and tissue microarray (TMA) multiplex immunofluorescence-stained image experiments (STAR Methods). Additionally, we leveraged a recent proteogenomic data from an independent CCRCC cohort3,34 to confirm the immune subtypes detected in CCRCC.
IHC validation experiments
We evaluated IHC staining data for CD8, CD4, and CD163 on adjacent tissue slices from a subset of 17 LSCC tumors from the CPTAC pan-cancer cohort.26 The IHC scores for these markers displayed strong concordance with the corresponding estimated cell type percentages (Figures S1A, S1B, Spearman’s correlation > 0.55, p value < 0.05). Next, we collected TMA images of CD8 IHC staining from a total of 60 LSCC tumors, including the aforementioned 17, in the study cohort (STAR Methods). We used these images to assess immune exclusion, defined as an enriched CD8 staining along the stroma-tumor interface (Figure S1C). Notably, we observed this characteristic in only 4 out of 60 tumors: 2 from CD8+/IFNG+ and 2 from fibroblast/TGF-β (Table S1). Given the limited number, we were unable to assess whether the immune exclusion pattern is a contributing factor to immune subtypes. Future research on a larger scale is warranted.
Finally, we leveraged the IHC staining image data of 4 GBM samples and 4 CCRCC tumors in this study, as provided by Clark et al.17 and Wang et al.,24 respectively. For both cancers, we confirmed higher abundance levels of macrophage and T cells markers in the CD8+/IFNG+ tumors (Figure S1A).
MRM and TMA validation experiment
We conducted an independent MRM experiment for 59 HNSCC tumors in the study cohort (STAR Methods). The analysis targeted five proteins upregulated in CD8+/IFNG+ to other subtypes and confirmed the significant upregulation (Figure S1D).
To validate the presence of stroma in the TME, we conducted a TMA multiplex immunofluorescence-stained image analysis on a subset of 64 LSCC in the study cohort (STAR Methods; Table S1). FFPE tissue sections were stained using antibodies against FAP and α-SMA. TMA data supported the significant upregulation of FAP in LSCC tumors from Fibroblast/TGF-β than from other subtypes (Figure S1E). Additionally, the combined density of FAP and α-SMA was the highest in tumors from Eosinophils/Endothelial, suggesting a highly heterogeneous and plastic state of these tumors.
Validation of immune subtypes in an independent CCRCC cohort
Analyzing proteogenomic data of 112 independent CCRCC tumors34 (STAR Methods), we detected all five subtypes identified within CCRCC in the CPTAC pan-cancer cohort (Figure S1F). Notably, the predominant subtype, CCRCC-endothelial remained as the largest subtype in the validation cohort, suggesting the reproducibility of these subtypes across cohorts.
Contrast with existing immune subtypes
We compared the proteogenomic-based immune subtypes with those identified by the TCGA pan-cancer study.5 After applying the TCGA immune subtype classification to CPTAC pan-cancer RNA-seq data (STAR Methods), we observed that CCRCC were allocated for the most part to the inflammatory subtype, HNSCC and OV to the interferon gamma dominant subtype, and GBM to the lymphocyte depleted subtype (Figures S2A and S2B). These results did not reveal the immune heterogeneity within cancers. On the other hand, our proteogenomic analysis suggested distinct immune profiles within each cancer, consistent with existing literature reporting diverse immune landscapes among these cancers.17,24,26 For example, in the CPTAC CCRCC study by Clark et al.,17 two subtypes of ‘‘cold’’ tumors were found: one characterized by low immune infiltration and the enrichment of endothelial cells, and another one characterized by the low presence of both stromal and immune cells. These results were confirmed by our pan-cancer classification, but they were not detected by TCGA classification. For GBM, we also observed significant (p values < 10−16) coherence between our pan-cancer immune subtypes and the GBM-specific immune subtypes (im1-im4) identified previously24 (Table S1).
On the other hand, compared with single-cancer studies, our pan-cancer immune subtype analysis allows the identification of new immune subtypes by borrowing information across different cancers. For instance, the CPTAC LUAD study26 failed to identify a subset of cold tumors with activation of interferon gamma signaling. The latter was also missed in the TCGA pan-cancer study,5 in which the interferon gamma dominant subtype contained a mixture of tumors from the CD8+/IFNG+ (more immunogenic) and CD8−/IFNG+ (less immunogenic) groups identified by our classification (Figure S2B).
Impact of DNA aberrations on immune subtypes
Association of mutation profiles with immune phenotypes
For a set of 470 frequently mutated genes in cancers,35 we assessed the association between their mutation profiles and immune phenotypes, including cell type proportions, immune pathway modules, and immune subtypes (STAR Methods). Overall, we identified 102 genes whose mutations showed significant association with at least one immune phenotype (Table S3; Figures 3A and 3B). Notably, STK11 mutation was positively associated with CD8−/IFNG+ and downregulated its RNA and protein expression in LUAD (p < 0.05, Figures 3A–3C, S3A, and S3B). Consistently, protein levels of STK11 were significantly reduced in CD8−/IFNG+ compared with other subtypes (Figure 3D). These findings suggest that STK11 may contribute to reduced immune infiltration in patients with activated interferon gamma signaling.36
Figure 3. Association of immune subtypes with DNA alterations$$PARABREAKHERE$$(A) Bar plot showing the total number of mutations per gene stratified by cancer.

(B) Pan-cancer association between mutation profiles and immune traits based on elastic-net regressions. Red and blue entries correspond to positive and negative coefficients in the regression model, respectively.
(C) Heatmaps showing, for each gene, the association between its mutation status and its RNA/protein expressions in each cancer. Colors in the heatmap correspond to log fold-change (log FC) of the expressions between mutant and wild-type samples. Significant associations (p value from two-sided Mann-Whitney U test < 0.05) are labeled with black dots.
(D) Heatmaps showing the association between protein/RNA expression and immune subtype. Colors in the heatmap represent signed −log10 Benjamini-Hochberg adjusted p values. Significant associations (adjusted p value < 10%) are labeled with black dots.
(E) Heatmap displaying pan-cancer association between CNV and immune traits. For each cancer, the bar plot on the top shows the proportion of samples with more than 50% of the genes depleted (blue) or amplified (red) in the corresponding chromosome region. The heatmap shows, for each chromosome, the number of genes whose copy-number values were positively or negatively associated with the immune axes, represented in red and blue, respectively.
(F) Manhattan plot summarizing the pan-cancer association between gene-level CNV data and the Wound Healing Proliferation module for selected chromosomes. The y axis shows −log10 p value from linear regression.
(G) Heatmap displaying, for each cancer, the pathways over-represented in the set of pProteins and eGenes. Significant enrichments at 10% FDR are displayed with a black dot for pProteins and a white square for eGenes.
On the other hand, we observed positive association between mutations of BAP1 and CASP8 and the highly immunogenic subtype, CD8+/IFNG+ (Figure 3B). Mutations in BAP1, which were the most frequent in CCRCC among all cancers (Figure 3A), were suggested to be pathogenic and promote CD8+ T cell infiltration in CCRCC.37 In our data, BAP1 mutation correlated with the downregulation of cognate RNA and protein expression in CCRCC (Figures 3C, S3A, and S3B). This is consistent with the previous observation that decreased BAP1 expression results in higher infiltration of immune cells.38 CASP8 is a critical player in the extrinsic apoptosis pathways. Its mutations have been suggested to help tumor cells escape from cytotoxic T cells, reflecting immune evasion mechanisms that follow immunological pressure.39 We found that both gene and protein expression of CASP8 were upregulated in CD8+/IFNG+ and CD8−/IFNG+, extending previous observations of increased cytolytic activity in tumors harboring defects in CASP839 (Figure 3D).
Moreover, our analysis revealed several mutations that were not previously linked to TME (Table S3). For example, mutation of AXIN1, a central component of the destruction complex in the Wnt/β-catenin signaling pathway, was associated with the presence of macrophages together with the fibroblast/TGF-β subtype. Mutations in KEAP1 and NFE2L2 were negatively associated with the IFNG module, endothelial, and CD8 T cell, while positively associated with the wound healing module. While the KEAP1-NFE2L2 pathway has not yet been directly associated with T cells, its role in regulating oxidative stress, metabolism, and inflammation supports that defects in this pathway can have indirect effects on T cell function. Overall, our analysis uncovered known and unexplored associations between mutations and several immune traits, some deserving further experimental follow-up.
Association of copy-number variation alterations with immune phenotypes
We examined the association between various immune phenotypes and gene-level copy-number variations (CNVs) and found Chr3p, 4p, 5p, and 9p enriched of such associations (Table S3, STAR Methods). Specifically, Chr3p contained the highest number of genes (n = 467) whose CNV were significantly (p < 0.001) associated with both CD8+ T cell and macrophage infiltration (Figure 3E). In the study cohort, the CCRCC tumors showed the highest percentage of Chr3p deletion (on average > 50% of genes on Chr3p had deletion in one tumor). Interestingly, it was reported that a subset of CCRCC patients with favorable prognosis were featured with elevated expression levels of CD8+ T cell effector markers as well as a low level of copy-number loss.40 This is consistent with our observation of a negative association between Chr3p deletion and CD8+ T cell signal as well as a positive association between CD8+ T cell infiltration and PFS of CCRCC patients (Figure 1D).
Another noteworthy region is 9p21, housing genes such as CDKN2A/B and MTAP, for which CNVs were significantly correlated with the wound healing proliferation module (Figure 3F). 9p21 loss was recently suggested to confer a cold TME and primary resistance to immune checkpoint therapy.41 Our observation supports the hypothesis that deletion of CDKN2A/B, MTAP and other genes in 9p21 may contribute to immune suppression in TME.
Besides CNV aberrations, we further assessed whether microsatellite instability (MSI) was associated with immune phenotypes (STAR Methods). We found that MSI high patients in CO were associated with higher infiltration of T cells and myeloid cells (Figure S3C; Table S3). This association was not identified for UCEC, another cancer enriched of MSI high patients (Table S3).
Germline DNA variations contribute to pan-cancer tumor microenvironment
Multiple studies suggested that germline genetic variants can play an important role in shaping TME,42,43 in addition to their more traditionally studied roles on cancer risk.44–46 To better understand the impact of germline variation, we performed quantitative trait loci (QTL) analyses using both gene expressions (eQTL) and protein expressions (pQTL) together with WGS data (STAR Methods) and revealed significant QTLs regulating genes (eGenes) and/or proteins (pProteins) (Table S3). Gene sets enriched among eGenes and pProteins included multiple immune pathways, such as complement and coagulation cascade, neutrophil degranulation, and cellular response to chemical stress (Table S3; Figure 3G).
Association of DNA methylation with immune subtypes
Based on gene-level DNA methylation (DNAm) data, we identified a collection of genes showing either pan-cancer association (Figure 4A) or cancer-specific association (Figure 4B) between their DNAm and immune subtypes (STAR Methods; Table S4). Especially, for a large number of genes, their DNA methylations were associated with CD8−/IFNG− in HNSCC (FDR < 10%) (Figure S4A). Note, the association between DNAm and immune subtypes were largely in the opposite directions compared with those between RNA/protein expressions and immune subtypes, as gene-level DNAm typically leads to downregulation of gene and protein expression.
Figure 4. Association of immune subtypes with DNA methylations.

(A) Heatmap illustrating DNAm associations with immune subtypes for a set of genes exhibiting significant associations in at least two cancers or in the pan-cancer analysis (STAR Methods). The color gradients represent the average (standardized) DNAm levels within tumors from each immune subtype stratified by cancers (left) or the average Z scores across tumors in each immune subtype across all cancers (right) for different omics (DNAm, RNA, and proteins). Significant associations (FDR < 10%) are labeled with black dots.
(B) Heatmap illustrating DNAm associations with immune subtypes as in (A) for the topmost significant genes whose DNAm was associated with immune subtypes in only one cancer (FDR < 10%).
(C) Diagram of the mediation analysis.
(D) Heatmap illustrating three association analyses for each gene in each cancer: COSMIC smoking signature vs. DNAm (left), DNAm vs. immune subtype (middle), and smoking-mediated DNAm vs. immune subtype (right) for a subset of genes with significant mediation effects. In addition, the Lung N column summarizes DNAm-smoking associations as reported by a previous study on normal human lung tissues.49 The genes shown in this panel were selected based on consistent association trends between DNAm and smoking in LUAD and in normal lung tissues (Lung N). Significant associations (FDR < 10%) are labeled with black dots.
(E) Volcano plots summarizing the association strengths in terms of Z scores (x axis) and signed p value (−log10 scale) (y axis) between DNAm and COSMIC
smoking signatures for the subset of genes considered in the mediation analysis.
(F) Boxplots showing the distributions of DNAm levels of PYCR1 across immune subtypes, considering the union of HNSCC, LUAD, and LSCC samples. p values from ANOVA test are reported (**p value <0.01; *p value < 0.05, ns, not significant).
Impact of smoking on immune subtype mediated through DNA methylation
Associations between tobacco use and epigenetics are well established and have been surveyed for multiple cancers.47–52 To study whether the impact of smoking on TME were mediated through epigenetic alterations, we performed a mediation analysis to identify smoking-related DNAm influencing TME in HNSCC, LSCC, and LUAD (Figure 4C, STAR Methods). Specifically, we first derived a somatic mutation-based smoking signature47,53 (Figure S4B). We subsequently focused on 160 genes whose DNAm showed association with immune subtypes in either the pan-cancer or cancer-specific analyses and identified significant mediation effect (FDR < 10%) for 69 genes (Table S4; Figures 4D and S4C). For 13 out of 69 genes, their DNAm’s were previously reported to be associated with smoking in normal lung tissue49 (Figure 4D). One gene of interest emerging from this analysis is PYCR1, whose DNAm was the most significantly associated with the smoking signature in both LUAD and HNSCC (Figure 4E). At the same time, DNAm levels of PYCR1 were higher in CD8+/IFNG+ and lower in CD8−/IFNG− across HNSCC, LUAD, and LSCC (Figure 4F). It has been suggested that the expression of PYCR1 may contribute to an immunosuppressive microenvironment54; together, our observations suggest a role for smoking-induced hypomethylation of PYCR1 in promoting this outcome.
Kinase activation in different immune subtypes
Phosphoproteomics data offer a unique opportunity to characterize kinase activation across different immune subtypes, revealing important TME mechanisms and suggesting potential targets to turn lowly immunogenic (cold) tumors into highly immunogenic (hot) tumors. To characterize the activity of kinases, we used two tools (STAR Methods): (1) the Kinase Library, which utilizes an experimentally derived global substrate-specificity atlas of the ser/thr kinome8; and (2) KEA3, which integrates knowledge about measured and predicted kinase-substrate phosphorylations, and kinase-gene co-expression from 20 databases.9
Based on the Kinase Library, we found that phosphosites upregulated in CD8+/IFNG+ were significantly enriched of substrates regulated by MAPKAPKs, IKKβ, and TBK1 (Figures 5A, S5A, and S5D; Table S5). This group of kinases was found to be activated consistently in CD8+/IFNG+ tumors across multiple cancers, including GBM, LSCC, and PDAC (Figure 5A). In a recent study, TBK1 was identified as an immune evasion gene, and targeting this kinase was found to enhance responses to PD-1 blockade.55 Our biochemistry-based approach independently identified a host of potential downstream targets of TBK1 that were enriched in the phosphosites upregulated in highly immunogenic tumors.
Figure 5. Associations of immune subtypes with kinases activities.

Kinases are reported as gene symbols followed by protein symbols in parenthesis.
(A) Left: bubble plot showing pathways associated with different immune subtypes based on RNA-seq and proteomics. Bubble color represents the differential mean of pathway score in each subtype compared with all other subtypes, while the bubble size illustrates the Benjamini-Hochberg adjusted p value (−log10 scale).
Middle: for each pathway, the plots show kinases whose activation was found differential across immune subtype at the pan-cancer level (adjusted p value < 10%) via the Kinase Library. The color of the bubble corresponds to the log2 frequency factor from the contingency table (log2 frequency factors [FF]), while the size of the bubble to the adjusted p value. Right: for some key kinases, the cancer-specific activation in CD8+/IFNG+ and CD8−/IFNG− are shown using similar bubble plot.
(B) Heatmap showing the associations between KEA3-based kinase activity and immune subtypes in each cancer for selected kinases. Significant associations (adjusted p value < 10%) are highlighted with a black dot. The columns on the left illustrate the overall associations between each kinase and immune subtypes from ANOVA test for each cancer. The columns on the right show the membership of kinases in immune-related pathways. The annotation track on the top illustrates whether adjacent normal tissue was considered for normalization (T/N) or not (T) when deriving KEA3 scores for each cancer.
Another group of kinases active in CD8+/IFNG+ were AKT kinases (Figures 5A, S5A, S5D; Table S5). The ser/thr kinase AKT is a central mediator of the PI3K signaling pathway and is known to play critical roles in the pathogenesis of multiple cancers.56,57 Collectively, the PI3K-AKT-mTOR signaling pathway is dysregulated in many cancers and has been the subject of targeted therapies to treat cancers. There is increasing evidence that this pathway can affect tumors as well as host immunity and therefore targeting this pathway might improve immunotherapeutic responses.58 Our analysis confirmed the activation of multiple kinases involved in the PI3K-AKT-mTOR signaling pathway such as AKTs, PDPK1 (PDK1), and SGKs in CD8+/IFNG+, the highly immunogenic tumors.
Moreover, for a subset of tyrosine kinases, enrichment analysis via KEA3 revealed their activation in CD8+/IFNG+ (Figures 5B, S5B, and S5D; Table S5). It is well known that tyrosine kinases are key regulators of signaling in the immune system; with Src family kinases such as LYN and HCK being more present in leukocytes and playing a critical role.59,60 The activation of these tyrosine kinases in CD8+/IFNG+ tumors was consistent across different cancers.
In contrast, cyclin-dependent kinases, including CDK1 and CDK2, were found to be activated in CD8−/IFNG− and CD8−/IFNG+ based on the Kinase Library (Figures 5A and S5A; Table S5), suggesting that a higher proportion of cells in these tumors are actively proliferating. Consistently, KEA3 analysis also suggested significantly higher activation of cell-cycle kinases (CDK1–6) in CD8−/IFNG− for CCRCC, GBM, HNSCC, LSCC, LUAD, and PDAC (Figures 5B and S5B; Table S5).
To understand whether the immune-related kinase activities were driven by DNA alterations, we screened for cis-regulation by mutation and/or CNV on kinase activities among relevant genes (STAR Methods). While no significant cis-regulation by mutation status was detected, we observed associations between some kinase activities and their CNVs (Table S5). For instance, ATR kinase activity was upregulated by its own CNV in HNSCC (Figure S5C). This kinase was more active in HNSCC with copy-number gains, which were also highly enriched with cold tumors. This observation aligns with the findings from a recent clinical trial where ATR was activated in conditions of DNA replication and ATR inhibitor treatment led to elevated immune responses.61
Kinase and transcription factor regulation relating to immune subtypes
We derived transcription factor (TF) activity scores for each tumor sample by applying ChEA362 on CPTAC pan-cancer RNA-seq data (STAR Methods) and further associated these TF scores with kinase activity scores to detect active cell signaling regulations in different immune subtypes. Specifically, we focused on CD8+/IFNG+ (hot) and CD8−/IFNG− (cold) and counted the number of tumors for which a pair of kinase and TF were both identified to be enriched (STAR Methods). Such co-occurrences were visualized in Figures S6A–S6D and 6A. We identified various modules of TFs regulated by kinases corresponding to different cell signaling pathways unique to either hot or cold tumors. Importantly, a set of immune-related TFs including STAT1, STAT5A,and CEBPB, were identified to be positively regulated by an immune module featuring specific tyrosine kinases (i.e., LYN and SYK), and concurrently negatively regulated by MYO3B and PDK1/3/4 from the glycolysis kinase module (Figure 6A; Table S6).
Figure 6. Kinase-TF regulation and cell-type-specific kinase activation.

(A) Kinase-TF modules from the top 1% scored kinase-TF pairs for hot and cold tumors. Arrowheads are assigned to consistent up- and down-regulations, and plungers to different signs of associations between kinases and TFs. Each module was labeled according to the most relevant pathway identified by Enrichr. Genes contained in the pathway are highlighted in bold.
(B) The diagram at the top depicts the proposed mechanism. The top bar plot (black) displays the number of genes overlapping between the sets of upregulated genes following each kinase CRISPR-Cas knockout and the experimentally determined targets of CEBPB from ENCODE ChIP-seq (STAR Methods). The middle bar plot (gray) shows the p values from Fisher’s exact test for testing whether the overlapping gene sets are significantly larger than what would occur by random chance. The bottom bar plot (blue) illustrates the enrichment of the kinase perturbation L1000 signatures for the Innate Immune System R-HSA-168249 Reactome pathway. The red lines indicate the level of 0.05. Cell line names are listed in parentheses, and their primary disease associations are: A549: lung cancer, AGS: gastric cancer, YAPC: pancreatic cancer, BICR6: head and neck cancer, A375: skin cancer.
(C) Cell-type-specific kinase activation via KEA3 and the Kinase Library. The color of the heatmap represents the signed p value (−log10 scale) from enrichment analysis. Red color represents activation in tumor cells; while blue color represents activation in immune/stromal cells. For kinases with a Pearson’s correlation between activity score and RNA expression higher than 0.2, we show the log2 fold-change (log2 FC) of gene expression between tumor cells and immune/stromal cells based on scRNA (right side). Significant associations (FDR < 10%) are displayed with a black dot. From top to bottom, we present deactivation in cold tumor cells, activation in cold tumor cells, deactivation in hot tumor cells, and activation in hot tumor cells.
(D) Differential kinase activity changes of FYN between different cell types (purple bars) and fold-changes of FYN gene expression between tumor cells and immune/stromal cells based on scRNA (light gray bars). The differential kinase activity results are displayed as signed p values (−log10 scale).
In order to computationally validate the negative association between the glycolysis kinase module and the immune TF module (Figures 6A and S6E), we leveraged the cell-line-based CRISPR-Cas knockouts L1000 data63 (STAR Methods). First, we found that knockout of PDK1/3/4 and MYO3B induced the expression of innate immune system related genes in several cellular contexts (Figure 6B). In addition, a key TF in the immune-related TF module, CEBPB, was identified as sharing many of its known downstream targets (from ENCODE ChIP-seq experiments) with the upregulated gene signatures resulting from the CRISPR-Cas knockouts of PDK1/3/4 and MYO3B. Finally, we observed significant overlap between targets of CEBPB and the upregulated gene sets upon PDK1/3/4 and MYO3B CRISPR-Cas knockouts in diverse cell lines (Figure 6B). This observation strongly suggests that PDK1/3/4 and MYO3B exert a suppressive influence on CEBPB activity. This analysis provides compelling evidence that the knockout of these glycolysis-related kinases leads to the upregulation of immune-related pathways.
Pan-cancer kinases and TFs activity scores and their relationship with immune subtypes can be interactively navigated via ProKAP12 and PhosNetVis.13
Cell-type-specific kinase activations relating to immune subtypes
Although the phosphoproteomics analyses presented in the previous sections provided valuable insights into kinase activation, it remains uncertain whether these activations occurred in tumor or in stromal/immune cells. This lack of cell type specificity hindered the interpretation of the results, particularly in identifying the kinases with different level of activation between tumor cells of highly immunogenic tumors (hot) compared with those from lowly immunogenic tumors (cold) or to other cell types. We thus implemented a customized analysis pipeline using BayesDeBulk to perform cell-type-specific differential analysis to screen for kinases with different level of activation between tumor cells from hot versus cold tumors, as well as immune/stroma cells (STAR Methods). As tumor cells of different cancers could employ different immune mechanisms, we conducted the analysis for each cancer individually. To increase the sample size for differential testing, we grouped different immune subtypes into the hot and cold groups: the hot group comprised CD8+/IFNG+, eosinophils/endothelial, and fibroblast/TGF-β; while the cold group included CD8−/IFNG−, CCRCC/endothelial, and brain/neuro. For simplicity, we refer to tumor cells in hot (cold) tumors as hot (cold) tumor cells. We then utilized BayesDeBulk to identify phosphosites differentially expressed across hot tumor cells, cold tumor cells and immune/stromal cells. Finally, we conducted kinase enrichment analyses via the Kinase Library and KEA3 for each cancer, identifying activated kinases corresponding to differential phosphosites (STAR Methods).
Although we analyzed different cancers separately, we found good consistency across cancers (Figures S6F–S6I). We then focused on 33 kinases which displayed consistent activation patterns across different cancers (Figure 6C, STAR Methods). In Figure 6C, we observed lower activation of tyrosine kinases (i.e., FYN, LYN, and LCK) in cold tumor cells compared with hot tumor cells and immune/stromal cells in multiple cancers (blue squares for the corresponding rows in Figure 6C; Table S6). However, for GBM, a higher activation of FYN was detected in cold tumor cells compared with both hot tumor cells and immune/stromal cells (Figure 6C). To validate these patterns, we leveraged a comprehensive database of single-cell RNA-seq (scRNA) data64 (STAR Methods). Since RNA-seq often does not reflect kinase activities, we only considered a subset of 11 genes whose kinase activity were positively correlated with their RNA expression based on bulk data (Pearson’s correlation > 0.20). To validate these findings, we performed differential analysis comparing tumor cells and immune/stromal cells within each cancer based on scRNA (STAR Methods). For 9 out of 11 kinases, scRNA was consistent with the inferred kinase activation. Specifically, we confirmed that, in GBM, FYN showed higher activation in tumor cells than in immune/stromal cells (Figures 6C and 6D; Table S6). These findings were consistent with prior literature that suggests the expression of FYN in glioma cells can decrease anti-glioma immune response, and its knockdown can reduce the proliferation of tumor cells.65,66
In addition, we observed higher activation of CDK19, CDK20, and PTK2 (FAK) in hot tumor cells compared with cold tumor cells and immune/stromal cells (Figure 6C; Table S6). Notably, FAK was found to be more activated in hot tumor cells for most cancers, and the differences were significant in LUAD and LSCC. FAK is well known to regulate different cellular processes in tumors such as tumor proliferation and invasion.67 In LSCC, the kinase activity of FAK was found to drive exhaustion of CD8+ T cells and the recruitment of regulatory T cells (Tregs), which can limit the effectiveness of immunotherapy.68 Consistently, through our kinase activity analysis, we found FAK to be activated in tumor cells of hot tumors which were enriched of CD8+ T cells as well as Tregs (Figure 1E).
Histopathology assessment of immune subtypes
We utilized digitally scanned tumor H&E images to build convolutional neural network models for predicting immune subtypes (STAR Methods). Due to the limited sample size, we focused on distinguishing between cold tumors (CD8−/INFG−) and hot tumors (CD8+/INFG+ and eosinophils/endothelial). We built ten tissue-specific models and one pan-cancer model using 4-fold split validation. The pan-cancer model was also evaluated for each cancer, separately. Comparative performance using the area under the receiver operating curve (AUC) (Figure 7A; Table S7) showed that the pan-cancer model performed better than tissue-specific models for most cancers. This might be due to the fact that tissue-specific models may not provide sufficient training diversity. On the other hand, when pooled together, relevant morphologic features in different tissues can be more effectively learned, suggesting that global immune morphologies distinguishing cold vs hot are generalizable to a pan-cancer level.
Figure 7. Histopathology assessment of immune subtypes.
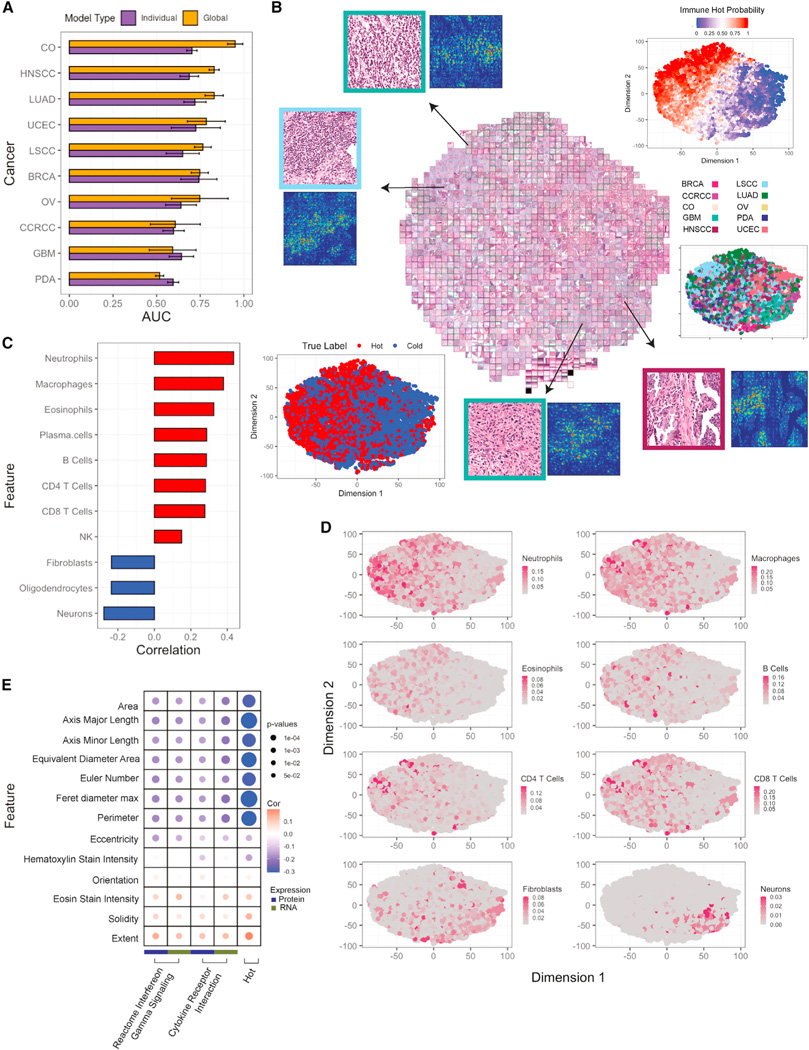
(A) Bar plots showing AUCs for predicting hot versus cold tumors based on histopathology images across different cancers. For each cancer, both single-cancer and pan-cancer models are reported. Error bars correspond to standard error across 4-fold tests.
(B) Based on pan-cancer model, imaging features are extracted from the penultimate layer and separated with tSNE clustering. The top-right plot shows the separation by the model’s prediction scores, and bottom-left is color-coded by the true label. Each point represents a different tile. Tiles are bordered with their respective cancer-type color. Selected tiles are zoomed in (top-left and bottom-right) to appreciate differences with immune infiltration.
(C) Bar plot reporting Pearson’s correlation between cell type fractions and image prediction probabilities.
(D) tSNE plot color-coded with the cell type scores.
(E) Bubble plot showing Pearson’s correlation between cellular morphology and cytokine expression pathways at a pan-cancer level. The size of the bubble corresponds to p value from correlation test.
To visually inspect the model’s discernment, we extracted latent features from the last convolutional layer and clustered image tiles using tSNE (Figure 7B). The resulting tSNE plot reveals a diagonal separation of cold and hot tiles, with no substantial clustering by cancer. To further understand the most important features influencing the model’s decision, we applied integrated-gradient-based saliency mapping to select tiles at the periphery of the tSNE plot. The result suggests that cell shape, size, and other nuclear features contributed to differentiating cold and hot tumors. Correlations between the cell type fractions and predictive probabilities show a positive association between known immune cells with images predicted as hot, and a negative association with non-immune cell types upon pathologist review (Figure 7C). Overlay of the tSNE plot with cell type fractions (Figure 7D) confirmed the concentration of immune cells in tiles from hot samples, and non-immune cells in tiles from cold samples.
Morphologies of neoplastic cells were also calculated (STAR Methods, Table S7) and correlated with cytokine expression signatures using Pearson’s correlation (Figures 7E and S7C). Notably, at a pan-cancer level, the area, axis major/minor lengths, diameter, and perimeter of neoplastic cells are inversely correlated with the cytokine expression pathways and cold/hot immune labels, suggesting that inflammatory cytokines may be limiting tumor cell growth, leading to smaller cellular area, diameter, and perimeter.
To further investigate the extent to which morphology images can distinguish different immune subtypes, we trained an additional pan-cancer model to predict 5 immune subtypes (excluding brain/neuro and CCRCC/endothelial). Across a 4-fold split, models performed well in predicting CD8+/INFG+, eosinophils/endothelial, and CD8−/INFG− (AUCs of 0.80, 0.72, 0.70, respectively), with suboptimal benchmarks for fibroblast/TGF-β and CD8−/INFG+ (AUCs of 0.66 and 0.62, respectively).
DISCUSSION
Proteomics and phosphoproteomics offer unique insights into key functional molecules underlying both immune infiltration and tumor immune evasion responses, which are often not fully appreciated by genomic approaches alone. We analyzed proteomic profiles along with matching genomic, epigenomic, and transcriptomic profiles of over 1,000 tumor samples across 10 cancers to comprehensively characterize the immune landscape of these tumors. These efforts were complemented by insights from histopathology, digital pathology, and clinical annotation of the patients.
To understand tissue function, we must understand its varied composition at the cellular level. We inferred the cell type compositions of all tumor samples based on both transcriptomics and proteomicss via BayesDeBulk,7 a deconvolution method that integrates proteogenomic data. The overall load of immune cells was linked to patient PFS outcomes in various cancers, including CCRCC, LUAD, PDAC, and CO (Figure 1D). Interestingly, the association directions differed across both tumor and immune cell types: for instance, increased CD8+ T cells were associated with superior PFS in CCRCC, LUAD, and PDAC, but inferior PFS in UCEC. This suggests that the clinical benefit of immune infiltration is dependent on the activation of oncogenic pathways in a tumor-specific manner.69 The functional state of the infiltrating immune cells likely also contributes.
Further combining the estimated cell type compositions with protein-based immune pathway activities, we identified multiple distinct pan-cancer immune subtypes. Some were shared across various cancers, suggesting common tumor-agnostic host immune reactions and evasion mechanisms. Specifically, our analysis revealed the distinction between tumors with low and high immune composition among those having active interferon gamma signaling (i.e., the CD8−/IFNG+ and CD8+/IFNG+ clusters), which was not evident in TCGA pan-cancer immune subtypes5 (Figure S1D). This distinction suggests potential intervening biology that limits CD8+ T cell infiltration despite the permissive IFNG+ cytokine activation axis. When we analyzed data from the phase III OAK clinical trial of lung cancer,30,33 we found a clear association between CD8+/IFNG+ tumors and significantly improved PFS following immune checkpoint treatment (Figure 2A). This finding lends support to the hypothesis that CD8+/IFNG+ infiltrates relate to favorable responses to immunotherapy across various cancers.6,70,71
We detected two cancer-specific subtypes, i.e., CCRCC-endothelial and Brain/Neurol. Ther. CCRCC/endothelial subtype has low immune infiltration, high percentage of endothelial cells, and was validated in an independent CCRCC cohort. GBM tumors in the Brain/Neuro subtype were lymphocyte-poor but had distinct cell type compositions compared with those allocated to the CD8−/IFNG−subtype. Specifically, the former showed enrichment of neurons, implying involvement of neurons in a subset of GBM tumors, as noted in a recent paper72 where some GBM tumor cells hijacked neuronal mechanisms for brain invasion. The Brain/Neuro subtype also exhibited upregulated oxidative phosphorylation and pyruvate metabolism, consistent with previous findings in less immunogenic GBM and pediatric brain tumors.22,24 Both CCRCC-endothelial and Brain/Neuro are of clinical interest as the responses of CCRCC and GBM to immune checkpoint inhibitor therapy is heterogeneous and no definitive biomarkers of benefit exist.73–75
Our immune subtypes differ substantially from those in the previous TCGA pan-cancer study.5 The latter captured less TME heterogeneity within each cancer (Figure S1C). Multiple factors underlie these differences. First, Thorsson et al.5 characterized cancer immunity across 30+ cancers; while we focused on 10 cancers. Thus, the two studies may capture the heterogeneity across cancers at different levels. Second, we derived immune subtypes based on estimated cell type composition in combination with immune pathway module scores, but Thorsson et al. considered only pathway activity information. Finally, preprocessing and normalization of the RNA/proteome data also impacted the analysis.
Influence of various biological and environmental factors on immune infiltration and evasion patterns is evidenced through significant associations between sex, race, and smoking status and pan-cancer immune subtypes. Screening for mutation, CNV, and methylation changes associated with immune subtypes and/or immune pathway activities further highlighted potential molecular alterations underlying immune evasion. For example, significant associations were detected between the CD8−/IFNG+ subtype and both STK11 mutations and low STK11 protein abundances. STK11 mutation confers primary resistance to PD-1/PD-L1 therapy.76–78 In addition, with methylation analysis, we identified roles for smoking-induced hypomethylation of PYCR1 in promoting an immunosuppressive microenvironment in LUAD and confirmed this with assessment of protein levels (Figure 4A).
By leveraging the CPTAC pan-cancer phosphoproteomics, we systematically characterized kinase activities associated with various immune evasion responses in tumors. Multiple kinases involved in the PI3K-AKT-mTOR signaling pathway were found activated in the CD8+/IFNG+ subtype among different tumors. Alternatively, cyclin-dependent kinases were more activated in CD8−/IFNG− and CD8−/IFNG+ subtypes, suggesting stimulated cell proliferation in cold tumors. Through multi-omics integration, we identified kinase-TF regulation across tumors. Activation of PDK1/3/4 and MYO3B co-occurred with the downregulation of immune module TFs (STAT1, STAT5A, and CEBPB) (Figure 6A). Further support for the suppressive effect of PDK1/3/4 and MYO3B on CEBPB and immune pathways stemmed from analyzing the cell-line-based CRISPR-Cas knockouts L1000 data (Figure 6B). PDK1–4 are oncogenic,79 while MYO3B is a class III myosin known as a selective transporter of receptors to the membrane. There is little evidence regarding MYO3B’s role in cancers and regulating the immune response. These kinases are potential targets for converting cold tumors into hot tumors, thus enhancing their responsiveness to immune-based treatments.
Further performing cell-type-specific analysis via BayesDeBulk, we detected a subset of kinases with different activities in tumor cells between highly immunogenic and lowly immunogenic tumors. For example, in multiple cancers including LUAD and LSCC, we noted upregulated kinase activation of PTK2 (FAK) in tumor cells of high-immunogenic tumors. These tumors demonstrated increased infiltration of regulatory T cells (Tregs). Prior work suggested that, in LSCC, FAK kinase activity drives the recruitment of Tregs and exhaustion of CD8+ T cells, which can limit the effect of immunotherapy.68 Our findings support the hypothesis that a PTK2 inhibitor could deplete Tregs and improve immunotherapy response in LSCC and other cancers. Further pre-clinical bench work is warranted to establish the clinical relevance of this discovery.
Lastly, we demonstrated that digitally acquired H&E images, when assessed and classified by convolutional neural networks, are predictive of lowly immunogenic versus highly immunogenic tumors. In addition, the classifier trained using the pan-cancer dataset outperformed those based on individual cancers in predicting immune subtypes, implying that the global immune morphologies distinguishing lowly immunogenic versus high-immunogenic tumors are generalizable to a pan-cancer level.
The discovery of a limited number of common immune subtypes across multiple cancers strongly implies shared pan-cancer mechanisms to adapt to, and evade, immune destruction, regardless of the specific diagnosis. This suggests the possibility for unified strategies to counteract immunotherapy resistance across various cancers and for the identification of predictive biomarkers.
Strength of the study
By employing a comprehensive multi-omics strategy coupled with advanced statistical modeling techniques, we obtained insights into TME in 1,000+ tumors. By jointly modeling 10 cancers, our analysis achieves superior power to detect mechanisms shared across. Compared with related works based on genomic data alone, the immune subtypes derived from proteogenomic data reveal increased meaningful heterogeneity within and across different cancers. The predictability of the tumor tissue image data for distinct immune subtypes defines the linkage between tumor morphology and molecular characteristics. Using phosphoproteomic profiles, we predicted targetable kinases associated with different immune phenotypes.
Limitation of the study
One limitation is the lack of detailed treatment information within the CPTAC pan-cancer cohort. This unknown treatment heterogeneity poses challenges for interpretation of the survival analysis. To address this limitation, we drew upon data from the phase III OAK trial of lung cancer30 to demonstrate the association between the CD8+/IFNG+ subtype and the response to immune checkpoint treatment. Future studies are warranted to explore the translational potential of immune subtypes.
Another challenge is the extensive sample heterogeneity, both within and across different cancers. With the CPTAC pan-cancer cohort (>1,000), we were more powered to reveal unique subtypes not detected in individual cancer studies. Nevertheless, we may not exhaustively identify every potential immune subtype present in these tumors. Also, tumors may exhibit a spectrum of immune infiltration that defies easy categorization into discrete subtypes.
Bulk total protein and phosphorylation signals are a convolution of those from tumor cells and immune cells. Therefore, we applied incisive deconvolution to dissect tumor-specific signaling from immune-related signaling, and obtain useful cellular level information (e.g., cell type compositions, and cell-type-specific differential kinase activities). We validated some results using independent single-cell RNA-seq datasets. However, transcriptomic data did not provide a direct read of activities for most kinase activities. More comprehensive validation efforts may require cellular level proteomic data, which is not currently available.
Despite the great depth of the CPTAC pan-cancer phosphoproteomic experiments, and the utilization of the Kinase Library,8 there remains a substantial gap as only the activities of serine/threonine kinases are observed. We thus utilized the KEA3 tool for broader kinase coverage, including protein-protein interaction and co-expression networks, to better infer the activities of tyrosine kinases and other ‘‘dark’’ kinases.80 Validation for some of these quantifications may require targeted proteomics experiments, especially for low-abundant PTMs.
Various antigens (neo-antigens, CT-antigens, and cancer-antigens) play a crucial role in shaping the immune activation landscape.81,82 However, relating these antigen activities to our immune subtypes remains a subject for future research.
CONSORTIUM
The members of the National Cancer Institute Clinical Proteomic Tumor Analysis Consortium for Pan-Cancer are François Aguet, Yo Akiyama, Eunkyung An, Shankara Anand, Meenakshi Anurag, Özgün Babur, Jasmin Bavarva, Chet Birger, Michael J. Birrer, Anna Calinawan, Lewis C. Cantley, Song Cao, Steven A. Carr, Michele Ceccarelli, Daniel W. Chan, Arul M. Chinnaiyan, Hanbyul Cho, Shrabanti Chowdhury, Marcin Cieslik, Karl R. Clauser, Antonio Colaprico, Daniel Cui Zhou, Felipe da Veiga Leprevost, Corbin Day, Saravana M. Dhanasekaran, Li Ding, Marcin J. Domagalski, Yongchao Dou, Brian J. Druker, Nathan Edwards, Matthew J. Ellis, Myvizhi Esai Selvan, David Fenyö , Steven M. Foltz, Alicia Francis, Yifat Geffen, Gad Getz, Michael A. Gillette, Tania J. Gonzalez Robles, Sara J.C. Gosline, Zeynep H. Gümüş, David I. Heiman, Tara Hiltke, Runyu Hong, Galen Hostetter, Yingwei Hu, Chen Huang, Emily Huntsman, Antonio Iavarone, Eric J. Jaehnig, Scott D. Jewell, Jiayi Ji, Wen Jiang, Jared L. Johnson , Lizabeth Katsnelson, Karen A. Ketchum, Iga Kolodziejczak, Karsten Krug, Chandan Kumar-Sinha, Alexander J. Lazar, Jonathan T. Lei, Yize Li, Wen-Wei Liang, Yuxing Liao, Caleb M. Lindgren, Tao Liu, Wenke Liu, Weiping Ma, D R Mani, Fernanda Martins Rodrigues, Wilson McKerrow, Mehdi Mesri, Alexey I. Nesvizhskii, Chelsea J. Newton, Robert Oldroyd, Gilbert S. Omenn, Amanda G. Paulovich, Samuel H. Payne, Francesca Petralia, Pietro Pugliese, Boris Reva, Ana I. Robles, Karin D. Rodland, Henry Rodriguez, Kelly V. Ruggles, Dmitry Rykunov, Shankha Satpathy, Sara R. Savage, Eric E. Schadt, Michael Schnaubelt, Tobias Schraink, Stephan Schürer, Zhiao Shi, Richard D. Smith, Xiaoyu Song, yizhe Song, Vasileios Stathias, Erik P. Storrs, Jimin Tan, Nadezhda v. Terekhanova, Ratna R. Thangudu, Mathangi Thiagarajan, Nicole Tignor, Joshua M. Wang, Liang-Bo Wang, Pei Wang, Ying Wang, Bo Wen, Maciej Wiznerowicz, Yige Wu, Matthew A. Wyczalkowski, Lijun Yao, Tomer M. Yaron, Xinpei Yi, Bing Zhang, Hui Zhang, Qing Zhang, Xu Zhang, Zhen Zhang, Qing Kay Li.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Pei Wang (pei.wang@mssm.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Raw proteomics data of the CPTAC Pan-Cancer cohort can be accessed via Proteomic Data Commons (PDC) at https://pdc.cancer.gov.
Raw genomics and transcriptomics files of the CPTAC Pan Cancer cohort are publically available via the Genomic Data Commons (GDC) Data Portal at https://portal.gdc.cancer.gov.
Processed genomic data with access control can be obtained via CDS through the NCI DAC approved, dbGaP compiled whitelists. Users can access the data for analysis through the Seven Bridges Cancer Genomics Cloud (SB-CGC) which is one of the NCI-funded Cloud Resource/platform for compute intensive analysis. Instructions for data access are as follows:
Create an account on CGC, Seven Bridges at https://cgc-accounts.sbgenomics.com/auth/register
Get approval from dbGaP to access the controlled study (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001287.v16.p6 )
Log into CGC to access Cancer Data Service (CDS) File Explore
Copy data into your own space and start analysis and exploration
Visit the CDS page on CGC to see what studies are available and instructions and guides to use the resources. (https://docs.cancergenomicscloud.org/page/cds-data)
Processed data without access control can be found at https://pdc.cancer.gov/pdc/cptac-pancancer. File names for different omics used in this paper are as follows:
Proteomic data: Proteome_UMich_SinaiPreprocessed_GENECODE34_v1.zip
Phosphoproteomic data: Phosphoproteome_UMich_SinaiPreprocessed_GENECODE34_v1.zip
RNAseq data: RNA_WashU_v1.zip
Methylation data: Methylation_MSSM_v1.zip
Mutation profile: PanCan_Union_Maf_Broad_WashU_v1.1.maf
CNV data: CNV_WGS_WashU_v1.zip. Note, WGS-based CNV data was obtained using the pipeline at https://github.com/ding-lab/BICSEQ2. In addition, for OV, CO and BR cancers, WGS data was not available; and CNV calling derived from the WXS data (CNV_WashU_v1.zip) was instead utilized. https://pdc.cancer.gov/pdc/cptac-pancancer
In this paper, we considered samples for which both RNAseq and proteomic data were measured. The full list of samples can be found in Table S1. All analysis results reported in this manuscript can be found in the supplementary tables. These results include cell type composition estimates, immune subtype labels, associations between DNA aberrations (i.e., mutation, CNV, WGS germline and methylation data) and immune subtypes, kinase activation in different immune subtypes, tumor cell-specific kinase activation inferences and histopathology assessment of immune subtypes.
All (inferred) kinase and transcript factor activity scores, as well as the clinical meta information of the cohort can be queried, visualized, and downloaded from an interactive ProKAP12 data portal: http://prokap.wanglab.cloud . Complete Pan Cancer kinase and transcription factor activity score tables can also be downloaded from https://pdc.cancer.gov/pdc/cptac-pancancer. Selected kinase and phosphosite regulatory networks can be queried, visualized, and downloaded from an interactive PhosNetVis13 data portal: https://gumuslab.github.io/PhosNetVis/cptac-vis.html.
Links to the original codes are listed in the key resources table.
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.
METHOD DETAILS
All the computational methods used in the paper are elaborated in the next section. Corresponding code is provided in the key resources table.
QUANTIFICATION AND STATISTICAL ANALYSIS
CPTAC Pan Cancer data preprocessing
CPTAC Pan Cancer data, including all genomics, epigenomics and proteomics data, were acquired and processed as described in 3, which provided a re-harmonized data freeze corresponding to the Pan-Cancer analysis of 10 tumor types. Briefly, during the re-harmonization, data were downloaded from the Genomics Data Commons (GDC) and the Proteomics Data Commons (PDC). Data for individual cohorts were processed separately using common computational pipelines and the same genome assembly and gene annotation (GENCODE V34 basic (CHR)).84 All omics data were mapped to the same set of primary protein isoforms.
Specifically, for proteomics and phosphoproteomics, raw files were searched and quantified through data generation pipeline from University of Michigan against harmonized GENCODE34 protein FASTA database, including MSFragger search engine,99 Philosopher toolkit version v4.0.1,100 and TMT-Integrator.101 Gene and phosphosite Intensities reported by the Michigan pipeline from the analysis of proteomics and phosphoproteomics data were normalized across cancers by median centering of the medians of reference intensities of each cancer. Phosphosite reannotation was performed to ensure consistent interpretation of the data across TMTs and cancers by BCM pipeline for pan-cancer multi-omics data harmonization.
We applied a customized preprocessing analysis pipeline to the abundance tables of the 10 cancers. The pipeline includes three steps: (1) identification and removal of outlier TMT multiplexes; (2) batch-effect correction across TMT-plexes; and (3) missing values imputation using DreamAI.83 Firstly, we identified outlier TMT multiplex data points by performing Intra TMT-plex T tests on the median-aligned intensity tables. For each cancer, we compared protein (or phosphosite) abundances within one TMT-plex to those in the remaining TMT-plexes using t-tests. Significant p-values indicated influences from artificial or technical factors, such as false peptide or protein identification in the TMT experiment output. We then removed the corresponding abundance measurements for the outlier protein-TMT-plex set, replacing them with ‘NA’. After removing outliers, we evaluated TMT-plex batch effects for each cancer by examining PC plots of the proteomic and phosphoproteomic abundance matrices. For datasets displaying significant batch effects (global proteomics data of CCRCC and phosphoproteomics data of ovarian cancer), we applied Combat102 to eliminate technical variation across TMT multiplexes. And for CCRCC, since tumor and normal sample sizes varied within each TMT-plex, we adjusted for tumor/normal tissue types during Combat correction. In the end, we applied DreamAI (https://github.com/WangLab-MSSM/DreamAI) on each of the tumor types separately. Imputation was done for the subset of proteins or phosphosites that quantified in at least 50% of samples in each data set. More details of the data pre-processing and harmonization steps are reported in.3
MRM experiment and data preprocessing
MRM experiment
We performed MRM (multiple reaction monitoring-mass spectrometry) experiment for a subset of 59 HNSCC samples with adequate sample materials. FFPE samples for MRM experiments were processed as described previously103 with the following modification: All sections were 5 μm tissue sections mounted on slides. Protein concentrations of lysates were measured in triplicate using Micro BCA Protein Assay Kit (Pierce, #23235) and stored at −80 ◦C until the day of digestion. A mix of cleavable stable isotope-labeled peptide standards was added to each lysate at 200 fmol/capture. 15–500 μg of protein from lysates was transferred to a deep-well plate for processing on an EpMotion 5057 (Eppendorf). Lysates were reduced in 30 mM TCEP for 30 minutes at 37 ◦C with shaking, followed by alkylation with 50 mM IAM at room temperature without shaking. Lysates were then diluted with 0.8 mL 200 mM TRIS before Lys-C endopeptidase was added at a 1:50 enzyme:substrate ratio by mass and incubated for 2 hours at 37 ◦C with mixing at 600 rpm (Thermomixer, EpMotion 5057). After 2 hours, sequencing grade trypsin was added at a 1:50 enzyme:substrate ratio. Digestion was carried out overnight at 37 ◦C with mixing at 600 rpm. After 16 hours, the reaction was quenched with formic acid (final concentration 1% by volume). Custom monoclonal antibodies were crosslinked on protein G beads (GE Sepharose, #28–9513-79), and peptide enrichment was performed using 1 μg antibody–protein G magnetic beads for each target as previously described.104
LC-MS was performed on the enriched samples with an Eksigent 425 nanoLC system (Eksigent Technologies) coupled to a 5500 QTRAP mass spectrometer (SCIEX) operated in the positive ion MRM mode. Peptides were loaded on a trap chip column (Reprosil C18-AQ, 0.5 mm × 200 μm, SCIEX, #804–00016) at 5 μL/minute for 3 minutes using mobile phase A (0.1% formic acid in water). The LC gradient was delivered at 300 nL/minute and consisted of a linear gradient of mobile phase B (90% acetonitrile and 0.1% formic acid in water) developed from 2–14% B in 1 minute, 14–34% B in 20 minutes, 34–90% B in 2 minutes, and reequilibration at 2% B on a 15 cm × 75 μm chip column (ChromXP 3C18-CL particles, 3 μm, SCIEX, #804–00001). Scheduled MRM transitions used a retention time window of 240 seconds, a desired cycle time of 1.5 seconds, a minimum of two transitions per peptide, and optimized collision energy values.
MRM data preprocessing
MRM data were analyzed using Skyline.86,105 Peak integrations were reviewed manually, and transitions from analyte peptides were confirmed by the same retention times and relative transition areas of the light peptides and heavy stable isotope-labeled peptides. Transitions with detected interferences were not used in the data analysis. Integrated raw peak areas were exported from Skyline and total intensity was calculated using peak area + background. Transitions were summed for each light/heavy pair and peak area ratios were obtained by dividing peak areas of light peptides by that of the corresponding heavy peptides. All measurements were filtered by the lower limit of quantification (LLOQ) determined from previous analytical characterization experiments (i.e., all measurements were required to be above the LLOQ).
MRM markers comprised 2 immune cell markers (CCL5, CD4) and 3 interferon signaling pathway markers (STAT1, IFIT1, and TAP2).
Immunohistochemistry (IHC) staining for immune cell markers
Tissue Microarrays (TMAs) were constructed from 72 LSCC cases that were available at the CPTAC Biospecimen Core Resource (BCR) with four 1.0 mm cores extracted compiled on two duplicate TMAs. Immunohistochemistry (IHC) staining for CD8 (clone 4B11, Bio-Rad, 1:35) was performed on Autostainer Link 48, Dako, Inc. For 60 out of the 72 tumors, at least one TMA image passed the QC and were successfully scored by the study pathologist (G. Hostetter). Specifically, modified H-Scores for CD8-stained TMA cores were adopted to capture in semi-quantitative manner ‘activity’ state of T-cells in the patient tumors with separate biopsies at baseline.87,106 Based on the CD8 H-Score as well as the spatial distribution of CD8+ cells, we screen for tumors with an ‘‘immune exclusion’’ feature, which was defined as an enriched CD8 staining along the stroma-tumor interface. We identified this feature in 4 out of 60 tumors: 2 from the CD8+/IFNG+ subtype and 2 from the Fibroblast/TGFBeta subtype (Figure S1C; Table S1). Moreover, we acquired the corresponding IHC scores for CD4, CD8 and CD163 from a previous study involving a subset of 17 LSCC tumors.26 The combined IHC scores of CD8, CD4 and CD163 of these 17 tumors were then used to evaluate the cell type percentage estimates of CD8+ T cells, CD4+ T Cells and Macrophages from the deconvolution analysis (Figure S1B).
We also obtained IHC images for 4 GBM and 4 CCRCC tumors from our prior studies, detailed in Wang et al.24 and Clark et al.,17 respectively. These IHC stains were performed at the Johns Hopkins Hospital clinical IHC laboratory using the autostainers (Ventana XT and Dako). Briefly, tissue sections (5-micron thickness) were incubated with primary antibodies following heat antigen retrieval. Antibody dilutions followed standard protocols or manufacturer recommendations. Immunostaining was developed using mouse-HRP and/or rabbit-AP polymer detection systems. Slides were counterstained with hematoxylin and dehydrated for permanent mounting, with inclusion of appropriate positive and negative controls.
Tissue Microarray (TMA) image scoring for FAP and α-SMA
To confirm the presence of fibroblast and stroma in the TME, we conducted TMA multiplex immunofluorescence-stained image analysis on a subset of 64 LSCC tumors with FFPE tissue slices from the same tumor blocks. FFPE tissue sections were cut at 4 μm and stained using antibodies against FAP (Fibroblast Activation Protein Alpha) and α-SMA (α-Smooth muscle actin), which were previously validated for immunohistochemistry. DAPI (4,6-Diamidino-2-phenylindole) was used as a counterstain. Each antibody was labeled with a specific fluorophore. The staining process was automated using the BOND-RX, model B3 (Leica Microsystems, Vista, CA, USA). A tyramine signal amplification system-based kit (OpalTM 7-color kit, Akoya/PerkinElmer, Waltham, MA; Cat#NEL797001KT) was used. The primary antibody was detected with a horseradish peroxidase (HRP)-conjugated secondary antibody. Upon introducing HRP, the fluorophore tyramide (Amplification Reagent) working solution was added to covalently label the epitope. Once the first labeling was complete, the tissue was prepared for detecting the next epitope. This process was repeated automatically. Positive and negative (autofluorescence) controls were used during each run. The multiplex immunofluorescence-stained tissues were imaged using the Vectra multispectral imaging system version 3.0 (Akoya Bioscience), which measured each fluorescence signal. Multispectral imaging involved capturing an image at low magnification (x10) through the full emission spectrum (10 nm increments between 420 to 720 nm). A trained pathologist selected a region of interest for scanning at high magnification using the Phenochart Software 1.0.9 (931 × 698 μm at 20x resolution). The development and optimization of this platform has been previously described.107
A spectral signature for each fluorophore was obtained using the Spectral unmixing library in the software (InForm™ 2.4.8, Akoya Bioscience) to separate the multispectral image into its individual fluorophores, which were then merged into a single image. Algorithms were trained to determine the cellular densities, and the final results were expressed as normalized densities of each cell phenotype (Table S1).
Estimation of tumor cell percentage, stromal and immune scores
ESTIMATE108 was utilized to infer immune and stromal scores based on RNA-seq data. Tumor cell percentage was estimated via TSNet109 based on gene expression data using immune and stromal signatures from108 as input.
Multi-omic based deconvolution
To estimate the fraction of different cell types in the tissue microenvironment, we performed a multi-omic based deconvolution integrating proteomic and RNAseq data via BayesDeBulk.7 By jointly analyzing both proteomic and gene expression data, BayesDeBulk achieves improved accuracy of the deconvolution results measured in terms of mean squared error between estimated and true cell-type fractions by 37% (42%) compared to other alternative methods applied to proteome (RNA) data alone. When comparing the multi-omic version of BayesDeBulk to its corresponding RNA-based deconvolution, the first one outperformed the latter by 18%.7
To perform the deconvolution, BayesDeBulk takes a list of cell-type specific markers for each cell type. For immune cells, such list was derived from the LM22 signature matrix.92 For this analysis, an aggregated version of the LM22 signature matrix was utilized. Specifically, we averaged the LM22 values mapping to different types of CD4 T Cells (i.e., Memory T Cells, Naϊve T Cells) to create a gene signature for CD4 T Cells. The same strategy was utilized for Dendritic cells, Macrophages, Natural Killers cells, Mast Cells and B Cells. For each pair of cell types, we considered a marker to be upregulated in the first cell type compared to the other cell type, if the corresponding value of the LM22 matrix for the first cell type was greater than 1,000 and 3 times the value of the other cell type. Besides immune cells, we considered endothelial and fibroblast cells. For GBM, neurons and oligodendrocytes were also considered. The list of cell-type specific markers for those additional cells were derived from the literature and defined as follows:
Fibroblast: CD36, PDGFRB, C5AR2, S100A4, CD70, PDPN, VIM, ITGA5, MME, PDGFRA, FAP, ACTA2,
Endothelial: PECAM1, VEGFA, KDR, CD34, ITGB1, CD74
Oligodendrocytes: MBP, CLDN11, PLP1UGT8, MOG, SOX10, ERMN, MAG, MOBP, IL1RAP, MYRF, OPALIN, APC, RTN4, GJC2, GJB1, GJC3, GPR17, PMP22, MPZL1, TRF, RAP1, GAL3ST1, MYO1D
Neurons: NCAM1, MAP2, RBFOX3, TUBB3, GRIN1
For BayesDeBulk estimation, 10000 Markov-Chain Monte Carlo iterations (MCMC) were performed. The estimated fractions were derived as the mean across MCMC iterations after discarding a burn-in of 1,000 iterations. This analysis was performed for each cancer, separately. Before performing this analysis, genes/proteins were normalized to z-score across tumor samples. For each patient, cell type fractions estimated by BayesDeBulk were normalized to sum to (1-tumor cell percentage); with the tumor cell percentage estimated by TSNet.109 This normalization guarantees that the total sum of non-malignant cells for each sample corresponds to (1-tumor cell percentage).
Association between different immune-axes and tumor types
We define immune-axes using the following metrics: tumor cell percentages, computed through TSNet109; the immune and stromal scores, computed via ESTIMATE108; and cell-type fractions, derived using BayesDeBulk.7 Each measurement of immune axes was standardized to have a mean 0 and standard deviation 1 across all tumor samples. Wilcoxon signed-rank test was performed to identify differential scores between one cancer versus all the others. P-values were adjusted via Bonferroni’s correction and only association with adjusted p-value < 1% were reported as significant. The heatmap in Figure 1B contains the average score of these immune axes for different cancers. Each measurement was first z-scored before calculating the average value for each cancer displayed in the heatmap.
Association between cell type fractions and survival
For each cancer, the association between cell type fractions and survival was assessed via univariate Cox proportional-hazards model without including any covariate. P-values were adjusted for multiple comparisons via Benjamini-Hochberg (BH) method.85 For this investigation, we employed progression-free survival (PFS) as the primary end points across all cancers except for CO, OV, and BR. In the cases of CO, OV, and BR, where PFS data was unavailable, we instead utilized overall survival in the analysis. Kaplen-Meier estimator was utilized in order to visualize the association with survival. To stratify patients into high- and low-infiltration of a particular cell type, the 1st and the 3rd quartiles of cell type fractions were utilized.
Immune related pathway signatures
To identify modules of immune related pathways activated in the whole CPTAC cohort, we curated a collection of 427 signatures from5,89 and gene sets in the Molecular Signatures Database (MSigDB – c8 collection).90,91 The analysis was performed in the same way for both global proteomics and RNA-seq data according to the following steps. First, gene or protein expression levels were standardized across samples for each tumor, separately. Then, the single-sample Mann-Whitney-Wilcoxon gene set test (mww-GST) was applied to calculate the normalized enrichment score (NES) for each signature, as previously described in 94. The activity matrix resulting from the previous step was used to calculate distances based on the spearman correlation values between every pair of signatures, across all tumors. Subsequently, the distances matrix was used to inform a consensus clustering between signatures (100 random samplings using 95% of signatures) and the optimal number of clusters was determined evaluating the relative change in area under the CDF curve for k=2 to 10. Signatures grouped together in the same cluster were further inspected in order to elucidate the biological significance of every module. Finally, for each sample we computed a score averaging the NES of signatures in each module. For the Wound Healing cluster, we only utilized two cell-cycle related signatures (i.e., Module11_Prolif_score and CHANG_CORE_SERUM_RESPONSE_UP). To investigate the correlation between protein and RNA-seq gene set activity the Spearman’s ρ statistic was used. Immune modules based on both RNAseq and proteome data are included in Table S1.
Consensus Clustering to Derive Immune Subtypes
Considering cell-type fractions derived via multi-omic deconvolution and proteomic-based signatures curated from the literature, consensus clustering was performed to identify groups of samples with the same immune/stromal characteristics. Consensus clustering was performed using the R packages ConsensusClusterPlus110 within the Bioconductor package CancerSubtypes.111 Specifically, 80% of the tumor samples were randomly subsampled without replacement and partitioned into seven major clusters using the K-means algorithm with Spearman’s correlation as metric.
Sensitivity analysis to assess the impact of estimation errors in the decomposition results on immune subtype clustering
To evaluate the impact of estimation errors in the decomposition results on the subtypes clustering, we have run additional computational experiments to assess the robustness of the clustering results. Specifically, we perturbed the cell-type fraction estimates and proteomic-based pathway scores by adding independent Gaussian noises with varying standard deviations (5%, 10% and 20% of the original standard deviation). We then evaluated how the clustering results might change based on the perturbed data matrices. This experiment was repeated 100 times. The Rand index between the original immune subtypes and the clusters derived from perturbed data matrices were above 0.87 with a median above 0.9 for all the SD levels, which indicates that the detected immune subtypes are rather robust to the variability in the cell-type fraction estimates.
TCGA Pan Cancer immune subtyping
Tumors were classified into the immune subtypes identified by the TCGA Pan Cancer analysis5 using ImmuneSubtypeClassifier R package.98
Association between immune subtypes and clinical variables
We examined the association between immune subtypes and demographic variables, such as sex, ancestry and smoking status, via logistic regression. We modeled the probability of a tumor being classified into a specific immune subtype as a function of the clinical variables of interest and the corresponding cancer type. For each demographic variable, only immune subtypes with at least 10 samples in one cancer were considered. Smoking status was categorized as Never smokers (including lifelong non-smokers) and Ever smokers including former and current smokers.
For each sample with germline WGS data, ancestry was annotated based on a Principal Component Analysis,3 and assigned as Ad-mixed American, African, East Asian, European or South Asian (Figure S2E). Immune subtypes were tested for association with ancestry only for the ancestry categories with the largest number of individuals based on the PCA groups, which included individuals of European and East Asian ancestries.
Besides the Pan Cancer analysis based on logistic regression models (results were shown in Figure 2), we also performed cancer specific association analysis for immune subtypes v.s. each of the demographic variables using Fisher Exact tests (results were shown in Figure S2).
Differential expression and pathway analysis across immune subtypes
Genes and proteins differentially expressed across the seven immune subtypes were identified based on all tumor samples. For each data type, every feature vector was normalized to z-score (i.e., mean 0 and sd 1). For each data type, the expression level of gene/protein was modeled as a linear function of immune subtypes. Table S1 shows upregulated and downregulated genes identified based on different data types. Considering genes that were up- and downregulated with Benjamini-Hochberg’s adjusted p value85 lower than 10%, Fisher’s exact test was implemented to derive enriched pathways (Table S2). For this analysis, pathways from the Reactome,93 KEGG112 and Hallmark113 databases were considered and as background the full list of gene/proteins observed under each data type was utilized. P-values from Fisher’s exact test were adjusted using Benjamini Hochberg’s correction. Table S2 shows summary statistics from this pathway analysis. To visualize differential pathway activity, pathway scores based on proteomics and RNAseq were computed via combined z-score using the R package GSVA.114 Bubble plots in Figures 2B, 2C, and 5A show the difference between the average pathway score for tumors belonging to a particular immune subtype and the average pathway score in tumors not contained in that immune subtype for a selection of differential pathway. Before computing the average, for each pathway, the score was normalized to z-score across all tumors (i.e., mean 0 and sd 1).
Validation of immune subtypes in an independent CCRCC cohort
We leveraged an independent proteogenomic data34 including 112 CCRCC tumor samples with available proteomics and RNAseq data to validate the CCRCC-endothelial subtype. The model was trained based on proteomics and RNAseq data for CCRCC samples in our cohort using the R package Pamr.115 Only proteins and genes overlapping between the two cohorts were considered to build a classifier. This number included 17632 genes and 2802 proteins. RNAseq data was log transformed in both cohorts. Each gene/protein was z-scored (mean zero and standard deviation one) in both training and testing data. The threshold parameter in the Pamr function was chosen by minimizing the classification error via cross validation based on the training data set. Notably, the predominant subtype, CCRCC-endothelial, which constituted 47% of CCRCC tumors in our study, remained as the largest subtype, accounting for 41% of cases in the validation cohort.
Association between immune subtypes and treatment response
We analyzed gene expression data from the phase III OAK clinical trial for lung cancer (NCT02008227).30 The OAK trial encompassed 425 non-small cell lung cancer patients who received immunotherapy (atezolizumab/MPDL3280A), with a median follow-up time of 21 months. For 344 out of the 425 patients, RNAseq profiling was performed on (pre-treatment) tumor tissues.33 With this data, we identified 75 tumors as CD8+/IFNG+ subtype based on a prediction model of immune subtypes trained using the CPTAC Pan Cancer RNAseq data. Specifically, the model was built based on lung cancer samples (including both LUAD and LSCC) in the Pan Cancer cohort using the R Cran package Pamr.115 Since the testing data contained only RNAseq data, the classifier was built based on gene expression data only. In particular, only genes overlapping between the training and testing data were considered (p=16898). RNA-seq data was log transformed and each gene was z-scored (mean zero and standard deviation one) in both training and testing data. The threshold parameter in the Pamr function was chosen by minimizing the classification error via cross validation based on the training data. The classification error resulting from the optimal threshold was about 8% considering 71 genes to train the model.
The same analysis was performed for 355 patients undergoing chemotherapy in the OAK clinical trial for which gene expression data was available.
Association between immune subtypes and mutation profiles
To characterize the association between mutation profiles and immune subtypes we built an elastic-net regularized model using the R package glmnet.116 The analysis was done considering a set of 470 oncogenic genes frequently mutated in cancer.35 Somatic variants were previously filtered to retain only not silent mutations with a variant allele frequency (VAF) greater than 0.5. The elastic-net model was used to model cell type fractions and proteomic modules as function of different mutations. To limit the effect of the enrichment of mutations in a particular cancer, the tumor type assignment was used as covariate. A cross-validation step was performed to select the best lambda, while the alpha value was set to 0.5. To select informative coefficients, a 1000-fold bootstrap sampling was computed. Finally, only genes with non-zero coefficients in at least 50% bootstraps were considered significant. Table S3 contains the average coefficient across bootstrap iterations for genes whose coefficients is non-zero in at least 50% bootstrap iterations.
Mutation-RNA/protein cis-regulation
For genes whose mutations were associated with immune subtypes, we conducted additional screening to assess cis-regulation of mutation effects on RNA and/or protein expression. Specifically, we tested for differential expression between mutated and wild-type, using the Mann-Whitney U test. Adjusted p-values were derived via Benjamini-Hochberg adjustment.
Association between immune subtypes and CNV
To characterize the association between copy number variation (CNV) and immune subtypes we trained a linear regression model using the lm function in R. The regression modeled log2 transformed CNV data as function of cell type fractions, immune subtypes and proteomic signature enrichments, and used the tumor type as covariate. The gene level log2 ratio of variation was derived from whole-genome sequencing profiling, except for BR, CO, and OV, for which the CNV was derived from whole-exome sequencing. Genes with a p-value from the model less than 0.001 were considered significant. The functional characterization of genes located on 3p arm and associated with CD8 T cells and macrophages was performed using an over representation analysis for Gene Ontology Biological Processes, as implemented in the ClusterProfiler R package.117
Association between immune subtypes and MSI
MSI scores were obtained from.3 Briefly, MSI scores were calculated by MSIsensor (https://github.com/ding-lab/msisensor) and interpreted as the percentage of microsatellite sites (with deep enough sequencing coverage) that have a lesion. Samples with an MSI score > 3.5 were classified as “MSI-High” and the rest were classified as “MSS”. An intermediate class with 1.0 <= score <= 3.5 were defined as MSI-Low. Association between each cell type fraction and MSI high/low status was assessed via two-sided t test (Figure S3C). For this analysis, only CO and UCEC cancers, the cancer types with at least 5 MSI-High samples, were considered.
Local impact of germline SNPs on RNA and protein expression in TME
Identification of germline genetic variants regulating local gene expression (eQTLs) and/or protein abundance (pQTLs) genome-wide
To identify eQTLs and pQTLs, we performed quantitative trait loci (QTL) analyses utilizing the MatrixeQTL package in the linear regression mode118 separately for each cancer. This included 103 CCRCC, 99 GBM, 110 HNSCC, 108 LSCC, 109 LUAD, 140 PDA and 95 UCEC samples. To identify genome-wide QTLs, we focused on WGS germline SNPs3 with minor allele frequency ≥ 5%. To control for potential confounding due to self-reported sex and/or ancestry, we included gender as well as ten principal components from the ancestry analysis3 as covariates. For eQTL analyses, we only included genes with TPM > 0.1 in at least 20% of samples. Specifically, in our QTL analyses we examined around 6 million SNPs for association with ~36,000 genes and 9,000 proteins (Table S3). Further, to control for outliers and allow cross-sample comparison, we quantile normalized and inverse normal transformed RNAseq data. Additionally, we included 15 PEER factors119 as covariates to eliminate the hidden determinants in the expression data. For pQTL analyses, we only included proteins with data in at least 20% of samples. We identified significant genes (eGenes) and proteins (pProteins) under germline genetic control of a SNP within 1 Mb (cis) of a transcription start site using FDR threshold of 1% (Table S3).
Gene-set enrichment analysis of genes and proteins
For each cancer, we performed an over representation analysis, using both eGenes and pProteins, as implemented in the ClusterProfiler R package117 (Table S3). Pathway were retrieved from the Reactome,93 KEGG112 and Hallmark113 databases using the msigdbr R package (https://igordot.github.io/msigdbr/). Significant results were derived using a cut-off for adjusted p-value of 0.1.
Association between immune subtypes and methylation profiles
Preprocessing of DNAm data
For each cancer, we first derived gene-level DNA methylation (DNAm) using the median beta-values of probes from the promotor and 5UTR regions of each gene, known to be associated with downregulation (silencing) of the gene expressions.3 These gene-level DNAm scores were then transformed into M-value data tables. Afterward, we conducted filtering by removing genes and then samples with >= 50% missing values. Outlier M-values beyond 4xIQR (inter-quantile range) of the median were truncated. Subsequently, we performed K-Nearest Neighbor imputation using the ‘knn.impute’ via the R package impute.120 The resulting data matrix contains DNAm for 16463 genes and 735 samples, distributed across seven cancers (103 CCRCC, 94 GBM, 105 HNSCC, 108 LSCC, 107 LUAD, 124 PDA, and 94 UCEC).
Identification of CIMP Subtype in DNAm
Exploratory clustering of methylation data of 16463 genes for 735 samples revealed 3 distinct subtypes characterized by an epigenome-wide pattern of low, medium or high levels of methylation. We identified these subtypes as CpG island methylator phenotypes (CIMP), which have been previously described for multiple cancers.121–123 While there was no significant association between CIMP and immune subtypes (p-value = 0.29, Fisher’s Exact Test, N = 735), we considered the substantial variations in DNAm across CIMP subtypes by including it as a covariate when assessing the association between DNAm with immune subtype in our subsequent analysis.
Association of DNAm with immune subtype
We focused on 11,610 autosomal genes and applied linear regression models to examine the relationship between their DNAm (gene-level M-value) and immune subtypes in each cancer separately. Specifically, for each cancer and gene, we first considered the subset of immune subtypes and CIMP subtype, represented by at least 5 tumor samples each. We derived covariate-adjusted M-values for these samples by employing a linear regression model that accounted for factors such as age, sex, smoking status (Never/Ever), CIMP subtype (High/Med/Low), immune subtypes, and the first 10 principal components from the ancestry analysis.3 Covariate-adjusted M-values were calculated by extracting from the M-values the estimated effects of each covariate, while retaining the effects related to immune subtype. Subsequently, we focused on immune subtypes with at least 10 samples to assess the association between them and covariate-adjusted M-values, utilizing a linear regression model without an intercept. To identify DNAm with similar association with immune subtypes across all cancers, combined p-values were derived based on the Cauchy combination test (CCT).97 The Pan Cancer combined scores (Figure 4A) are the Z-scores corresponding to the combined p-value, where the sign of the combined score corresponds to the sign of the median Z-score for each DNAm across the different cancers. The Benjamini-Hochberg correction was used to adjust combined p-values to account for multiple comparisons.85
Mediation analysis to test for effects of smoking on immune subtypes as mediated by DNAm
Mediation analysis was performed for a subset of the genes found to be associated with immune subtypes, including (1) 73 genes with a Pan Cancer combined association (FDR values < 0.01), as well as (2) genes with a cancer-specific association (FDR < 0.10) in HSNCC (N = 85), LUAD (N =4), or LSCC (N =5). For investigation of methylation mediated effects due to smoking, we used the COSMIC smoking signature (SBS4), a somatic mutation signature associated with tobacco smoking, that has been detected in head/neck and lung tissues.124 The associations between this smoking signature and self-reported smoking status are illustrated in Figure S4B for all three cancers.
For each cancer, we focused on CIMP and immune subtype categories with a minimum of 5 samples. Also, samples with missing values in any of the variables were excluded. We adjusted gene level methylation M-values according to CIMP Subtypes and the top 10 PCAs representing the ancestry genomic backgrounds3 by regressing the M-values against these covariates and obtaining the residuals.
Then for the qth gene, we perform the mediation analysis using the below model:
where is the probability for the sample to fall in the immune subtype; represents the adjusted gene-level methylation score of the gene in the sample; is the SBS4 smoking signature value of the sample; and are unknown parameters. In this model, a significant non-zero estimate of suggests an association between smoking and the immune subtype through the methylation changes of the qth gene. The Divide-Aggregate Composite-null Test (DACT) was used to test for significance of the mediated effect.125 The DACT function https://rdrr.io/github/zhonghualiu/DACT/src/R/DACT.R with correction parameter set to JC was used to obtain bias-corrected p-values for the mediated effect.
We also compared the association direction between DNAm and smoking in the CPTAC cohort and that from a previous study of the normal human lung.49 Among the genes with significant mediation effects, those showed consistent DNAm v.s. smoking associations in both LUAD and normal lung tissues were shown in Figure 4D, while the rest were shown in Figure S4C. Note, the p-values from the regression analysis for assessing the associations between DNAm and smoking were adjusted using the Bonferroni correction.
Kinase activity in different immune subtypes via the Kinase Library
Pan Cancer association between phosphorylation abundance and immune subtypes
For this analysis, we consider sites with less than 95% missing values across the ten cancers. Each phosphosite abundance was adjusted by the global abundance of the corresponding protein and the cancer indicator via linear regression. Then, the residuals of this linear regression were modeled as a linear function of the immune subtypes. For each site, only immune subtypes with at least 5 observations were considered into the model. P-values from linear regression were adjusted for multiple comparisons via Benjamini-Hochberg adjustment. Only associations with an adjusted p-value less than 10% were considered significant. Pan Cancer level association analysis results can be found in Table S5.
Cancer-specific association between phosphorylation abundance and immune subtypes
For each cancer, we consider sites with less than 80% missing values. Each phosphosite abundance was adjusted by the abundance of the corresponding protein. Then, the residuals of this linear regression were modeled as a linear function of the immune subtypes. For each site, only immune subtypes with at least 5 observations were considered into the model. P-values from linear regression were adjusted for multiple comparisons via Benjamini-Hochberg adjustment. To control for variability in association values between different cancers, we used the top and bottom 10% of the sites (ranked based on nominal p-values) as positively and negatively associated sites, respectively, and the middle 80% as non-associated sites, as an input to the Kinase Library enrichment analysis. Single-cancer association analysis results can be found in Table S5.
Kinase enrichment via the Kinase Library
Based on the list of differential sites derived following the procedure illustrated in the previous section, kinase enrichment was performed. Full description of the substrate specificities atlas of the Ser/Thr kinome can be found in.8 The phosphorylation sites detected in this study were scored by all the characterized kinases (303 S/T kinases), and their ranks in the known phosphoproteome score distribution were determined (percentile-score). For every non-duplicate, singly phosphorylated site, kinases that ranked within the top-15 kinases for the S/T kinases were considered as biochemically predicted kinases for that phosphorylation site. Toward assessing a kinase motif enrichment, we compared the percentage of phosphorylation sites for which each kinase was predicted among the top 10% positively and the top 10% negatively associated phosphorylation sites with each relevant signature, versus the percentage of biochemically favored phosphorylation sites for that kinase within the set of un-associated sites (i.e., those not falling into the top 10% positively or negatively associated sets). Contingency tables were corrected using Haldane correction. Statistical significance was determined using one-sided Fisher’s exact test, and the corresponding p-values were adjusted using the Benjamini-Hochberg procedure. Then, for every kinase, the most significant enrichment side (upregulated or downregulated) was selected based on the adjusted p-value and presented in the bubble plots. Bubble plots were generated with size and color strength representing the adjusted p-values and frequency factors (FF) respectively, only displaying significant kinases (adjusted p-value <= 0.1). Kinases that were significant (adjusted p-value <= 0.1) for both upregulated and downregulated analysis were plotted using the parameters of the more significant site, but were also outlined with a yellow outer-circle.
Derivation of kinase activity scores for each tumor sample via KEA3
For each cancer, we standardized the phosphosite abundance data by subtracting the average abundance of each phosphosite in the normal adjacent tissue (NAT) samples from its abundance in each tumor sample. Subsequently, we scaled these values by the standard deviation of the phosphosite abundances across tumor samples. In the case of 4 cancers (BRCA, GBM, PDA, UCEC) without any NAT samples, we standardized the abundance of each phosphosite across tumor samples to z-score (i.e., mean 0 and standard deviation 1). Then, based on the standardized phosphosite abundance matrices, for each tumor sample, we applied kinase enrichment analysis on the sets of proteins corresponding to the top (bottom) 500 phosphosites with the highest (lowest) abundances using KEA3 Appyter.9
Since rank-scores obtained from the KEA3 enrichment analysis were impacted by many factors beyond kinase activation levels in the samples (e.g., the number of known substrates for each kinase), we utilized a permutation procedure to further normalize the KEA3 rank scores to obtain meaningful interpretation of kinase activation. Specifically, we randomly sampled 5000 independent protein sets of size 500 from the 8305 proteins represented in the Pan Cancer phosphoproteomics. For each set, we performed KEA3 analyses and recorded the resulting rank-scores for each kinase. By aggregating the rank-scores of a given kinase across all 5000 sets, we obtained a null distribution representing the expected rank-score distribution of the kinase under the assumption of no activation (as the query sets were randomly selected). This enabled us to compare the observed rank-scores from the real dataset against their null distributions and identify kinases with significant activation. We normalized the observed rank-scores of each kinase by calculating the reversed z-scores:
where and are the mean and standard deviation of the permutation null distribution of the kinase. The normalized rank-scores, which we refer to as kinase activity scores, reflected the activation levels of each kinase in tumor samples with higher values indicating higher activity.
Association between KEA3 kinase activity scores and immune subtypes
To investigate the association between kinase activity scores and immune subtypes, we conducted two analyses. First, we examined the global variation of kinase activity scores across immune subtypes within each cancer using an ANOVA test. Second, we assessed the effect of each immune subtype on kinase activity scores using a linear regression model with kinase activity scores as the response variable and immune subtypes as predictors. Both analyses were stratified by cancer. ANOVA test p-values and sub-type-specific coefficients/significance from linear regression models are reported in Table S5. To account for multiple testing, we further adjusted p-values for each cancer using the Benjamini-Hochberg (BH) method.85
Cis-regulation between KEA3 kinase activity scores and mutation/CNV
For the 40 kinases included in Figure 5B, we tested whether their kinase activity scores were influenced by the mutation/CNV of the corresponding genes using linear regression for each cancer, separately. Specifically, we modeled KEA3 kinase activity scores as function of mutation status, gene-level CNV, age, sex and tumor cell percentage. Especially, for CNV levels, we were interested in detecting significantly positive coefficients in the regression models and utilized p-values from one sided test for this purpose. We further derived family wise error rate (FWER) by adjusting p-values via Bonferroni correction for each cancer separately. Coefficients with FWER<0.1 were considered significant cis-regulations.
Cell type-Specific Kinase Activation
We implemented a novel analysis pipeline to perform cell-type specific differential analysis. Specifically, we used BayesDeBulk to estimate the phosphosite abundances in tumor cells of high-immunogenic tumors, in tumor cells of low-immunogenic tumors and in immune/stromal cells. This analysis was performed for each cancer separately, since phosphoproteomics in tumor cells might be different across cancers. To improve the sample sizes for differential testing, we combined different immune subtypes to form the Hotand Cold groups. Hot group included the immune subtypes with higher immune composition such as CD8+/IFNG+, Eosinophils/Endothelial and Fibroblast/TGFBeta; while the Cold group included CD8-/IFNG-, CCRCC/Endothelial and Brain/Neuro. Given the Hot- and Cold-group assignments, the abundance of the jth phosphosite for sample was modeled as the following function:
with being the estimated tumor cell percentage for sample ; an indicator function equal to 1 if the ith sample belongs to the Hot group and 0 otherwise; being an indicator function equal to 1 if the ith sample belong to the Cold group and 0 otherwise;, and being the abundance of the jth phosphosite in tumor cells of the Hot tumors, Cold tumors and that in immune/stromal cells, respectively. Note, here, we assumed that phosphosite abundance distributions in immune/stromal cells were the same across different tumors. Tumor cell percentages were considered as fixed and estimated via TSNet (Table S1). BayesDeBulk was utilized to estimate the parameters of this model, i.e., , and for each phosphosite j. For BayesDeBulk estimation, 10000 Markov-Chain Monte Carlo iterations (MCMC) were considered. The estimated parameters were derived as the median across the MCMC iterations after discarding a burn-in of 1,000 iterations. Once parameters of interest were estimated, we performed inference using their 95% CIs derived from the MCMC iterations. For example, we claimed a phosphosite to be significantly higher in the tumor cell of Hot tumors compared to the other cell groups (i.e., tumor cells of the Cold tumors and immune/stromal cells) if the 95% CI of was larger than that of and . In summary, for each cancer, we derived the following four cell type-specific differential lists of phosphosites (Table S6):
- Phosphosites upregulated in tumor cells of Hot tumors (Up-Tumor_Hot):
- Phosphosites downregulated in tumor cells of Hot tumors (Down-Tumor_Hot):
- Phosphosites upregulated in tumor cells of Cold tumors (Up-Tumor_Cold):
- Phosphosites downregulated in tumor cells of Cold tumors (Down-Tumor_Cold):
For each of the four lists of phosphosites, kinase enrichment was then performed via KEA3 and the Kinase library as explained in the following sections.
Cell type specific kinase activation by KEA3 and the Kinase Library
Considering the list of differential phosphosites derived as described in the previous section, we performed kinase enrichment analysis via KEA3 and the Kinase Library. KEA39 enrichment analysis was performed via KEA3 Appyter. We also employed a permutation strategy to derive the normalized KEA3 scores (see next section for further details). Significance of KEA3 rank scores was assessed by calculating the proportion of values smaller than the observed KEA3 from the permutation-based null distributions (Table S6). Kinase enrichment analysis was also performed via the Kinase Library using the same strategy described in detail in the previous sections.
In Figure 6C, we reported 33 kinases meeting the following criteria: (1) their differential activation-pattern between different cell categories (i.e., hot tumor cells, cold tumor cells and immune/stromal cells) was consistent across at least 8 out of the 10 tumor types; (2) showed consistent significant differential activation/deactivation patterns (FDR<10%) in at least 2 tumor types based on either KEA3 or the Kinase Library results; and (3) exhibited differential activation patterns exclusively in either hot or cold tumor cells. Those criteria were used to identify kinases with significant and consistent activation trends observed in hot and cold tumor cells across different cancers. Results for the full list of kinases are reported in Table S6.
Permutation procedure to derive p-values for KEA3 rank scores
Due to the property of enrichment tests, distributions of the KEA3 enrichment rank scores are affected by the number of proteins (phosphosites) in the input list to KEA3. Thus, to generate faithful null distributions of KEA3 rank scores using the aforementioned permutation procedure, it is necessary to match the size of the random phosphosite set to that of the DE (differentially expressed) phosphosites from the cell-type specific analysis. For this purpose, we created a collection of baseline distributions by randomly sampling protein sets of varying sizes (i.e., n = 10, 50, 100, 150, 200, 250, 300, 400, 500) from the complete protein list (8305 proteins). To estimate the baseline distribution for a specific size, we used the distributions of the closest two sets (smaller and larger) in the sequence. This estimation approach significantly reduces the computational burden while maintaining accuracy.
Specifically, for a given differential list of phosphosites, if matches one of the values in the sequence (n = 10, 50, 100, 150, 200, 250, 300, 400, 500), we will use the null distribution corresponding to normalize KEA3 ranks and to infer significance. For smaller than 10 or greater than 500, we will use the distribution of size 10 or 500. Otherwise, the closest sizes in the sequence and with will be used.
Mean and standard deviation were calculated from the 5000 random samples with size and separately, denoted as and . Then, the estimated baseline mean and standard deviation of size was calculated as:
Finally, the normalized kinase activity scores are derived with the same procedure described in Derivation of kinase activity scores for each tumor sample via KEA3.
Similarly, the p-value of corresponding kinase ranks from KEA3 was calculated from the distribution of size and , separately. This was achieved by calculating the proportion of baseline ranks less than the observation on the same kinase, i.e., p = mean(random sampled ranks < observed rank), and followed by the estimation of p-value from the baseline distribution of size as follows:
Normalized kinase activity scores and p-values for all the cell type specific differential phosphoprotein lists are provided in Table S6.
Single-cell validation of cell-type specific kinase activation
We collected data from 1,587,530 single cells, spanning 10 cancers in the CPTAC cohort. These cells were grouped by sample, of which the number of samples per cancer varied considerably (i.e., single digits for OV to over 150 for LUAD). For each patient sample, we had already pre-annotated all cell types, and as such we reduced these down into three simpler groups for analysis: malignant, immune/stromal, and epithelial-normal. Gene expression profiles were then averaged together across those three groups to create pseudo-bulk expression profiles for each sample, which were then used downstream for comparative differential expression analysis. For each cancer subtype and each given kinase, we performed a differential expression analysis in the form of an independent t-test between pseudo-bulk averaged log normalized expression values across malignant vs. immune/stromal groups. Log fold-factors (fold-changes) were also calculated.
Transcription factor regulator analysis
Firstly, to standardize the tumor RNAseq expression data for each cancer, we employed the same approach as used for creating kinase activity scores based on KEA3 by normalizing the data with respect to the matched RNA data from the corresponding NAT (please see section Derivation of kinase activity scores for each tumor sample via KEA3). For the subset of cancers where NAT samples were not available, each gene was standardized to have mean 0 and standard deviation 1. We then performed transcription factor (TF) enrichment analysis by applying ChEA3 on the sets of the top 500 and bottom 500 genes with the highest and lowest expression levels, respectively, for each tumor sample.62 ChEA3 enrichment rank scores were normalized using the same permutation procedure utilized to normalize the KEA3 rank scores of tumor samples.
For a given kinase-TF pair, we assigned a score to each tumor sample as follows:
If either the kinase or the transcription factor (TF) was not ranked among the top 30 for the tumor, the kinase-TF pair score of this sample was set to zero.
If both the kinase and the TF were ranked in the top 30 for the tumor, the kinase-TF pair score was calculated as: (31 – [TF Rank])/30 +(31 - [Kinase Rank])/30.
Since there were two sets of ranks (upregulated and downregulated) for each feature (kinase or TF) for each tumor sample, we generated four sets of kinase-TF pair scores for each sample to measure:
Co-upregulation of the kinase-TF pair;
Co-downregulation of the kinase-TF pair;
Co-occurrence of TF-upregulation and Kinase-downregulation for the Kinase-TF pair;
Co-occurrence of TF-downregulation and Kinase-upregulation for the Kinase-TF pair.
We then focused on the Hot tumors from the CD8+/IFNG+ subtype (n = 175) and the Cold tumors from the CD8-/IFNG-subtype (n = 306), and derived the average kinase-TF pair scores (for each of the four sets of kinase-TF scores) within the Hot and Cold tumor groups, separately.
Next, for each of the four sets of kinase-TF scores, we extracted the top 1% of kinase-TF pairs with the highest scores (Table S6). Pairs only present in the top 1% sets of Hot tumors were assigned a value of 2; pairs that were only present in top 1% sets of Cold tumors were assigned a value of −2; and pairs that were present in both the top 1% for Hot tumors and cold tumors were assigned a value of 1. The resulting matrices were used to construct four heatmaps shown in Figure S6A.
Finally, to produce the summary bipartite graphs (Figure 6A), clusters were identified from the heatmaps manually. Kinases and TFs common in multiple clusters across the heatmaps were grouped and connections were drawn between groups to indicate inferred activation (up-up or down-down) or inhibition (down-up or up-down) in Hot, Cold, or both groups based on the condition from which the cluster was extracted from. Each cluster was submitted to Enrichr126 and the most relevant enriched term was used to label the cluster.
Validation of transcription factor regulation via L1000 database
CEBPB targets were sourced from the ENCODE ChIP-seq gene set library (ENCODE_TF_ChIP-seq_2015) downloaded from Enrichr.126 CEBPB targets appearing in at least two sets were retained resulting in 4767 genes. Processed gene set signatures pertaining to PDK1, PDK3, PDK4, and MYO3B L1000127 CRISPR/Cas knockouts were sourced from SigCom LINCS.63 The signatures in SigCom LINCS are computed using the Characteristic Direction method.88 The overlap of each signature with the CEBPB targets was assessed with the Fisher exact test and those with significant overlap were retained. The overlapping genes between significant signatures and the CEBPB targets were submitted to Enrichr128 for analysis against the Reactome_2022 library and those significantly enriched for ‘Innate Immune System R-HSA-168249’ with it appearing in the top three returned terms were retained. Additionally, overlapping genes appearing in at least half (n=5) of the retained signatures were extracted and included in the diagram.
Histopathology Assessment of Immune Subtypes
Data Pre-processing
Histopathology images are scanned at a maximum depth of 20x resolution and segmented into smaller tiles of 299 by 299 pixels with an overlapping area of 49 pixels between each tile, at the 10x, 5x, and 2.5x resolution and geographically linked such that the model always views tiles in the same spatial region. Tiles with excess white space (>60%) or previous annotations marked by pathologists were removed. To account for differences in staining procedures by different institutions, color normalization is performed using Vahadane’s method. Slides from the same patient were divided into training, validation, and test sets with 4-fold splitting. Histopathology images are cut into smaller tiles of 299 by 299 pixels with an overlapping area of 49 pixels between each tile. Tiles of 10x, 5x, and 2.5x resolutions from the same geographical region were fed as a multi-input set to output a single categorical outcome. To avoid overfitting, images were augmented with random 90◦ rotation, random vertical and horizontal flip, random change to hue, brightness, contrast, and saturation.
Model training
The multi-resolution architecture is based on Xception and modified to incorporate a global average pooling layer, dropout layer, and a fully dense predictive layer. The architecture is opened to include 3 branches, such that tiles of 10x, 5x, and 2.5x resolutions of the same region are forward passed together. Models were optimized with Adam, initial learning rate set to 0.001, and loss calculated using sparse categorical crossentropy weighted by tumor type frequency in the training set. Early stopping of training is employed when the validation loss does not improve for two epochs. Both Individual cancer models and a single pan-cancer model were trained to assess the extent that global morphologies correlate with immune infiltration across multiple tumor types. In addition, 4-fold validation was performed to accurately assess model performance.
Performance Evaluation and Model Visualization
Performance is evaluated with Receiver Operating Characteristic (ROC) curves at the per-tile and per-patient level. Metrics for per-patient level are obtained by averaging the respective tiles’ metrics belonging to that patient. Latent imaging features are extracted from the final convolution layer and visualized with tSNE clustering. To identify morphology patterns that were important for the model’s decision-making process, integrated-gradient based saliency maps were applied. The gradients of each class score were calculated with respect to the input layer, the magnitude of difference is used to generate a heatmap overlaying the original input image. Larger differences signify greater network activation and subsequently more important regions of interest.
Tumor Morphology features based on H&E
We devised a custom methodology for the segmentation and classification of neoplastic cells, employing a two-step procedure. Initially, cells were segmented at the whole-slide level utilizing the Cellpose cyto model.129 Subsequently, a straightforward Convolutional Neural Network (CNN) model was developed, which underwent training on the Pannuke dataset.130 This dataset was partitioned into an 80:10:10 ratio for training, validation, and testing purposes, respectively. The trained CNN model was then subjected to comprehensive evaluation on all cells identified by the Cellpose cyto model. The primary objective was to accurately discern neoplastic cells within this set. The Pannuke dataset encompasses five distinct cell types: neoplastic, inflammatory, connective, dead, and non-neoplastic epithelial cells. Tumor cells are defined as having >=50% probability for neoplastic class. Segmented masks encapsulating the contour and cellular morphology attributes of each cell were extracted, leveraging the skimage.measure.regionprops function. Features extracted include human-interpretable measurements (ex. area, perimeter, eccentricity) and other abstract features (ex. inertia, moments etc.). The Hematoxylin and Eosin (H&E) staining intensity was quantified by converting RGB color space to HED using skimage.color.rgb2hed and averaging the values at each channel level. These measurements were aggregated at the slide level, Pearson correlated with cytokine expression pathway scores at the patient level, and significance tested using scipy.stats.pearsonr, which performs a test of the null hypothesis that the underlying sample distributions are uncorrelated and normally distributed. These results are attached as a supplementary table (Table S7).
Supplementary Material
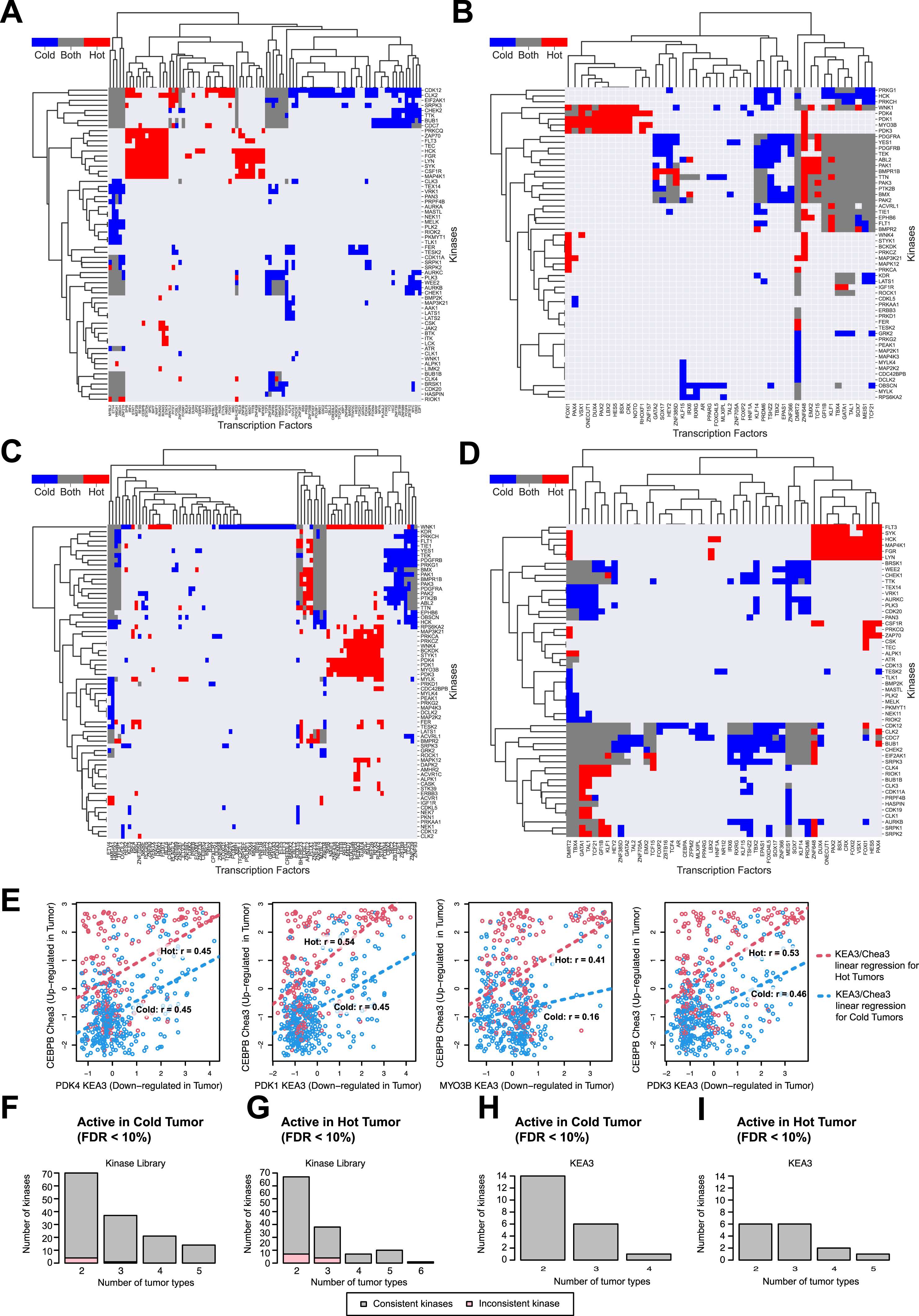{kind=link}
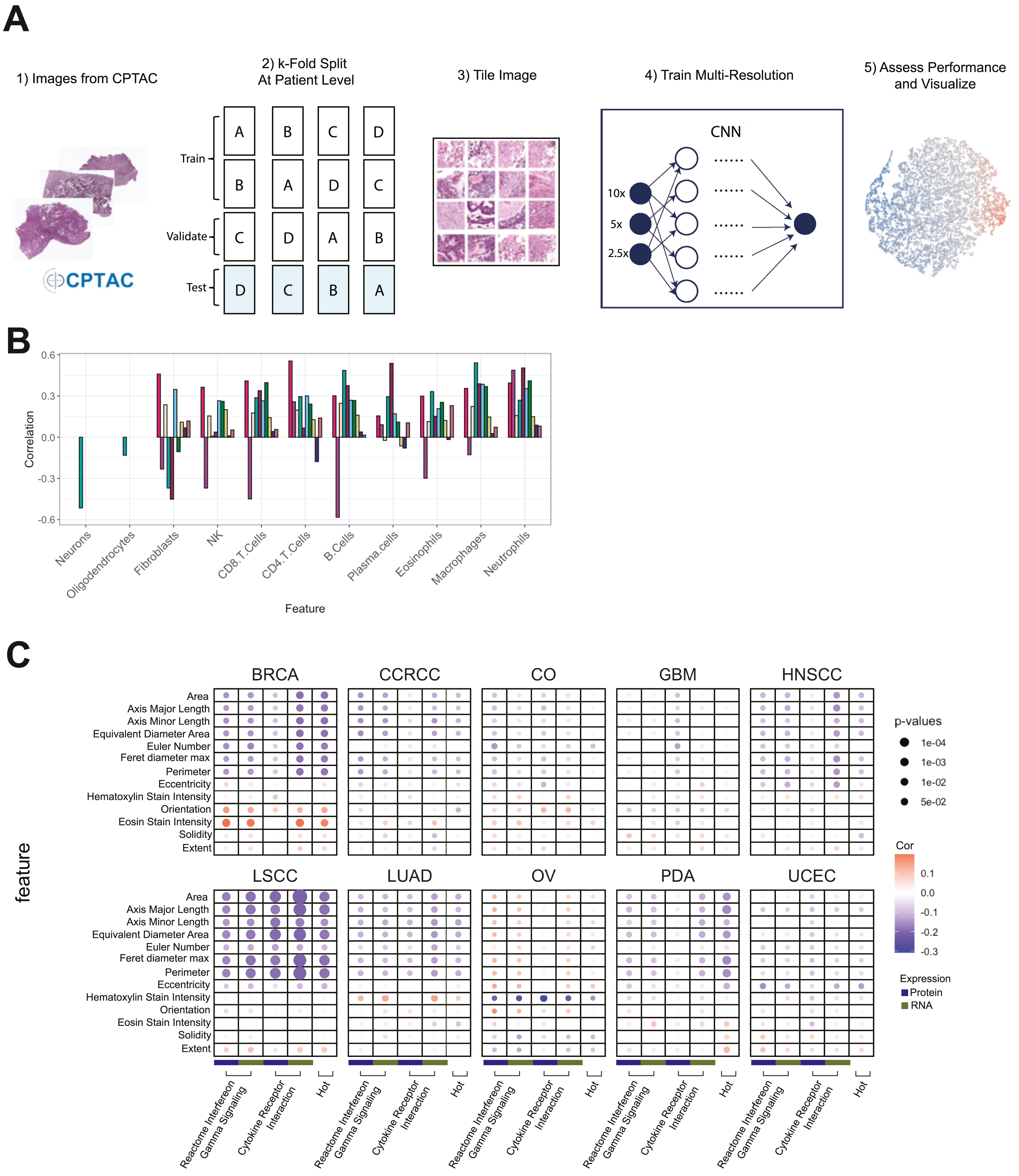{kind=link}
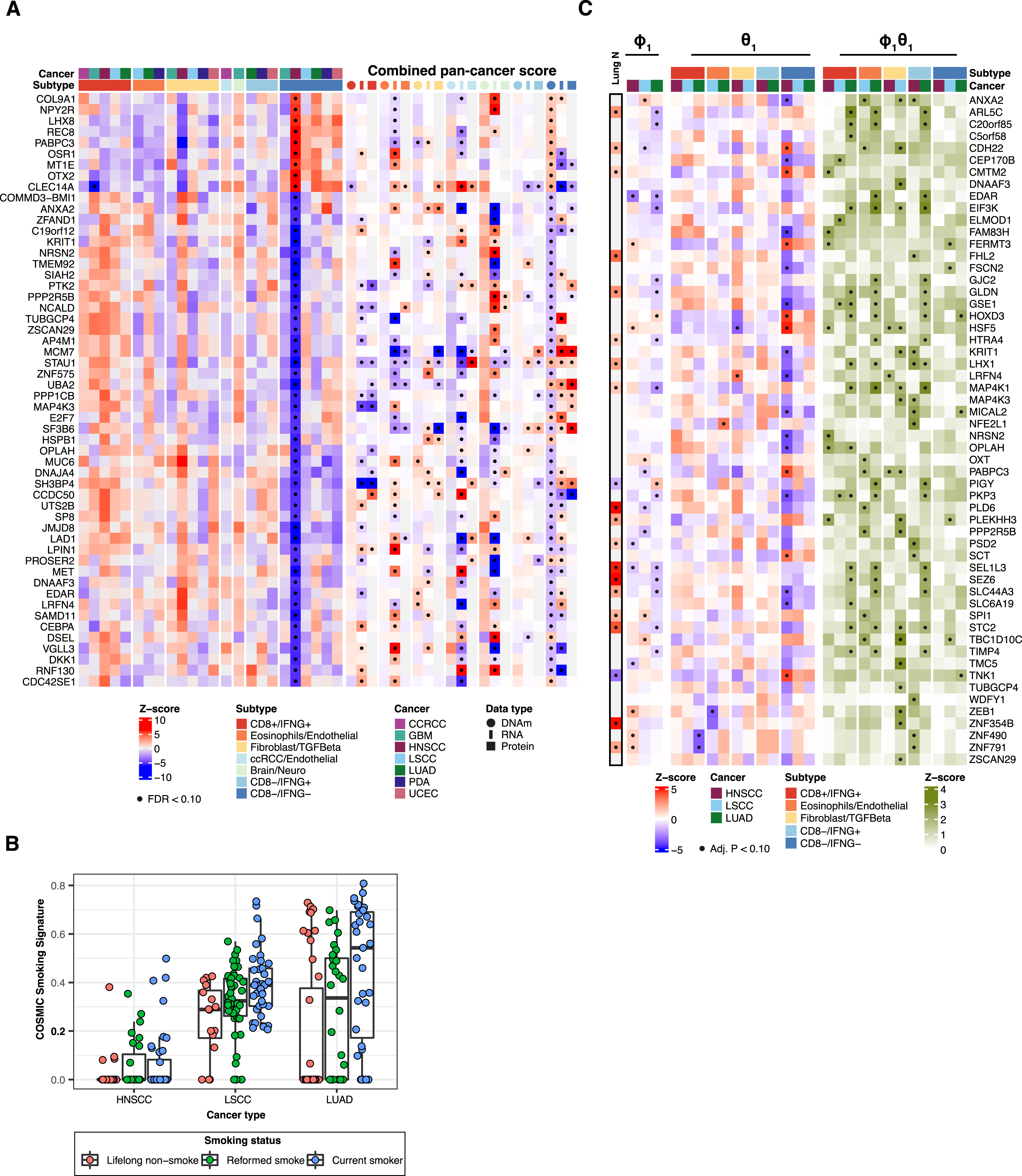{kind=link}
{kind=link}
{kind=link}
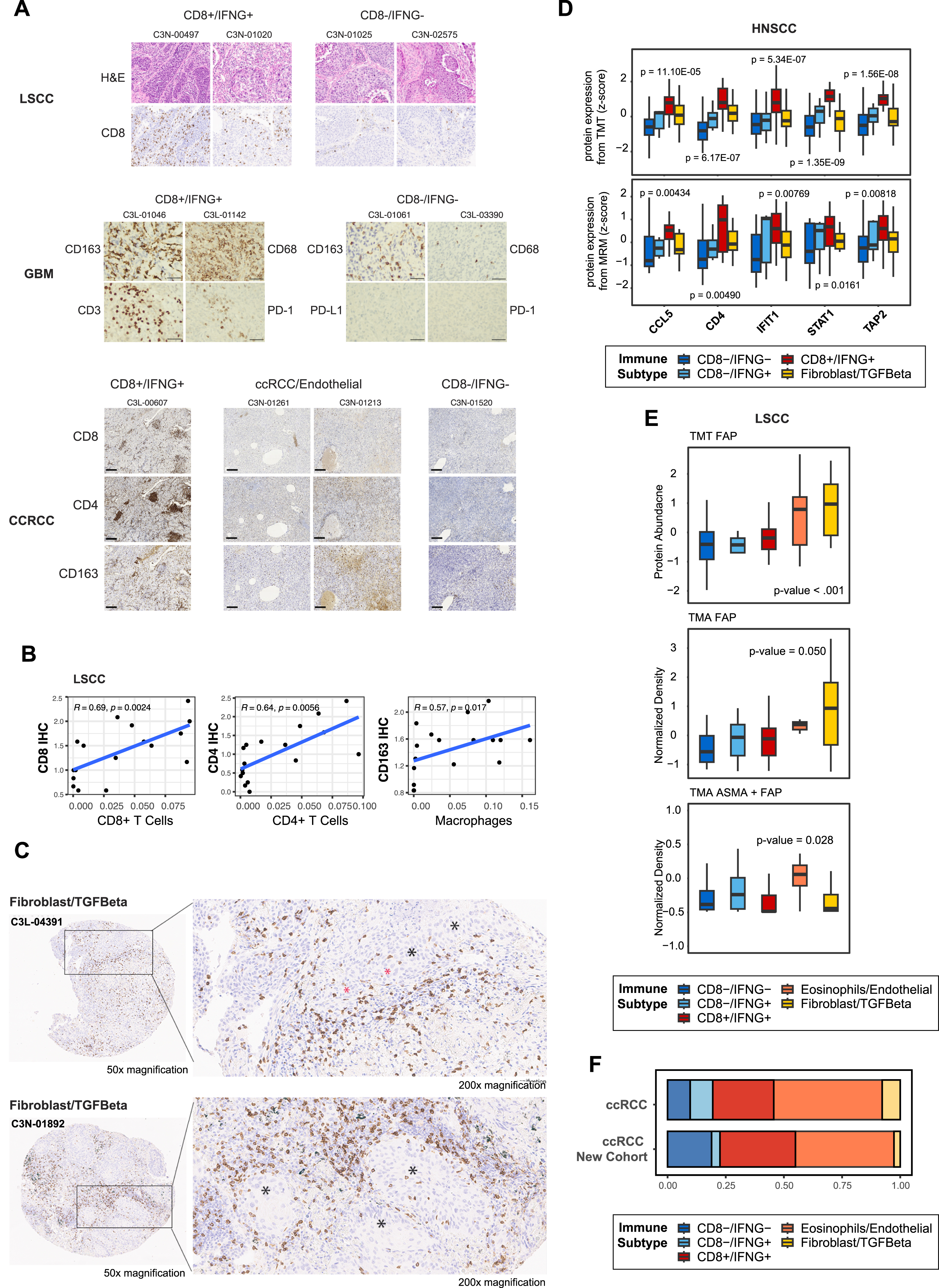{kind=link}
{kind=link}
KEY RESOURCES TABLE
Highlights.
Proteogenomics reveals seven immune subtypes spanning 10 cancer types
DNA alterations associate with immune subtypes and affect proteomic profiles
Kinase activation in immune subtypes suggests potential therapeutic targets
Digital pathology reveals infiltrating cells associated with immune subtypes
ACKNOWLEDGMENTS
The Clinical Proteomic Tumor Analysis Consortium (CPTAC) is supported by the National Cancer Institute of the National Institutes of Health under award numbers U24CA210955, U24CA210985, U24CA210986, U24CA210954, U24CA210967, U24CA210972, U24CA210979, U24CA210993, U01CA214114, U01CA214116, U01CA214125, U24CA271114, and U24CA270823. This project has also been funded in part with federal funds from the National Cancer Institute of the National Institutes of Health under contract no. HHSN261201500003I, task order no. HHSN26100064. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government. Additional funding support was provided by NIH awards U24CA224260, U24CA264250, R33CA263705, P30ES017885, T32GM136542, 1F30CA265288, and F30CA271622; HHMI Gilliam Fellowship GT15758; and Associazione Italiana Ricerca sul Cancro (AIRC) under IG 2018−ID. 21846 and AIRC 5 per Mille 2018−ID.21073. The Pacific Northwest National Laboratory is operated for the DOE by Battelle Memorial Institute under contract DE-AC05–76RL01830.
Footnotes
DECLARATION OF INTERESTS
R. Sebra is currently a paid consultant and equity holder at GeneDx. L.C.C. is a founder and member of the board of directors of Agios Pharmaceuticals; is a founder and receives research support from Petra Pharmaceuticals; has equity in and consults for Cell Signaling Technologies, Volastra, Larkspur, and 1 Base Pharmaceuticals; and consults for Loxo-Lilly. J.L.J. has received consulting fees from Scorpion Therapeutics and Volastra Therapeutics. T.M.Y. is a co-founder and stockholder of DeStroke.
DECLARATION OF GENERATIVE AI AND AI-ASSISTED TECHNOLOGIES IN THE WRITING PROCESS
During the preparation of this work the authors used ChatGPT to improve English grammar. After using this tool/service, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2024.01.027.
REFERENCE
- 1.Hanahan D, and Weinberg RA. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 2.Hiam-Galvez KJ, Allen BM, and Spitzer MH. (2021). Systemic immunity in cancer. Nat. Rev. Cancer 21, 345–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y, Dou Y, Da Veiga Leprevost F, Geffen Y, Calinawan AP, Aguet F, Akiyama Y, Anand S, Birger C, Cao S, et al. (2023). Proteogenomic data and resources for pan-cancer analysis. Cancer Cell 41, 1397–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng L, Qin S, Si W, Wang A, Xing B, Gao R, Ren X, Wang L, Wu X, Zhang J, et al. (2021). Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 374, abe6474. [DOI] [PubMed] [Google Scholar]
- 5.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, et al. (2018). The Immune Landscape of Cancer. Immunity 48, 812–830.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bagaev A, Kotlov N, Nomie K, Svekolkin V, Gafurov A, Isaeva O, Osokin N, Kozlov I, Frenkel F, Gancharova O, et al. (2021). Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 39, 845–865.e7. [DOI] [PubMed] [Google Scholar]
- 7.Petralia F, Krek A, Calinawan AP, Charytonowicz D, Sebra R, Feng S, Gosline S, Pugliese P, Paulovich A, Kennedy JJ, et al. (2023). BayesDeBulk: A Flexible Bayesian Algorithm for the Deconvolution of Bulk Tumor Data. Preprint at bioRxiv. 10.1101/2021.06.25.449763. [DOI] [Google Scholar]
- 8.Johnson JL, Yaron TM, Huntsman EM, Kerelsky A, Song J, Regev A, Lin TY, Liberatore K, Cizin DM, Cohen BM, et al. (2023). An atlas of substrate specificities for the human serine/threonine kinome. Nature 613, 759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuleshov MV, Xie Z, London ABK, Yang J, Evangelista JE, Lachmann A, Shu I, Torre D, and Ma’ayan A. (2021). KEA3: improved kinase enrichment analysis via data integration. Nucleic Acids Res. 49, W304–W316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hong R, Liu W, DeLair D, Razavian N, and Fenyo D. (2021). Predicting endometrial cancer subtypes and molecular features from histopathology images using multi-resolution deep learning models. Cell. Rep Med. 2, 100400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang JM, Hong R, Demicco EG, Tan J, Lazcano R, Moreira AL, Li Y, Calinawan A, Razavian N, Schraink T, et al. (2023). Deep learning integrates histopathology and proteogenomics at a pan-cancer level. Cell Rep. Med. 4, 101173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calinawan A, Ma W, Evangelista JE, Reva B, Petralia F, Ma’ayan A, and Wang P. (2021). CPTAC Pancancer Phosphoproteomics Kinase Enrichment Analysis with ProKAP Provides Insights into Immunogenic Signaling Pathways. Preprint at bioRxiv. 10.1101/2021.11.05.450069. [DOI] [Google Scholar]
- 13.Turhan B, Peradejordi IF, Chandrasekar S, Kalaycı S, Johnson J, Bouhaddou M, and Gümüş ZH. PhosNetVis: a web-based tool for kinase enrichment analyses and interactive 3D network visualizations of phosphoproteomics data. 10.48550/arXiv.2402.05016. [DOI] [Google Scholar]
- 14.Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE, Zhou JY, Petyuk VA, Chen L, Ray D, et al. (2016). Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 166, 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, et al. (2016). Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim S, et al. (2014). Proteogenomic characterization of human colon and rectal cancer. Nature 513, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clark DJ, Dhanasekaran SM, Petralia F, Pan J, Song X, Hu Y, da Veiga Leprevost F, Reva B, Lih TM, Chang HY, et al. (2019). Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 179, 964–983.e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vasaikar S, Huang C, Wang X, Petyuk VA, Savage SR, Wen B, Dou Y, Zhang Y, Shi Z, Arshad OA, et al. (2019). Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 177, 1035–1049.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dou Y, Kawaler EA, Cui Zhou D, Gritsenko MA, Huang C, Blumenberg L, Karpova A, Petyuk VA, Savage SR, Satpathy S, et al. (2020). Proteogenomic Characterization of Endometrial Carcinoma. Cell 180, 729–748.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gillette MA, Satpathy S, Cao S, Dhanasekaran SM, Vasaikar SV, Krug K, Petralia F, Li Y, Liang WW, Reva B, et al. (2020). Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 182, 200–225.e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu Y, Pan J, Shah P, Ao M, Thomas SN, Liu Y, Chen L, Schnaubelt M, Clark DJ, Rodriguez H, et al. (2020). Integrated Proteomic and Glycoproteomic Characterization of Human High-Grade Serous Ovarian Carcinoma. Cell Rep. 33, 108276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petralia F, Tignor N, Reva B, Koptyra M, Chowdhury S, Rykunov D, Krek A, Ma W, Zhu Y, Ji J, et al. (2020). Integrated Proteogenomic Characterization across Major Histological Types of Pediatric Brain Cancer. Cell 183, 1962–1985.e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krug K, Jaehnig EJ, Satpathy S, Blumenberg L, Karpova A, Anurag M, Miles G, Mertins P, Geffen Y, Tang LC, et al. (2020). Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell 183, 1436–1456.e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang LB, Karpova A, Gritsenko MA, Kyle JE, Cao S, Li Y, Rykunov D, Colaprico A, Rothstein JH, Hong R, et al. (2021). Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 39, 509–528.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang C, Chen L, Savage SR, Eguez RV, Dou Y, Li Y, da Veiga Leprevost F, Jaehnig EJ, Lei JT, Wen B, et al. (2021). Proteogenomic insights into the biology and treatment of HPV-negative head and neck squamous cell carcinoma. Cancer Cell 39, 361–379.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Satpathy S, Krug K, Jean Beltran PM, Savage SR, Petralia F, Kumar-Sinha C, Dou Y, Reva B, Kane MH, Avanessian SC, et al. (2021). A proteogenomic portrait of lung squamous cell carcinoma. Cell 184, 4348–4371.e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao L, Huang C, Cui Zhou D, Hu Y, Lih TM, Savage SR, Krug K, Clark DJ, Schnaubelt M, Chen L, et al. (2021). Proteogenomic characterization of pancreatic ductal adenocarcinoma. Cell 184, 5031–5052.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abdelfattah N, Kumar P, Wang C, Leu JS, Flynn WF, Gao R, Baskin DS, Pichumani K, Ijare OB, Wood SL, et al. (2022). Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nat. Commun. 13, 767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ando A, Hashimoto N, Sakamoto K, Omote N, Miyazaki S, Nakahara Y, Imaizumi K, Kawabe T, and Hasegawa Y. (2019). Repressive role of stabilized hypoxia inducible factor 1alpha expression on transforming growth factor beta-induced extracellular matrix production in lung cancer cells. Cancer Sci. 110, 1959–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, Gadgeel SM, Hida T, Kowalski DM, Dols MC, et al. (2017). Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 389, 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marx N, Mach F, Sauty A, Leung JH, Sarafi MN, Ransohoff RM, Libby P, Plutzky J, and Luster AD. (2000). Peroxisome proliferator-activated receptor-gamma activators inhibit IFN-gamma-induced expression of the T cell-active CXC chemokines IP-10, Mig, and I-TAC in human endothelial cells. J. Immunol. 164, 6503–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Le Menn G, and Neels JG. (2018). Regulation of Immune Cell Function by PPARs and the Connection with Metabolic and Neurodegenerative Diseases. Int. J. Mol. Sci. 19, 1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patil NS, Nabet BY, Müller S, Koeppen H, Zou W, Giltnane J, Au-Yeung A, Srivats S, Cheng JH, Takahashi C, et al. (2022). Intratumoral plasma cells predict outcomes to PD-L1 blockade in non-small cell lung cancer. Cancer Cell 40, 289–300.e4. [DOI] [PubMed] [Google Scholar]
- 34.Li Y, Lih TM, Dhanasekaran SM, Mannan R, Chen L, Cieslik M, Wu Y, Lu RJ, Clark DJ, Ko1odziejczak I, et al. (2023). Histopathologic and proteogenomic heterogeneity reveals features of clear cell renal cell carcinoma aggressiveness. Cancer Cell 41, 139–163.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Gonçalves E, Barthorpe S, Lightfoot H, et al. (2016). A Landscape of Pharmacogenomic Interactions in Cancer. Cell 166, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Z, Ding B, Xu J, Mao K, Zhang P, and Xue Q. (2020). Relevance of STK11 Mutations Regarding Immune Cell Infiltration, Drug Sensitivity, and Cellular Processes in Lung Adenocarcinoma. Front. Oncol. 10, 580027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Friedhoff J, Schneider F, Jurcic C, Endris V, Kirchner M, Sun A, Bolnavu I, Pohl L, Teroerde M, Kippenberger M, et al. (2023). BAP1 and PTEN mutations shape the immunological landscape of clear cell renal cell carcinoma and reveal the intertumoral heterogeneity of T cell suppression: a proof-of-concept study. Cancer Immunol. Immunother. 72, 1603–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Masclef L, Ahmed O, Estavoyer B, Larrivée B, Labrecque N, Nijnik A, and Affar EB. (2021). Roles and mechanisms of BAP1 deubiquitinase in tumor suppression. Cell Death Differ. 28, 606–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rooney MS, Shukla SA, Wu CJ, Getz G, and Hacohen N. (2015). Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu X, Jiang D, Liu H, Lu X, Lv D, and Liang L. (2021). CD8(+) T Cell-Based Molecular Classification With Heterogeneous Immunogenomic Landscapes and Clinical Significance of Clear Cell Renal Cell Carcinoma. Front. Immunol. 12, 745945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han G, Yang G, Hao D, Lu Y, Thein K, Simpson BS, Chen J, Sun R, Alhalabi O, Wang R, et al. (2021). 9p21 loss confers a cold tumor immune microenvironment and primary resistance to immune checkpoint therapy. Nat. Commun. 12, 5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Esai Selvan M, Onel K, Gnjatic S, Klein RJ, and Gümüş ZH. (2023). Germline rare deleterious variant load alters cancer risk, age of onset and tumor characteristics. NPJ Precis. Oncol. 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sayaman RW, Saad M, Thorsson V, Hu D, Hendrickx W, Roelands J, Porta-Pardo E, Mokrab Y, Farshidfar F, Kirchhoff T, et al. (2021). Germline genetic contribution to the immune landscape of cancer. Immunity 54, 367–386.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Esai Selvan M, Zauderer MG, Rudin CM, Jones S, Mukherjee S, Offit K, Onel K, Rennert G, Velculescu VE, Lipkin SM, et al. (2020). Inherited Rare, Deleterious Variants in ATM Increase Lung Adenocarcinoma Risk. J. Thorac. Oncol. 15, 1871–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Esai Selvan M, Klein RJ, and Gümüş ZH. (2019). Rare, Pathogenic Germline Variants in Fanconi Anemia Genes Increase Risk for Squamous Lung Cancer. Clin. Cancer Res. 25, 1517–1525. [DOI] [PubMed] [Google Scholar]
- 46.Klein RJ, and Gümüş ZH. (2022). Are polygenic risk scores ready for the cancer clinic?-a perspective. Transl. Lung Cancer Res. 11, 910–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, Totoki Y, Fujimoto A, Nakagawa H, Shibata T, et al. (2016). Mutational signatures associated with tobacco smoking in human cancer. Science 354, 618–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bakulski KM, Dou J, Lin N, London SJ, and Colacino JA. (2019). DNA methylation signature of smoking in lung cancer is enriched for exposure signatures in newborn and adult blood. Sci. Rep. 9, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, Guan W, Xu T, Elks CE, Aslibekyan S, et al. (2016). Epigenetic Signatures of Cigarette Smoking. Circ. Cardiovasc. Genet. 9, 436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shui IM, Wong CJ, Zhao S, Kolb S, Ebot EM, Geybels MS, Rubicz R, Wright JL, Lin DW, Klotzle B, et al. (2016). Prostate tumor DNA methylation is associated with cigarette smoking and adverse prostate cancer outcomes. Cancer 122, 2168–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Teschendorff AE, Yang Z, Wong A, Pipinikas CP, Jiao Y, Jones A, Anjum S, Hardy R, Salvesen HB, Thirlwell C, et al. (2015). Correlation of Smoking-Associated DNA Methylation Changes in Buccal Cells With DNA Methylation Changes in Epithelial Cancer. JAMA Oncol. 1, 476–485. [DOI] [PubMed] [Google Scholar]
- 52.Yu C, Jordahl KM, Bassett JK, Joo JE, Wong EM, Brinkman MT, Schmidt DF, Bolton DM, Makalic E, Brasky TM, et al. (2021). Smoking Methylation Marks for Prediction of Urothelial Cancer Risk. Cancer Epidemiol. Biomarkers Prev. 30, 2197–2206. [DOI] [PubMed] [Google Scholar]
- 53.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, et al. (2020). The repertoire of mutational signatures in human cancer. Nature 578, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wei X, Zhang X, Wang S, Wang Y, Ji C, Yao L, and Song N. (2022). PYCR1 regulates glutamine metabolism to construct an immune-suppressive microenvironment for the progression of clear cell renal cell carcinoma. Am. J. Cancer Res. 12, 3780–3798. [PMC free article] [PubMed] [Google Scholar]
- 55.Sun Y, Revach OY, Anderson S, Kessler EA, Wolfe CH, Jenney A, Mills CE, Robitschek EJ, Davis TGR, Kim S, et al. (2023). Targeting TBK1 to overcome resistance to cancer immunotherapy. Nature 615, 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Manning BD, and Toker A. (2017). AKT/PKB Signaling: Navigating the Network. Cell 169, 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Revathidevi S, and Munirajan AK. (2019). Akt in cancer: Mediator and more. Semin. Cancer Biol. 59, 80–91. [DOI] [PubMed] [Google Scholar]
- 58.O’Donnell JS, Massi D, Teng MWL, and Mandala M. (2018). PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin. Cancer Biol. 48, 91–103. [DOI] [PubMed] [Google Scholar]
- 59.Johnson P, and Cross JL. (2009). Tyrosine phosphorylation in immune cells: direct and indirect effects on toll-like receptor-induced proinflammatory cytokine production. Crit. Rev. Immunol. 29, 347–367. [DOI] [PubMed] [Google Scholar]
- 60.Kolanus W, Romeo C, and Seed B. (1993). T cell activation by clustered tyrosine kinases. Cell 74, 171–183. [DOI] [PubMed] [Google Scholar]
- 61.Yap TA, Tan DSP, Terbuch A, Caldwell R, Guo C, Goh BC, Heong V, Haris NRM, Bashir S, Drew Y, et al. (2021). First-in-Human Trial of the Oral Ataxia Telangiectasia and RAD3-Related (ATR) Inhibitor BAY 1895344 in Patients with Advanced Solid Tumors. Cancer Discov. 11, 80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Keenan AB, Torre D, Lachmann A, Leong AK, Wojciechowicz ML, Utti V, Jagodnik KM, Kropiwnicki E, Wang Z, and Ma’ayan A. (2019). ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 47, W212–W224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Evangelista JE, Clarke DJB, Xie Z, Lachmann A, Jeon M, Chen K, Jagodnik KM, Jenkins SL, Kuleshov MV, Wojciechowicz ML, et al. (2022). SigCom LINCS: data and metadata search engine for a million gene expression signatures. Nucleic Acids Res. 50, W697–W709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Charytonowicz D, Brody R, and Sebra R. (2023). Interpretable and context-free deconvolution of multi-scale whole transcriptomic data with UniCell deconvolve. Nat. Commun. 14, 1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Comba A, Dunn PJ, Argento AE, Kadiyala P, Ventosa M, Patel P, Zamler DB, Núñez FJ, Zhao L, Castro MG, et al. (2020). Fyn tyrosine kinase, a downstream target of receptor tyrosine kinases, modulates antiglioma immune responses. Neuro. Oncol 22, 806–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Comba A, Dunn PJ, Argento AE, Kadiyala P, Ventosa M, Patel P, Zamler DB, Nunez FJ, Zhao L, Castro MG, et al. (2019). TMIC-62. FYN, an effector of oncogenic receptor tyrosine kinases signaling in glioblastoma, inhibits anti-glioma immune responses: implications for immunotherapy. Neuro-Oncology 21. vi261–vi261. [Google Scholar]
- 67.Webb ER, Dodd GL, Noskova M, Bullock E, Muir M, Frame MC, Serrels A, and Brunton VG. (2023). Kindlin-1 regulates IL-6 secretion and modulates the immune environment in breast cancer models. eLife 12, 7554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Serrels A, Lund T, Serrels B, Byron A, McPherson RC, von Kriegsheim A, Gómez-Cuadrado L, Canel M, Muir M, Ring JE, et al. (2015). Nuclear FAK controls chemokine transcription, Tregs, and evasion of anti-tumor immunity. Cell 163, 160–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roelands J, Hendrickx W, Zoppoli G, Mall R, Saad M, Halliwill K, Curigliano G, Rinchai D, Decock J, Delogu LG, et al. (2020). Oncogenic states dictate the prognostic and predictive connotations of intratumoral immune response. J. Immunother. Cancer 8, 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zaitsev A, Chelushkin M, Dyikanov D, Cheremushkin I, Shpak B, Nomie K, Zyrin V, Nuzhdina E, Lozinsky Y, Zotova A, et al. (2022). Precise reconstruction of the TME using bulk RNA-seq and a machine learning algorithm trained on artificial transcriptomes. Cancer Cell 40, 879–894.e16. [DOI] [PubMed] [Google Scholar]
- 72.Venkataramani V, Yang Y, Schubert MC, Reyhan E, Tetzlaff SK, Wißmann N, Botz M, Soyka SJ, Beretta CA, Pramatarov RL, et al. (2022). Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell 185, 2899–2917.e31. [DOI] [PubMed] [Google Scholar]
- 73.Sobel R, Zabelle S, and Scharf SM. (1987). Increased prevalence of strongly positive tuberculin skin reactions in children from a desert agricultural community in Israel. Pediatr. Infect. Dis. J. 6, 766–768. [PubMed] [Google Scholar]
- 74.Monteiro FSM, Soares A, Rizzo A, Santoni M, Mollica V, Grande E, and Massari F. (2023). The role of immune checkpoint inhibitors (ICI) as adjuvant treatment in renal cell carcinoma (RCC): A systematic review and meta-analysis. Clin. Genitourin. Cancer 21, 324–333. [DOI] [PubMed] [Google Scholar]
- 75.Hu W, Liu H, Li Z, Liu J, and Chen L. (2022). Impact of molecular and clinical variables on survival outcome with immunotherapy for glioblastoma patients: A systematic review and meta-analysis. CNS Neurosci. Ther. 28, 1476–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, Schrock AB, Hartmaier RJ, Trabucco SE, Gay L, et al. (2018). STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 8, 822–835. 2159–8290.CD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Negrao MV, Lam VK, Reuben A, Rubin ML, Landry LL, Roarty EB, Rinsurongkawong W, Lewis J, Roth JA, Swisher SG, et al. (2019). PD-L1 Expression, Tumor Mutational Burden, and Cancer Gene Mutations Are Stronger Predictors of Benefit from Immune Checkpoint Blockade than HLA Class I Genotype in Non-Small Cell Lung Cancer. J. Thorac. Oncol. 14, 1021–1031. [DOI] [PubMed] [Google Scholar]
- 78.Malhotra J, Ryan B, Patel M, Chan N, Guo Y, Aisner J, Jabbour SK, and Pine S. (2022). Clinical outcomes and immune phenotypes associated with STK11 co-occurring mutations in non-small cell lung cancer. J. Thorac. Dis. 14, 1772–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Atas E, Oberhuber M, and Kenner L. (2020). The Implications of PDK1–4 on Tumor Energy Metabolism, Aggressiveness and Therapy Resistance. Front. Oncol. 10, 583217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Berginski ME, Moret N, Liu C, Goldfarb D, Sorger PK, and Gomez SM. (2021). The Dark Kinase Knowledgebase: an online compendium of knowledge and experimental results of understudied kinases. Nucleic Acids Res. 49, D529–D535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yi X, Liao Y, Wen B, Li K, Dou Y, Savage SR, and Zhang B. (2021). caAtlas: An immunopeptidome atlas of human cancer. iScience 24, 103107. 10.1016/j.isci.2021.103107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li Y, Porta-Pardo E, Tokheim C, Bailey MH, Yaron TM, Stathias V, Geffen Y, Imbach KJ, Cao S, Anand S, et al. (2023). Pan-cancer proteogenomics connects oncogenic drivers to functional states. Cell 186, 3921–3944.e25. 10.1016/j.cell.2023.07.014. [DOI] [PubMed] [Google Scholar]
- 83.Ma W, Kim S, Chowdhury S, Li Z, Yang M, Yoo S, Petralia F, Jacobsen J, Li JJ, Ge X, et al. (2021). DreamAI: algorithm for the imputation of proteomics data. Preprint at bioRxiv. 10.1101/2020.07.21.214205. [DOI] [Google Scholar]
- 84.Frankish A, Carbonell-Sala S, Diekhans M, Jungreis I, Loveland JE, Mudge JM, Sisu C, Wright JC, Arnan C, Barnes I, et al. (2023). GENCODE: reference annotation for the human and mouse genomes in 2023. Nucleic Acids Res. 51, D942–D949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Benjamini Y, and Hochberg Y. (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B Methodol. 57, 289–300. [Google Scholar]
- 86.Pino LK, Searle BC, Bollinger JG, Nunn B, MacLean B, and Mac-Coss MJ. (2020). The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 39, 229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jang HJ, Hostetter G, Macfarlane AW, Madaj Z, Ross EA, Hinoue T, Kulchycki JR, Burgos RS, Tafseer M, Alpaugh RK, et al. (2023). A Phase II Trial of Guadecitabine plus Atezolizumab in Metastatic Urothelial Carcinoma Progressing after Initial Immune Checkpoint Inhibitor Therapy. Clin. Cancer Res. 29, 2052–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Clark NR, Hu KS, Feldmann AS, Kou Y, Chen EY, Duan Q, and Ma’ayan A. (2014). The characteristic direction: a geometrical approach to identify differentially expressed genes. BMC Bioinformatics 15, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Caruso FP, Garofano L, D’Angelo F, Yu K, Tang F, Yuan J, Zhang J, Cerulo L, Pagnotta SM, Bedognetti D, et al. (2020). A map of tumor-host interactions in glioma at single-cell resolution. Giga-Science 9, giaa109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, and Mesirov JP. (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen B, Khodadoust MS, Liu CL, Newman AM, and Alizadeh AA. (2018). Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 1711, 243–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, Sidiropoulos K, Cook J, Gillespie M, Haw R, et al. (2020). The reactome pathway knowledgebase. Nucleic Acids Res. 48, D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Frattini V, Pagnotta SM, Tala, Fan JJ, Russo MV, Lee SB, Garofano L, Zhang J, Shi P, Lewis G, et al. (2018). A metabolic function of FGFR3-TACC3 gene fusions in cancer. Nature 553, 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Abadi M, Barham P, Chen J, Chen Z, Davis A, Dean J, Devin M, Ghemawat S, Irving G, Isard M, et al. (2016). TensorFlow: a system for large-scale machine learning. Proceedings of the 12th USENIX conference on Operating Systems Design and Implementation (USENIX Association), 265–283. [Google Scholar]
- 96.Szegedy C, Vanhoucke V, Ioffe S, Shlens J, and Wojna Z. (2016). Rethinking the Inception Architecture for Computer Vision. 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV, USA, 2016, pp. 2818–2826. 10.1109/CVPR.2016.308. [DOI] [Google Scholar]
- 97.Liu Y, and Xie J. (2020). Cauchy combination test: a powerful test with analytic p-value calculation under arbitrary dependency structures. J. Am. Stat. Assoc. 115, 393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gibbs DL. (2020). Robust classification of Immune Subtypes in Cancer. Preprint at bioRxiv. 10.1101/2020.01.17.910950. [DOI] [Google Scholar]
- 99.Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, and Nesvizhskii AI. (2017). MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 14, 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.da Veiga Leprevost F, Haynes SE, Avtonomov DM, Chang HY, Shanmugam AK, Mellacheruvu D, Kong AT, and Nesvizhskii AI. (2020). Philosopher: a versatile toolkit for shotgun proteomics data analysis. Nat. Methods 17, 869–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Djomehri SI, Gonzalez ME, da Veiga Leprevost F, Tekula SR, Chang HY, White MJ, Cimino-Mathews A, Burman B, Basrur V, Argani P, et al. (2020). Quantitative proteomic landscape of metaplastic breast carcinoma pathological subtypes and their relationship to triple-negative tumors. Nat. Commun. 11, 1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Johnson WE, Li C, and Rabinovic A. (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- 103.Kennedy JJ, Whiteaker JR, Schoenherr RM, Yan P, Allison K, Shipley M, Lerch M, Hoofnagle AN, Baird GS, and Paulovich AG. (2016). Optimized Protocol for Quantitative Multiple Reaction Monitoring-Based Proteomic Analysis of Formalin-Fixed, Paraffin-Embedded Tissues. J. Proteome Res. 15, 2717–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Whiteaker JR, Lundeen RA, Zhao L, Schoenherr RM, Burian A, Huang D, Voytovich U, Wang T, Kennedy JJ, Ivey RG, et al. (2021). Targeted Mass Spectrometry Enables Multiplexed Quantification of Immunomodulatory Proteins in Clinical Biospecimens. Front. Immunol. 12, 765898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ. (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Topper MJ, Anagnostou V, Marrone KA, Velculescu VE, Jones PA, Brahmer JR, Baylin SB, and Hostetter GH. (2023). Derivation of CD8+ T cell infiltration potentiators in non-small-cell lung cancer through tumor microenvironment analysis. iScience 26, 107095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Parra ER, Jiang M, Solis L, Mino B, Laberiano C, Hernandez S, Gite S, Verma A, Tetzlaff M, Haymaker C, et al. (2020). Procedural Requirements and Recommendations for Multiplex Immunofluorescence Tyramide Signal Amplification Assays to Support Translational Oncology Studies. Cancers (Basel) 12, 12020255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, et al. (2013). Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Petralia F, Wang L, Peng J, Yan A, Zhu J, and Wang P. (2018). A new method for constructing tumor specific gene co-expression networks based on samples with tumor purity heterogeneity. Bioinformatics 34, i528–i536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wilkerson MD, and Hayes DN. (2010). ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xu T, Le TD, Liu L, Su N, Wang R, Sun B, Colaprico A, Bontempi G, and Li J. (2017). CancerSubtypes: an R/Bioconductor package for molecular cancer subtype identification, validation and visualization. Bioinformatics 33, 3131–3133. [DOI] [PubMed] [Google Scholar]
- 112.Kanehisa M, and Goto S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P. (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hänzelmann S, Castelo R, and Guinney J. (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tibshirani R, Hastie T, Narasimhan B, and Chu G. (2002). Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl. Acad. Sci. USA 99, 6567–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Friedman J, Hastie T, and Tibshirani R. (2010). Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 33, 1–22. [PMC free article] [PubMed] [Google Scholar]
- 117.Yu G, Wang LG, Han Y, and He QY. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shabalin AA. (2012). Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics 28, 1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Stegle O, Parts L, Piipari M, Winn J, and Durbin R. (2012). Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc. 7, 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hastie T, Tibshirani R, Narasimhan B, and Chu G. (2021). impute: impute: Imputation for microarray data. R package version 1.66.0. https://bioconductor.org/packages/release/bioc/manuals/impute/man/impute.pdf. [Google Scholar]
- 121.Nazemalhosseini Mojarad E, Kuppen PJ, Aghdaei HA, and Zali MR. (2013). The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterol. Hepatol. Bed Bench 6, 120–128. [PMC free article] [PubMed] [Google Scholar]
- 122.Malta TM, de Souza CF, Sabedot TS, Silva TC, Mosella MS, Kalkanis SN, Snyder J, Castro AVB, and Noushmehr H. (2018). Glioma CpG island methylator phenotype (G-CIMP): biological and clinical implications. Neuro. Oncol 20, 608–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Teodoridis JM, Hardie C, and Brown R. (2008). CpG island methylator phenotype (CIMP) in cancer: causes and implications. Cancer Lett. 268, 177–186. [DOI] [PubMed] [Google Scholar]
- 124.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liu Z, Shen J, Barfield R, Schwartz J, Baccarelli AA, and Lin X. (2022). Large-Scale Hypothesis Testing for Causal Mediation Effects with Applications in Genome-wide Epigenetic Studies. J. Am. Stat. Assoc. 117, 67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK, et al. (2017). A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 171, 1437–1452.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A. (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Stringer C, Wang T, Michaelos M, and Pachitariu M. (2021). Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods 18, 100–106. [DOI] [PubMed] [Google Scholar]
- 130.Gamper J, Koohbanani NA, Benes K, Graham S, Jahanifar M, Khurram SA, Azam A, Hewitt K, and Rajpoot N. (2020). PanNuke Dataset Extension, Insights and Baselines. Preprint at arXiv. 10.48550/arXiv.2003.10778. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw proteomics data of the CPTAC Pan-Cancer cohort can be accessed via Proteomic Data Commons (PDC) at https://pdc.cancer.gov.
Raw genomics and transcriptomics files of the CPTAC Pan Cancer cohort are publically available via the Genomic Data Commons (GDC) Data Portal at https://portal.gdc.cancer.gov.
Processed genomic data with access control can be obtained via CDS through the NCI DAC approved, dbGaP compiled whitelists. Users can access the data for analysis through the Seven Bridges Cancer Genomics Cloud (SB-CGC) which is one of the NCI-funded Cloud Resource/platform for compute intensive analysis. Instructions for data access are as follows:
Create an account on CGC, Seven Bridges at https://cgc-accounts.sbgenomics.com/auth/register
Get approval from dbGaP to access the controlled study (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001287.v16.p6 )
Log into CGC to access Cancer Data Service (CDS) File Explore
Copy data into your own space and start analysis and exploration
Visit the CDS page on CGC to see what studies are available and instructions and guides to use the resources. (https://docs.cancergenomicscloud.org/page/cds-data)
Processed data without access control can be found at https://pdc.cancer.gov/pdc/cptac-pancancer. File names for different omics used in this paper are as follows:
Proteomic data: Proteome_UMich_SinaiPreprocessed_GENECODE34_v1.zip
Phosphoproteomic data: Phosphoproteome_UMich_SinaiPreprocessed_GENECODE34_v1.zip
RNAseq data: RNA_WashU_v1.zip
Methylation data: Methylation_MSSM_v1.zip
Mutation profile: PanCan_Union_Maf_Broad_WashU_v1.1.maf
CNV data: CNV_WGS_WashU_v1.zip. Note, WGS-based CNV data was obtained using the pipeline at https://github.com/ding-lab/BICSEQ2. In addition, for OV, CO and BR cancers, WGS data was not available; and CNV calling derived from the WXS data (CNV_WashU_v1.zip) was instead utilized. https://pdc.cancer.gov/pdc/cptac-pancancer
In this paper, we considered samples for which both RNAseq and proteomic data were measured. The full list of samples can be found in Table S1. All analysis results reported in this manuscript can be found in the supplementary tables. These results include cell type composition estimates, immune subtype labels, associations between DNA aberrations (i.e., mutation, CNV, WGS germline and methylation data) and immune subtypes, kinase activation in different immune subtypes, tumor cell-specific kinase activation inferences and histopathology assessment of immune subtypes.
All (inferred) kinase and transcript factor activity scores, as well as the clinical meta information of the cohort can be queried, visualized, and downloaded from an interactive ProKAP12 data portal: http://prokap.wanglab.cloud . Complete Pan Cancer kinase and transcription factor activity score tables can also be downloaded from https://pdc.cancer.gov/pdc/cptac-pancancer. Selected kinase and phosphosite regulatory networks can be queried, visualized, and downloaded from an interactive PhosNetVis13 data portal: https://gumuslab.github.io/PhosNetVis/cptac-vis.html.
Links to the original codes are listed in the key resources table.
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.