

Summary
The composition of the human microbiome profoundly impacts human well-being. However, the mechanisms underlying microbiome maturation are poorly understood. The nasal microbiome is of particular importance as a source of many respiratory infections. Here, we performed a large sequencing and culture-based analysis of the human nasal microbiota from different age groups. We observed a significant decline of pathogenic bacteria before adulthood, with an increase of the commensal Staphylococcus epidermidis. In seniors, this effect was partially reversed. Mechanistically, many S. epidermidis isolates stimulated nasal epithelia in vitro to produce antimicrobial peptides, killing pathogenic competitors. S. epidermidis itself proved highly resistant, owing to its exceptional capacity to form biofilms. We further confirm that high peptide-producing and biofilm-forming S. epidermidis strains can outcompete pathogenic bacteria in vivo. Our study identifies a pivotal role of S. epidermidis in healthy maturation of nasal microbiome, at least in part by symbiotic cooperation with innate host defense.
Keywords: Microbiome, nose, innate immunity, antimicrobial peptides, immune evasion, biofilm, Staphylococcus epidermidis
Introduction
Over the last decade, the human microbiome has received much attention and the importance of microbiome composition for human health has become abundantly clear. Many studies have reported correlations between microbiome composition and various physiological characteristics and disease manifestations (Chen et al., 2018; Clemente et al., 2012; Gilbert et al., 2016). Importantly, the colonizing microbiota of human epithelia, which include those of the intestine, the skin, and the nose, are subject to control by mucosal immune defense mechanisms (Allaire et al., 2018; Chen et al., 2018; Hariri and Cohen, 2016) and the composition of those microbial communities has been shown to change considerably during immune maturation in early life (Bomar et al., 2018; Capone et al., 2011; Mika et al., 2015; Stewart et al., 2018; Yatsunenko et al., 2012). Furthermore, lack of microbiome maturation has been associated with disease (Bjorksten et al., 2001; Stokholm et al., 2018). However, the mechanisms that lead to the development of a heathy microbiome are poorly understood.
In comparison to the gut and skin microbiomes, that of the human nose has remained understudied. Yet, the nasal microbiota are of particular concern as the nostrils may harbor pathogens, such as Staphylococcus aureus or Moraxella catarrhalis, that can mount severe respiratory and associated systemic infections (Krismer et al., 2017; Murphy and Parameswaran, 2009; Pettigrew et al., 2012; Rawlings et al., 2013). While many studies have determined the nasal microbiome in populations of a specific age, mostly children, changes in the nasal microbiome in direct comparison of individuals from all periods of a human life have not yet been investigated (Bomar et al., 2018). Only one study investigated microbiomes of the skin and nares in children and young adults, emphasizing significant changes in the nasal microbiome during puberty, but was quite limited regarding the number of analyzed participants (total, n=28) (Oh et al., 2012). Furthermore, several studies associated presence of specific microbial taxa with respiratory diseases (Bomar et al., 2018; Bosch et al., 2017; Hasegawa et al., 2016; Teo et al., 2015). Generally, however, previous studies addressing the nasal microbiome were limited to 16S rRNA sequencing and thus mostly to differences between larger-order taxa, essentially precluding species-specific conclusions (Bomar et al., 2018).
A major genus present in the nose and often associated with nasal microbiome changes is the genus Staphylococcus (Bomar et al., 2018). This genus includes the important human pathogen S. aureus, nasal colonization with which in about one fourth of the population is associated with disease development (von Eiff et al., 2001; Wertheim et al., 2005). Several studies have specifically focused on correlations between nasal carriage of S. aureus and that of other organisms, as addressed by culture-based analysis of S. aureus growth combined with 16S rRNA-sequencing based determination of the nasal microbiome (Liu et al., 2015; Yan et al., 2013). Other staphylococcal species, although sometimes involved in opportunistic infections, are generally beneficial and normal components of the human epithelial microbiota (Grice and Segre, 2011; Otto, 2004, 2009). Almost all of these belong to the coagulase-negative staphylococci (CNS), the predominant member of which is Staphylococcus epidermidis (Otto, 2009). Notably, the genus Staphylococcus exemplifies, by including beneficial but also pathogenic species, that correlative 16S rRNA sequencing-based microbiome studies, which seldom reach below the genus level, are prone to miss important biological relationships.
In the present study, we investigated the nasal microbiota of a total of 467 healthy volunteers from three different age groups by 16S rRNA-sequencing in addition to culture-based methods. We report that the human nasal microbiota change significantly from child- to adulthood, resulting in pathogen exclusion, an effect partially reversed during senescence. Our results indicate a key role of the beneficial commensal S. epidermidis in this development, and we provide a mechanistic explanation for this association.
Results
Development of the human nasal microbiome with age
To analyze the evolution of the nasal microbiota during a human lifetime, we first determined the nasal microbiomes of 155 children (5.45 ± 0.50 years old), 171 young adults (19.47 ± 0.73 years old), and 141 seniors (82.50 ± 8.29 years old) by 16S rRNA sequencing. Analysis of the ten major microbial phyla revealed that the composition of the human nasal microbiome changed significantly with age and microbial diversity decreased upon entry into adulthood (Fig. 1A-C). In contrast, nasal microbiomes did not differ significantly by gender (Fig. 1D). Compared to the other two groups, presence of Proteobacteria was particularly pronounced in children and highly variable, while other major phyla were reduced in relative abundance.
Figure 1. Development of the human nasal microbiome with age.
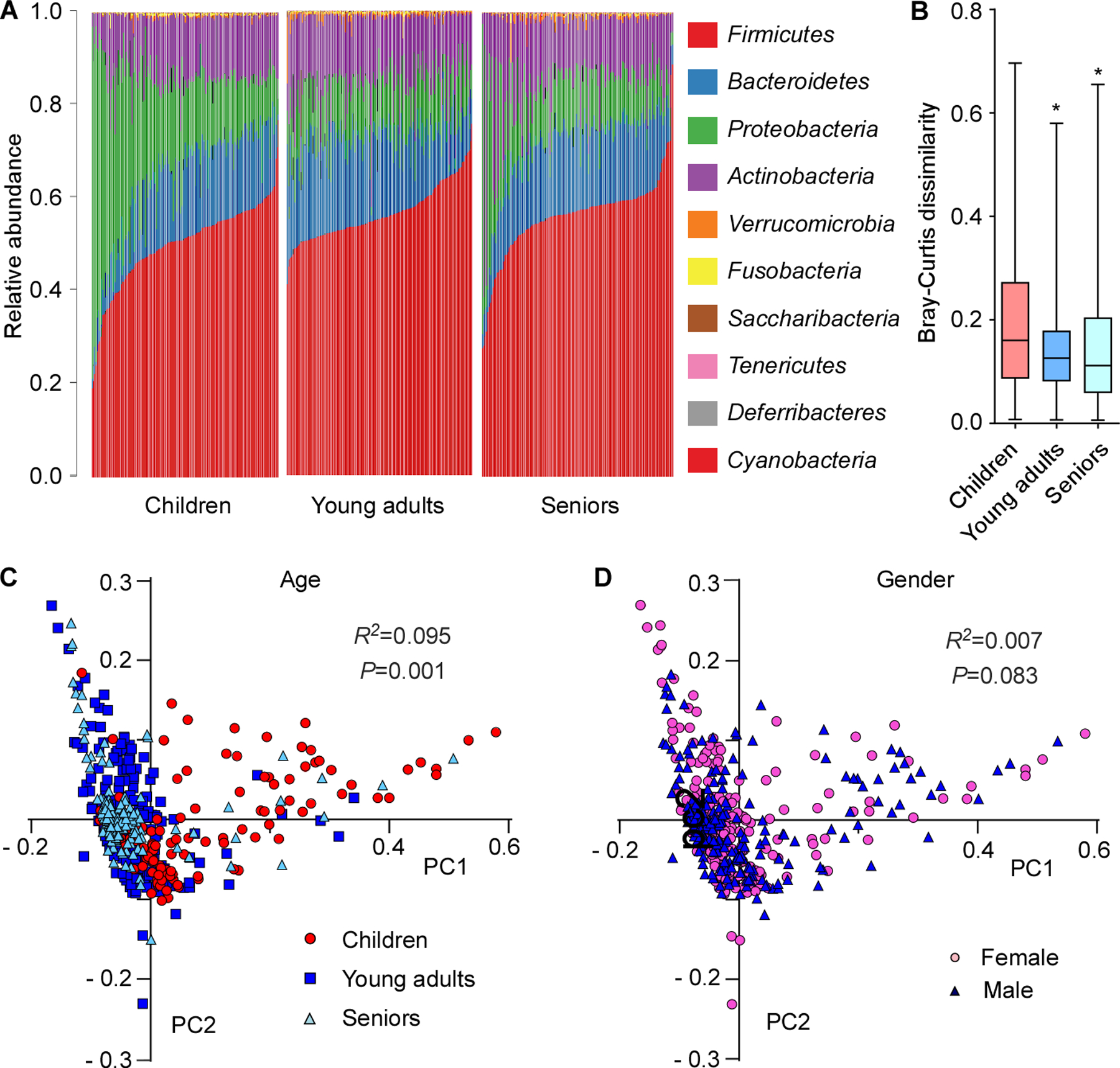
(A) Relative abundance of the ten major phyla in investigated individuals by age group. (B) Bray-Curtis dissimilarity of the age groups (phylum level). Statistical analysis is by 1-way ANOVA and Tukey’s post-test. *, P<0.0001. Whisker boxes are drawn from the first to third quartiles. Error bars show minima and maxima. (C) Principal coordinate analysis by age group. (D) Principal coordinate analysis by gender. (C,D) Statistical analysis is by Analysis of Similarities (ANOSIM) test.
Inverse correlation between Staphylococcus and opportunistic pathogens in the nasal microbiome
Analysis on the genus level also revealed considerable differences between age groups (Fig. 2A,B). One striking example was the difference between the dominance of Moraxella in children and the virtual absence of that genus from the other two age groups. In contrast, Staphylococcus gained considerably in presence during the transition to adulthood (~ 4.4 times) and was the predominant genus in young adults (Fig. 2B,C). With Moraxella belonging to the Proteobacteria, this change is most likely associated with the observed difference in abundance of this phylum between age groups. A further considerable difference between the children and young adult groups was the decline in Dolosigranulum (~ 2.4 times), a genus that only contains the opportunistic pathogen Dolosigranulum pigrum, which can cause upper respiratory tract infections, nosocomial pneumonia, and septicemia (Hoedemaekers et al., 2006; Lecuyer et al., 2007). In the senior group, the increase in Staphylococcus and decrease in Dolosigranulum observed in young adults was again partially reversed. Finally, some of the other main genera showed significant changes comparing age groups, but these were much smaller in extent (Fig. S1).
Figure 2. Correlation of abundance of Staphylococcus and other genera in the nasal microbiome.
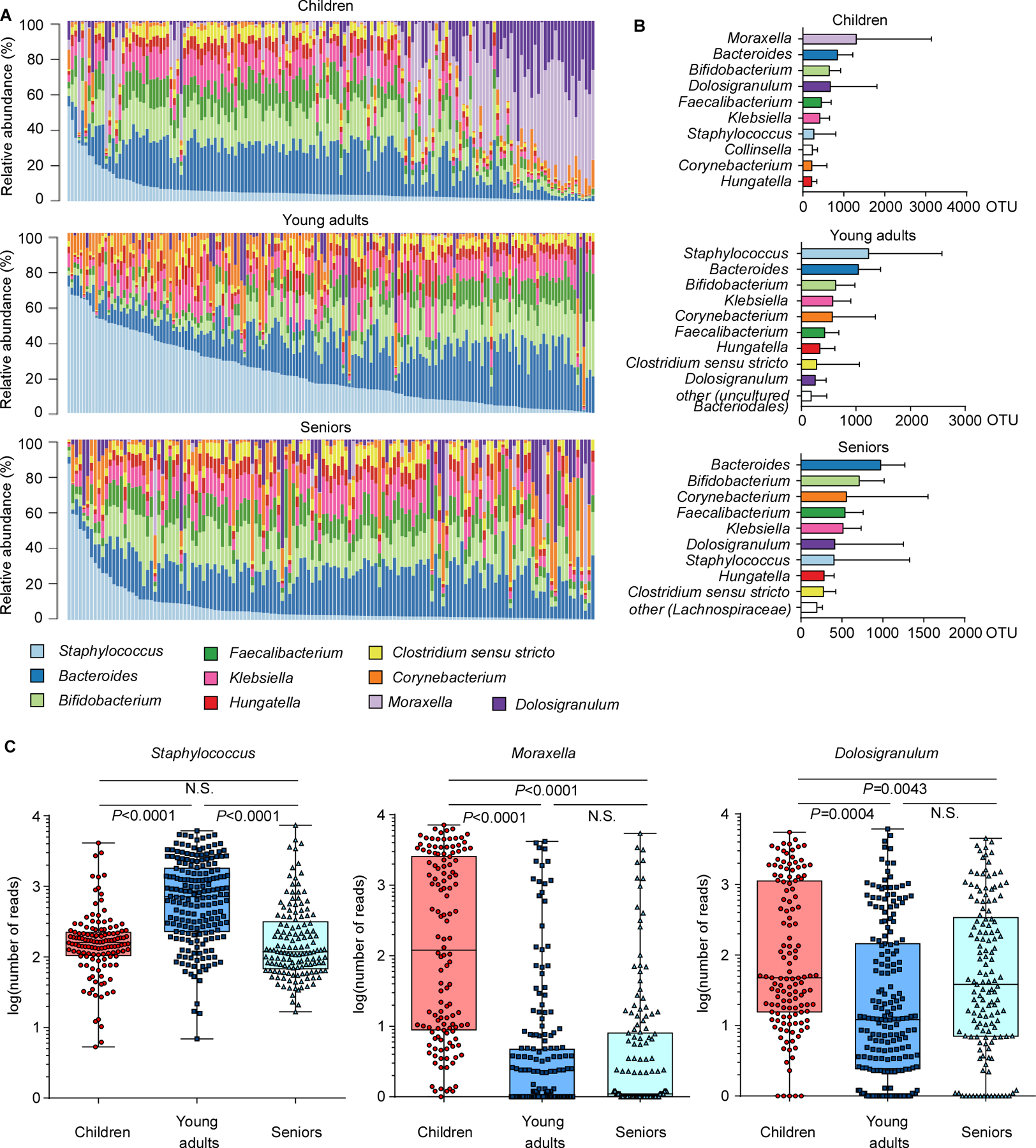
(A) Relative abundance of major genera in the nasal microbiome of single individuals in the three analyzed age groups. (B) Abundance of major genera in the three age groups. Bars depict means; error bars SD. (C) Abundance of Staphylococcus, Moraxella, and Dolosigranulum in the three age groups, in single individuals. See Fig. S1 for other main genera. Whisker boxes are drawn from the first to third quartiles. Error bars show minima and maxima. Statistical evaluation is by 1-way ANOVA with Tukey’s post-test. Data were also analyzed by ANCOM, by which all three genera showed significant differences between groups (no post tests are available with this analysis; see Supplemental Data Set 2 for ANCOM results). The difference in the abundance of each OTU across different samples or groups was compared based on the subset of 10,000 random reads for each sample.
Culture-based analysis of S. epidermidis abundance and inverse correlation with presence of pathogens in the nasal microbiota
On the basis of our sequencing data, we hypothesized that the decrease in pathogens in adult nostrils might be due to the increase in Staphylococcus bacteria. To further investigate this hypothesis, and because identification below the genus level is often difficult by 16S rRNA sequencing analysis, we complemented our study with culture-based identification (Fig. 3, Supplemental Data Set 1). To that end, we grew samples on tryptic soy broth (TSB) agar, a growth medium used generally for a wide range of bacteria, under aerobic conditions. Analysis of the relative abundance of staphylococcal species in the nostrils revealed general predominance of S. epidermidis, which was significantly more pronounced in young adults and to a lesser extent, seniors, than in children (Fig. 3A), suggesting that this species may be involved in the observed microbiome shifts.
Figure 3. Culture-based analysis of S. epidermidis abundance and inverse correlation with presence of pathogens in the nasal microbiota.
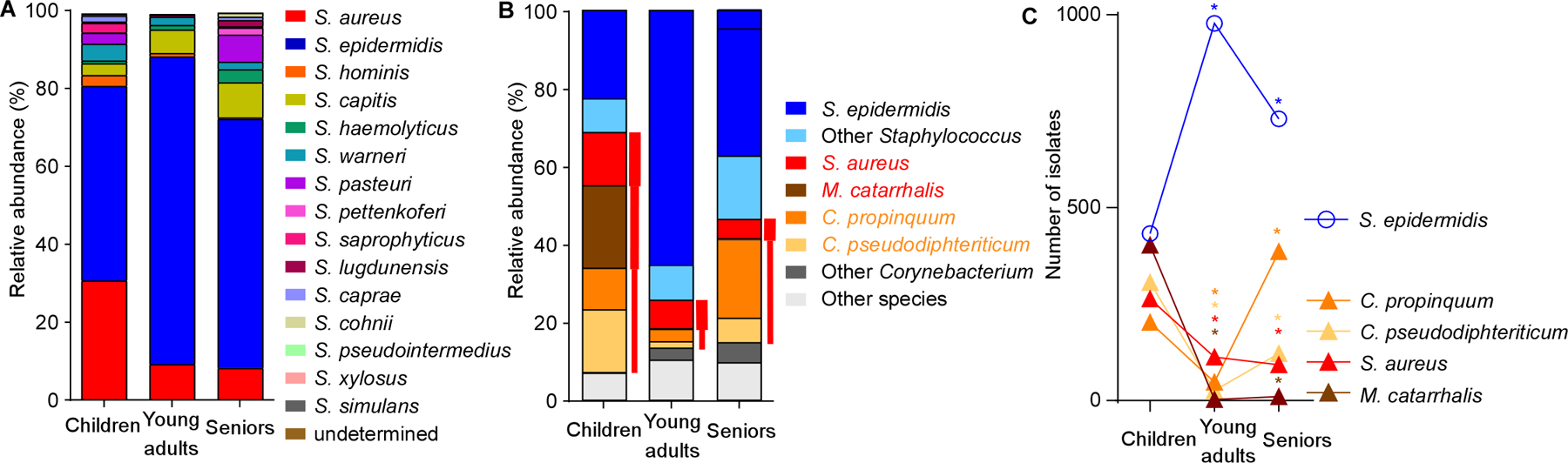
(A) Relative abundance of staphylococcal species (by culture-based quantification) in the nasal microbiota in the three age groups. (B) Relative abundance of all culturable bacteria in the three age groups. Red font and bars depict opportunistic respiratory pathogens, with bar thickness depicting relative pathogenic potential. (C) Inverse correlation of abundance of S. epidermidis and respiratory pathogens in the nasal microbiota in the three age groups. Statistical analysis is by Fisher’s exact test (comparing presence of a species in the young adult versus children and senior versus young adult groups). *, P<0.0001.
Among all nasal isolates, of which the vast majority was culturable and identifiable (> 99%), the five most frequently isolated were S. epidermidis, S. aureus, Moraxella catarrhalis, Corynebacterium propinquum, and Corynebacterium pseudodiphteriticum (Fig. 3B). The latter four species all represent opportunistic respiratory pathogens (Murphy and Parameswaran, 2009; Van Roeden et al., 2015; von Eiff et al., 2001; Yang et al., 2018). Notably, abundance of S. epidermidis was inversely correlated with that of each of those species (Fig. 3C). While S. epidermidis significantly increased in presence during adolescence, presence of the other four species declined. In seniors, significant decrease of the presence of S. epidermidis was paralleled by significant increase of some of those species, while presence of others remained low. With S. epidermidis dominating among staphylococci, and M. catarrhalis representing the vast majority (93.3%) of Moraxella isolates (Supplemental Data Set 1), these culture-based results are in good accordance with those obtained by sequencing. Our culture-based study has the limitation that the used growth medium and aerobic conditions do not allow for the growth of all species of bacteria. Altogether, our data indicate that the nasal microbiota in young adults contain fewer pathogens than those of children, and to a more limited degree, seniors. Furthermore, our results suggest a role of S. epidermidis in shaping the human nasal microbiome by exerting a broad pathogen exclusion effect.
To analyze whether the observed differences in microbial composition of the nostrils are associated with a specific subtype of S. epidermidis, we typed 300 randomly selected isolates (100 per age group). While we observed considerable heterogeneity in all age groups, the presence of sequence types (STs) 59 and 89 significantly increased, while that of ST130 decreased with age (Fig. 4). Interestingly, we found no ST2 among the commensal S. epidermidis isolates, which confirms previous studies that have found that this specific ST is associated with infection (Lee et al., 2018; Li et al., 2009).
Figure 4. Evolutionary relationships within and differences between age groups of S. epidermidis nasal isolates.
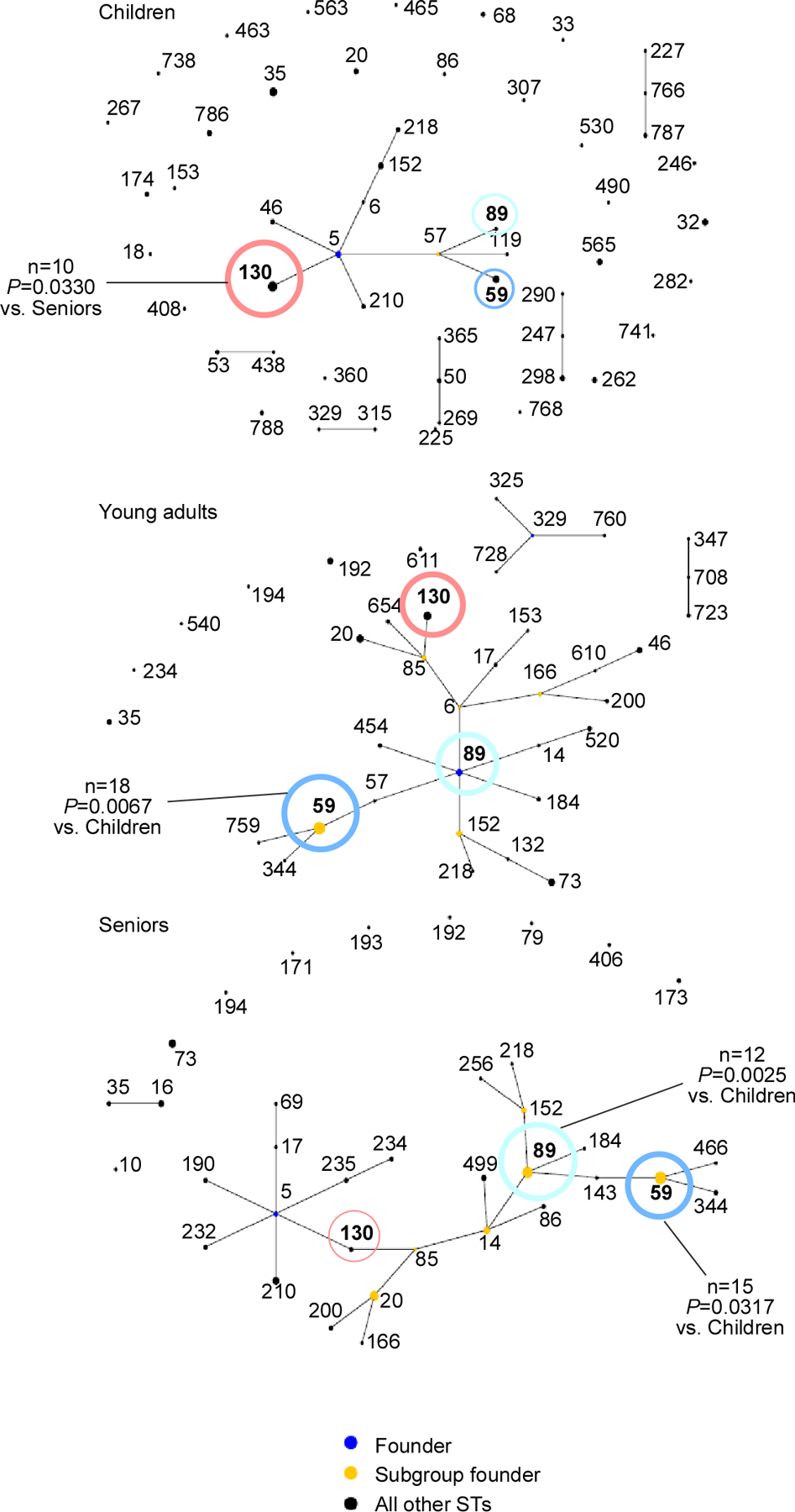
S. epidermidis isolates were typed by MLST and evolutionary relationship was computed using eBURST. Abundant STs are noted in boldface. The important STs for which statistically significant differences in abundance between age groups were detected are colored: ST59, blue; ST89, turquoise; ST130, red. Dot sizes correspond to abundance. Comparisons between age groups for specific STs were performed by contingency analyses using Fisher’s exact test (number of isolates of analyzed ST versus number of remaining isolates in the two groups to be compared).
Symbiotic mechanism of S. epidermidis-mediated pathogen exclusion on the nasal epithelium
Next, we set out to investigate a potential mechanism underlying the S. epidermidis-associated differences in the abundance of pathogens and opportunistic pathogens in the nasal microbiota. While colonization of specific bacteria may be affected by inter-bacterial interactions (Nakatsuji et al., 2017; Piewngam et al., 2018; Sassone-Corsi et al., 2016), broad effects as those found in the present study are generally believed to be mainly impacted by the interaction with host defenses (Chen et al., 2018). Nevertheless, because S. epidermidis nasal isolates from healthy adults have been reported to show widespread production of bacteriocins (Janek et al., 2016), which if occurring preferentially in that age group may explain our observations, we tested concentrated culture filtrates of all 2241 nasal in addition to 107 S. epidermidis clinical infection isolates for antimicrobial activity using the Kirby-Bauer disk-diffusion method (Bauer et al., 1959) (Fig. S2). We detected antimicrobial activity only in two isolates toward S. aureus, and never toward D. pigrum or M. catarrhalis. These results indicating rare bacteriocin production in S. epidermidis are in accordance with earlier studies that have used deferred antagonism assays (Skalka, 1992). However, they are at odds with those from the abovementioned study, which used stamps of live bacteria on test plates (Janek et al., 2016), and another study that measured growth of test strains in liquid culture with spent culture filtrates (Nakatsuji et al., 2017). When we used those two methods with our isolates, we obtained results very similar to those previously described: in the assay used by Janek et al. we detected frequent activities against M. catarrhalis and D. pigrum as opposed to rare activity against S. aureus, while we found widespread inhibitory effects, with similar test strain dependence, using the liquid culture method employed by Nakatsuji et al. (Fig. S2). The assay used by Janek et al. may be more sensitive than the disk diffusion method in detecting low-level production of antimicrobial activities, but we believe that the observed discrepancies are also due at least in part to the fact that those methods, which use live bacteria, may yield false positives owing to nutrient competition or other non-bacteriocin-mediated effects. Particularly the liquid culture method is expected to be considerably influenced by the limited availability of nutrients in spent as compared to the fresh media control. Furthermore, the fact that Gram-positive bacteriocins are known to be generally less active against Gram-negative bacteria (such as M. catarrhalis), due to their outer membrane (Prudencio et al., 2015), is at odds with the finding of widespread activity particularly against M. catarrhalis. Notably, also with the methods employed by Janek et al. and Nakatsuji et al., we did not detect significant differences in frequency of antimicrobial activity between the age groups (Fig. S2). Consequently, direct competition by bacteriocins is not a likely general explanation for the broad pathogen exclusion effects we observed during the evolution of the nasal microbiome with age.
We then turned to investigating the interaction with host defenses, as the human immune system, including its innate arm, is known to show age-dependent maturation and deterioration processes (Kampmann and Jones, 2015; Metcalf et al., 2015; Simon et al., 2015). Immune mechanisms that govern asymptomatic epithelial colonization mainly comprise secretion of antimicrobial peptides (AMPs) (Diamond et al., 2009; Diamond et al., 2000). Lai et al. showed that S. epidermidis, but not a selection of other bacteria, induces production of human beta-defensins (hBD) 2 and 3 in normal human keratinocytes in a TLR2-dependent manner (Lai et al., 2010). However, the other bacteria tested in that study were not pathogenic members of the nasal microbiota. Therefore, we first extended on that previous study by measuring induction of AMPs by S. epidermidis versus the bacterial species that arose as the most important competing pathogenic nasal colonizers in our microbiome study. Also, we used a human nasal keratinocyte cell line to better mimic the situation in the human nasal epithelium. In addition to that of hBD3, we included measurement of expression of LL37, another important AMP in skin (Doss et al., 2010). Finally, we distinguished between colonizing nasal isolates of S. epidermidis and isolates from infection (Table S1). We found that S. epidermidis isolates stimulated production of LL37 and hBD3 to a strongly and significantly higher extent than the other tested bacteria, which were largely devoid of such capacity (Fig. 5A,B). This phenotype was more pronounced in commensal than infectious S. epidermidis, and particularly prominent in ST59 isolates. Induction by S. epidermidis commensal isolates of hBD3 and LL37 production was strongly correlated (R2=0.8027; P<0.0001), suggesting a common mechanism. Thus, S. epidermidis isolates that colonize the nose, but not competing members of the human nasal microbiota and less so other S. epidermidis isolates, trigger AMP production in nasal human keratinocytes.
Figure 5. Symbiotic mechanism of S. epidermidis-mediated pathogen exclusion on the nasal epithelium – in vitro tests.
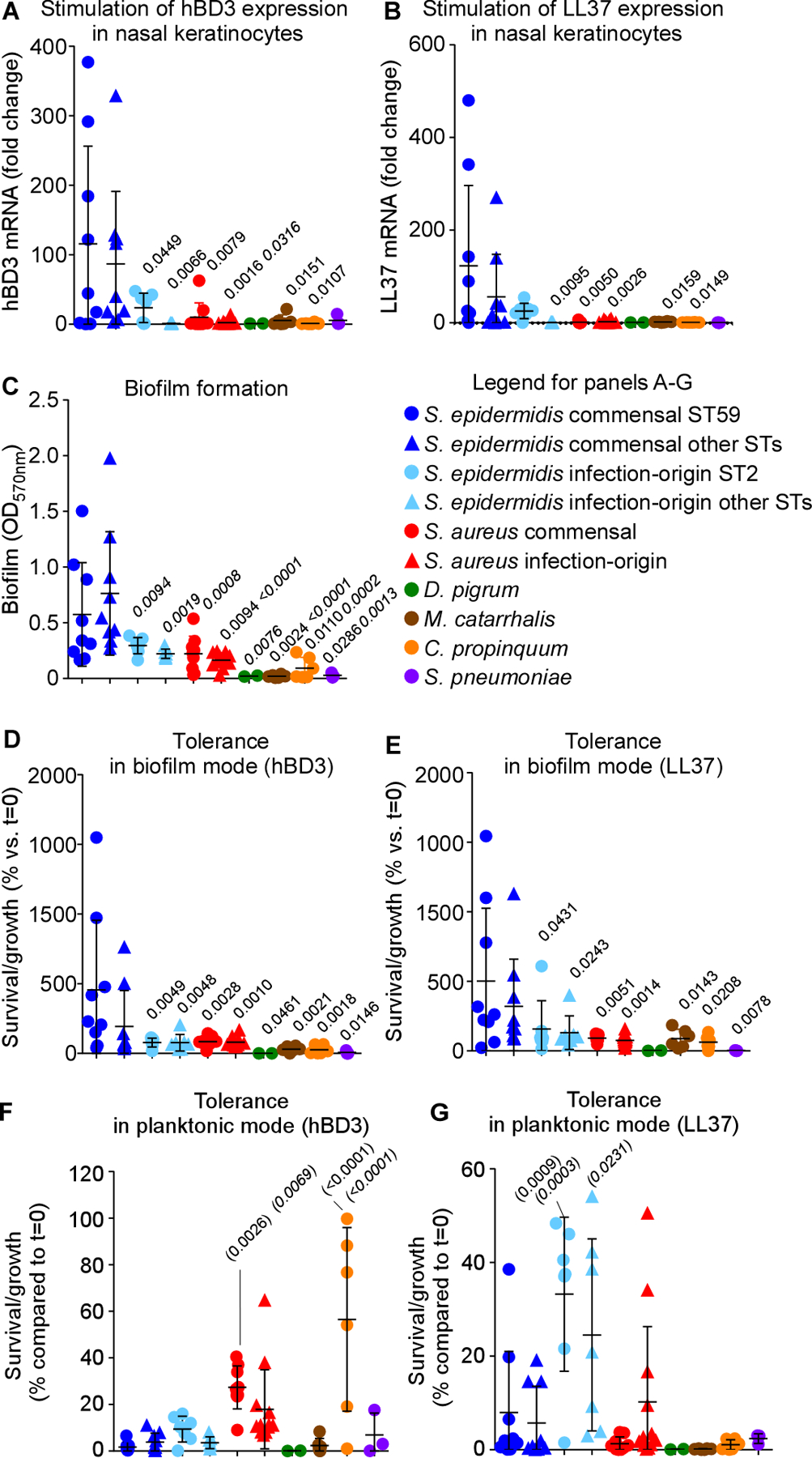
(A,B) Stimulation of expression of hBD3 (A) and LL37 (B) in nasal epithelial cells by S. epidermidis commensal and infection-origin isolates (with separate analysis of the major STs), commensal and infection-origin isolates of S. aureus, and other major opportunistic respiratory pathogens. See Table S1 for isolate selection. (C) Biofilm formation by the same isolates in TSB. (D,E) Tolerance to the AMPs hBD3 (D) and LL37 (E) by the same isolates in biofilm mode of growth (TSB). (F,G) AMP tolerance by the same isolates in planktonic mode of growth (TSB). See Fig. S3 for results in SNM3. (A-G) Three biological replicates were measured for every strain. The depicted data represent the average for each strain. Statistical analysis is by 1-way ANOVA with Dunnett’s post-tests versus data obtained with the S. epidermidis commensal ST59 (regular font P values) and S. epidermidis commensal other STs (italic font P values). Error bars show means ± SD. Experiments depicted in panels A-C were repeated four times, and those in panels D-G twice, with similar results.
Immune stimulation as a bacterial strategy to compete with other microorganisms only makes biological sense if the stimulatory bacteria are protected from the ensuing immune attacks. In the previous study by Lai et al., S. epidermidis appeared protected from induced keratinocyte lysate, while the lysate was inhibitory toward one strain of S. aureus and one of S. pneumoniae (Lai et al., 2010). However, the range of these experiments was very limited in terms of strains and species used, the comparative effects were minor, and the experiments did not directly test sensitivity to AMPs. Furthermore, these authors did not address the mechanism of protection. Therefore, we analyzed isolates of S. epidermidis and competing pathogenic nasal bacteria for their capacity to resist hBD3 and LL37. Given that biofilm formation is a known AMP resistance mechanism (Otto, 2006) and bacteria are believed to grow in a biofilm-like manner during epithelial colonization (Otto, 2014), we hypothesized that different tolerance phenotypes may be due to biofilm formation. Thus, we performed these analyses in planktonic (free-floating) and biofilm modes of growth. S. epidermidis showed significantly increased biofilm formation (Fig. 5C) and capacity to resist killing by AMPs in biofilm, but not in planktonic mode, as compared to M. catarrhalis, D. pigrum, S. aureus, C. propinquum, and Streptococcus pneumoniae (Fig. 5D-G). Using a growth medium that was composed to mimic the situation in human nostrils (synthetic nasal medium SNM3) (Krismer et al., 2014), we achieved similar results (Fig. S3). AMP tolerance and biofilm formation were strongly correlated (TSB, for LL37, R2=0.7478; P<0.0001; for hBD3, R2=0.6494; P<0.0001; SNM3, for LL37, R2=0.6957; P<0.0001; for hBD3, R2=0.7867; P<0.0001). Nasal commensal isolates of S. epidermidis had significantly more pronounced biofilm formation and associated AMP resistance than infection isolates, in accordance with the notion that biofilm formation is important for colonization. Altogether, these results indicated that S. epidermidis nasal isolates have specific capacity to induce major immune defenses on human nasal epithelia to which they are themselves highly resistant due to increased capacity for biofilm formation.
To provide in-vivo confirmation for this notion, we selected four representative commensal S. epidermidis strains with either high biofilm-forming and high AMP induction capacity, high biofilm-forming and low AMP induction capacity, low biofilm-forming and high AMP induction capacity, or low biofilm-forming and low AMP induction capacity, in addition to an infection-origin isolate with the moderate biofilm-forming and low AMP induction capacity we found to be characteristic for infection isolates (Table S2). The strong biofilm former/strong AMP inducer strain achieved significantly higher nasal colonization in mice and in-vivo induction of transcription of the CRAMP mouse homologue of LL-37 than all other strains, reflecting in-vitro results and emphasizing dependence on both these phenotypes for successful in-vivo colonization (Fig. 6A,B, Table S2).
Figure 6. Symbiotic mechanism of S. epidermidis-mediated pathogen exclusion on the nasal epithelium – in vivo tests.
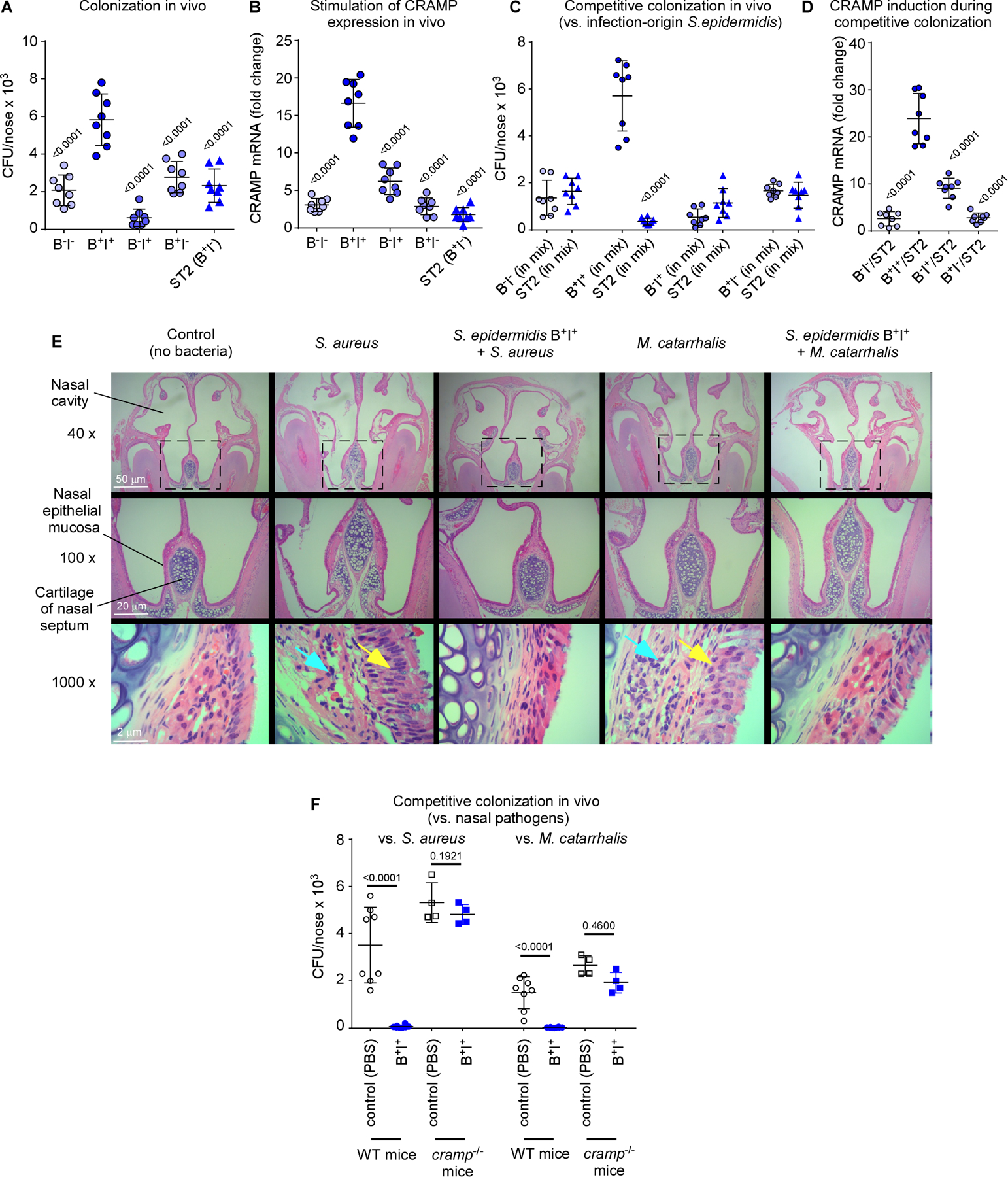
(A) Nasal colonization in mice of selected S. epidermidis commensal strains (B−I−, weak biofilm former and low AMP induction capacity; B+I+, strong biofilm former and high AMP induction capacity; B−I+, weak biofilm former and high AMP induction capacity; B+I−, strong biofilm-former and low AMP induction capacity, see Table S2), and an infection-origin isolate with the B+I− phenotype characteristic for that group. Inocula, 1 × 107 CFU, once a day for 4 days, CFU determination 6 days after first instilment. (B) Induction of cramp gene in the same experiment as in panel A. (C) Competitive nasal colonization with the four selected commensal S. epidermidis strains and the infection-origin ST2 strain (1:1 inocula, 1 × 107 CFU each, instilments and CFU determination as described for panel A). (D) Induction of cramp gene in the same experiment as in panel C. (E,F) Competitive nasal colonization with B+I+ S. epidermidis and S. aureus or M. catarrhalis (S. epidermidis, 1 × 107 CFU every day over four days; S. aureus, 1 × 108 CFU at day 6; M. catarrhalis, 4 × 109 CFU at day 6). (E) Histological examination of noses. Yellow arrows, proliferation of cilia in nasal epithelial mucosa; blue arrows, destruction of epithelial submucosa. (F) CFU. (A-D, F) n=8. (B,D) Three measurements were taken for determination of CRAMP mRNA expression. The shown data represent the average for every mouse. (A,B,D) Statistical analysis is by 1-way ANOVA with Dunnett’s post-tests versus data obtained with with B+I+. (C,F) Statistical analysis is by unpaired t-tests. Error bars show means ± SD.
In direct support of the idea that commensal S. epidermidis outcompetes niche competitors by stimulating AMPs to which it is itself tolerant due to biofilm formation, only the strong biofilm former/strong AMP inducer, but not the other strains, outcompeted the infection-origin S. epidermidis strain in vivo (Fig. 6C) and led to significantly higher associated CRAMP induction (Fig. 6D). Furthermore, that strain efficiently outcompeted the two pathogenic bacteria S. aureus and M. catarrhalis in vivo and led to decreased signs of infection caused by these pathogens (Fig. 6E,F). Notably, this effect disappeared in cramp−/− mice (Fig. 6F), which lack production of CRAMP, demonstrating the need for AMP induction for the competitive effect and providing direct support for the in-vivo relevance of our model.
Discussion
There is a very limited number of studies that have analyzed the development of the human nasal microbiome in different age groups and a general paucity of studies that have attempted to provide mechanistic explanations for microbiome diversity during a human life, considering changing immune maturity and its impact on host-microbiome interaction. The present study included a large number of isolates from three age groups - children, young adults, and seniors, which were chosen to represent ages with not yet developed, mature, and declining immune status, respectively. We found significant changes in the composition of the human nasal microbiome between those age groups. The most interesting aspects were the generally high abundance of S. epidermidis as well as a significant increase of S. epidermidis presence in the young adult group, which was associated with a significant decrease in the presence of a series of opportunistic pathogens, including S. aureus or M. catarrhalis. These changes were confirmed by culture-based analysis. Our 16S rRNA sequencing and culture-based analyses of the human nasal microbiota has limitations, similar to all such analyses, which include the bias of the chosen oligonucleotide pair and the absence of strictly anerobic bacteria in the aerobically grown cultures. Certain genera, such as Propionibacterium (Cutibacterium) may therefore be underrepresented in our analysis.
Our microbiota analyses suggested a potential mechanism by which S. epidermidis leads to pathogen exclusion in the nose in possible concert with the host immune system, given that it was most pronounced in the young adult group. Studies investigating the impact of S. epidermidis on the host’s immune system have focused mostly on innate immunity. One study has described TLR2-mediated stimulation of hBD2 and hBD3 expression in human keratinocytes by S. epidermidis (Lai et al., 2010), while another recent study suggested that S. epidermidis also moderates that effect via induction of the host regulator protein A20, decreasing hBD2 expression (Simanski et al., 2019). According to another study, a further pathway exists that involves dendritic and T-cells, resulting in increased production of S100-type antimicrobial peptides by keratinocytes, as shown when the skin of live mice was associated with S. epidermidis (Naik et al., 2015). Finally, S. epidermidis was shown to stimulate interferon-γ expression, resulting in increased immunity against influenza virus in a TLR2-independent manner (Kim et al., 2019).
In our study, we detected stimulation of gene expression of hBD3 and LL37, both key AMPs in human skin, when human keratinocytes were exposed to live S. epidermidis. Notably, we compared AMP-stimulatory activity among a series of different S. epidermidis strains as well a series of opportunistic pathogens, many of which we found to have decreased abundance in the nasal microbiota of young adults. Stimulatory activity was detected with commensal S. epidermidis isolates, but to a much lesser degree, S. epidermidis infection isolates or isolates of the used opportunistic pathogens. These findings thus showed that commensal S. epidermidis strains have an almost unique capacity to stimulate AMP production in human keratinocytes.
In one of the studies mentioned above, association of mouse skin with S. epidermidis led to a reduction of subsequent colonization with another, albeit eukaryotic microorganism (Candida albicans), suggesting a mechanism of pathogen exclusion, which given the increase of S100 AMP gene expression observed by the authors may be explained as AMP-mediated (Naik et al., 2015). In the study by Lai et al. previous application of S. epidermidis led to decreased skin infection by group A Streptococcus subsequently injected into the skin (Lai et al., 2010). Of note, no study that we are aware of has provided a plausible explanation of how the observed immune-stimulatory activity leads to a competitive advantage of the stimulatory microorganism in the context of the human microbiome, as such an advantage would require a mechanism of increased AMP resistance. Furthermore, an experimental setup of subsequent colonization or infection cannot adequately analyze directly competitive phenomena. Finally, the involvement of AMPs in the exclusion phenomenon was not directly tested in vivo in those studies. In our study, we analyzed biofilm-forming capacity as a known AMP resistance mechanism (Otto, 2006) and detected significantly higher biofilm formation among the S. epidermidis as compared to all other isolates, which was significantly correlated with AMP resistance. Furthermore, using a mouse competitive nasal colonization model, we could directly show stimulation by S. epidermidis of LL37 (CRAMP) gene expression in the noses of mice as well as competitive exclusion by S. epidermidis of S. aureus and M. catarrhalis, which using LL37 (CRAMP)-negative mice could be directly linked to AMP expression. These results are consistent with a model in which S. epidermidis achieves pathogen exclusion by stimulation of AMPs in the nose to which it is itself resistant owing to pronounced biofilm formation.
The factors that stimulate AMP expression and are, according to our and two other studies (Lai et al., 2010; Naik et al., 2015), unique to or predominantly expressed in S. epidermidis, so far remain unknown. In a recent study, the previously described immune-stimulatory phenomena involving T-cells were linked to N-formylated peptides (Linehan et al., 2018), but these are produced by virtually all bacteria and can hardly account for the observed differences between S. epidermidis and the other studied isolates. On the other hand, the group that reported the TLR2-mediated effects recently presented a novel lipopeptide structure as allegedly underlying AMP stimulation (Li et al., 2013). However, immune-stimulatory phenomena were predominantly shown with a synthesized form of that molecule and a rigorous structure elucidation as well as genetic evidence for production of and link to the AMP stimulation by that molecule in S. epidermidis were not provided. Altogether, we thus believe that the S. epidermidis-specific AMP-stimulating factors are still elusive and their identification warrants thorough future investigation.
In summary, we here describe changes in the composition and healthiness of the human nasal microbiome that reflect increase and decrease of immune efficiency with age. Furthermore, we discovered a mechanism of symbiotic interplay between a member of the human microbiota and innate host defense that contributes to pathogen exclusion and microbiome stabilization and helps host defenses overcome the difficult problem of distinction between beneficial and harmful colonizers. Finally, our findings attribute a key function to S. epidermidis in preventing pathogen overgrowth in the nose and thus, subsequent infections of the respiratory tract and other organs.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further requests for information may be directed to Min Li (ruth_limin@126.com) or Michael Otto (motto@niaid.nih.gov). Bacterial isolates are available from Min Li (ruth_limin@126.com). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects
This study was approved by the ethics committee of Renji Hospital, School of Medicine, Shanghai Jiaotong University, Shanghai, China (protocol 2017001). All individuals/legal guardians provided informed consent. Three groups of healthy volunteers were recruited from the community. In total, 158 children (5–6 years old), 210 young adults (18–20 years old), and 158 seniors (50–90 years old), who had resided in the community of Shanghai, China, were enrolled for the research. Among the subjects passing restriction criteria, there were 214 males and 248 females. No significant differences in microbiome composition were found according to gender (see Fig. 1D). The senior group contained a larger age span than the other two groups. This cohort likely includes a mix of pre/perimenopausal and postmenopausal women who differ physiologically. Routine physical examination and health status enquiries were carried out to exclude unhealthy volunteers. The exclusion criteria for the study were as follows: abnormal physical examination results, pregnancy or breast-feeding, diabetes or other chronic metabolic diseases, various cancers, history of rhinitis or other chronic nostril diseases, current or prior chronic skin disorders, carriage of medical devices, contact with the hospital environment in the last six months preceding enrollment, use of any antimicrobials within one month preceding enrollment. Nasal swabs were collected from the mucosal surface internal to the limen nasi of each participant. Swabs were immediately submerged in 1 ml sterile saline for further use.
Mice
6-week-old female C57BL/6J or B6.129X1-Camptm1Rlg/J mice (Jackson Laboratories) were used for the experiments. They were provided with food and water ad libitum. Littermates of the same sex were randomly assigned to experimental groups. Researchers were not blinded as for group allocation of animals. All animal work was approved by the ethics committee of Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China.
Bacteria
Bacteria were generally clinical isolates from this study. See Table S1 for all bacterial strains used in in-vitro and in-vivo experiments. They were grown in tryptic soy broth (TSB) at 37 °C, with the exceptions outlined in the respective methods section.
Cell lines
The human nasal epidermal cell line RPMI2650 (ATCC) was cultured in DMEM (Dulbecco's modified eagle medium, GIBCO 11965–084) with 10% fetal bovine serum (FBS, GIBCO 10100147) at 37 °C with 5% CO2. The cell line is derived from a squamous cell carcinoma of the nasal septum in a male patient.
METHOD DETAILS
DNA extraction and 16S rRNA gene sequencing
Nostril swabs were submerged in 1 ml sterile saline and vortexed for 2 min. 500-μl samples from each swab were centrifuged at 13,000 × g for 10 min, 4 °C, upon which the pellets were dissolved in Buffer ALT (QIAamp DNA Mini Kit, Qiagen 51306) with lysozyme (1.25 mg/ml, Sigma L6876) and lysostaphin (25 μg/ml, Sigma L4402) and incubated for 30 min at 37 °C. Then, DNA extraction was performed according to the protocol of the QIAamp DNA Mini Kit. 16S-rRNA gene libraries were generated by PCR from purified genomic DNA with primers 341F (5’-CCTACGGGNBGCASCAG-3’) and 805R (5’-GACTACHVGGGTATCTAATCC-3’), which amplify the hypervariable V3 and V4 regions of the 16S rRNA gene. As all primer combinations used for microbiome studies, these primers underrepresent some species. The specific combination used here was reported, for example, to underrepresent the genus Propionibacterium (Gohl et al., 2016). The amplicons were further amplified and sequenced on the Illumina Hiseq 2500 platform. A total of 12773 to 464276 sequencing reads were obtained from 16S rRNA amplicons for each nasal sample.
Sequence analysis
The FastX tool kit (Gordon and Hannon, 2012) was used to trim sequencing data, and sequencing reads with a Phred base quality above 25 and read length longer than 30 were kept for analysis. Paired-end read pairs were assembled using Flash software (Magoc and Salzberg, 2011). USEARCH 8.0 implemented in QIIME was used for detection and filtration of chimeric sequences (Edgar et al., 2011). The lengths of amplified sequences obtained after splicing were between 400 and 500 base pairs for all samples. Samples with reads of less than 10,000 were excluded. Finally, 467 samples were amplified successfully, which were from 155 children [91 male, 64 female, 5.45 ± 0.50 (S.D.) years old], 171 young adults [76 male, 95 female, 19.47 ± 0.73 (S.D.) years old], and 141 seniors [47 male, 94 female, 82.50 ± 8.29 (S.D.) years old].
Quality-trimmed sequences were then clustered into operational taxonomic units (OTUs) at a similarity level of 99% by using the Ucluster algorithm implemented in QIIME (Caporaso et al., 2010). Taxonomic assignments were performed using the RDP classifier with a confidence cutoff of 0.8 and the Silva database (https://www.arb-silva.de/documentation/release-128/) as a reference (Wang et al., 2007). For each sample, a subset of 10,000 random reads were picked to construct the OTU abundance table. Taxonomic classification from the phylum to genus level for the same 10,000 reads was performed with the RDP Classifier based on the Silva database. To estimate sampling saturation, rarefaction and Shannon-Wiener curves were generated (Fig. S4). Beta diversity indices [Principal coordinate analysis (PCoA) and Bray-Curtis dissimilarity] were calculated at a 97%-similarity cutoff by QIIME. Based on phylum-level composition, beta diversity was computed using the functions of “vegdist” and “cmdscale” implemented in the VEGAN package in R (Dixon, 2009). The difference in the abundance of each OTU across different samples or groups was compared based on the subset of 10,000 random reads for each sample.
Culture-based analyses
For bacterial cultures, nostril swabs were submerged in 1 ml sterile saline and vortexed for 2 min. 100-μl samples from each swab were plated on 5% sheep blood agar and incubated at 37 °C for 24 h. In almost all swab samples (518/526), 20 colonies or less grew, and all colonies were subjected to species identification. In the eight swab samples that grew more than 20 colonies, 20 colonies were randomly selected. Species identification was performed using MALDI-TOF-MS (Bruker Daltonics, Bremen, Germany). To that end, a single clone was spotted onto the steel target plate, 1 μl 10% formic acid (Sigma F0507) was added to the bacteria and dried for 5 min at 75 °C. The spot was then overlaid with 1 μl MALDI matrix [a saturated solution of a-cyano-4-hydroxycinnamic acid (Sigma 70990) in 50% acetonitrile / 2.5% trifluoroacetic acid]. After drying, the plate was subjected to the MALDI-TOF MS system for analysis. The spectrum was obtained in linear positive-ion mode range from 2000 to 20,000 Da. Each spot was measured manually on five different positions by using 1000 laser shots at 25 Hz in groups of 40 shots. The MALDI Bruker Biotyper 3.0 software and library (Bruker Daltonics) were used for spectra analysis. PCR amplification and 16S rRNA sequencing using primers F: 5’-AGTTTGATCCTGGCTCAG-3’ and R: 5’-GGTTACCTTGTTACGACTT-3’ was used to identify isolates that failed to be confirmed by MALDI-TOF-MS. All identified bacterial isolates are listed in Supplemental Data Set 1.
MLST typing and eBURST algorithm
MLST was performed using the primer sequences for the PCR amplification of the seven housekeeping genes arcC, aroE, gtr, mutS, pyrR, tpiA, and yqiL. The number of alleles and STs was determined using the online MLST database (http://www.pubmlst.org). Previously unknown STs were deposited to the MLST database. In total, we obtained 2141 S. epidermidis isolates from nostril swabs, of which we randomly selected 100 isolates from each age group. The eBURST algorithm (http://eburst.mlst.net) was used to infer the evolutionary relation of S. epidermidis isolates.
Screening for antimicrobial activity
All 2141 S. epidermidis nasal isolates and 107 S. epidermidis isolates, which represent all the non-duplicate isolates obtained during the study period (2017) from inpatients with invasive S. epidermidis infections at Renji hospital, were used for the agar diffusion (Kirby-Bauer) antimicrobial activity assay. S. epidermidis isolates were cultured in TSB at 37 °C with shaking at 180 rpm for 12 h. Culture filtrates were obtained by centrifugation and sterilized using 0.22-μM filters. For each sample, 100-μl culture filtrate (four times 25 μl) was dropped on sterile filter paper disks. The dried filter papers were placed on tryptic soy agar (TSA) agar plates inoculated with the relevant test strains (M. catarrhalis and S. aureus) or Todd-Hewitt broth agar (THA) plates with D. pigrum and incubated at 37 °C for 24 h. To prepare M. catarrhalis and S. aureus test plates, autoclaved TSA was cooled to 45 °C and inoculated with the test strains to an OD600 = 0.01 for M. catarrhalis and 0.001 for S. aureus. For D. pigrum plates, autoclaved THA was cooled to 45 °C and inoculated with the test strain to an OD600 = 0.2. After mixing the medium with the cells, plates of 50 ml volume were poured.
For the live bacteria stamp assay according to Janek et al. (Janek et al., 2016), 2-μl cultures of live bacterial stocks were dropped onto test plates prepared as above, and incubated for the same time and conditions. For the liquid culture test according to Nakatsuji et al. (Nakatsuji et al., 2017), 1 x 104 CFU of the respective test strains were mixed with the obtained culture filtrates and grown for 24 h (for S. aureus and M. catarrhalis) or 72 h (for D. pigrum).
Isolate selection for biofilm and AMP studies
The bacterial isolates used for biofilm and AMP studies are listed in Table S1. The selection of isolates was as follows. For commensal S. epidermidis, we randomly selected three isolates per age group of the most abundant ST, ST59, as well as three isolates per group from other STs from nostril isolates obtained in this study. For infectious S. epidermidis, we randomly selected seven isolates of the predominant infectious ST2, from a strain collection of ST2 isolates published previously (Du et al., 2013), and seven isolates from other STs. For commensal S. aureus, we randomly selected three isolates per age group from nostril isolates obtained in this study. For clinical S. aureus, we selected isolates from our strain collection at Renji hospital representing the major community- and healthcare-associated methicillin-resistant S. aureus (MRSA) STs in China (STs 398, 59, 239, and 5). For C. propinquum and M. catarrhalis, we randomly selected two isolates each per age group from nostril isolates obtained in this study. For S. pneumoniae, we selected one isolate from the nostril isolates obtained in this study, one ATCC strain, and one clinical isolate from Renji hospital. For D. pigrum, we could only obtain four isolates from two people, and selected one from each. D. pigrum was grown in Todd-Hewitt broth (THB), all other bacteria were grown in tryptic soy broth (TSB) or synthetic nasal medium (SNM3) (Krismer et al., 2014), as indicated.
qRT-PCR
The human nasal epidermal cell line RPMI2650 (ATCC) was cultured in Dulbecco’s Modified Eagle’s medium (DMEM, GIBCO) with 10% fetal bovine serum. Cells were seeded to a confluence of up to 70% in 24-well plates. After stimulation with 104 CFU of live bacteria for 24 h, we harvested cells and analyzed gene expression by real-time Quantitative Reverse Transcription-PCR (qRT-PCR). Total RNA was isolated from cells using a Qiagen RNeasy kit (Qiagen 74106) according to the manufacturer’s instructions. After treatment with a genomic DNA wipeout buffer (Qiagen 205311), approximately 1 μg of total RNA was reverse-transcribed with a Reverse Transcription Kit (Qiagen 205311), and the obtained cDNA was used as a template for qRT-PCR using SYBR-green PCR reagent (Roche). The primers used are listed in Table S3. The GADPH gene was used as a housekeeping gene control. Reactions were performed in a MicroAmp Optical 96-well reaction plates using a 7500 Sequence Detector (Applied Biosystems).
Semi-quantitative biofilm assays
Overnight cultures of bacterial strains were diluted 1:1,000 into fresh TSB. For biofilm formation in SNM3, overnight cultures of bacterial strains grown in TSB were collected, washed twice with SNM3, and adjusted to OD600 = 1.0 in SNM3. Then, the diluted cultures were pipetted into sterile 96-well flat-bottom tissue culture plates and incubated at 37 °C for 24 h. Culture supernatants were gently removed, and wells were washed with phosphate-buffered saline (PBS). Biofilms at the bottom of the wells were treated with Bouin fixative for 1 h. Then, the fixative was gently removed, wells were washed with PBS, and stained with 0.4% (wt/vol) crystal violet. Biofilm formation was measured with a MicroELISA autoreader (BioTeK, USA) at 570 nm.
Killing assays
For planktonic bacteria: (TSB) Bacteria were grown to OD600 = 2.0, cultures were harvested, washed twice with 10 mM sodium phosphate buffer (pH 6.5) including 100 mM NaCl, and resuspended in the same buffer to a final concentration of 106 in each sample. (SNM3) Cultures grown at 37 °C for 24 h in SNM3 were harvested and adjusted to 2 × 107 CFU/ml. Then, AMPs were added to final concentrations of 75 μg/ml (hBD3, GenScript, USA) or 180 μg/ml (LL37, GenScript, USA), and samples were incubated at 37 °C for 3 h. Then, appropriate dilution series were plated on TSB agar plates. Plates were incubated at 37 °C for 24 h, after which colonies were counted.
For biofilm: (TSB) Bacterial cultures were adjusted to OD600 = 0.1 in TSB with 0.5% glucose. (SNM3) Precultures grown overnight in TSB were collected, washed twice with SNM3, and adjusted to OD600 = 1.0 in SNM3. Bacteria were then cultured in 96-well flat-bottom microtiter plates for 24 h. The culture media were removed carefully and AMPs (300 μg/ml hBD3 or 720 μg/ml LL37 in 100 μl sodium phosphate buffer, pH 7.2) were added to the biofilm. After 48 h, the biofilms were harvested carefully in 900 μl sodium phosphate buffer, pH 7.2, sonicated for 5 min, and vortexed for 15 min. Appropriate dilution series of the samples were plated on TSB agar plates. Plates were incubated at 37 °C for 24 h, after which colonies were counted.
Mouse nasal colonization model
Based on the observed levels of biofilm formation and AMP induction in epithelial cells, we selected four commensal S. epidermidis strains (ST59-N44-2: biofilm+, induction+; ST59-N2-2-1: biofilm+, induction−; ST59-N9-2: biofilm−, induction+; and ST59-NO105-4: biofilm−, induction−) and one infection-origin S. epidermidis strain (ST2-2011-413: biofilm+, induction−) for in-vivo experiments. Overnight bacterial cultures were diluted 1:100 in fresh TSB and grown for 4 h at 37 °C. Bacteria were harvested by centrifugation at 4000 × g for 10 min, washed twice in sterile PBS, and suspended in PBS. 6-week-old female C57BL/6J mice (n=8 mice/isolate) were anesthetized by intraperitoneal injection with Avertin (Sigma T48402), and 20-μl aliquots containing 1×107 CFU were instilled intranasally drop-wise once every day over 4 days, equally split between the two nostrils. The animals were sacrificed at the 6th day and the noses were surgically removed. Half of the noses were homogenized and used for RNA extraction using a Qiagen RNeasy kit (Qiagen 74106) according to the manufacturer’s instructions. The reverse transcription protocol was performed as described above (qRT-PCR protocol). The primers used are listed in Table S3. The other half of the noses were homogenized in 0.5 ml of PBS on ice using a manual homogenizer (Tiangen), the homogenized tissues were diluted and then plated on TSB agar for CFU determination.
Competitive nasal colonization model
The bacteria were prepared as above. The experiment was performed in 9 groups with n=8 mice/group. Groups consisted of mice instilled with 1×107 CFU of one of the selected commensal S. epidermidis, 1×107 CFU of the selected infection-origin S. epidermidis, or 1×107 CFU each of one of the selected commensal S. epidermidis mixed with the infection-origin S. epidermidis in a 1:1 mix. 6-week-old female C57BL/6J mice were anesthetized by intraperitoneal injection with Avertin, and 20-μl aliquots containing 1×107 CFU were instilled intranasally drop-wise once every day over 4 days, equally split between the two nostrils. The animals were sacrificed on day 6 after instilment and the noses were surgically removed. Half of the noses were homogenized and used for the detection of cramp expression. The other half of the noses were homogenized on ice in 0.5 ml of PBS using a manual homogenizer (Tiangen), the homogenized tissues were diluted and plated on TSB agar with or without ampicillin (100 μg/ml) or kanamycin (50 μg/ml) for strain distinction and determination of CFU. (The infection-origin S. epidermidis strain is resistant to both ampicillin and kanamycin (AmpR, KanR), in contrast to the commensal S. epidermidis isolates).
For competitive colonization with S. aureus or M. catarrhalis, the high biofilm (biofilm +) and strong AMP inducer (induction +) S. epidermidis strain ST59-N44-2 was used. 20-μl aliquots containing 1×107 CFU of S. epidermidis or PBS only as control was pipetted slowly into the nares of C57BL/6J mice (n=8 mice/group) or B6.129X1-Camptm1Rlg/J mice (n=4 mice/group) (Jackson Laboratories) every day over four days. On day 6 after the first installation of S. epidermidis, a 20-μl aliquot containing 1 × 108 CFU of S. aureus ST239-09-770 or 4 × 109 CFU of M. catarrhalis N3-5-2 was intranasally instilled drop-wise equally between the two nostrils. The mice were sacrificed at day 7 for M. catarrhalis or day 8 for S. aureus. The noses were removed immediately and homogenized in 0.5 ml of PBS. Then the homogenized tissues were diluted and plated on TSB blood agar for determination of S. aureus or on chocolate blood agar with or without vancomycin for determination of M. catarrhalis CFU. S. aureus was distinguished by hemolysis on blood agar. M. catarrhalis is naturally resistant to vancomycin, and also distinguishable by specific colony characteristics.
Histological analysis
Mice were killed by lethal anesthesia and the noses were harvested immediately. The nasal cavity was removed by one cut along the orbital bone between the eyes and one cut in the distal hard palate at the level of the first molar teeth. Excess soft tissue was removed, and the nose was rinsed in PBS, decalcified in 10% EDTA for at least 3 weeks, and then embedded in paraffin. The hematoxylin & eosin (H&E) staining was performed as previously described (Chamanza and Wright, 2015).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using GraphPad Prism Version 6.05 using 1-way or 2-way ANOVA, linear regression, or Fisher’s exact test, as appropriate, or using QIIME (statistical analysis presented in Fig. 1C,D). In ANOVAs, Tukey post-tests were used, which correct for multiple comparisons using statistical hypothesis testing. All error bars show the mean and standard deviation (SD). All replicates are biological. Random selections were performed using a random number generator [Rand function of Perl5 (http://www.perl.org)]. The number of replicates or animals used is given in the figure legends. For AMP and biofilm studies, the bacterial strains used are detailed in Table S1.
DATA AND CODE AVAILABILITY
Microbiome (16S rRNA) sequencing data were deposited to the NCBI Sequence Read Archive (SRA) database under the accession code PRJNA508588. All other data generated or analyzed during this study are included in this published article or in the supplementary information files.
Supplementary Material
Supplemental Data Set 1 – All identified bacterial isolates. Related to Figure 3.
Supplemental Data Set 2 – ANCOM results. Related to Figure 1.
Acknowledgments
This study was supported by the innovative research team of high-level local universities in Shanghai (to M. L and Qian L.), National Natural Science Foundation of China, grant numbers 81873957 and 81861138043 (to M. L.), 81772139 (to Qian L.), and 81701975 (to Qingyun L.), the Foundation for Innovative Research Groups of the National Natural Science Foundation of China, grant number 81421001 (to M. L.), the Shanghai Health System Talents Training Program, grant numbers 2017BR001 (to M. L.), and the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (NIAID), U.S. National Institutes of Health (NIH), project number ZIA AI000904 (to M. O.).
Footnotes
Declaration of interests
The authors declare no competing interests.
References
- Allaire JM, Crowley SM, Law HT, Chang SY, Ko HJ, and Vallance BA (2018). The Intestinal Epithelium: Central Coordinator of Mucosal Immunity. Trends Immunol. 39(9), 677–696. Published online 2018/05/03 DOI: 10.1016/j.it.2018.04.002. [DOI] [PubMed] [Google Scholar]
- Bjorksten B, Sepp E, Julge K, Voor T, and Mikelsaar M (2001). Allergy development and the intestinal microflora during the first year of life. J Allergy Clin Immunol. 108(4), 516–520. Published online 2001/10/09 DOI: 10.1067/mai.2001.118130. [DOI] [PubMed] [Google Scholar]
- Bomar L, Brugger SD, and Lemon KP (2018). Bacterial microbiota of the nasal passages across the span of human life. Curr Opin Microbiol. 41, 8–14. Published online 2017/11/21 DOI: 10.1016/j.mib.2017.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch A, de Steenhuijsen Piters WAA, van Houten MA, Chu M, Biesbroek G, Kool J, Pernet P, de Groot PCM, Eijkemans MJC, Keijser BJF, et al. (2017). Maturation of the Infant Respiratory Microbiota, Environmental Drivers, and Health Consequences. A Prospective Cohort Study. Am J Respir Crit Care Med. 196(12), 1582–1590. Published online 2017/07/01 DOI: 10.1164/rccm.201703-0554OC. [DOI] [PubMed] [Google Scholar]
- Capone KA, Dowd SE, Stamatas GN, and Nikolovski J (2011). Diversity of the human skin microbiome early in life. J Invest Dermatol. 131(10), 2026–2032. Published online 2011/06/24 DOI: 10.1038/jid.2011.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 7(5), 335–336. Published online 2010/04/13 DOI: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamanza R, and Wright JA (2015). A Review of the Comparative Anatomy, Histology, Physiology and Pathology of the Nasal Cavity of Rats, Mice, Dogs and Non-human Primates. Relevance to Inhalation Toxicology and Human Health Risk Assessment. J Comp Pathol. 153(4), 287–314. Published online 2015/10/16 DOI: 10.1016/j.jcpa.2015.08.009. [DOI] [PubMed] [Google Scholar]
- Chen YE, Fischbach MA, and Belkaid Y (2018). Skin microbiota-host interactions. Nature. 553(7689), 427–436. Published online 2018/01/25 DOI: 10.1038/nature25177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente JC, Ursell LK, Parfrey LW, and Knight R (2012). The impact of the gut microbiota on human health: an integrative view. Cell. 148(6), 1258–1270. Published online 2012/03/20 DOI: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond G, Beckloff N, Weinberg A, and Kisich KO (2009). The roles of antimicrobial peptides in innate host defense. Curr Pharm Des. 15(21), 2377–2392. Published online 2009/07/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond G, Legarda D, and Ryan LK (2000). The innate immune response of the respiratory epithelium. Immunol Rev. 173, 27–38. Published online 2000/03/17. [DOI] [PubMed] [Google Scholar]
- Dixon P (2009). VEGAN, a package of R functions for community ecology. J Veg Sci. 14(6), 927–930. [Google Scholar]
- Doss M, White MR, Tecle T, and Hartshorn KL (2010). Human defensins and LL-37 in mucosal immunity. J Leukoc Biol. 87(1), 79–92. Published online 2009/10/08 DOI: 10.1189/jlb.0609382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Zhu Y, Song Y, Li T, Luo T, Sun G, Yang C, Cao C, Lu Y, and Li M (2013). Molecular analysis of Staphylococcus epidermidis strains isolated from community and hospital environments in China. PLoS One. 8(5), e62742. Published online 2013/05/16 DOI: 10.1371/journal.pone.0062742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, and Knight R (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 27(16), 2194–2200. Published online 2011/06/28 DOI: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert JA, Quinn RA, Debelius J, Xu ZZ, Morton J, Garg N, Jansson JK, Dorrestein PC, and Knight R (2016). Microbiome-wide association studies link dynamic microbial consortia to disease. Nature. 535(7610), 94–103. Published online 2016/07/08 DOI: 10.1038/nature18850. [DOI] [PubMed] [Google Scholar]
- Gohl DM, Vangay P, Garbe J, MacLean A, Hauge A, Becker A, Gould TJ, Clayton JB, Johnson TJ, Hunter R, et al. (2016). Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat Biotechnol. 34(9), 942–949. Published online 2016/07/28 DOI: 10.1038/nbt.3601. [DOI] [PubMed] [Google Scholar]
- Gordon A, and Hannon G, 2012. FastX-toolkit. [Google Scholar]
- Grice EA, and Segre JA (2011). The skin microbiome. Nat Rev Microbiol. 9(4), 244–253. Published online 2011/03/17 DOI: 10.1038/nrmicro2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariri BM, and Cohen NA (2016). New insights into upper airway innate immunity. Am J Rhinol Allergy. 30(5), 319–323. Published online 2016/09/24 DOI: 10.2500/ajra.2016.30.4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K, Mansbach JM, Ajami NJ, Espinola JA, Henke DM, Petrosino JF, Piedra PA, Shaw CA, Sullivan AF, Camargo CA Jr., et al. (2016). Association of nasopharyngeal microbiota profiles with bronchiolitis severity in infants hospitalised for bronchiolitis. Eur Respir J. 48(5), 1329–1339. Published online 2016/11/02 DOI: 10.1183/13993003.00152-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoedemaekers A, Schulin T, Tonk B, Melchers WJ, and Sturm PD (2006). Ventilator-associated pneumonia caused by Dolosigranulum pigrum. J Clin Microbiol. 44(9), 3461–3462. Published online 2006/09/07 DOI: 10.1128/JCM.01050-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janek D, Zipperer A, Kulik A, Krismer B, and Peschel A (2016). High Frequency and Diversity of Antimicrobial Activities Produced by Nasal Staphylococcus Strains against Bacterial Competitors. PLoS Pathog. 12(8), e1005812. Published online 2016/08/05 DOI: 10.1371/journal.ppat.1005812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampmann B, and Jones CE (2015). Factors influencing innate immunity and vaccine responses in infancy. Philos Trans R Soc Lond B Biol Sci. 370(1671). Published online 2015/05/13 DOI: 10.1098/rstb.2014.0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Jo A, Jeon YJ, An S, Lee KM, Yoon SS, and Choi JY (2019). Nasal commensal Staphylococcus epidermidis enhances interferon-lambda-dependent immunity against influenza virus. Microbiome. 7(1), 80. Published online 2019/05/31 DOI: 10.1186/s40168-019-0691-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krismer B, Liebeke M, Janek D, Nega M, Rautenberg M, Hornig G, Unger C, Weidenmaier C, Lalk M, and Peschel A (2014). Nutrient limitation governs Staphylococcus aureus metabolism and niche adaptation in the human nose. PLoS Pathog. 10(1), e1003862. Published online 2014/01/24 DOI: 10.1371/journal.ppat.1003862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krismer B, Weidenmaier C, Zipperer A, and Peschel A (2017). The commensal lifestyle of Staphylococcus aureus and its interactions with the nasal microbiota. Nat Rev Microbiol. 15(11), 675–687. Published online 2017/10/13 DOI: 10.1038/nrmicro.2017.104. [DOI] [PubMed] [Google Scholar]
- Lai Y, Cogen AL, Radek KA, Park HJ, Macleod DT, Leichtle A, Ryan AF, Di Nardo A, and Gallo RL (2010). Activation of TLR2 by a small molecule produced by Staphylococcus epidermidis increases antimicrobial defense against bacterial skin infections. J Invest Dermatol. 130(9), 2211–2221. Published online 2010/05/14 DOI: 10.1038/jid.2010.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecuyer H, Audibert J, Bobigny A, Eckert C, Janniere-Nartey C, Buu-Hoi A, Mainardi JL, and Podglajen I (2007). Dolosigranulum pigrum causing nosocomial pneumonia and septicemia. J Clin Microbiol. 45(10), 3474–3475. Published online 2007/08/10 DOI: 10.1128/JCM.01373-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JYH, Monk IR, Goncalves da Silva A, Seemann T, Chua KYL, Kearns A, Hill R, Woodford N, Bartels MD, Strommenger B, et al. (2018). Global spread of three multidrug-resistant lineages of Staphylococcus epidermidis. Nat Microbiol. 3(10), 1175–1185. Published online 2018/09/05 DOI: 10.1038/s41564-018-0230-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Lei H, Li Z, Li H, Wang Y, and Lai Y (2013). A novel lipopeptide from skin commensal activates TLR2/CD36-p38 MAPK signaling to increase antibacterial defense against bacterial infection. PLoS One. 8(3), e58288. Published online 2013/03/09 DOI: 10.1371/journal.pone.0058288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Wang X, Gao Q, and Lu Y (2009). Molecular characterization of Staphylococcus epidermidis strains isolated from a teaching hospital in Shanghai, China. J Med Microbiol. 58(Pt 4), 456–461. Published online 2009/03/11 DOI: 10.1099/jmm.0.007567-0. [DOI] [PubMed] [Google Scholar]
- Linehan JL, Harrison OJ, Han SJ, Byrd AL, Vujkovic-Cvijin I, Villarino AV, Sen SK, Shaik J, Smelkinson M, Tamoutounour S, et al. (2018). Non-classical Immunity Controls Microbiota Impact on Skin Immunity and Tissue Repair. Cell. 172(4), 784–796 e718. Published online 2018/01/24 DOI: 10.1016/j.cell.2017.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CM, Price LB, Hungate BA, Abraham AG, Larsen LA, Christensen K, Stegger M, Skov R, and Andersen PS (2015). Staphylococcus aureus and the ecology of the nasal microbiome. Sci Adv. 1(5), e1400216. Published online 2015/11/26 DOI: 10.1126/sciadv.1400216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoc T, and Salzberg SL (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 27(21), 2957–2963. Published online 2011/09/10 DOI: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf TU, Cubas RA, Ghneim K, Cartwright MJ, Grevenynghe JV, Richner JM, Olagnier DP, Wilkinson PA, Cameron MJ, Park BS, et al. (2015). Global analyses revealed age-related alterations in innate immune responses after stimulation of pathogen recognition receptors. Aging Cell. 14(3), 421–432. Published online 2015/03/03 DOI: 10.1111/acel.12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mika M, Mack I, Korten I, Qi W, Aebi S, Frey U, Latzin P, and Hilty M (2015). Dynamics of the nasal microbiota in infancy: a prospective cohort study. J Allergy Clin Immunol. 135(4), 905–912 e911. Published online 2015/02/01 DOI: 10.1016/j.jaci.2014.12.1909. [DOI] [PubMed] [Google Scholar]
- Murphy TF, and Parameswaran GI (2009). Moraxella catarrhalis, a human respiratory tract pathogen. Clin Infect Dis. 49(1), 124–131. Published online 2009/06/02 DOI: 10.1086/599375. [DOI] [PubMed] [Google Scholar]
- Naik S, Bouladoux N, Linehan JL, Han SJ, Harrison OJ, Wilhelm C, Conlan S, Himmelfarb S, Byrd AL, Deming C, et al. (2015). Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature. 520(7545), 104–108. Published online 2014/12/30 DOI: 10.1038/nature14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuji T, Chen TH, Narala S, Chun KA, Two AM, Yun T, Shafiq F, Kotol PF, Bouslimani A, Melnik AV, et al. (2017). Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci Transl Med. 9(378). Published online 2017/02/24 DOI: 10.1126/scitranslmed.aah4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Conlan S, Polley EC, Segre JA, and Kong HH (2012). Shifts in human skin and nares microbiota of healthy children and adults. Genome Med. 4(10), 77. Published online 2012/10/12 DOI: 10.1186/gm378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M (2004). Virulence factors of the coagulase-negative staphylococci. Front Biosci. 9, 841–863. Published online 2004/02/10. [DOI] [PubMed] [Google Scholar]
- Otto M (2006). Bacterial evasion of antimicrobial peptides by biofilm formation. Curr Top Microbiol Immunol. 306, 251–258. Published online 2006/08/17. [DOI] [PubMed] [Google Scholar]
- Otto M (2009). Staphylococcus epidermidis--the 'accidental' pathogen. Nat Rev Microbiol. 7(8), 555–567. Published online 2009/07/18 DOI: 10.1038/nrmicro2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M (2014). Physical stress and bacterial colonization. FEMS Microbiol Rev. 38(6), 1250–1270. Published online 2014/09/13 DOI: 10.1111/1574-6976.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettigrew MM, Laufer AS, Gent JF, Kong Y, Fennie KP, and Metlay JP (2012). Upper respiratory tract microbial communities, acute otitis media pathogens, and antibiotic use in healthy and sick children. Appl Environ Microbiol. 78(17), 6262–6270. Published online 2012/07/04 DOI: 10.1128/AEM.01051-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piewngam P, Zheng Y, Nguyen TH, Dickey SW, Joo HS, Villaruz AE, Glose KA, Fisher EL, Hunt RL, Li B, et al. (2018). Pathogen elimination by probiotic Bacillus via signalling interference. Nature. 562(7728), 532–537. Published online 2018/10/12 DOI: 10.1038/s41586-018-0616-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings BA, Higgins TS, and Han JK (2013). Bacterial pathogens in the nasopharynx, nasal cavity, and osteomeatal complex during wellness and viral infection. Am J Rhinol Allergy. 27(1), 39–42. Published online 2013/02/15 DOI: 10.2500/ajra.2013.27.3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassone-Corsi M, Nuccio SP, Liu H, Hernandez D, Vu CT, Takahashi AA, Edwards RA, and Raffatellu M (2016). Microcins mediate competition among Enterobacteriaceae in the inflamed gut. Nature. 540(7632), 280–283. Published online 2016/11/01 DOI: 10.1038/nature20557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simanski M, Erkens AS, Rademacher F, and Harder J (2019). Staphylococcus epidermidis-induced Interleukin-1 Beta and Human Beta-defensin-2 Expression in Human Keratinocytes is Regulated by the Host Molecule A20 (TNFAIP3). Acta Derm Venereol. 99(2), 181–187. Published online 2018/10/18 DOI: 10.2340/00015555-3073. [DOI] [PubMed] [Google Scholar]
- Simon AK, Hollander GA, and McMichael A (2015). Evolution of the immune system in humans from infancy to old age. Proc Biol Sci. 282(1821), 20143085. Published online 2015/12/25 DOI: 10.1098/rspb.2014.3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalka B (1992). Bacteriocin typing of staphylococci. Acta Vet Brno. 61, 179–187. [Google Scholar]
- Stewart CJ, Ajami NJ, O'Brien JL, Hutchinson DS, Smith DP, Wong MC, Ross MC, Lloyd RE, Doddapaneni H, Metcalf GA, et al. (2018). Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature. 562(7728), 583–588. Published online 2018/10/26 DOI: 10.1038/s41586-018-0617-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokholm J, Blaser MJ, Thorsen J, Rasmussen MA, Waage J, Vinding RK, Schoos AM, Kunoe A, Fink NR, Chawes BL, et al. (2018). Maturation of the gut microbiome and risk of asthma in childhood. Nat Commun. 9(1), 141. Published online 2018/01/13 DOI: 10.1038/s41467-017-02573-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo SM, Mok D, Pham K, Kusel M, Serralha M, Troy N, Holt BJ, Hales BJ, Walker ML, Hollams E, et al. (2015). The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell Host Microbe. 17(5), 704–715. Published online 2015/04/14 DOI: 10.1016/j.chom.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Roeden SE, Thijsen SF, Sankatsing SU, and Limonard GJ (2015). Clinical relevance of Corynebacterium pseudodiphtheriticum in lower respiratory tract specimens. Infect Dis (Lond). 47(12), 862–868. Published online 2015/07/28 DOI: 10.3109/23744235.2015.1070962. [DOI] [PubMed] [Google Scholar]
- von Eiff C, Becker K, Machka K, Stammer H, and Peters G (2001). Nasal carriage as a source of Staphylococcus aureus bacteremia. Study Group. N Engl J Med. 344(1), 11–16. Published online 2001/01/04 DOI: 10.1056/NEJM200101043440102. [DOI] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, and Cole JR (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 73(16), 5261–5267. Published online 2007/06/26 DOI: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, and Nouwen JL (2005). The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis. 5(12), 751–762. Published online 2005/11/29 DOI: 10.1016/S1473-3099(05)70295–4. [DOI] [PubMed] [Google Scholar]
- Yan M, Pamp SJ, Fukuyama J, Hwang PH, Cho DY, Holmes S, and Relman DA (2013). Nasal microenvironments and interspecific interactions influence nasal microbiota complexity and S. aureus carriage. Cell Host Microbe. 14(6), 631–640. Published online 2013/12/18 DOI: 10.1016/j.chom.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Kruse RL, Lin WV, and Musher DM (2018). Corynebacteria as a cause of pulmonary infection: a case series and literature review. Pneumonia (Nathan). 10, 10. Published online 2018/10/17 DOI: 10.1186/s41479-018-0054-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. (2012). Human gut microbiome viewed across age and geography. Nature. 486(7402), 222–227. Published online 2012/06/16 DOI: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data Set 1 – All identified bacterial isolates. Related to Figure 3.
Supplemental Data Set 2 – ANCOM results. Related to Figure 1.
Data Availability Statement
Microbiome (16S rRNA) sequencing data were deposited to the NCBI Sequence Read Archive (SRA) database under the accession code PRJNA508588. All other data generated or analyzed during this study are included in this published article or in the supplementary information files.