

Abstract
The foremost aim of this thermodynamic study was to evaluate the pharmacokinetics (PK) and pharmacodynamics (PD) profiles of linagliptin (LG), rabeprazole sodium (RS), and their 1:1 formed complex by interacting with bovine serum albumin (BSA) at physiological pH 7.4. The molecular interactions of these ligands with the desired biomolecule were substantiated by the spectral quelling of fluorescence intensity of BSA. The fluorescent test and molecular docking revealed that the quenching mechanism was a spontaneous and exothermic static process, and the protein gained its secondary structure due to the interactions. The spectroscopic method was exercised to determine the thermodynamic factors that supported the interactions mediated by van der Waals forces and hydrogen bonds. The activation energy of the formed complex was higher than its precursor drugs while interacting with BSA, and the energy transformation profiles were studied by UV‐fluorescence overlaid curves according to Förster resonance energy transfer (FRET) theory. The double log plot verified that these ligands bound with protein at a 1:1 ratio, which was confirmed by the approximately estimated values of the binding parameters. The drastically lower value of the binding constant of the formed complex suggested the lower half‐life as well as its triggered elimination rate from the cardiovascular system, which may be an initial indicator of the reduced hypoglycemic property of linagliptin. Moreover, the UV–vis and synchronous fluorescence spectroscopic methods affirmed the conformational changes of the BSA due to drug‐protein complexation and polarity alterations in the microenvironment of disparate chromophores of the biomolecule.
Keywords: fluorescence quenching, linagliptin, lower half‐life, molecular docking, rabeprazole sodium, synchronous fluorescence, UV–vis spectroscopy
ABBREVIATIONS
- 3D
three‐dimensional
- BSA
bovine serum albumin
- FRET
Forster resonance energy transfer
- LG
linagliptin
- PD
pharmacodynamics
- PDB
protein data bank
- Phe
phenylalanine
- PK
pharmacokinetics
- PPI
proton pump inhibitor
- RS
rabeprazole sodium
- SA
serum albumin
- Trp
tryptophan
- Tyr
tyrosine
- UV
ultraviolet
1. INTRODUCTION
In living organisms, protein plays a vital role by participating in various biological processes. Serum albumins (SAs), which account for 50%–65% of the total protein, are the highest soluble and affluent protein present in the circulatory system, involved in the binding, and act as carriers of ligands or responsible for the movement of various drugs and biomolecules. 1 , 2 Since most of the administered medications reversibly interact with serum proteins, they are transported in the systemic circulation primarily as protein complexes. In this manner, drug–protein interaction also has immense biological importance and has been found to play a critical role in pharmacology. 3 The bound drug works as a depot while the free drug contributes to the desired pharmacological activity. The drug stability and toxicity in the chemotherapeutic process can also be significantly affected by this drug–protein interaction. Therefore, the drug–SA interaction modeling study is imperative to describe the direct association between the structure and activity of the drug. 4 , 5
Moreover, many mandatory thermodynamic data or others, structural characteristics, or basic highlights of small molecules can be established from the investigation of interactions between drug particles and protein. Hence insight into these interactions of a new ligand with protein is a significant step for the drug development process. 6 As an in vitro model, bovine serum albumin (BSA) is widely used for studying drug–protein interaction since its molecular composition is almost 76% similar to that of human SA. 7 Besides, the drug–protein interaction causes the interference of the binding of other drugs due to conformational changes or overlap of the binding sites. Thus, this particular study plays a dominant role in construing the potential drug–drug interactions. 4
The conventional fluorescence spectroscopic method is frequently used to perform the thermodynamic study between the drugs and proteins. The technique is applied to measure the fluorescence intensity at the maximum emission wavelength of the protein before and after the addition of ligands. It is commonly utilized to calculate the reaction mechanisms, binding parameters, and distances between energy donor and acceptor molecules, other detailed information with the thermodynamic factors. 8 , 9 Due to the high sensitivity, this fluorescence spectroscopy is also a beneficial tool to extrapolate the drug–drug interaction, primarily considering the predicted half‐life of the formed complex and the precursor molecules. 4 , 9
Linagliptin (LG) is a potent dipeptidyl peptidase‐4 inhibitor that works against diabetic mellitus (type II) by enhanced insulin production. It features a nonlinear pharmacokinetics (PK), concentration‐dependent protein binding, limited renal clearance, and no mandatory dosage adjustment criteria that revealed a particular type of PK/PD model. 10
Rabeprazole sodium (RS), a well‐known drug as a proton pump inhibitor (PPI), blocks the secretion of gastric acid from parietal cells through the inhibition of H+/K+‐ATPase system. 11 It is a prodrug, activated in the acidic pH, prescribed to treat a common disease peptic ulcer and gastro‐oesophageal reflux disorder. About 90% of the drug is excreted in the urine, basically as thioether carboxylic acid, as well as its metabolites of glucuronide and mercapturic acid. 12 As LG prescribed diabetic patients may take RS, the thermodynamic study of the developed complex between the two drugs is essential to interpret the plausible drug–drug interaction. 13
This current research's primary purpose was to analyze the bindings of both drugs and their 1:1 complex (LG‐RS) with BSA by molecular docking and fluorescence quenching strategies. By calculating the Stern–Volmer constants, the binding constants, and the binding points at three different temperatures at physiological conditions, the detection and characterization of the binding moods of these two drugs and LG‐RS complex with BSA were investigated. The energy transfer profile of these ligands with the biomolecule was also established by the Förster resonance energy transfer (FRET) theory. Most importantly, thermodynamic factors such as change of enthalpy (∆H), entropy (∆S), and free energy (∆G) were calculated to assess the binding profiles of these ligand–protein complexes. The geometric coordination of these drugs while binding with protein was studied to confirm the three‐dimensional (3D) interacting profile by utilizing the molecular docking model. In order to investigate the conformational changes of BSA owing to the interactions, the synchronous fluorescence and ultraviolet (UV)‐vis spectroscopic approaches were conducted at room temperature through maintaining physiological conditions.
2. MATERIALS AND METHODS
2.1. Instruments
F‐7000 spectrophotometer (Hitachi, Japan) equipped with 1 cm quartz cell was utilized for taking the fluorescence spectroscopy of the samples. Some other analytical tools like a UV–visible spectrophotometer (UV 1800; Shimadzu, Japan), an electric balance (AS 220.R2; Shimadzu), a pH meter (Orion Star A111), a sonicator (Ultrasons Medi. Li), a thermostatic water bath (Unitronic Orbital, Spectra, Spain), etc., were utilized for the analysis.
2.2. Drugs and reagents
From ACI Pharmaceuticals Ltd. (Dhaka, Bangladesh), we accepted LG (potency: 99.44%) and RS (potency: 96.65%) as gift samples. Potassium phosphate dibasic (K2HPO4), potassium di‐hydrogen orthophosphate (KH2PO4), phosphoric acid 85%, acetonitrile, methanol, ethanol, etc., were of analytical grade and supplied by CARS, University of Dhaka (Dhaka, Bangladesh). A mother solution of BSA was prepared in phosphate buffer at blood pH 7.4.
2.3. Preparation of pH 7.4 buffer solutions
Note that 65 ml of 0.01 M KH2PO4 was added with 235 ml of 0.01 M K2HPO4 to yield the targeted pH buffer solution. The pH 7.4 was adjusted by adding two to three drops of concentrated sodium hydroxide. Then the prepared buffer solution was diluted with DM water to 1000 ml. 13
2.4. Preparation of 100 ml of 0.1 mM stock solutions
Note that 47.25 mg of LG or 38.24 mg of RS was dissolved in DM water to make 0.1 mM mother solution of LG and RS, respectively. The final volume was adjusted to 100 ml by the same solvent. Then the prepared stock solutions were kept in the sonicator for 10 min to make proper solutions. The stock solutions were diluted with the freshly prepared buffer solution to get the desired concentrations during the experiment. 13
2.5. Synthesis and characterizations of the drug–drug complex
The LG‐RS complex was synthesized at a ratio of 1:1 in a water bath at 37 ± 2°C for 24 h according to the described method by Hossain et al. 13 Then the synthesized complex was characterized by various spectroscopic and chromatographic methods. 13
2.6. Molecular docking
The widely used software was carried out to establish the best‐fitted model, geometric orientation, molecular rotation, binding affinity, graphical visualization, and mechanisms of interactions of the drugs with BSA. The molecular docking was done by utilizing PyRx software to discover the best‐fitted model of the ligands with BSA. The ID of 3D BSA crystal structure was PDB: 3v03, and that was used for molecular docking. The designed protein was collected from the protein data bank (PDB; https://www.rcsb.org/structure/3v03), and the geometric structures of the ligands were downloaded from the link https://pubchem.ncbi.nlm.nih.gov/. The prominent binding sites were pointed out prior to the docking analysis. Furthermore, the computational analysis of LG and RS with the protein was examined by using PyRx, and the analysis of the result was interpreted by running BIOVIA Discovery Studio 4.5. PyMol was used to remove any unexpected ligands and water molecules from the protein. The energy minimization through conjugate gradient strategy, a fundamental process to clean and prepare chemically correct and optimized macromolecule, was performed to eradicate unwanted residues of the protein by utilizing Swiss PDB viewer 4.1. Then the cleaned and minimized protein and SDF form of ligands were imported in PyRx software for docking by utilizing AutoDock Vina. The energy minimized protein and optimized drug structures were identified as macromolecule and ligand, respectively, using Vina Wizard. After selecting ligand and macromolecule, AutoDock Tools (ADT) was run to convert pdb into pdbqt format using molecular analysis of drug–protein interaction. The computational docking strategy utilizing the AutoDock Vina protocol anticipated the potential interactive affinity between the ligand and BSA. The maximum docking grid box was set as the most probable active binding sites of the whole biomolecule, where the grid‐mapping was fixed as Vina search space at center X: 64.25, Y: 25.82, Z: 32.74, and dimensions 142.70, 66.12, and 90.28 Å along with X‐, Y‐, and Z‐axis, respectively. The remaining parameters during the docking analysis were according to default settings, and the spacing of the grid point was 0.375 Å that illustrated a simulation environment. After completing docking, the best docking scores and drug–protein interactions were collected in an excel sheet. The binding affinity of the drugs to the target molecule, BSA, was enumerated utilizing binding free energy. A postulation was fixed as the lower the binding energy values, the higher the binding affinity of the ligands to the protein. The unit was expressed as kcal/mole to represent the binding affinity revealed by the negative docking score. Finally, PyMol 2.3 and BIOVIA Discovery Studio version 4.5 were adopted to visualize and detect the noncovalent interaction between the drugs and protein.
2.7. Fluorescence quenching
The fluorescence spectra were conducted at three different temperatures, 298, 308, and 318 K, maintained by using a circulating water bath. The aqueous drug concentrations ranged from 1 to 10 mM, while BSA concentration was fixed at 0.025% (w/v) BSA solution. Most of the 200–500 nm range of fluorescence emission spectra were reported at two distinct excitation wavelengths, 280 , and 293 nm. 7 In this current research, the prepared sample was scanned and repeated three times to record the emission fluorescence spectrum at around 340 nm. The experimental error was less than 1%, which endorsed the reproducibility of the collected data.
The protein, consisting of tryptophan (Trp), tyrosine (Tyr), and phenylalanine (Phe), is considered to have intrinsic fluorescence property. The frequency of its intrinsic fluorescence also varies, either quenching or de‐quenching with the ligands when interacting with other molecules such as drugs or any kind of ligands. 14 When the residues of the protein interact with any drug or ligand, the intrinsic fluorescence generally changes with the drug's variation of concentrations. Consequently, the fluorescence technique is often considered a useful method to interpret the binding mechanisms of ligands' interactions with proteins. Quenching of fluorescence intensity reduces the fluorescence quantum of a fluorophore exhorted by various sorts of interactions, such as excited state and ground‐state molecular reactions, energy transformation, and collisional quenching. Molecular interactions of a biomolecule with ligands cause quenching of fluorescence strength of the fluorophore. 8 Commonly, the mechanism of fluorescence quenching can be explained under three classifications: (i) static quenching, which is occurred due to ground‐state complexation between the quencher and the fluorophore; (ii) dynamic quenching, which is resulted from the collision between the molecules in the transition to the excited state; and (iii) combination of the two mechanisms, which is caused by both collision and complexation with the similar quencher. 15 , 16 , 17 In general, the dynamic and static quenching mechanisms can be identified based on the correlation between the quenching constant and the temperature or the excited state lifetime. 17 In static quenching mechanism, the quenching constant is reduced with temperature, whereas the value of quenching constant is augmented with temperature, which is called dynamic quenching. The fluorophore and the ligand interact during the exciting lifetime phase in the energetic quelling process, whereas inert quenching approach indicates complex formation between the fluorophore and quencher. 15 To investigate the interaction property, the fluorescence quenching data can be established by the Stern–Volmer relationship 16 :
where F 0 is fluorescence intensity before the quencher is added, and F is fluorescence intensity after it is reacted. [Q] is associated with quencher concentration with units of mol/L. Hence, Stern–Volmer equation can be utilized to find out Ksv values by linear regression of the plot of F 0/F against [Q]. kq is considered as the bimolecular quenching rate constant with units of L mol–1s–1. τ 0 is the average lifetime of the pure SA. The mean fluorescence lifetime value was collected from the previously published literature, τ 0 = 10–8 s. 17 By utilizing the average lifetime, the quenching rate constant is determined by the following relationship:
The magnitude of activation energy required for the quenching process can be utilized to elucidate the binding mechanism of the ligands with the protein. In order to determine the apparent activation energy, the following Arrhenius equation can be used 18 :
Here, kq is known as the quenching rate constant and Ea denotes the quenching process's activation energy. R and T are familiar as the gas constant and absolute temperature, respectively, where A is known as the preexponential factor.
2.8. Thermodynamic parameters
The thermodynamic process is considered responsible for complex formation. The temperature‐dependent thermodynamic parameters are engaged to further investigate the acting forces of ligands or drugs with protein. 19 The binding forces acting upon ligand–protein complexation may be classified as hydrophobic forces, electrostatic interactions, Van der Waals interactions, and hydrogen bonds. 16 To determine the interaction forces between the drug and BSA, the thermodynamic parameters, which are considered the prerequisites for determining binding nature, can be calculated from the following van ’t Hoff equation 20 :
where Ka is the constant at the corresponding temperature equivalent to the Stern–Volmer quenching constants (Ksv ). Both enthalpy change (∆H) and entropy change (∆S) were computed from the slope and intercept value found from the curve of ln Ksv against 1/T. The following relationship can be used to calculate the appropriate change of free energy (∆G) during the thermodynamic quenching process:
The negative sign of ∆G can demonstrate the spontaneous interaction process. When both enthalpy change and entropy change are positive, that is, ∆H > 0, ∆S > 0, hydrophobic interaction occurs, when both of them are negative, that is, ∆H < 0, ∆S < 0, van der Waals forces and hydrogen bonds are creditworthy for the processes, when enthalpy change is negative, and entropy change is positive, that is, ∆H < 0, ∆S > 0, only electrostatic forces are influencing for the interactions. 16
2.9. Binding parameters
The affinity of a ligand to a receptor is one of the imperative indicators for assessing the ligand's pharmacological activity, which is dependent upon the binding parameters of the ligand upon interaction with the biomolecule. In the ligands' adhering process to a set of active binding sites of protein, the binding constants and the number of binding sites play a significant role. Generally, the higher the value of the binding constant dictates the drug's higher affinity to the protein. According to the postulation, the fluorescence of BSA was quenched by these molecules (LG, RS, and their 1:1 formed complex) in the manner of a dynamic process that can be thought the formation of a nonfluorescent complexation. In this regard, the binding parameters can be obtained by utilizing the following formula described by Kou et al. 20 :
where Kb and n describe the constant of binding to a site and the number of binding per molecule, respectively. [D]0 and [P]0 are the total concentration of drug (ligand) and protein, respectively. Both of the parameters (Kb and n) can be computed from the intercept and slope values of the regression line found from log(F 0–F)/F versus log{[D]0 – n [P]0(F 0 – F)/F} plot. It is important to state that the change of binding affinity was enumerated by applying the following relationship 21 , 22 :
Here, Kb 1 and Kb 2 indicate the binding constants of the pure drug and drug–drug complex, respectively.
2.10. Energy transfer and binding distance
The transformation of energy occurs when the distance between protein and ligand remains below 8 nm. 22 According to FRET theory, it will be possible when the emission spectrum of protein overlaps enough to the acceptor's absorption spectrum. The distance between protein and ligand can be estimated by utilizing the following equations 23 , 24 :
where E is the efficiency of energy transfer, r is the distance between protein and ligand, R 0 represents the critical distance when it is found at 50% transfer efficiency and can be determined by the following equation 25 :
Here, k 2 is known as the orientation factor, and it is supposed to be 2/3 for this experiment as a random factor. φ and n are supposed to be the quantum yield of the amino acid residue of the protein sequence and the medium's refractive index. J is expressed as the integral overlap area between the emission of donor and absorption of acceptor spectral curves, which might be calculated by the following formula 26 :
where ϵ(λ) and F(λ) are known as the molar absorption coefficient of the acceptor and fluorescence intensity of BSA of the donor on the wavelength λ, respectively.
2.11. Conformational changes of BSA
Although the binding of the ligands with the BSA was affirmed by the molecular docking and change of intrinsic fluorescence intensity or quenching approaches, it is still a question about whether the interactions affected the molecular structure and microenvironment of the biomolecule. Therefore, the UV–vis absorptions and synchronous fluorescence spectroscopic analyses were further investigated for the protein's conformational changes. 14
UV–vis spectra measurements: The UV–vis spectra of the pure BSA and LG were recorded by utilizing UV‐1800 Spectrophotometer (Shimadzu) equipped with a 10 mm quartz cell. The wavelength range was 220–400 nm, and the sample medium was simulated with the physiological condition at pH 7.4.
Synchronous fluorescence spectroscopy: The synchronous fluorescence spectra were collected from an F‐7000 spectrophotometer (Hitachi) equipped with a 10 mm quartz cell at physiological pH 7.4. The BSA solution concentration was steady at 10 µM, where the ligand concentrations varied from 5 to 100 µM. The difference between emission and excitation wavelengths (Δλ = λ em – λ ex) was set fixed at 60 and 15 nm during the scanning process to observe the spectral property of Trp and Tyr, respectively. 17 The temperature was controlled at 300 K for both amino acid residues characterization by synchronous fluorescence spectroscopy.
2.12. Data analysis and software
PyRx, PyMol 2.3, Swiss PDB viewer 4.1, and Discovery Studio 4.5 were utilized for molecular docking of the ligand‐biomolecule interactions. Microsoft Excel (MS Excel, 2010) was used for data processing and analysis.
3. RESULTS AND DISCUSSIONS
3.1. Molecular docking
The fluorescence spectroscopic and molecular docking approaches are widely used to establish useful thermodynamic data and the ligand‐protein interactive mechanisms. Trp fluorescence examines the multifunctional binding characteristics of BSA with any ligand or drug while interactive molecular configurations can be designed by the molecular modeling study. Trp has two interacting residues in BSA: Trp‐134 in domain I and Trp‐212 in domain II. 27 The energy minimization of the SA protein was done by utilizing PyMol and Swiss PDB viewer 4.1. The docking between the drugs and the protein was run by AutoDock Vina in PyRx software. The combined file obtained from PyMol was analyzed for both 2D and 3D structures by using Discovery Studio 4.5. The screening strategy and scoring function were applied to get the top docking score during the computational modeling study. The highest negative docking scores for the LG‐BSA complex and RS‐BSA complex were –9.5 and –7.9 kcal/mol, respectively, tabulated in Table 1, which revealed the highest binding affinity of the ligands toward the protein. The best‐fitted docking model and the geometric orientation of LG and RS with BSA are shown in Figures 1 and 2, respectively. The simulated docking designs illustrated the preferred binding site and more favorable hydrophobic pockets of BSA during complexation. The ligand was surrounded and bonded with many amino acid residues through mainly hydrophobic interactions. A clear view from Figure 1b and c was sketched from LG‐BSA interaction that the ligand was surrounded in chain B by Arg 458 (4.43, 4.53, and 3.87A°), Ala 193 (4.45, 5.29, and 4.74A°), Leu 189 (4.23 and 5.34A°), Asp 108 (3.53A°), Pro 110 (4.81A°), and Ile 455 (5.22A°) through various interactions like electrostatic and hydrophobic interactions. The 3D structures of LG were interacted with the protein in Figure 1d–f and showed the aromatic (edge vs. face), hydrogen bond (donor vs. acceptor), and hydrophobic (positive vs. negative) side pockets of the target molecules. Similarly, the docking models of RS with BSA were delineated in Figure 2a–f with 3D, 2D, aromatic, hydrogen bond, and hydrophobic interactions, where the drug was surrounded by Val 215 and 481, Leu 326 and 346, Lys 211, Ala 212, Arg 208, and Glu 353 via hydrogen bond and hydrophobic interactions (Table 1).
TABLE 1.
Noncovalent interactions of linagliptin (LG) and rabeprazole sodium (RS) with bovine serum albumin (PDB ID: 3v03)
| Drug Code | Structure (3D) | Binding affinity (kcal/mol) | Hydrogen bond (AA…Ligand) | Hydrophobic interaction (AA…Ligand) | Electrostatic interaction (AA…Ligand) |
|---|---|---|---|---|---|
| LG |
|
–9.5 | ASP108 (3.53) Carbon‐hydrogen bond | ARG458 (4.53) Pi‐Cation | |
| PRO110 (4.81) Alkyl | ARG458 (3.87) Pi‐Cation | ||||
| LEU189 (4.23) Alkyl | |||||
| LEU189 (5.34) Alkyl | |||||
| ILE455 (5.22) Pi‐Alkyl | |||||
| ALA193 (4.45) Alkyl | |||||
| ALA193 (5.29) Pi‐Alkyl | |||||
| ALA193 (4.74) Pi‐Alkyl | |||||
| ARG458 (4.43) Pi‐Alkyl | |||||
| RS |
|
–7.9 |
GLU353 (2.26) C‐O…H‐N ARG (2.24) O‐H…O = S |
VAL215 (5.13) Pi‐Alkyl VAL481 (4.82) Alkyl LYS211 (4.46) Amide Pi‐Stacked ALA212 (3.72) Pi‐sigma ALA212 (3.80) Pi‐sigma ALA212 (3.63) Alkyl LEU326 (5.48) Pi‐Alkyl LEU346 (4.14) Alkyl |
Abbreviations: AA, amino acid; ALA, alanine; ARG, arginine; ASP, asparate; GLU, glutamate; LEU, leucine; LG, linagliptin; Lys, Lysine; PRO, proline; RS, rabeprazole sodium; VAL, valine.
FIGURE 1.
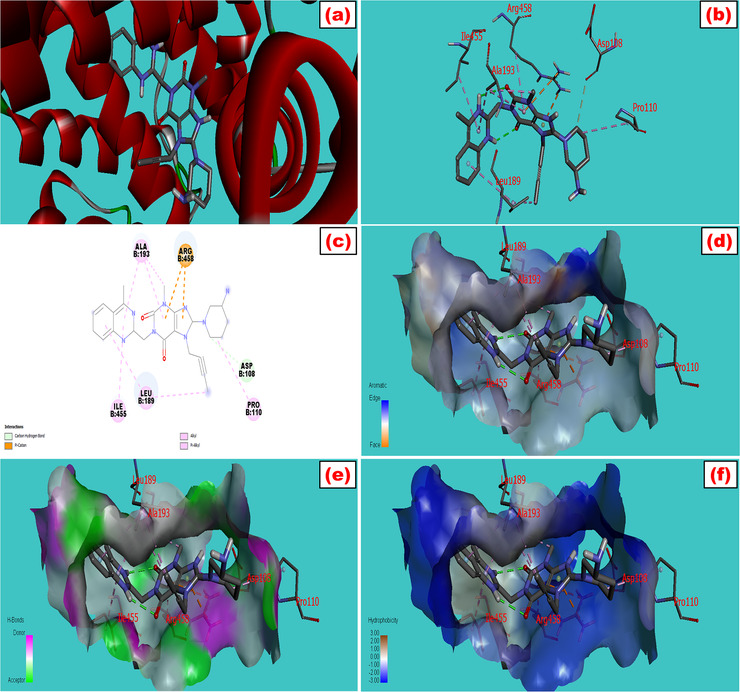
Molecular docking model of bovine serum albumin with (a) linagliptin which showed the best fitted geometric orientation of the ligand with protein. (b and c) 3D and 2D docking models of linagliptin with BSA, respectively. (d–f) The aromatic, hydrogen bond, and hydrophobic interactions across multiple side pockets of the protein, respectively. The various types of bonding were shown during interactions with their bond lengths. The SDF 3D conformer of the drug and PDB format of BSA (PDB: 3v03) were downloaded from PubChem and protein data bank, respectively. The ligand was docked with the protein by utilizing a set of extensive software like PyRx, PyMol 2.3, and Discovery Studio 4.5 to study the best‐fitted molecular orientation of LG with BSA. AutoDock Tools (ADT) and Swiss PDB viewer 4.1 were used to convert pdb into pdbqt format and to minimize the energy
FIGURE 2.
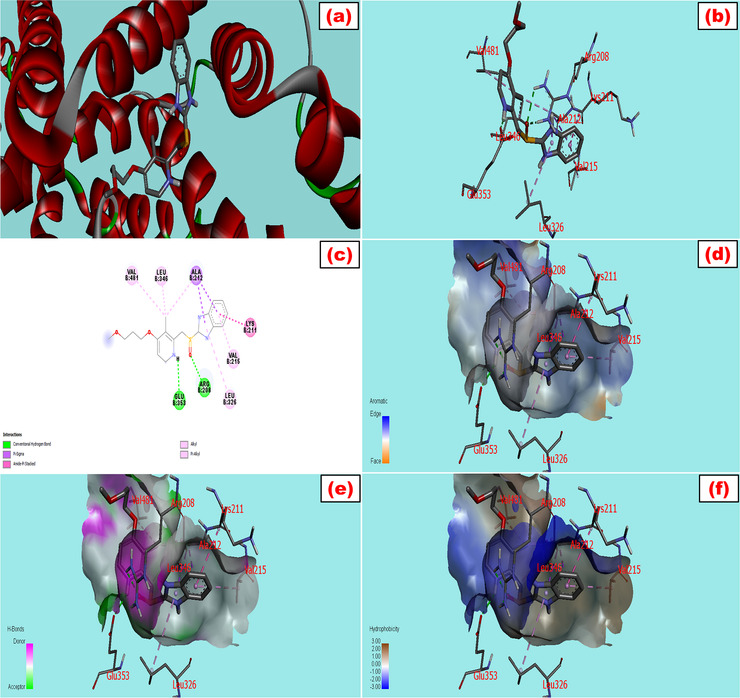
Molecular docking model of bovine serum albumin with (a) rabeprazole, which showed the best fitted geometric orientation of the ligand with protein. (b and c) 3D and 2D docking models of rabeprazole with BSA, respectively. (d–f) The aromatic, hydrogen bond, and hydrophobic interactions across multiple side pockets of the protein, respectively. The various types of bonding were shown during interactions with their bond lengths
3.2. Fluorescence quenching mechanism
Some amino acids (Trp, Tyr, Phe) located in the peptide chain play the major contributions to the fluorescence of the protein. 14 Trp residue displays the principal role at the maximum excitation (λ ex = 285 nm) and emission wavelengths (λ em = 350 nm). Tyr also plays 1% of the relative intensity of Trp at the 275 nm excitation and 303 nm emission wavelengths, while Phe contributes the lowest fluorescence intensity of the protein. 28 Trp residue showed the maximum contribution due to its maximum excitation, and emission wavelengths are analogous to the excitation and emission wavelengths (λ ex = 285 nm, and λ em = 355 nm, respectively) of BSA. 28
In order to ascertain the participation of both Trp and Tyr residues during the interactions of serum protein with these ligands, we conducted fluorescence spectroscopic analyses at the excitation wavelengths at 280 and 293 nm to sort out different quenching patterns. The quenching of BSA fluorescence intensity at both wavelengths indicated that the Tyr residue along with Trp residue contributed to quench the fluorescence intensity of the protein, which was endorsed by the distinct quenching characteristics of the biomolecule due to the interactions of two different amino acid residues (Figure 3). The transfer of fluorophores from high to low polarity environments usually causes spectral shifts (10–20 nm) in the spectrum of excitation and emission of the drugs as the intrinsic fluorescence emissions are environmentally sensitive. 29 , 30 It was obvious that the fluorescence of BSA protein was quenched after adding the ligands with the protein solutions, which was the outcome of a static ligand–protein complex formation. BSA fluorescence quenching spectra with different concentrations of LG, RS, and their 1:1 complex were measured under physiological conditions. A wide band centered at around 340 nm was found by observing the spectral curves, as shown in Figure 3. It was clearly viewed that the presence of individual drug alone or complex of drugs leads to a regular quenching in fluorescence intensity with an insignificant hypsochromic shift by 344 nm → 341 nm for the LG‐BSA system (Figure 3a), 339 nm → 335 nm for RS‐BSA system (Figure 3d), and 336 nm → 333 nm for (LG‐RS)‐BSA (Figure 3g) system in the maximum emission wavelength. The Stern‐Volmer plots for quenching BSA fluorescence by LG, RS, and their 1:1 complex at different temperatures were shown in the inset of Figure 3a–i. The corresponding Stern–Volmer quenching constants (Ksv ) and quenching rate constants (kq ) are listed in Table 2. The findings prognosticated that these drugs or their complex were likely to quench BSA fluorescence as a static quenching mechanism because Ksv values shortened with rising temperatures. Moreover, the enumerated kq values were less than the maximum quenching constant for a diffusion‐controlled quenching process (2 × 10 10 L/mol s), which endorsed the interpreted static quenching mechanism. The present interpretation is likely to the reports of the previous studies. 23 , 27
FIGURE 3.
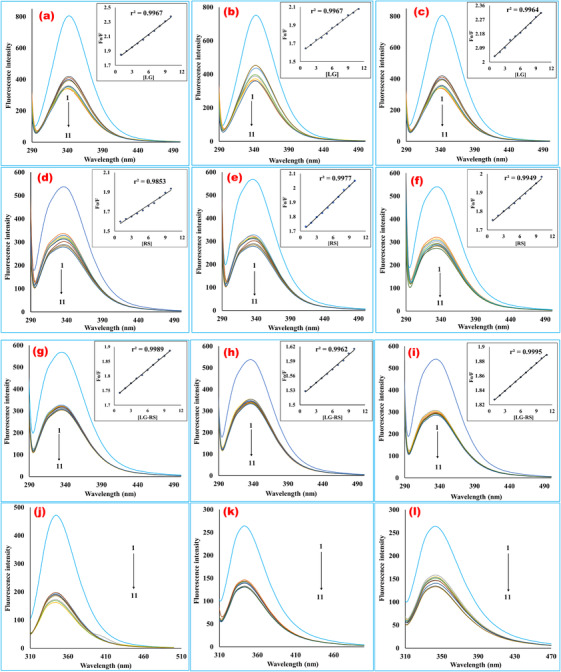
Fluorescence emission spectra of bovine serum albumin in the presence of various concentrations of linagliptin (a‐c), rabeprazole sodium (d‐f), and their 1:1 complex (g‐i) (T = 298, 308, and 318 K, respectively, for each ligand, at 280 nm excitation wavelength). (j–l) The fluorescence emission spectra of bovine serum albumin in the presence of various concentrations of linagliptin (j), rabeprazole sodium (k), and their 1:1 complex (l) at 293 nm excitation wavelength at room temperature. The curves 1–11 indicate the concentrations of 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 mM, respectively. The concentration of bovine serum albumin was fixed at 0.025% for each fluorescence scanning. The plots contain fluorescence intensity in the Y‐axis, and wavelength (nm) in the X‐axis. The fluorescence intensity of the macromolecule was quenching during the rising concentrations of the ligands in a regular trend. The emission spectra were scanned in the range of 200–500 nm at 280 and 293 nm excitation wavelengths. The interaction media was maintained at physiological pH 7.4 by phosphate buffer. The inset a–i bears BSA quenching Stern–Volmer plots with increased concentrations of linagliptin (a‐c), rabeprazole sodium (d‐f), and their 1:1 formed complex (g‐i) at 298, 308, and 318 K temperatures. The curves were plotted by inputting data of F 0/F in the Y‐axis and the quencher concentrations (1–10 mM) in the X‐axis. From the slope value of the regression line, the Stern–Volmer constant was computed. The quenching rate constant (kq ) was extrapolated from Ksv divided by the average lifetime of the protein (LG, linagliptin; RS, rabeprazole sodium)
TABLE 2.
Stern–Volmer quenching constants of the linagliptin‐bovine serum albumin, rabeprazole sodium‐bovine serum albumin, and 1:1 drug‐drug complex‐bovine serum albumin systems at three different temperatures (pH 7.4)
| System | T (K) | 1/T (K–1) | Ksv (L/mol) | kq (×1010 L·mole–1 ·s –1) | r 2 |
|---|---|---|---|---|---|
| LG‐BSA | 298 | 0.0033 | 57.8 | 0.578 | 0.9967 |
| 308 | 0.0032 | 44.5 | 0.445 | 0.9967 | |
| 318 | 0.0031 | 30.8 | 0.308 | 0.9964 | |
| RS‐BSA | 298 | 0.0033 | 37.8 | 0.378 | 0.9853 |
| 308 | 0.0032 | 36.2 | 0.362 | 0.9977 | |
| 318 | 0.0031 | 25.2 | 0.252 | 0.9949 | |
| (LG‐RS)‐BSA | 298 | 0.0033 | 16.2 | 0.162 | 0.9989 |
| 308 | 0.0032 | 9.62 | 0.096 | 0.9962 | |
| 318 | 0.0031 | 6.32 | 0.063 | 0.9995 |
Abbreviations: BSA, bovine serum albumin; LG, linagliptin; RS, rabeprazole sodium.
Note: The Stern–Volmer quenching constants (Ksv ) and quenching rate constants (kq ) were calculated by applying the Stern–Volmer equation. The value of Ksv was found from the slope obtained from the linear regression line of F 0/F versus quencher concentrations, and kq was estimated from the ratio of Stern–Volmer quenching constants and average lifetime of macromolecule (Ksv /τ 0).
3.3. Analysis of thermodynamic parameters
The primary aim of the thermodynamic research was to establish the interaction mechanism of the ligand with the protein and identify the force responsible for the interaction. The thermodynamic factors are the crucial tools to elucidate the binding mood of the drug‐biomolecule interaction. The thermodynamic parameters clearly endorse the forces liable for the interaction of the ligand with the protein. The van ’t Hoff plot was constructed from ln kq versus 1/T, which is displayed in Figure 4a. From the slope and intercept of the regression line, the thermodynamic parameters were calculated at three different temperatures, 298, 308, and 318 K. Finally, all the estimated thermodynamic factors (∆H, ∆S, and ∆G) are enlisted in the Table 3.
FIGURE 4.
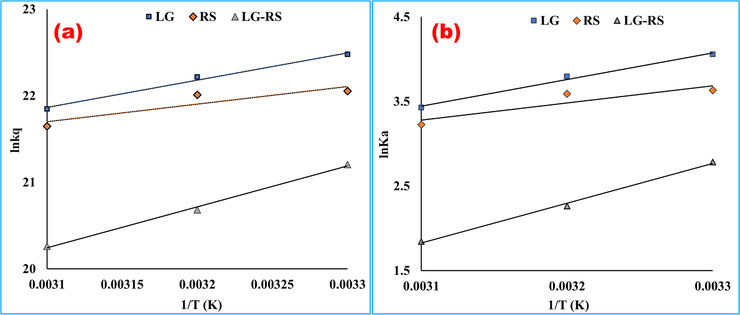
Arrhenius plot (a) and van ’t Hoff plot (b) for the interaction of bovine serum albumin with linagliptin, rabeprazole sodium, and their 1:1 complex at pH 7.4. According to the Arrhenius equation, the activation energy, Ea was computed by using the slope of (a ) [Ea = (–RT) × slope] plotted by imputing ln kq in the Y‐axis and 1/T in the X‐axis. The slope of (b ) was utilized to calculate the difference of enthalpy from the van ’t Hoff equation [∆H = (–RT) × slope], and the intercept value was utilized to measure the entropy change [∆S = R × intercept] (LG, = linagliptin; RS, rabeprazole sodium)
TABLE 3.
Thermodynamic parameters of linagliptin‐bovine serum albumin, rabeprazole sodium‐bovine serum albumin, and 1:1 drug–drug complex‐bovine serum albumin systems at three different temperatures (pH 7.4)
| System | T (K) | 1/T (K–1) | r 2 | Ea (kJ/mol) | ∆H (kJ/mol) | ∆S (J/mol/K) | ∆G (kJ/mol) |
|---|---|---|---|---|---|---|---|
| LG‐BSA | 298 | 0.0033 | –26.15 | –26.15 | –52.44 | –10.52 | |
| 308 | 0.0032 | 0.9906 | –26.15 | –26.15 | –52.44 | –9.99 | |
| 318 | 0.0031 | –26.15 | –26.15 | –52.44 | –9.47 | ||
| RS‐BSA | 298 | 0.0033 | –16.84 | –16.84 | –24.96 | –9.40 | |
| 308 | 0.0032 | 0.8290 | –16.84 | –16.84 | –24.96 | –9.15 | |
| 318 | 0.0031 | –16.84 | –16.84 | –24.96 | –8.90 | ||
| (LG+RS)‐BSA | 298 | 0.0033 | –39.28 | –39.28 | –106.06 | –7.67 | |
| 308 | 0.0032 | 0.9961 | –39.28 | –39.28 | –106.06 | –6.61 | |
| 318 | 0.0031 | –39.28 | –39.28 | –106.06 | –5.55 |
Abbreviations: BSA, bovine serum albumin; LG, linagliptin; RS, rabeprazole sodium.
Note: The listed thermodynamic factors like change of enthalpy (∆H) and entropy (∆S) were unique in all three temperatures. Both parameters (∆H and ∆S) were calculated from the slope and intercept obtained from the linear regression line (ln Ka vs. 1/T) plotted by utilizing the van ’t Hoff equation. The temperature‐dependent parameter, free energy change (∆G), was interpolated by utilizing the obtained thermodynamic parameters (∆H and ∆S) at three different temperatures. The value of ∆G for all the systems was varied due to temperature variation. According to the Arrhenius equation, the activation energy (Ea ) was calculated from plotted the regression line (ln kq vs. 1/T). The activation energy was constant despite the temperature rising. The observed Ea value of the drug–drug complex was found higher than the mother compounds.
From the Arrhenius plot (Figure 4b), the activation energy of the quenching mechanism was calculated from the slope of the straight line, and the values of the different systems are presented in Table 3. It was noticed that the activation energy of their 1:1 complex with BSA was less than the activation energy values of their individual interactions. The different activation energy of the formed complex indicated the distinct binding affinity to the protein.
The interaction process was spontaneous, which was confirmed by the negative sign of ∆G (Table 3). The positive ∆H and ∆S values alluded that the bindings of LG, RS, and their 1:1 formed complex with BSA were mainly van der Waals forces driven, where hydrogen bond played a major role in the reaction. 26 Such bindings were exothermic reactions due to the negative change in enthalpy associated with the temperature increase in Ksv values. 14 From the thermodynamic viewpoint, all the calculated parameters showed one thing obviously that these pure drugs or their formed complex bound with the protein strongly and showed quenching property regularly with increasing concentrations of the ligands. In physiological or slightly basic pH, the nonpolar alkyl group and the tertiary ammonium group (‐NH) of the indole ring of the Trp residue interacted with the ligands via der Waals forces, or hydrogen bonding, which endorsed the findings from the docking analysis. However, the strong binding effects with the highest affinity of the drugs or their complex with the Trp residue mainly caused the fluorescence quenching of BSA. 28
3.4. Analysis of binding parameters
The pharmacological activity of a therapeutic target has an outright correlation with its binding characteristics to the target macromolecule. The ligand‐protein interaction acts as a simulated model to delineate the overall cascade of the chemotherapeutic process. The binding energy or strength represented by the binding constant (Kb ) is a prerequisite parameter to interpret the bioavailability in the circulatory system and the time required to reach the target sites of the drug. 14 A plot of log(F 0 – F)/F versus log{[D]0 ‐ n[P]0(F 0 – F)/F} (Figure 5) was constructed, and the binding constants (Kb ) and the number of binding sites (n) were calculated from its slope and intercept value for each system, and these binding parameters are listed in Table 4. The tabular values signified an increase in the binding constant with the increase in temperature (except the BSA‐RS system), but the n values were found close to one and remained almost constant. These results indicated the interacting mole ratio of these systems, and it was confirmed 1:1 from n values. From this result, it can be concluded that one mole of drug interacted with each mole of protein.
FIGURE 5.
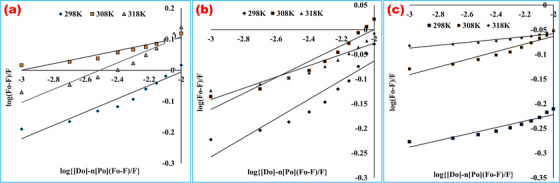
Double‐log plots for binding constant and binding points for (a) linagliptin‐bovine serum albumin, (b) rabeprazole sodium‐bovine serum albumin, and (c) linagliptin‐rabeprazole sodium‐bovine serum albumin systems at 298, 308, and 318 K temperatures. Log (F 0 – F)/F and log{[D]0 – n [P]0(F 0 – F)/F} were expressed in the Y‐ and X‐axes, respectively. From the slope value, the number of the binding sites (n) and the antilogarithm values of intercept indicated the binding constant of the ligand
TABLE 4.
Binding constant and number of binding sites of linagliptin‐bovine serum albumin, rabeprazole sodium‐bovine serum albumin, and 1:1 drug–drug complex‐bovine serum albumin systems at different temperatures
| System | pH | T (K) | r 2 | Kb (L/mol) | logK b | n |
|---|---|---|---|---|---|---|
| LG‐BSA | 7.4 | 298 | 0.9237 | 2.65 | 0.424 | ˜1.0 |
| 308 | 0.9210 | 2.05 | 0.313 | ˜1.0 | ||
| 318 | 0.9109 | 3.41 | 0.533 | ˜1.0 | ||
| RS‐BSA | 7.4 | 298 | 0.8709 | 2.11 | 0.325 | ˜1.0 |
| 308 | 0.9109 | 2.10 | 0.324 | ˜1.0 | ||
| 318 | 0.8924 | 1.58 | 0.201 | ˜1.0 | ||
| (LG‐RS)‐BSA | 7.4 | 298 | 0.890 | 0.12 | –0.905 | ˜1.0 |
| 308 | 0.915 | 1.23 | 0.092 | ˜1.0 | ||
| 318 | 0.901 | 1.00 | 0.0005 | ˜1.0 |
Abbreviations: BSA, bovine serum albumin; LG, linagliptin; RS, rabeprazole sodium
Note: The binding parameters (Kb = binding constant and n = number of binding sites) were obtained from the linear relationship between log{(F 0 – F)/ F } and log{[D]0 – n[P]0(F 0 – F)/F} for all systems at three different temperatures at pH 7.4. The binding constants (Kb ) for the drug–drug complex with BSA were found smaller than the parent compounds. The number of binding sites (n) was almost steady and approximately one.
3.5. Effect of drug–drug complexation on protein binding
The appearance of one ligand during the binding process of another ligand with protein has a significant effect on the binding mode and affinity of the drug to the protein. The presence of one drug or ion may alter the binding strength or affinity of the other drug with the macromolecule. 21 , 22 The binding affinity and binding mode of the ligand are highly temperature dependent. The effects of drug–drug complexation on binding constant and binding affinity were also observed from Table 4.
The binding affinity of RS decreased theoretically by 379, 71.60, and 99.75% at 298, 308, and 318 K temperatures, respectively, due to complexation with LG. On the other hand, the binding affinity of LG decreased significantly by 313, 70.60, and 99.90% at the three aforementioned temperatures, respectively, due to the interaction with RS. In addition, it was also investigated that the binding constants of both ligands at the three temperatures decreased drastically. This might be occurred due to the structural modification of the ligands while interacting between themselves or the significant conformational changes of the macromolecule during the binding process. The lower binding constant of the formed complex indicated the lower protein binding capacity. It can trigger its higher elimination rate, which can be considered as an indicator of a shorter half‐life or lower bioavailability than the parent compound LG. The lower protein binding affinity of the formed complex may construe lower potency and significantly lower the hypoglycemic effect of LG. This hypothetical interpretation of the fluorescence spectroscopic study was validated through in vivo research conducted by Hossain et al. 13
3.6. Energy transfer profile
FRET mechanism demonstrates how the energy transmits from donor to acceptor. The overlaid emission curves of BSA with the absorption spectra of LG, RS, and their formed complex were displayed in Figure 6. The curve overlaid by the absorption spectrum and fluorescence spectrum dictated the proof of a higher probability of energy transformation between the biomolecule and the ligands. In this study, the theoretical values k 2 = 2/3, n = 1.34, and Ф = 0.15 were collected from previously published concepts, 23 , 30 and the calculated areas from the curves of Figure 6 are listed in Table 5. The calculated average distances of these ligands from protein were observed below 7 nm, which indicated that the energy transfer occurred with a high possibility from BSA to drug molecules or their formed complex. 8 As temperature variation has not outright correlation on the nonradioactive energy transformation, 9 this study established the energy transfer profile at one temperature (298 K). Moreover, it is vital to note here that the calculated distance between the protein and ligand (r) was larger than the value of critical distance (R 0) located at 50% energy transfer efficiency, which indicated the fluorescence quenching mechanism of the biomolecule was an obvious static process mediated by these three ligands. 23 This interpretation is consistent and further supporting proof of the findings of this current study.
FIGURE 6.
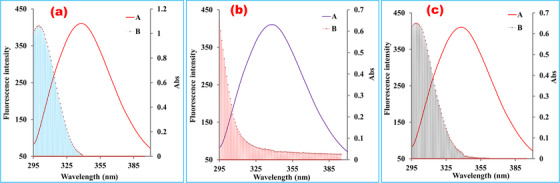
Overlap spectra of bovine serum albumin with (a) linagliptin, (b) rabeprazole sodium, and (c) their 1:1 formed complex. The enclosed area was formed between (A) bovine serum albumin fluorescence and (B) UV absorption spectra. The primary and secondary Y‐axes were expressed as fluorescence intensity and UV absorbance, respectively, while the X‐axis was denoted by the wavelength (nm). The energy transformation from the donor to the ligand can be confirmed by the sufficient overlap between the two curves. The curves of the fluorescence spectrum of the bovine serum albumin and absorption spectrum of the ligand overlaid that indicated the higher possibility of the existence of energy transfer between donor and acceptor molecules
TABLE 5.
Parameters related to energy transfer of bovine serum albumin with linagliptin, rabeprazole sodium, and their 1:1 complex at room temperature
| Ligand | E (%) | J (×10–18 cm3 L mol–1) | R 0 (nm) | r (nm) |
|---|---|---|---|---|
| LG | 14.53 | 4.737 | 0.71 | 0.95 |
| RS | 27.46 | 3.981 | 0.69 | 0.81 |
| LG‐RS | 27.81 | 4.99 | 0.72 | 0.84 |
Abbreviations: BSA, bovine serum albumin; E, efficiency of energy transfer; LG, linagliptin; RS, rabeprazole sodium.
Note: r represents the distance between protein and ligand, and R 0 represents the critical distance found at 50% transfer efficiency. According to the Förster theory, the distance between ligand and protein was tabulated precisely. For all the systems, the observed values of distance were <8 nm.
3.7. Analysis of conformational changes of BSA
Analysis of UV–vis spectra: UV–vis absorption spectroscopy, a very useful and simple strategy, can be easily applicable to study the complex formation of a protein with a small molecule and investigate the anatomical modifications of the complex structure. 14 , 15 The characteristics UV absorption peak of BSA often changes due to the interactions of ligand, and this property is utilized to analyze the structural conformation of the protein. In this experiment, the UV–vis absorption spectra of the fixed concentrated BSA, in the absence and presence of LG (concentrations were varied from 5 to 100 µM), were recorded at physiological pH 7.4 buffer solution, and the spectra were exhibited in Figure 7. The serum protein showed a strong absorption peak around 279 nm due to the presence of a particular functional group (phenyl group) of some aromatic amino acids (Trp, Tyr, and Phe) because the peak appeared from the π–π* electronic transitions. However, with gradual addition upon the ligand, the peak started to shift at a higher wavelength (red‐shift or bathochromic effect), and the peaks intensified by increasing the concentrations of the ligand, which probably indicated a complex formation between the protein and ligand. Moreover, the complexation between the ligand and protein (LG–BSA) resulted in a shift of 279–293 nm of the band of the spectra (Figure 7) and indicated such interaction of the ligand with the serum protein that lead to the loosening and unfolding of the protein. Consequently, the hydrophobicity of the microenvironment of the biomolecule was decreased. 15 , 31 Finally, this result revealed that the fluorescence quenching of the serum protein was mainly occurred due to the complex formation of the ligand.
FIGURE 7.
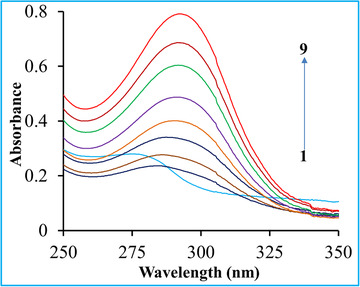
Effect of linagliptin on UV–vis absorption spectra of bovine serum albumin. C BSA = 10 µM that was fixed, and curves 1–9 indicated the UV–vis absorption spectra BSA upon addition of linagliptin (0, 5, 10, 20, 30, 40, 60, 80, 100 µM)
3.7.1. Analysis of synchronous fluorescence spectra
Synchronous fluorescence spectrometry, first introduced by Lloyd, 9 has several merits like better selectivity, higher sensitivity, and lesser interference compared to conventional fluorescence spectroscopy. Besides, this technique shows spectral simplicity, spectral bandwidth reduction, and avoiding different perturbing effects, and can also be applied for the simultaneous determination of compounds in a multicomponent mixture. The method is very useful and simply applicable to investigate the microenvironment of various amino acid residues by estimating the maximum emission wavelength (λ em) shift. 9 , 15 This approach is used to probe the conformational changes of BSA because of its several fundamental characteristic information of polarity alterations in the vicinity of the disparate chromophore molecules. If the interval between emission and excitation wavelength (Δλ) is 60 nm, the synchronous fluorescence spectra will characterize the Trp residues of the protein. On the other side, if the Δλ is 15 nm, the spectra will provide the characteristic information of Tyr residues. 17
Synchronous fluorescence spectra for BSA in the absence and presence of LG at various concentrations (5, 10, 20, 30, 40, 60, 80, 100 µM) were displayed at Δλ = 60 and 15 nm in Figure 8. From the synchronous spectral analysis of Figure 8, it was affirmed that the emission intensity of both residues (Tyr and Trp) was successively decreased upon the addition of the drug. It was stated that there was a slight blue shift (278–273 nm) for the Tyr residues (Figure 8a), which indicated the structural changes of Tyr was occurred by a more hydrophobic environment with decreased polarity. In contrast, a notable red‐shift of Trp residues was observed (Figure 8b) in maximum emission wavelength, which implied a less hydrophobic microenvironment was available during the interaction between the BSA and the drug, and the augmentation of the stretching extent of the peptide chain. 15 In addition, it might be concluded that the quenching band of the protein during interaction with the drug was more obvious and regular at Δλ 15 nm than Δλ 60 nm, which demonstrated that the ligand was more adjacent to the Tyr residues than the Trp residues.
FIGURE 8.
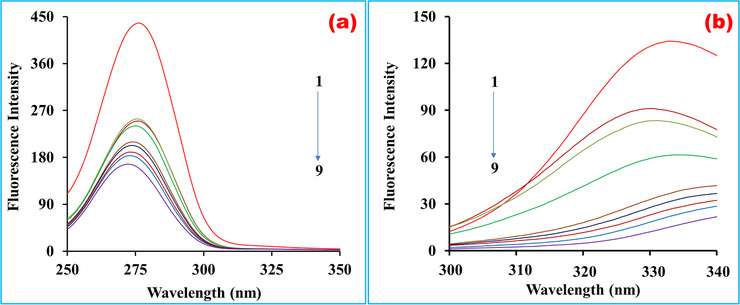
Synchronous fluorescence spectra of BSA solutions (C BSA = 10 µM, T = 300 K, pH 7.4). Curves 1–9 indicated the spectra where the concentrations of linagliptin were 0, 5, 10, 20, 30, 40, 60, 80, and 100 µM, respectively. (a) Δλ = 15 nm and (b) Δλ = 60 nm
4. CONCLUSIONS
This research provided detailed information on the binding mode and binding affinity of LG, RS, and their 1:1 formed complex with BSA. The interaction mechanisms of these ligands with the serum protein were explored by fluorescence quenching, synchronous fluorescence, UV–vis spectroscopy, FRET theory, and molecular docking approaches. These methods concluded that the bindings of these drugs or their 1:1 complex with BSA were absolutely exothermic static reactions. The thermodynamic factors indicated the interactions were spontaneous processes where van der Waals forces and hydrogen bond played a crucial role in the interactions. The binding parameters indicated one mole of drug interacted with one mole of BSA where the binding constant revealed the shorter half‐life of the drug complex as well as the primary assumption of decreased hypoglycemic effect of LG due to complexation with RS. The molecular modifications of the BSA by these drugs were experimented with, and the geometric orientations of these ligands in the core part of the protein were also established by molecular docking. The UV–vis and synchronous fluorescence spectroscopic techniques demonstrated that the conformational changes of the BSA were occurred due to the ligand–protein complexation and the polarity variations in the vicinity of the disparate chromophores of the biomolecule. Numerically, the activation energy of their 1:1 complex with BSA was higher than the activation energy values of their individual interactions, and the FRET theory confirmed that the energy transformation has occurred from Trp residue of BSA to the ligands with high probability.
CONFLICT OF INTEREST
The authors have declared no conflict of interest.
AUTHOR CONTRIBUTIONS
Md. Jamal Hossain conceptualized and conducted the entire lab work. He also analyzed the raw data, interpreted the research findings, and compiled the original manuscript. Moreover, he addressed all the reviewers' insightful and critical questions and edited the manuscript several times during the peer‐review process. Md. Zakir Sultan and Md. Ruhul Kuddus supervised the research work. Mohammad A. Rashid provided logistic support. All the authors reviewed and approved the final version of the manuscript for submission and subsequent publication.
ACKNOWLEDGMENTS
The authors are grateful to the Center for Advanced Research in Sciences (CARS) and the Department of Pharmaceutical Chemistry, University of Dhaka, Dhaka‐1000, Bangladesh, for logistical support.
Hossain J, Sultan Z, Rashid MA, Kuddus R. Interactions of linagliptin, rabeprazole sodium, and their formed complex with bovine serum albumin: Computational docking and fluorescence spectroscopic methods. Anal Sci Adv. 2021;2:480–494. 10.1002/ansa.202000153
REFERENCES
- 1. Wang H, Shi H, Pang J, et al. Studies on the interaction between triptolide and bovine serum albumin (BSA) by spectroscopic and molecular modeling methods. Afr J Tradit Complement Altern Med. 2016;13:121‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cheng Z, Liu R, Jiang X. Spectroscopic studies on the interaction between tetrandrine and two serum albumins by chemometrics methods. Spectrochim Acta‐Part A Mol Biomol Spectrosc. 2013;115:92‐105. [DOI] [PubMed] [Google Scholar]
- 3. Shahabadi N, Hadidi S. Molecular modeling and spectroscopic studies on the interaction of the chiral drug venlafaxine hydrochloride with bovine serum albumin. Spectrochim Acta‐Part A Mol Biomol Spectrosc. 2014;122:100‐106. [DOI] [PubMed] [Google Scholar]
- 4. Zhang Y, Shi S, Huang K, et al. Effect of Cu2+ and Fe3+ for drug delivery: decreased binding affinity of ilaprazole to bovine serum albumin. J Luminesc. 2011;131:1927‐1931. [Google Scholar]
- 5. Sun S, Zhou B, Hou H, et al. Studies on the interaction between oxaprozin‐E and bovine serum albumin by spectroscopic methods. Int J Biol Macromol. 2006;39:197‐200. [DOI] [PubMed] [Google Scholar]
- 6. Reshma, Vaishanav SK, Yadav T, et al. Antidepressant drug‐protein interactions studied by spectroscopic methods based on fluorescent carbon quantum dots. Heliyon. 2019;5:e01631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hossain MJ, Sultan MZ, Rashid MA, Kuddus MR. In vitro interactions of secnidazole and its iron (ii), copper (ii) complexes with bovine serum albumin by fluorescence quenching method. Bangladesh Pharm J. 2020;23:1‐9. [Google Scholar]
- 8. Li G, Liu BS, Zhang Q, et al. Investigation on the effect of fluorescence quenching of bovine serum albumin by cefoxitin sodium using fluorescence spectroscopy and synchronous fluorescence spectroscopy. Luminescence. 2016;31:1054‐1062. [DOI] [PubMed] [Google Scholar]
- 9. Zhang L, Liu B, Li Z, et al. Comparative studies on the interaction of cefixime with bovine serum albumin by fluorescence quenching spectroscopy and synchronous fluorescence spectroscopy. Luminescence. 2014;30:686‐692. [DOI] [PubMed] [Google Scholar]
- 10. Graefe‐mody U, Retlich S, Friedrich C, et al. Clinical pharmacokinetics and pharmacodynamics of linagliptin. Clin Pharmacokinet. 2012;51:411‐427. [DOI] [PubMed] [Google Scholar]
- 11. Sawant RL, Hadawale SD, Dhikale GK, et al. Spectrophotometric methods for simultaneous estimation of rabeprazole sodium and aceclofenac from the combined capsule dosage form. Pharm Methods. 2011;2:193‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dash RP, Rais R, Srinivas NR. Stereoselective and nonstereoselective pharmacokinetics of rabeprazole—an overview. Xenobiotica. 2018;48:422‐432. [DOI] [PubMed] [Google Scholar]
- 13. Hossain MJ, Sultan MZ, Rashid MA, Kuddus MR. Does rabeprazole sodium alleviate the anti‐diabetic activity of linagliptin? Drug‐drug interaction analysis by in vitro and in vivo methods. Drug Res. 2020;70:519‐527. [DOI] [PubMed] [Google Scholar]
- 14. Suryawanshi VD, Walekar LS, Gore AH, et al. Spectroscopic analysis on the binding interaction of biologically active pyrimidine derivative with bovine serum albumin. J Pharm Anal. 2016;6:56‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Q, Huang CR, Jiang M, et al. Binding interaction of atorvastatin with bovine serum albumin: Spectroscopic methods and molecular docking. Spectrochim Acta A Mol Biomol Spectrosc. 2016;156:155‐163. [DOI] [PubMed] [Google Scholar]
- 16. Wang BL, Pan DQ, Zhou KL, Lou YY, Shi JH. Multi‐spectroscopic approaches and molecular simulation research of the intermolecular interaction between the angiotensin‐converting enzyme inhibitor (ACE inhibitor) benazepril and bovine serum albumin (BSA). Spectrochim Acta A Mol Biomol Spectrosc. 2019;212:15‐24. [DOI] [PubMed] [Google Scholar]
- 17. Zhang YF, Zhou KL, Lou YY, Pan DQ, Shi JH. Investigation of the binding interaction between estazolam and bovine serum albumin: multi‐spectroscopic methods and molecular docking technique. J Biomol Struct Dyn. 2017;35:3605‐3614. [DOI] [PubMed] [Google Scholar]
- 18. Qiong WU, Chaohong LI, Yanjun HU, et al. Study of caffeine binding to human serum albumin using optical spectroscopic methods. Sci China Ser B: Chem. 2009;52:2205‐2212. [Google Scholar]
- 19. Abdelhameed AS. Insight into the interaction between the HIV‐1 integrase inhibitor elvitegravir and bovine serum albumin: A spectroscopic study. J Spectrosc. 2015. 10.1155/2015/435674. [DOI] [Google Scholar]
- 20. Kou SB, Lin ZY, Wang BL, Shi JH, Liu YX. Evaluation of the binding behavior of olmutinib (HM61713) with model transport protein: insights from spectroscopic and molecular docking studies. J Mol Struct. 2020;1224. 10.1016/j.molstruc.2020.129024 [DOI] [Google Scholar]
- 21. Peng M, Shi S, Zhang Y. The influence of Cd2+, Hg2+ and Pb2+ on taxifolin binding to bovine serum albumin by spectroscopic methods: with the viewpoint of toxic ions/drug interference. Environ Toxicol Pharmacol. 2011;33:327‐333. [DOI] [PubMed] [Google Scholar]
- 22. Hossain MJ, Rashid MA, Sultan MZ. Transition metal chelation augments the half‐life of secnidazole: molecular docking and fluorescence spectroscopic approaches. Drug Res. 2020;70:582‐593. [DOI] [PubMed] [Google Scholar]
- 23. Shen GF, Liu TT, Wang Q, et al. Spectroscopic and molecular docking studies of binding interaction of gefitinib, lapatinib and sunitinib with bovine serum albumin (BSA). J Photochem Photobiol B Biol. 2015;153:380‐390. [DOI] [PubMed] [Google Scholar]
- 24. Buddanavar AT, Nandibewoor ST. Multi‐spectroscopic characterization of bovine serum albumin upon interaction with atomoxetine. J Pharm Anal. 2017;7:148‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shi J, Pan D, Jiang M, et al. Binding interaction of ramipril with bovine serum albumin (BSA): insights from multi‐spectroscopy and molecular docking methods. J Photochem Photobiol B Biol. 2016;164:103‐111. [DOI] [PubMed] [Google Scholar]
- 26. Ang CW, Ang ZW, Hao JZ. Study of the interaction of carbamazepine with bovine serum albumin by fluorescence quenching method. Anal Sci. 2006:22:435‐438. [DOI] [PubMed] [Google Scholar]
- 27. Ojha B, Das G. Role of hydrophobic and polar interactions for BSA‐amphiphile composites. Chem Phys Lipids. 2011;164:144‐150. [DOI] [PubMed] [Google Scholar]
- 28. Yang J, Deng S, Liu Z, et al. Fluorescence quenching of serum albumin by rifamycin antibiotics and their analytical application. Luminescence. 2007;22:559‐566. [DOI] [PubMed] [Google Scholar]
- 29. Aleksić MM, Kapetanović V. An overview of the optical and electrochemical methods for detection of DNA‐drug interactions. Acta Chim Slov. 2014;61:555‐573. [PubMed] [Google Scholar]
- 30. Shi J, Pan D, Wang X, et al. Characterizing the binding interaction between antimalarial artemether (AMT) and bovine serum albumin (BSA): spectroscopic and molecular docking methods. J Photochem Photobiol B Biol. 2016;162:14‐23. [DOI] [PubMed] [Google Scholar]
- 31. Pan X, Qin P, Liu R, Wang J. Characterizing the interaction between tartrazine and two serum albumins by a hybrid spectroscopic approach. J Agric Food Chem. 2011;59:6650‐6656. [DOI] [PubMed] [Google Scholar]