

Abstract
It is difficult for beginners to learn and use amplicon analysis software because there are so many software tools to choose from, and all of them need multiple steps of operation. Herein, we provide a cross‐platform, open‐source, and community‐supported analysis pipeline EasyAmplicon. EasyAmplicon has most of the modules needed for an amplicon analysis, including data quality control, merging of paired‐end reads, dereplication, clustering or denoising, chimera detection, generation of feature tables, taxonomic diversity analysis, compositional analysis, biomarker discovery, and publication‐quality visualization. EasyAmplicon includes more than 30 cross‐platform modules and R packages commonly used in the field. All steps of the pipeline are integrated into RStudio, which reduces learning costs, keeps the flexibility of the analysis process, and facilitates personalized analysis. The pipeline is maintained and updated by the authors and editors of WeChat official account “Meta‐genome.” Our team will regularly release the latest tutorials both in Chinese and English, read the feedback from users, and provide help to them in the WeChat account and GitHub. The pipeline can be deployed on various platforms, and the installation time is less than half an hour. On an ordinary laptop, the whole analysis process for dozens of samples can be completed within 3 h. The pipeline is available at GitHub (https://github.com/YongxinLiu/EasyAmplicon) and Gitee (https://gitee.com/YongxinLiu/EasyAmplicon).
Keywords: amplicon, bioinformatics, microbiome, pipeline, visualization, metagenome
EasyAmplicon is a user‐friendly, cross‐platform, and community‐supported pipeline for amplicon data analysis. It has most of the modules for data processing and visualization in microbiome research. The pipeline is maintained and updated regularly. We encourage users to contribute appropriate code.

Highlights
EasyAmplicon is a user‐friendly, cross‐platform, and community‐supported pipeline for amplicon data analysis.
It has most of the modules for data processing and visualization in microbiome research.
The pipeline is maintained and updated regularly, and we encourage users to contribute appropriate code.
INTRODUCTION
The rapid development of high‐throughput sequencing technologies in the past 20 years has promoted an increasingly deeper exploration of the crucial roles of microbiome in humans [1–5], animals [6–8], plants [9–11], and the environment [12–14]. Most of them were driven by amplicon sequencing (such as 16S rDNA sequencing of bacteria or archaea, eukaryotic 18S rDNA or internal transcribed spacer, and nitrogen‐fixing prokaryote's nifH gene) and had profiled the taxonomic composition of the microbiome in various environments [15–17].
Wet laboratory operations of amplicon sequencing are now standardized, and most operations are implemented by specialized biotechnology companies or sequencing centers. However, bioinformatics analyses of amplicon data are still challenging, and the existence of overwhelming software, methods, and algorithms brings difficult choices for beginners. The popular amplicon analysis pipelines include mothur [18], USEARCH [19], and QIIME [20], all of which have been cited over 10,000 times. However, they still have obvious shortcomings, such as a lack of downstream statistical analyses and visualization solutions, higher time costs, and being limited to specified operating systems. Some online analysis webservers are easy to use, such as Qiita [21], MGnify [22], and gcMeta [23], but they also have several limitations, such as slow upload speed, long waiting/running time, and few adjustable parameters, which make it impossible to conduct customized analyses [24, 25].
The lack of an easy‐to‐use and flexible amplicon analysis pipeline seriously restricts researchers to understand the data analysis process and hinders the development of this field. Therefore, we developed an easy‐to‐use, open‐source, and cross‐platform amplicon analysis pipeline—EasyAmplicon. It can be used in both command‐line mode and interactive mode in RStudio. Currently, it provides more than 20 visualization styles and generates publication‐quality figures easily. The open‐source code could facilitate reproducible analysis and allow personalized modification. In addition, it also generates standard input for the most popular software, such as STAMP [26], LEfSe [27], PICRUSt 1 & 2 [28, 29], BugBase [30], FAPROTAX [31], ImageGP[32], and iTOL [33]. EasyAmplicon provides a free, reproducible, and personalized solution for amplicon analysis, which could be an amazing software tool for microbiome research.
RESULTS
Overview of EasyAmplicon pipeline
EasyAmplicon is an integrated pipeline for amplicon data analysis and visualization on a laptop or server, and it provides various tables and figures to explore underlying biological interpretations. This pipeline is easy to install on Windows, MacOS, and Linux systems. The installation method is described in detail in the Methods section or available at https://github.com/YongxinLiu/EasyAmplicon. For the test data, comprising 18 samples and 50,000 PE250 reads per sample, the complete analysis could be finished within about 3 h with a peak memory footprint of less than 4 gigabytes (CPU: 2 cores, 2.1 GHz).
The EasyAmplicon is an end‐to‐end pipeline. It starts with raw reads and ends with data tables and publication‐quality figures (Figure 1). It mainly comprises three steps: dimensionality reduction, analysis, and visualization & statistics (Figure 1). All the related software is easy to install (Table 1), and we provide a batch download package to accelerate pipeline deployment.
Figure 1.
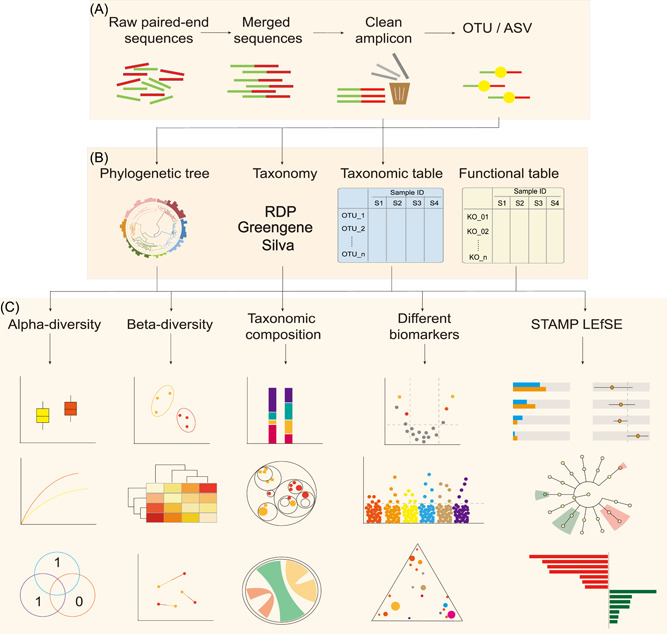
Pipeline of EasyAmplicon for analyzing paired‐end amplicon sequences. (A) Dimensionality reduction: processing raw sequencing reads into feature tables. (B) Analysis: providing phylogenetic analysis, taxonomic classification, functional prediction, and alpha‐ and beta‐diversity calculations. (C) Statistics and visualization: generating publication‐quality figures and performing statistical tests for biological interpretations. ASVs, amplicon sequence variants; OTU, operational taxonomic units.
Table 1.
Software and packages included in EasyAmplicon
| Software | Function in the pipeline | Website |
|---|---|---|
| Git for Windows | Provides Linux Shell like environment in Windows | http://gitforwindows.org/ |
| R | Statistical processing and data visualization | https://www.r-project.org |
| RStudio | Integrated development environment for R and Shell | https://posit.co/ |
| VSEARCH | A fast, free, and cross‐platform pipeline for amplicon sequencing analysis [34] | https://github.com/torognes/vsearch |
| USEARCH | Processes sequences and calculates alpha‐ and beta‐diversities [19] | http://www.drive5.com/usearch/ |
| SeqKit | Toolkit for FASTA or FASTQ file manipulation [35] | https://github.com/shenwei356/seqkit |
| ggplot2 | R package for data visualization | https://github.com/tidyverse/ggplot2 |
| ggClusterNet | R package for microbiome network visualization [36] | https://github.com/taowenmicro/ggClusterNet/ |
| vegan | R package for alpha and beta diversity analysis | https://cran.r-project.org/package=vegan |
| ggraph | Creates layout for tree map and circle packing chart | https://github.com/thomasp85/ggraph |
| circlize | Circular visualization [37] | https://github.com/jokergoo/circlize |
Running the pipeline in command line or R markdown mode
First, we open the pipeline file “pipeline.sh” using RStudio. After setting the working directory, the analysis process could be run step‐by‐step with only a mouse clicking the “Run” button. For users to conduct their own analysis, only the raw sequencing data and sample metadata are needed, and the following analysis would be processed by EasyAmplicon. If RStudio is not applicable, we can copy and paste the scripts into the pipeline.sh and run them in any Shell environment (such as a terminal in Linux/Mac either locally or remotely, or Git bash in Windows). All the related software and packages are listed in Table 1. All the figures are saved in PDF format by default, and some examples are shown in Figures 2 and 3.
Figure 2.
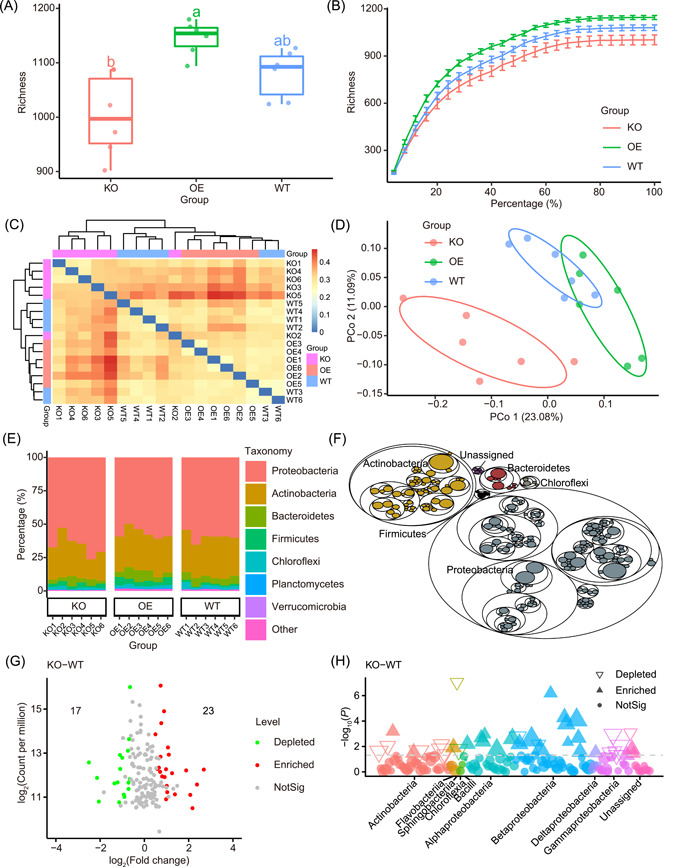
Examples of publication‐quality visualizations. (A) Boxplot showing alpha diversity in richness metrics among groups. Different letters indicate statistical significance among groups (p < 0.05, ANOVA, Tukey HSD). The horizontal bars within boxes represent medians. The tops and bottoms of the boxes represent the 75th and 25th percentiles, respectively. The upper and lower whiskers extend to data no more than 1.5× the interquartile range from the upper and lower edge of the box, respectively. (B) Rarefaction curve of richness shows that features reach saturation stage with increasing sequencing depth. Each vertical bar represents standard error. (C) Heatmap based on Bray−Curtis dissimilarity. (D) Principal coordinate analysis (PCoA) of Bray−Curtis dissimilarity. (E) Stacked bar plot of taxonomic composition in grouped samples at phylum level. (F) Tree map of taxonomic composition. (G) Volcano plot showing significantly differential abundance taxa between KO and WT groups. (H) Manhattan plot showing different features and related taxa between KO and WT groups. The numbers of replicated samples in this figure are as follows: in KO (n = 6), OE (n = 6), and WT (n = 6). KO, knock‐out; OE, overexpression; WT, wild‐type.
Figure 3.
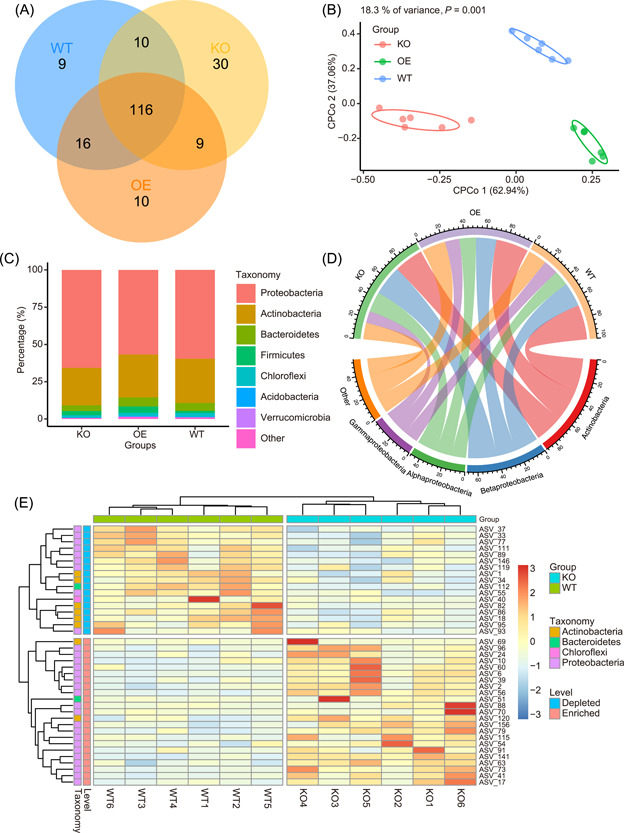
Supplementary examples of publication‐quality visualizations to Figure 2. (A) Venn diagram showing common and unique ASVs (relative abundance >0.1%) among three groups. (B) Constrained principal coordinate analysis (CPCoA) of three groups. (C) Stacked plot of average relative abundance at phylum level of three groups. (D) Circle plot of average relative abundance at phylum level of three groups. (E) Heatmap showing significantly different ASVs between KO and WT groups (Wilcoxon test, p < 0.05). ASVs, amplicon sequence variants; KO, knock‐out; WT, wild‐type.
To make statistics and visualization of microbiome data more personalized, users can open the “Tutorial.Rmd” document in RStudio and then modify the details of the figures, such as item order, color scheme, legend layout, and so on. It can even generate a publish‐ready combo figure (Figures 2 and 3) and a reproducible HTML format report (Tutorial.html).
Third‐party software supporting
EasyAmplicon does not cover all the functions required for microbiome analysis. There are currently some mainstream and very distinctive microbiome analysis tools, such as STAMP [26], LEfSe [27], PICRUSt 1 & 2 [28, 29], BugBase [30], FAPROTAX [31], and iTOL [33]. However, some input files are difficult to prepare for users without bioinformatics backgrounds. In EasyAmplicon, a lot of scripts are used to prepare input for all the above software tools. The example visualizations, STAMP (Figure 4A), LEfSe (Figure 4B), BugBase (Figure 4C), and iTOL (Figure 4D) are shown. As for the most popular QIIME 2 pipeline, the intermediate files generated by EasyAmplicon can be imported into QIIME 2, and the output files from QIIME 2 can also be imported into EasyAmplicon for downstream analyses.
Figure 4.
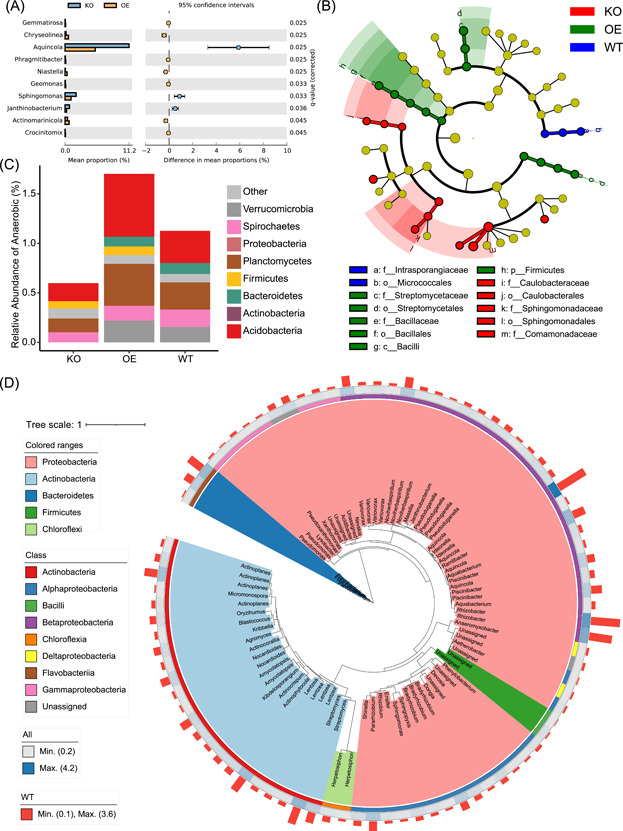
Visualizations generated by third‐party software using the intermediate files of EasyAmplicon. (A) Extended error bar plot at genus level in WT and KO groups by STAMP. (B) Cladogram showing biomarkers in each group by LEfSe. (C) Percentage of BugBase annotated anaerobic bacteria at the phylum level. (D) Phylogenetic tree of 86 ASVs (relative abundance > 0.2%). The tree background is colored by Phylum. The outer strip represents different classes. The heatmap represents the average relative abundance of all samples. The bar plot represents the relative abundance of the WT group. ASVs, amplicon sequence variants; KO, knock‐out; WT, wild‐type.
Anticipated results
EasyAmplicon provides multiple visualization styles for amplicon data analysis. For alpha diversity (within‐sample diversity), the boxplot is the best way to visualize the data and compare each group (Figure 2A), and the different letters represent significant differences (p < 0.05, ANOVA, Tukey HSD test). Rarefaction analysis reveals that the features reach the saturation stage with increasing sequencing depth, and lines and error bars represent the mean and standard error, respectively (Figure 2B). If you want to examine the unique or common features among samples or groups, the Venn diagram is a good way to show this pattern (Figure 3A). As for beta diversity, a heatmap based on Bray−Curtis dissimilarity would be a good visualization method. The colored grouping labels show how the samples cluster (Figure 2C).
DISCUSSION
At present, for amplicon analysis, the most popular pipelines are QIIME [20] and QIIME 2 [15], which have been cited 54,900 times (Google Scholar, January 4, 2023). However, the two pipelines have some disadvantages that limit their use in microbiome analysis, such as a too‐large installation package, no support for the Windows system, and a lack of publication‐quality visualization. EasyAmplicon is trying to solve the above problems.
Currently, this is just the first version of EasyAmplicon. Runtime and memory usage depend on the data set size. The current version has been used by more than thousands of users and formally cited 36 times by the end of 2022 (searching “EasyAmplicon” in Google Scholar). The authors and core team of the WeChat account “Meta‐genome” will update the pipeline in time. The scripts for correlation, network analysis [38, 39], random forest [40, 41], machine learning [42], deep learning [43], transfer learning [44], and source track [45–47] analyses are ongoing and will be included in the pipeline soon. A webserver version like MicrobiomeAnalyst [48, 49] will be set up in the future. More general command‐line scripts and visualization styles are still in development, and they will be available in a new version of the pipeline. Anyone who is interested in this project is welcome to contribute scripts pertaining to analysis methods, visualization styles, and other issues mentioned in the GitHub repository.
CONCLUSION
In summary, the EasyAmplicon pipeline provides an efficient, cross‐platform framework for amplicon analysis. Additionally, more than 20 predefined analysis and visualization solutions are provided for the multidimensional exploration of your data and for generating publication‐quality figures. Additionally, EasyAmplicon provides some utilities to be integrated with other widely used software for various needs.
METHODS
Quick start of EasyAmplicon
EasyAmplicon is coded mainly in Shell bash and R language and could be run in command‐line (terminal) mode or RStudio interactive mode. It is recommended to be deployed on a Windows system (with the installation of Git for Windows) and run in RStudio, especially for researchers without programming knowledge and skills. In addition, it also supports MacOS and Linux. To install it, please follow the instructions available at https://github.com/YongxinLiu/EasyAmplicon. Some dependent software and packages are listed in Table 1, and they are integrated for easy installation. The analysis process mainly includes three steps, as shown in Figure 1. To prove its practicability, we provide a demo data set that contains 18 samples belonging to three groups, and each sample is rarefied to 50,000 reads. This example data set is a subset of our previously published data (CRA001464) [50] (rarefied example data are deposited in GSA https://ngdc.cncb.ac.cn/gsa/, with accession ID: CRA002352).
Dimensionality reduction (from sequences to tables)
The accepted input includes paired‐end or single‐end/merged sequences (fastq format), clean amplicons (fasta format), and even the intermediate files generated by other pipelines, as shown in Figure 1. Most amplicons are sequenced on the Illumina HiSeq. 2500 or NovaSeq. 6000 platform in paired‐end 250 bp mode. Typically, the pipeline starts with paired‐end reads in fastq format and merges them to get single‐end sequences. Primers and barcodes are cut, and then low‐quality reads are filtered out to get clean amplicons. These steps are performed mainly using vsearch [34] or USEARCH [19] (Figure 1A). The clean amplicons of 16S rDNA can be directly mapped to the reference database GreenGenes [51], and the closed‐reference operational taxonomic units (OTUs) table can be generated, which can be used as the input of PICRUSt to predict the potential functions [28, 52], and can be used as the input of BugBase for phenotypic prediction [30]. Alternatively, clean amplicons are usually clustered into OTUs (97% similarity) or denoised into amplicon sequence variants (ASVs) in de novo mode. Finally, the clean amplicons will be mapped to the de novo identified OTUs/ASVs to generate a feature table. Representative sequences can be used to construct a phylogenetic tree and perform taxonomic annotation (Figure 1A).
Analysis (from big tables to small tables)
Feature tables are milestone outputs of the dimensionality reduction step. We can use the feature tables and phylogenetic tree to calculate all kinds of alpha‐ and beta‐diversity metrics. The feature tables with taxonomy annotations can be used to collapse into a specific taxonomic level and discover biomarkers at all taxonomic levels (Figure 1B). In addition, EasyAmplicon provides many glue scripts to generate input files for other widely used tools, such as QIIME 2 [15], STAMP [26], and LEfSe [27].
Statistics and visualization (from tables to figures)
EasyAmplicon can generate visualizations for alpha diversity, beta diversity, taxonomic composition, and biomarkers along with related statistical tables (Figure 1C and 2; Table 2), and these publication‐quality graphs include a box‐plot, scatter plot, stacked bar plot, and heatmap. In addition, the output of EasyAmplicon can be imported into STAMP [26], or LEfSe [27] for biomarker identification, and visualized in an extended error bar plot or Cladogram, respectively.
Table 2.
Summary of main visualization functions in EasyAmplicon
| Plot | Script | Description | References |
|---|---|---|---|
| Boxplot | alpha_boxplot.R | The plot shows data distribution in each group. Dots represent each sample, and labeled letters indicate statistical significance among groups | [10] |
| Rarefaction curve | alpha_rare_curve.R | Richness of rarefied samples or groups from 1% to 100% | [10] |
| Venn diagram | sp_vennDiagram.sh | Visualizes common and unique elements among 2−5 groups | [53, 54] |
| Ordination scatter plot |
beta_pcoa.R beta_cpcoa.R |
Shows the results of dimensionality reduction | [55] |
| Heatmap |
sp_pheatmap.sh compare_heatmap.sh |
Shows distance or similarity matrix and deferential abundance features | [50, 56] |
| Stack plot | tax_stackplot.R | Shows taxonomic and functional composition in each sample or group | [57] |
| Cricular plot | tax_circlize.R | Shows taxonomic composition | [58] |
| Circle packing chart | tax_maptree.R | Shows the relations and relative abundance of taxonomic hierarchy | [59] |
| Volcano plot | compare_volcano.R | The dots in the plot show abundance and fold changes between two groups | [50] |
| Manhattan plot | compare_manhattan.sh | Shows taxonomy, abundance, and pattern between two groups | [60] |
AUTHOR CONTRIBUTIONS
Yong‐Xin Liu, Tao Wen, Tong Chen, Lei Chen, and Tengfei Ma developed the pipeline, performed the analysis, and wrote the manuscript. Xiaofang Li, Maosheng Zheng, Xin Zhou, Liang Chen, Xubo Qian, Jiao Xi, Hongye Lu, Huiluo Cao, Xiaoya Ma, Bian Bian, Pengfan Zhang, Jiqiu Wu, Ren‐You Gan, Baolei Jia, Linyang Sun, Zhicheng Ju, and Yunyun Gao have tested the pipeline, suggested pipeline amendments, and revised the manuscript. Tao Wen, Tong Chen, and Tengfei Ma write the codes for several visualization styles. Tong Chen improved the pipeline and designed the framework of visualization. Xin Zhou contributed to the VSEARCH analysis pipeline document. Yong‐Xin Liu, Tao Wen, Tong Chen, Xin Zhou, Xubo Qian, and Liang Chen contributed to the scripts and developed the user documentation. All authors have read the final manuscript and approved it for publication.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (U21A20182), and the Youth Innovation Promotion Association CAS (2021092).
Liu, Yong‐Xin , Chen Lei, Ma Tengfei, Li Xiaofang, Zheng Maosheng, Zhou Xin, Chen Liang, et al. 2023. “EasyAmplicon: An Easy‐To‐Use, Open‐source, Reproducible, and Community‐Based Pipeline for Amplicon Data Analysis in Microbiome Research.” iMeta, 2, e83. 10.1002/imt2.83
Yong‐Xin Liu, Lei Chen, and Tengfei Ma contributed equally to this work.
Contributor Information
Yong‐Xin Liu, Email: liuyongxin@caas.cn.
Tao Wen, Email: taowen@njau.edu.cn.
Tong Chen, Email: chent@nrc.ac.cn.
DATA AVAILABILITY STATEMENT
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive [61] in the National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA002352) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa. EasyAmplicon is freely available and implemented in Shell and R, and easy to install. The step‐by‐step protocols can be found at GitHub https://github.com/YongxinLiu/EasyAmplicon or Gitee https://gitee.com/YongxinLiu/EasyAmplicon. All the data of the figures can be downloaded in GitHub, or supplementary tables.
REFERENCES
- 1. Proctor, Lita M. , Creasy Heather H., Fettweis Jennifer M., Lloyd‐Price Jason, Mahurkar Anup, Zhou Wenyu, Buck Gregory A., et al. 2019. “The Integrative Human Microbiome Project.” Nature 569: 641–48. 10.1038/s41586-019-1238-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zuo, Tao , Wu Xiaojian, Wen Weiping, and Lan Ping. 2021. “Gut Microbiome Alterations in COVID‐19.” Genomics, Proteomics & Bioinformatics 19: 679–88. 10.1016/j.gpb.2021.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen, Lianmin , Wang Daoming, Garmaeva Sanzhima, Kurilshikov Alexander, Vich Vila Arnau, Gacesa Ranko, Sinha Trishla, et al. 2021. “The Long‐Term Genetic Stability and Individual Specificity of the Human Gut Microbiome.” Cell 184: 2302–2315. 10.1016/j.cell.2021.03.024 [DOI] [PubMed] [Google Scholar]
- 4. Qin, Junjie , Li Ruiqiang, Raes Jeroen, Arumugam Manimozhiyan, Burgdorf Kristoffer Solvsten, Manichanh Chaysavanh, Nielsen Trine, et al. 2010. “A Human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing.” Nature 464: 59–65. 10.1038/nature08821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Falony, Gwen , Joossens Marie, Vieira‐Silva Sara, Wang Jun, Darzi Youssef, Faust Karoline, Kurilshikov Alexander, et al. 2016. “Population‐Level Analysis of Gut Microbiome Variation.” Science 352: 560–64. 10.1126/science.aad3503 [DOI] [PubMed] [Google Scholar]
- 6. Stewart, Robert D. , Auffret Marc D., Warr Amanda, Walker Alan W., Roehe Rainer, and Watson Mick. 2019. “Compendium of 4,941 Rumen Metagenome‐Assembled Genomes for Rumen Microbiome Biology and Enzyme Discovery.” Nature Biotechnology 37: 953–61. 10.1038/s41587-019-0202-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang, Peng , Zhang Yan, Xiao Kangpeng, Jiang Fan, Wang Hengchao, Tang Dazhi, Liu Dan, et al. 2018. “The Chicken Gut Metagenome and the Modulatory Effects of Plant‐Derived Benzylisoquinoline Alkaloids.” Microbiome 6: 211. 10.1186/s40168-018-0590-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xiao, Liang , Estellé Jordi, Kiilerich Pia, Ramayo‐Caldas Yuliaxis, Xia Zhong kui, Feng Qiang, Liang Suisha, et al. 2016. “A Reference Gene Catalogue of the Pig Gut Microbiome.” Nature Microbiology 1: 16161. 10.1038/nmicrobiol.2016.161 [DOI] [PubMed] [Google Scholar]
- 9. Liu, Yong‐Xin , Qin Yuan, and Bai Yang. 2019. “Reductionist Synthetic Community Approaches in Root Microbiome Research.” Current Opinion in Microbiology 49: 97–102. 10.1016/j.mib.2019.10.010 [DOI] [PubMed] [Google Scholar]
- 10. Zhang, Jingying , Liu Yong‐Xin, Zhang Na, Hu Bin, Jin Tao, Xu Haoran, Qin Yuan, et al. 2019. “ NRT1.1B is Associated with Root Microbiota Composition and Nitrogen Use in Field‐Grown Rice.” Nature Biotechnology 37: 676–84. 10.1038/s41587-019-0104-4 [DOI] [PubMed] [Google Scholar]
- 11. Xi, Jiao , Ding Zanbo, Xu Tengqi, Qu Wenxing, Xu Yanzhi, Ma Yongqing, Xue Quanhong, Liu Yongxin, and Lin Yanbing. 2022. “Maize Rotation Combined with Streptomyces Rochei D74 to Eliminate Orobanche Cumana Seed Bank in the Farmland.” Agronomy 12: 3129. 10.3390/agronomy12123129 [DOI] [Google Scholar]
- 12. Nayfach, Stephen , Roux Simon, Seshadri Rekha, Udwary Daniel, Varghese Neha, Schulz Frederik, Wu Dongying, et al. 2021. “A Genomic Catalog of Earth's Microbiomes.” Nature Biotechnology 39: 499–509. 10.1038/s41587-020-0718-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu, Yong‐Xin , Chen Tong, Li Danyi, Fu Jingyuan, and Liu Shuang‐Jiang. 2022. “iMeta: Integrated Meta‐Omics for Biology and Environments.” iMeta 1: e15. 10.1002/imt2.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li, Wenjun , Wang Likun, Li Xiaofang, Zheng Xin, Cohen Michael F., and Liu Yong‐Xin. 2022. “Sequence‐Based Functional Metagenomics Reveals Novel Natural Diversity of Functioning CopA in Environmental Microbiomes.” Genomics, Proteomics & Bioinformatics. 10.1016/j.gpb.2022.08.006 [DOI] [PubMed] [Google Scholar]
- 15. Bolyen, Evan , Rideout Jai Ram, Dillon Matthew R., Bokulich Nicholas A., Abnet Christian C., Al‐Ghalith Gabriel A., Alexander Harriet, et al. 2019. “Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2.” Nature Biotechnology 37: 852–57. 10.1038/s41587-019-0209-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang, Jingying , Liu Yong‐Xin, Guo Xiaoxuan, Qin Yuan, Garrido‐Oter Ruben, Schulze‐Lefert Paul, and Bai Yang. 2021. “High‐Throughput Cultivation and Identification of Bacteria from the Plant Root Microbiota.” Nature Protocols 16: 988–1012. 10.1038/s41596-020-00444-7 [DOI] [PubMed] [Google Scholar]
- 17. Liu, Yong‐Xin , Qin Yuan, Guo Xiaoxuan, and Bai Yang. 2019. “Methods and Applications for Microbiome Data Analysis.” Hereditas (Beijing) 41: 845–826. 10.16288/j.yczz.19-222 [DOI] [PubMed] [Google Scholar]
- 18. Schloss, Patrick D. , Westcott Sarah L., Ryabin Thomas, Hall Justine R., Hartmann Martin, Hollister Emily B., Lesniewski Ryan A., et al. 2009. “Introducing Mothur: Open‐Source, Platform‐Independent, Community‐Supported Software for Describing and Comparing Microbial Communities.” Applied and Environmental Microbiology 75: 7537–41. 10.1128/aem.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Edgar, Robert C. 2010. “Search and Clustering Orders of Magnitude Faster than BLAST.” Bioinformatics 26: 2460–61. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 20. Caporaso, J. Gregory , Kuczynski Justin, Stombaugh Jesse, Bittinger Kyle, Bushman Frederic D., Costello Elizabeth K., Fierer Noah, et al. 2010. “QIIME allows Analysis of High‐Throughput Community Sequencing Data.” Nature Methods 7: 335–36. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gonzalez, Antonio , Navas‐Molina Jose A., Kosciolek Tomasz, McDonald Daniel, Vázquez‐Baeza Yoshiki, Ackermann Gail, DeReus Jeff, et al. 2018. “Qiita: Rapid, Web‐Enabled Microbiome Meta‐Analysis.” Nature Methods 15: 796–98. 10.1038/s41592-018-0141-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mitchell, Alex L. , Almeida Alexandre, Beracochea Martin, Boland Miguel, Burgin Josephine, Cochrane Guy, Crusoe Michael R., et al. 2020. “MGnify: The Microbiome Analysis Resource in 2020.” Nucleic Acids Research 48: D570–D578. 10.1093/nar/gkz1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shi, Wenyu , Qi Heyuan, Sun Qinglan, Fan Guomei, Liu Shuangjiang, Wang Jun, Zhu Baoli, et al. 2019. “gcMeta: A Global Catalogue of Metagenomics Platform to Support the Archiving, Standardization and Analysis of Microbiome Data.” Nucleic Acids Research 47: D637–D648. 10.1093/nar/gky1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu, Yong‐Xin , Qin Yuan, Chen Tong, Lu Meiping, Qian Xubo, Guo Xiaoxuan, and Bai Yang. 2021. “A Practical Guide to Amplicon and Metagenomic Analysis of Microbiome Data.” Protein & Cell 12: 315–30. 10.1007/s13238-020-00724-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Qian, Xu‐Bo , Chen Tong, Xu Yi‐Ping, Chen Lei, Sun Fu‐Xiang, Lu Mei‐Ping, and Liu Yong‐Xin. 2020. “A Guide to Human Microbiome Research: Study Design, Sample Collection, and Bioinformatics Analysis.” Chinese Medical Journal 133: 1844–55. 10.1097/cm9.0000000000000871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parks, Donovan H. , Tyson Gene W., Hugenholtz Philip, and Beiko Robert G.. 2014. “STAMP: Statistical Analysis of Taxonomic and Functional Profiles.” Bioinformatics 30: 3123–24. 10.1093/bioinformatics/btu494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Segata, Nicola , Izard Jacques, Waldron Levi, Gevers Dirk, Miropolsky Larisa, Garrett Wendy S., and Huttenhower Curtis. 2011. “Metagenomic Biomarker Discovery and Explanation.” Genome Biology 12: R60. 10.1186/gb-2011-12-6-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Langille, Morgan G. I. , Zaneveld Jesse, Caporaso J. Gregory, McDonald Daniel, Knights Dan, Reyes Joshua A., Clemente Jose C., et al. 2013. “Predictive Functional Profiling of Microbial Communities Using 16S rRNA Marker Gene Sequences.” Nature Biotechnology 31: 814–21. 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Douglas, Gavin M. , Maffei Vincent J., Zaneveld Jesse R., Yurgel Svetlana N., Brown James R., Taylor Christopher M., Huttenhower Curtis, and Langille Morgan G. I.. 2020. “PICRUSt2 for Prediction of Metagenome Functions.” Nature Biotechnology 38: 685–88. 10.1038/s41587-020-0548-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ward, Tonya , Larson Jake, Meulemans Jeremy, Hillmann Ben, Lynch Joshua, Sidiropoulos Dimitri, and Spear John R., et al. 2017. "BugBase Predicts Organism‐Level Microbiome Phenotypes." bioRxiv : 133462. 10.1101/133462 [DOI] [Google Scholar]
- 31. Louca, Stilianos , Parfrey Laura Wegener, and Doebeli Michael. 2016. “Decoupling Function and Taxonomy in the Global Ocean Microbiome.” Science 353: 1272–77. 10.1126/science.aaf4507 [DOI] [PubMed] [Google Scholar]
- 32. Chen, Tong , Liu Yong‐Xin, and Huang Luqi. 2022. “ImageGP: An Easy‐To‐Use Data Visualization Web Server for Scientific Researchers.” iMeta 1: e5. 10.1002/imt2.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Letunic, Ivica , and Bork Peer. 2021. “Interactive Tree Of Life (iTOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation.” Nucleic Acids Research 49: W293–W296. 10.1093/nar/gkab301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rognes, Torbjørn , Flouri Tomáš, Nichols Ben, Quince Christopher, and Mahé Frédéric. 2016. “VSEARCH: A Versatile Open Source Tool for Metagenomics.” PeerJ 4: e2584. 10.7717/peerj.2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shen, Wei , Le Shuai, Li Yan, and Hu Fuquan. 2016. “SeqKit: A Cross‐Platform and Ultrafast Toolkit for fasta/q File Manipulation.” PloS One 11: e0163962. 10.1371/journal.pone.0163962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wen, Tao , Xie Penghao, Yang Shengdie, Niu Guoqing, Liu Xiaoyu, Ding Zhexu, Xue Chao, et al. 2022. “ggClusterNet: An R Package for Microbiome Network Analysis and Modularity‐Based Multiple Network Layouts.” iMeta 1: e32. 10.1002/imt2.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gu, Zuguang , Gu Lei, Eils Roland, Schlesner Matthias, and Brors Benedikt. 2014. “Circlize Implements and Enhances Circular Visualization in R.” Bioinformatics 30: 2811–12. 10.1093/bioinformatics/btu393 [DOI] [PubMed] [Google Scholar]
- 38. Lv, Zhiyao , Dai Rui, Xu Haoran, Liu Yongxin, Bai Bo, Meng Ying, Li Haiyan, et al. 2021. “The Rice Histone Methylation Regulates Hub Species of the Root Microbiota.” Journal of Genetics and Genomics 48: 836–43. 10.1016/j.jgg.2021.06.005 [DOI] [PubMed] [Google Scholar]
- 39. Wang, Jinfeng , Zheng Jiayong, Shi Wenyu, Du Nan, Xu Xiaomin, Zhang Yanming, Ji Peifeng, et al. 2018. “Dysbiosis of Maternal and Neonatal Microbiota Associated with Gestational Diabetes Mellitus.” Gut 67: 1614–25. 10.1136/gutjnl-2018-315988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yuan, Jun , Wen Tao, Zhang He, Zhao Mengli, Penton C. Ryan, Thomashow Linda S., and Shen Qirong. 2020. “Predicting Disease Occurrence with High Accuracy Based on Soil Macroecological Patterns of Fusarium Wilt.” The ISME Journal 14: 2936–50. 10.1038/s41396-020-0720-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li, Xiaoqing , Zheng Jiayong, Ma Xiuling, Zhang Bing, Zhang Jinyang, Wang Wenhuan, Sun Congcong, et al. 2021. “The Oral Microbiome of Pregnant Women Facilitates Gestational Diabetes Discrimination.” Journal of Genetics and Genomics 48: 32–39. 10.1016/j.jgg.2020.11.006 [DOI] [PubMed] [Google Scholar]
- 42. Qu, Kaiyang , Guo Fei, Liu Xiangrong, Lin Yuan, and Zou Quan. 2019. “Application of Machine Learning in Microbiology.” Frontiers in Microbiology 10: 827. 10.3389/fmicb.2019.00827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hernández Medina, Ricardo , Kutuzova Svetlana, Nielsen Knud Nor, Johansen Joachim, Hansen Lars Hestbjerg, Nielsen Mads, and Rasmussen Simon. 2022. “Machine Learning and Deep Learning Applications in Microbiome Research.” ISME Communications 2: 98. 10.1038/s43705-022-00182-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang, Nan , Cheng Mingyue, and Ning Kang. 2022. “Overcoming Regional Limitations: Transfer Learning for Cross‐regional Microbial‐Based Diagnosis of Diseases.” Gut gutjnl‐2022‐328216. 10.1136/gutjnl-2022-328216 [DOI] [PMC free article] [PubMed]
- 45. Wang, Jinfeng , Jia Zhen, Zhang Bing, Peng Lei, and Zhao Fangqing. 2020. “Tracing the Accumulation of In Vivo Human Oral Microbiota Elucidates Microbial Community Dynamics at the Gateway to the GI Tract.” Gut 69: 1355–56. 10.1136/gutjnl-2019-318977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Knights, Dan , Kuczynski Justin, Charlson Emily S., Zaneveld Jesse, Mozer Michael C., Collman Ronald G., Bushman Frederic D., Knight Rob, and Kelley Scott T.. 2011. “Bayesian Community‐Wide Culture‐Independent Microbial Source Tracking.” Nature Methods 8: 761–63. 10.1038/nmeth.1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shenhav, Liat , Thompson Mike, Joseph Tyler A., Briscoe Leah, Furman Ori, Bogumil David, Mizrahi Itzhak, Pe'er Itsik, and Halperin Eran. 2019. “FEAST: Fast Expectation‐Maximization for Microbial Source Tracking.” Nature Methods 16: 627–32. 10.1038/s41592-019-0431-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dhariwal, Achal , Chong Jasmine, Habib Salam, King Irah L., Agellon Luis B., and Xia Jianguo. 2017. “MicrobiomeAnalyst: A Web‐Based Tool for Comprehensive Statistical, Visual and Meta‐Analysis of Microbiome Data.” Nucleic Acids Research 45: W180–W188. 10.1093/nar/gkx295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chong, Jasmine , Liu Peng, Zhou Guangyan, and Xia Jianguo. 2020. “Using MicrobiomeAnalyst for Comprehensive Statistical, Functional, and Meta‐Analysis of Microbiome Data.” Nature Protocols 15: 799–821. 10.1038/s41596-019-0264-1 [DOI] [PubMed] [Google Scholar]
- 50. Huang, Ancheng C. , Jiang Ting, Liu Yong‐Xin, Bai Yue‐Chen, Reed James, Qu Baoyuan, Goossens Alain, et al. 2019. “A Specialized Metabolic Network Selectively Modulates Arabidopsis Root Microbiota.” Science 364: eaau6389. 10.1126/science.aau6389 [DOI] [PubMed] [Google Scholar]
- 51. McDonald, Daniel , Price Morgan N., Goodrich Julia, Nawrocki Eric P., DeSantis Todd Z., Probst Alexander, Andersen Gary L., Knight Rob, and Hugenholtz Philip. 2011. “An Improved Greengenes Taxonomy With Explicit Ranks for Ecological and Evolutionary Analyses of Bacteria and Archaea.” The ISME Journal 6: 610–18. 10.1038/ismej.2011.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zheng, Maosheng , Zhou Nan, Liu Shufeng, Dang Chenyuan, Liu Yong‐Xin, He Shishi, Zhao Yijun, Liu Wen, and Wang Xiangke. 2019. “N2O and NO Emission from a Biological Aerated Filter Treating Coking Wastewater: Main Source and Microbial Community.” Journal of Cleaner Production 213: 365–74. 10.1016/j.jclepro.2018.12.182 [DOI] [Google Scholar]
- 53. Chen, Tong , Zhang Haiyan, Liu Yu, Liu Yong‐Xin, and Huang Luqi. 2021. “EVenn: Easy To Create Repeatable and Editable Venn Diagrams and Venn Networks Online.” Journal of Genetics and Genomics 48: 863–66. 10.1016/j.jgg.2021.07.007 [DOI] [PubMed] [Google Scholar]
- 54. Gao, Chun‐Hui , Yu Guangchuang, and Cai Peng. 2021. “ggVennDiagram: An Intuitive, Easy‐to‐Use, and Highly Customizable R Package to Generate Venn Diagram.” Frontiers in Genetics 12: 706907. 10.3389/fgene.2021.706907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen, Qingwen , Jiang Ting, Liu Yong‐Xin, Liu Haili, Zhao Tao, Liu Zhixi, Gan Xiangchao, et al. 2019. “Recently Duplicated Sesterterpene (C25) Gene Clusters in Arabidopsis thaliana Modulate Root Microbiota.” Science China Life Sciences 62: 947–58. 10.1007/s11427-019-9521-2 [DOI] [PubMed] [Google Scholar]
- 56. Gu, Zuguang . 2022. “Complex Heatmap Visualization.” iMeta 1: e43. 10.1002/imt2.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang, Jingying , Zhang Na, Liu Yong‐Xin, Zhang Xiaoning, Hu Bin, Qin Yuan, Xu Haoran, et al. 2018. “Root Microbiota Shift in Rice Correlates with Resident Time in the Field and Developmental Stage.” Science China Life Sciences 61: 613–21. 10.1007/s11427-018-9284-4 [DOI] [PubMed] [Google Scholar]
- 58. Xu, Jun , Zhang Jie‐Ni, Sun Bo‐Hui, Liu Qing, Ma Juan, Zhang Qian, Liu Yong‐Xin, Chen Ning, and Chen Feng. 2022. “The Role of Genotype and Diet in Shaping Gut Microbiome in a Genetic Vitamin A Deficient Mouse Model.” Journal of Genetics and Genomics 49: 155–64. 10.1016/j.jgg.2021.08.015 [DOI] [PubMed] [Google Scholar]
- 59. Carrión, Víctor J. , Perez‐Jaramillo Juan, Cordovez Viviane, Tracanna Vittorio, de Hollander Mattias, Ruiz‐Buck Daniel, Mendes Lucas W., et al. 2019. “Pathogen‐Induced Activation of Disease‐Suppressive Functions in the Endophytic Root Microbiome.” Science 366: 606–12. 10.1126/science.aaw9285 [DOI] [PubMed] [Google Scholar]
- 60. Zgadzaj, Rafal , Garrido‐Oter Ruben, Jensen Dorthe Bodker, Koprivova Anna, Schulze‐Lefert Paul, and Radutoiu Simona. 2016. “Root Nodule Symbiosis in Lotus Japonicus Drives the Establishment of Distinctive Rhizosphere, Root, and Nodule Bacterial Communities.” Proceedings of the National Academy of Sciences of the United States of America 113: E7996–E8005. 10.1073/pnas.1616564113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chen, Tingting , Chen Xu, Zhang Sisi, Zhu Junwei, Tang Bixia, Wang Anke, Dong Lili, et al. 2021. “The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types.” Genomics, Proteomics & Bioinformatics 19: 578–83. 10.1016/j.gpb.2021.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive [61] in the National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA002352) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa. EasyAmplicon is freely available and implemented in Shell and R, and easy to install. The step‐by‐step protocols can be found at GitHub https://github.com/YongxinLiu/EasyAmplicon or Gitee https://gitee.com/YongxinLiu/EasyAmplicon. All the data of the figures can be downloaded in GitHub, or supplementary tables.