

Abstract
The compartment niche is the main reason behind the shifts in endophytic bacterial communities. Long‐term organic greenhouse exerted limited influence on the variations of endophytic bacterial communities. Organic greenhouse and root had more complex co‐occurrence networks than conventional greenhouse and stem, respectively. Cultivable method results found that Protecbacteria, Bacteriodes, and Actinobacteria are the dominant phyla in the endophytes.

INTRODUCTION
Greenhouse vegetable production in China has exploded in 2014, with a yield of 260 million tons, representing 35% of the overall vegetable production accounts, and tomato (Solanum lycopersicum L.) as an important economic vegetable reached 55 million tons in 2019 [1]. Organic farming production excludes inorganic fertilizers and operates on the basis of green resource cycling in an ecological manner by applying bio‐fertilizer instead of pesticide or fertilizer, which delivered greater ecological services, reduced adverse impacts on the environment, and increased sustainability of agricultural systems compared to conventional farming systems [2, 3].
Intriguingly, a lot of evidence has shown that a positive link between the microbiome and plant health by plant growth promoting rhizosphere (PGPR), which could enhance the nutrition metabolism and resistance to biological stress as plant's second genome [4]. Various compartment niches (such as roots, stems, leaves, flowers, and fruits) are inhabited by a large number of highly diverse microorganisms (i.e., the plant microbiome), including bacteria, fungi, archaea, protists, and so on, together with the host plant form a “symbiotic totality,” interacting and coevolving together [5, 6]. The plant microbiome plays an important role in plant growth and development, nutrient absorption, biotic and abiotic stress resistance, and so on [7]. Endophytes normally completed their whole life cycle within host plants without occurring plant diseases, and their multiproduction might be influenced by host plants and the growing environment [8]. In addition, plant bacteria can provide protection against fungal pathogen diseases by directly promoting plant growth or antibiotic production, nutrient solubilization and transportation, and nitrogen fixation [9, 10].
Existing studies have shown that soil and plant microbiome are determined by many factors, including plant genetic diversity, growth period, soil physical and chemical properties, and soil nutrient status [11, 12]. The majority of plant‐associated microbiomes acted as biofertilizers [13], illustrating that the bacterial communities associated with crops are meaningful to unearth potentially beneficial candidates for biologically controlling crop diseases [14].
In recent years, cultivation‐independent approaches, especially metagenomics approaches, have allowed a comprehensive analysis of bacterial diversity from various types of plants, including agronomic crops, dicotyledon, and herb plants [7, 9, 14, 15]. Nonetheless, previous studies on microorganisms mostly focused on rhizosphere soil bacterial quantity and community diversity, but the root and stem bacteria under long‐term greenhouse trials are largely unknown. Exploring the endophytic bacterial communities will benefit exploring the mechanisms of selectivity adaption in different niches and identifying potential biologic control or promoting growth microbiome candidates in agricultural production [16, 17].
The principal objective of this study was (1) to assess how host (compartment niche) and environmental factors (organic farming or conventional farming) interactively shape tomato endophytic microbiome assemblies and concurrence patterns based on long‐term field trials since 2002, and (2) to isolate the potential beneficial tomato microbiomes as a candidate for agricultural inoculants. This study will help to provide comprehensive insight into the tomato endophytic bacterial communities cultivated in a greenhouse agroecosystem and provide useful information for using the beneficial microbiome in agricultural production.
RESULTS
Soil physicochemical factors and taxonomy of tomato bacteria shift among different developmental stages in tomato
To explore the variation of microbiota in long‐term organic (ORG) and conventional (CON) systems, the experiments were procedure in two semi‐round arched greenhouses (Figure 1A). The change of physicochemical parameters during the key crop season with different agricultural practices is shown in Figure 1B–D. The total nitrogen (TN) concentration increased both in the organic and conventional greenhouses, but the TN concentration was higher in the organic greenhouse than that of the conventional greenhouse (Student's t test, p < 0.001) NH4 +–N showed a similar trend with TN, and the concentration of ammonia nitrogen increased with the plant development (Student's t test, p < 0.001), and the ammonium concentration in the organic greenhouse was higher than that of conventional greenhouse (Student's t test, p < 0.001). The soil organic matter content decreased from 54.31 to 33.85 g/kg in the organic greenhouse and from 50.44 to 29.65 g/kg in the conventional greenhouse (Student's t test, p < 0.001). See Supporting Information: Table S1 for the results of the two‐way analysis of variance (ANOVA) for soil properties as affected by agricultural manipulation (treat), growing phase (time), and the interaction (treat × time).
Figure 1.

Experimental design, soil chemical properties, and microbiota taxonomy of tomato root and stem. (A) The experimental design demonstrates a treatment and control setup for the organic greenhouse, niche selection, and crop phases. (B, D) Differences in soil major chemical properties for four phases of growth in plants (T1–T4), including (B) soil total nitrogen, (C) soil NH4+–N, and (D) soil organic content. **Significant dramatically (p < 0.01). (E) Changes in the endophytic microbiome composition during the field experiment at the phylum level.
A total of 648 operational taxonomy units (OTUs) and 2,014,992 high‐quality, nonplastid sequences were acquired (Supporting Information: Figure S1). The taxonomic analysis found that the tomato bacteria mainly comprised four phyla (Figure 1F), among which Proteobacteria was the most abundant (40.63%), followed by Firmicutes (21.76%), Actinobacteria (20.3%), and Bacteroidetes (8.7%). In addition, the relative abundance of the top 30 most abundant genera is shown in Supporting Information: Figure S2. The dominant genera belonged to Weissella, Bacillus, Mesorhizobium, and Chryseobacterium.sp in the endophytes. The abundance of Firmicutes in the bacterial community was decreased and the abundance of Proteobacteria increased significantly, especially after the fruiting phase in the root endophytes (Supporting Information: Figure S3), and a similar phenomenon occurred in stem endophytic bacterial communities: the relative abundance of Arthrobacter.sp, Rhizobium.sp gradually increased, while the Bacillus.sp decreased dramatically during the late crop season (Figure 1F and Supporting Information: Figure S2).
Plant organs shaped tomato microbiome assembly more strongly than agricultural farming system factors
Results suggested that compartment niche had a strong effect on bacterial diversity (Shannon index), and roots have higher Shannon diversity than stems. Shannon index was higher in the late growth phase than in the early growth phase; however, it had no difference between different manipulation (Figure 2A–C). See Supporting Information: Figure S4 for Chao1 index. Results based on Bray–Curtis distance and ADONIS analysis of the complete data set suggested that the variation of the bacterial community derived from compartment‐niche in the organic greenhouse (R 2 = 0.076, p = 0.001) (Figure 2D). Moreover, bacterial communities in the conventional greenhouses were similar, of which the variations in microbial communities were mainly explained by compartment niche (R 2 = 0.056, p = 0.021) (Figure 2E,F). Nonmetric multidimensional scaling (NMDS) showed that bacteria in four crop development phases separated clusters in the first two coordinate axes (Figure 2G, R 2 = 0.106, p = 0.002). Root and stem samples grouped with organic, conventional agricultural practice did not form a distinct cluster (R 2 = 0.04, p = 0.135 and R 2 = 0.02, p = 0.59, for root and stem, respectively, Figure 2H,I).
Figure 2.

(A–C) Shannon index of the microbiota in the tomato microbiome. (A) Different agricultural manipulation, (B) different compartment niches, (C) different growth phases. (D–I) Nonmetric multidimensional scaling (NMDS) ordinations based on Bray–Curtis distances of endophytic bacterial communities. (D) For all samples, (E, F) NMDS in each compartment niche. (E) Root and stem microbiota in the organic farming system. (F) Root and stem microbiota in the conventional farming system. (G) In different growth phases. (H, I) NMDS in each agricultural farming system. (H) Root microbiota in the different farming systems. (I) Stem microbiota in the different farming systems. CONR, conventional system root endophytes, CONS, conventional system stem endophytes, ORGR, organic system root endophytes; ORGS, organic system stem endophytes.
Biomarker taxa and potential function of bacterial microbiome among different farming manipulation
The LDA effect size (LEfSe) analysis demonstrated that Flavobacterium, Sphingomonadales, xanthomnadaccae, and Rhizobiom were enriched in the organic tomato root, while Bacteriodes were contained in the conventional tomato root. Enterobacteriaceae are enriched in the organic stem, and corynbacterium is enriched in the conventional stem (Figure 3A,B). Bacterial function prediction analyses for these dominant taxa showed that the top four abundant functional groups (accounting for >75% of total groups) included lipid metabolism and signal transduction. Little function discrepancy was found between the organic and conventional tomato endophytic bacteria; however, a greater relative abundance of energy metabolism functional group was observed in root samples (3.8%) than that in stem (0.8%) samples (Figure 3C,D).
Figure 3.
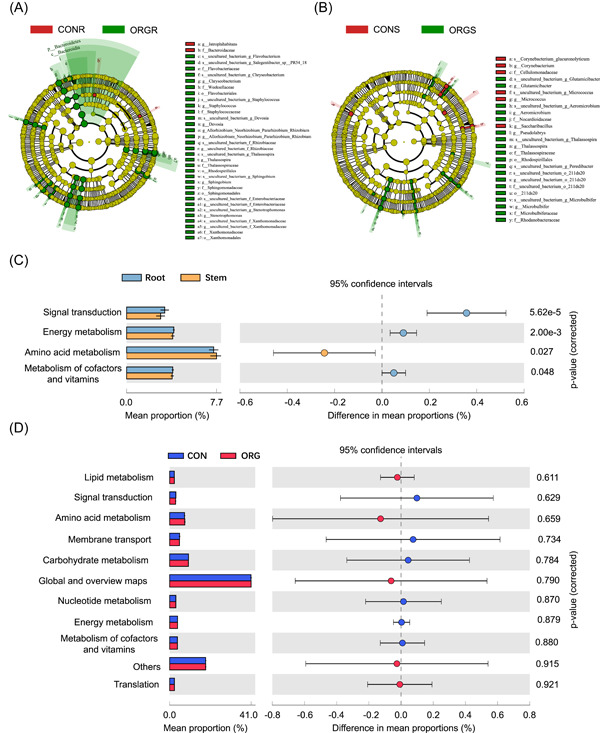
Organic (A) and conventional system (B) biomarkers in the different treatments based on the LEfSe analysis. Different colors represent different treatments and the circles from inside to outside correspond with phylum and genus. The color‐coded one within the cladogram denotes the taxa with a significantly higher relative abundance in the treatment as analyzed by the Kruskal–Wallis test with p < 0.05 and a logarithmic LDA score >3.5. Genera with a relative abundance of less than 0.1% were not included. (C) KEGG categories (L2 level) differ significantly between root and stem based on Welch's t test. (D) KEGG categories (L2 level) differ significantly between ORG and CON based on Welch's t test.
Network associations and microbial hubs in different treatments
To explore the effect of organic greenhouse and niche selection, the crop phase is not the main factor of our concern. Thus, four co‐occurrence networks were constructed (conventional system root endophytes, conventional system stem endophytes, organic system root endophytes, and organic system stem endophytes) merged by four plant crop phases (Figure 4A–D). The topological features of the co‐occurrence network are shown in Figure 4E and Supporting Information: Table S2, and details concerning the bacteria classified as nodes in the connector and module hub area can be found in Supporting Information: Table S3. The taxonomic composition of the networks showed no difference among the four treatments, with the majority of nodes belonging to Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria, respectively. Network complexity gradually decreased from organic greenhouse to conventional greenhouse. The average connectivity degree of roots and stems in the organic greenhouse was 30.172 and 6.215, while that in the conventional greenhouse decreased by 19.415 and 5.893. All the nodes with Zi ≥ 2.5 or Pi ≥ 0.62 were determined as the keystone species, that is, nodes in the area of connectors (0.24%), module hubs (0.65%), and network hubs (0%) played a crucial role in the co‐occurrence networks. Among them, the connectors and module hubs of phyla Proteobacteria (e.g., Rhizobiaceae, Enterobacteriaceae, Sphingomonadaceae, and Pseudomonadaceae), Bacteroidetes (e.g., Sphingobacteriaceae), and Actinobacteria (e.g., Anaerolinea) were accounted for 85%, 1%, and 0.5%, respectively (see Supporting Information: Table S3). RDA analysis was performed to reveal that soil TN was considered the major driver, which led to a tomato endophytic bacterial community shift (Figure 4F).
Figure 4.

Visualized networks of microbial co‐occurrence in different treatments. (A) CONR, (B) CONS, (C) ORGR, (D) ORGS. (E) Zi–Pi plot shows the distribution of OTUs based on their topological roles with different treatments. (F) The relationship between the bacterial community, soil physical, chemical properties, and agricultural practice. The redundancy analyses (RDA) of the correlation between the bacterial community (genus level) and key factors using forward selections. The blue lines represent significant factors and the red lines represent the family level. CONR, conventional system root endophytes; CONS, conventional system stem endophytes; ORGR, organic system root endophytes. ORGS, organic system stem endophytes; OTU, operational taxonomy unit.
To further characterize the compartment niche selection effect on tomato microbiota, and the key functional bacteria for TN identified based on the network analysis, microbial networks were generated (Figure 5A,B). More root bacteria were beneficial for soil organic formation than the stem. Network complexity gradually decreased from root to stem (with an average degree of 5.59 in the root and 4.30 in the stem); (see Supporting Information: Table S4). Results also demonstrated that most of the nodes (99.11%) were in the peripheral region, and their connections were primarily connected to the nodes in the module, at the same time, nodes in the area of connectors (1.8%), module hubs (2.2%), and network hubs (0%) played a crucial role in the co‐occurrence networks. Among them, the connectors and module hubs of phyla Proteobacteria (e.g., Rhizobiaceae, Enterobacteriaceae, Sphingomonadaceae, and Pseudomonadaceae), Bacteroidetes (e.g., Sphingobacteriaceae), and Bacteroides (e.g., Sphingobacteriaceae) were accounted for 77% and 21%, respectively (see Supporting Information: Table S3).
Figure 5.

(A, B) Correlation network visualized the interaction patterns between soil physiochemical (eg., pH, total nitrogen, NH4+–N, and organic content) and related bacterial based on class level (i.e., bacterial community with relative incidence >20% and relative abundance >0.01% and strong correlations “|r| > 0.6, p < 0.05,” only strong correlations “p < 0.05” were showed) in (A) root and (B) stem. The red and green lines denote significantly positive and negative correlations, respectively (p < 0.05). (C) Zi–Pi plot shows the distribution of operational taxonomy units (OTUs) based on their topological roles representing an OTU with a different niche.
Cultivable isolation endophytic bacterial diversity
We isolated a total of 663 bacteria by the cultural‐dependent method, but after 16S sequence alignment to remove redundancy, we obtained 60 bacteria from a different genus. Results showed that the number of endophytic bacterial increased with a plant growing, ranging from 4.21 to 7.45 (log10 CFU/g.FW), reaching a peak in the fruiting phase (Figure 6A). Organic agricultural practice and conventional practice nearly almost have no influence on the number of endophytic bacteria, and the isolated number of bacteria was more in roots than that in stems (p < 0.01, Figure 6A). Bacteroidetes and Proteobacteria were dominant endophytic bacterial phylum, specifically, Bacillus is more prevalent in the organic farming greenhouse than that in the conventional farming greenhouse (Figure 6B).
Figure 6.

(A) The isolation frequency of the tomato bacteria with different farming systems and compartment niches. (B) Taxonomy evolutionary tree of the cultivatable endophytic bacteria.
DISCUSSION
Effects of compartment niche and agricultural regimes on the tomato endophytic microbial community
In this study, a greenhouse survey demonstrated that tomato endophytic microbiomes were assembled largely driven at the compartment niche perspective, but had insignificant influence from fertilization environmental practice. Numerous studies indicated that the plant microbiome is shaped largely by host selection, such as genetics and organ compartment niche, while environmental factors (i.e., fertilizer regimes) exerted limited influence [18]. The lower diversity in the endosphere than that in the rhizosphere and soil is likely due to the host selective effect [19]. Endophytic bacterial showed environmental insensitivity, while rhizosphere soil microbial communities fluctuated over the course of plant development. This may be due to changes in root exudate profiles having a more significant influence on rhizosphere soil microbiome over growth phases [20]. Han et al. [20, 21] have shown that the tomato bacterial communities in different compartment niches are significantly different, with host selection sequentially increased from soils to epiphytes to endophytes, and the successful colonization of microorganisms in different parts of plants requires the ability to overcome host immunity and abiotic pressures in different microhabitats. Similar results were observed in sugarcane [22], rice [23], and corn [24] associated microbiome, that is, compartment niche played a key role either in the composition of endophyte or epiphyte [25]. Xia et al. [26] revealed the relative contributions of host and environmental factors to maize [27], wheat [28], and barley [29] microbiome community construction, confirming that crop microbiome community construction was mainly shaped by site niche and host species, while location (and site‐related soil and climate factors) or fertilization practices had little effect.
Assemblies and maintenance of tomato endophytic bacterial microbiomes
Co‐occurrence patterns of bacterial communities of different agricultural practices were revealed in this study, and organic regimes established a more complicated network than conventional practices, and root networks are more complex than stem networks in both agriculture practices. Furthermore, the analysis of network correlation to soil chemical properties indicated that the positive and negative effects of organic on bacterial community up to 50% of the time, whereas TN accounted for 40.4% and 59.6% in root and stem, respectively. Similar to the soil microbiome study findings by Owens, S. M et al. [30]. The TN accounted for the positive effect of almost 70% (Figure 5), highlighting the importance of TN in driving bacterial community composition and function [31, 32]. In addition, the KEGG categories (L2 level) indicated that the amino acid metabolism of the microbial community in the stem niche was significantly higher than that in the root, while the signal transduction, energy metabolism, and metabolism of cofactors and vitamins were lower than root, and these results may cause less impact of stem bacterial community in soil organic content compared with root. Moreover, the regulation of soil TN may be more important for remodeling the root microbial community [33].
Keystone taxa for tomato microbiome
Several biomarkers were identified at the genus level in different treatments based on Lefse analysis. Results showed that Bacillales were enriched in the organic tomato, while Enterobacteriales and Actinobacteria were enriched in the conventional tomato (LDA > 3.5, p < 0.05, Figure 3A,B). Surprisingly, more taxonomy discrepancies occurred between root and stem in this study (Supporting Information: Figure S4). Zhao et al. [34] found that plant roots recruited a higher proportion of Proteobacteria and Bacteroides, which played a key role in modulating nutrition intake [35], pathogen suppression [36], and plant tolerance to stress [37]. In addition, the KEGG metabolism pathway (L2 level) of compartment niche is insignificantly different among root and stem (Figure 3C). The tomato root microbiome responds to environmental stresses by regulating specific metabolic pathways, such as amino acid metabolism and carbohydrate metabolism functions, and the activity of signal transduction was significantly improved to increase the resistance to adverse environments [38] (Figure 3D). This study revealed that the organ compartment niche was a key driver shaping endophytic bacteria and root microbiome but not that in the stem. Cultivable isolation further demonstrated that most of the dominant taxa belonged to Proteobacteria and Firmicutes, and the majority of biocontrol microbiome belonged to this phylum, having antagonism against soil‐borne diseases [39].
CONCLUSION
Comprehensive analysis of the relative contribution of compartment niche and environmental factors to the tomato endophytic microbiome assembly in this study. The findings revealed that compartment niche can drive community‐scale shifts in tomato endophytic bacterial composition; however, the agricultural practice had little influence on endophytic bacterial communities. Organic practices remodeled a more complicated network than conventional practices, and root endophytic bacterial networks are more complex than that stem networks. The amino acid metabolism of the microbial community in the stem was significantly higher than that in the root, while the signal transduction, energy metabolism, and metabolism of cofactors and vitamins were lower than root. Proteobacteria and Firmicutes are dominant taxonomies in the cultivable endophytic bacteria, and Bacillus is more highly abundant in organic farming.
In conclusion, this study provides a comprehensive view of compartment niche and agricultural practice factors shaping the tomato endophytic bacterial communities under greenhouse conditions. These efforts will provide important information for the incorporation of beneficial bacteria into tomato agricultural production.
METHODS
Greenhouse trial and sample collection
Long‐term trials have been conducted in Quzhou county, Hebei province (36°52′N, 115°01′E) since 2002. Both the ORG and CON systems were part of this experiment. There were two semi‐round arched greenhouses (52 m in length and 7 m in width for each greenhouse) as shown in Figure 1A. Crop cultivars, irrigation, and tillage schemes were the same in all two systems in the same growing season, and a previous study had documented the agricultural management in each greenhouse in detail [40]. ORG manipulation was carried out in accordance with the International Federation of Organic Agriculture Movements' (IFOAM) guidelines, employing compost obtained from chicken and cow dung, as well as biocontrol for plant protection (i.e., Sticky yellow paper traps and insect net to control pests, mechanical removal of diseased plants and ridging planting to control plant diseases, mechanical/mulch to suppress weeds, etc.) for plant protection. The CON system used fertilizers, insecticides, and poultry to produce greenhouse vegetables locally [41, 42].
Tomato samples (Solanum lycopersicum cultivar “Money Marker”) were collected from organic and conventional greenhouses at four‐time points (i.e., seedling phase, flowering phase, fruiting phase, and harvesting phase), and both roots and stems parts were sampled. Each treatment had five replications; thus 40 root and 40 stem samples were sampled in total (Figure 1A). R2A agar plate (Reasoner's 2A agar) is a culture medium, which is a nutritionally rich media supporting the growth of fast‐growing bacteria [43, 44].
Measurements of soil chemical properties
Soil pH was measured using a pH meter and a soil‐to‐water ratio of 1:5 (PHS‐3C). The elemental analyzer was used to measure soil chemical properties, such as TN and organic content (Vario EL III‐Elementer), NH4+–N was analyzed by Flow Analysis Instrument (AutoAnalyzer3). Soil chemical properties were recorded by Excel (v.2019) and significance was tested by ANOVA in SPSS (v.19).
DNA extraction and 16S ribosomal RNA (rRNA) gene amplification
Plant samples were syndicated in Diagenode Bioruptor at a low frequency for physical removal of microscopic soil aggregates and attached microbes, and all samples were chilled on ice before further storage at −80°C. Surface sterilization was performed as previously described [45, 46]. No bacterial growth was detected after inoculating the roots on the R2A agar plate at 30°C for 7 days, as well as no amplification of the 16S rRNA gene was observed when final wash water as template DNA, and sterilization was qualified.
Total microbial community DNA from 80 samples was extracted using a Fast DNA spin Kit for soil (MP, Biomedicals). The bacterial 16S rRNA gene fragments were amplified with the universal primers 799 F (5′‐GTGCCAGCMGCCGCGGTAA‐3′) and 1193 R (5′‐CCCCGYCAATTCMTTTRAGT‐3′) fused with unique barcode [47]. Gel‐purified polymerase chain reaction products were mixed with equal molar following illumine sequencing using the platform of Hiseq. 2500.
Bioinformatic and statistical analysis
All sequence reads were assigned to each sample based on primer and barcodes and the technical regions were trimmed for the following analyses. The quality‐filtered and demultiplexed sequences (length > 300 bp, without ambiguous base “N,” and average base quality score >30) were used for further analyses. Generation of the taxonomic OTU was performed as previously described [48]. Classification of representative OTUs were performed with RDP naïve bayes classifier version by the UNOISE algorithm [49, 50]. All sequences under the accession number PRJCA010522 in this study, and the raw sequencing reads were deposited in the Genome Sequence Archive (GSA) (CRA007512) [51].
The microbiome composition was ordinated by NMDS using Bray–Cutis distance, and the effect of different factors, including sampling time, treatment, and plant status, on microbial community dissimilarity was tested by ADONIS using the vegan package in R (http://cran.r-project.org/) [52]. Redundant and canonical correlation analyses were performed to display the relationships between microbial communities and environmental variables using the Canoco 5 [53]. OTU present in more than 60% of samples were retained for the network analysis to avoid possible biases, and the network topological parameters were calculated in Cytoscape v 3.8.0 and Gpehi v 0.9.2 [54]. Linear discriminant analysis and a significance test were used to explore the most discriminating OTUs using LEfSe and DESeq2, respectively [55]. Three screening criteria were used: linear discriminant analysis with a score of ≥3.5 (health condition relative to diseased condition); fold change ≥2 (health condition relative to diseased condition); significance test with p < 0.05. PICRUSt v 1.1.0 (http://picrust.github.io/picrust/) was used to investigate the tomato endophytic bacteria's functional genes, and KEGG functional annotation was used to obtain information on the annotation of OTUs at each KEGG function level. STAMP v 2.1.3 was used for statistical hypothesis tests and exploratory plots using Welch's t test to compare the differences between the two groups.
Isolation of tomato root and stem bacteria
Samples were washed twice with sterilized PBST buffer (Na2HPO4 1.42 g/L; KH2PO4 0.24 g/L; NaCl 8 g/L; KCl 0.2 g/L; 0.01% Triton X‐100, pH 7.4) with shaking (180 rpm) for 1 h at 30°C. The roots and stems were continued to be washed until all dirt particles were removed after centrifugation at 1000 rpm at 4°C, and removing epiphytic bacteria as much as possible. Then the clean roots and stems were cut up into pieces and a subsample of root tissue, typical of the entire plant system, including fresh roots as well as older root tissues, was collected and utilized for bacteria isolation. All bacteria were isolated by picking colonies, and each isolate was randomly selected by Sanger sequencing analysis, and sequence reads were aligned using BLASTn1, and the closest match was identified [56].
AUTHOR CONTRIBUTIONS
Zeyu Zhang and Ji Li: Designed the research, implemented the experiment, writing—original draft, data curation, writing—review, and editing. Zeyu Zhang and Yang Sean Xiao: performed the data analysis. Yang Sean Xiao: Revised figures and tables, writing—review, and editing. Yabin Zhan, Zengqiang Zhang, Youzhou Liu, Yuquan Wei, and Ting Xu: Writing—review and editing. All authors contributed to the article and approved the submittedversion.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supplementary information.
Supplementary information.
ACKNOWLEDGMENTS
This study was financially supported by the National Natural Science Foundation of China (201503010311304). We appreciate the editor's and reviewers' comments on the draft of this manuscript.
Zhang, Zeyu , Xiao Yang Sean, Zhan Yabin, Zhang Zengqiang, Liu Youzhou, Wei Yuquan, Xu Ting, and Li Ji. 2022. “Tomato microbiome under long‐term organic and conventional farming.” iMeta 1, e48. 10.1002/imt2.48
Zeyu Zhang and Yang Sean Xiao contributed equally to this work and should be considered co‐first authors.
DATA AVAILABILITY STATEMENT
All data analyzed during this study are included in this published article and its Supporting Information files. The data sets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: PRJCA010522, and CRA007512 in the GSA. Supporting Information materials (figures, tables, scripts, graphical abstract, slides, videos, Chinese translated version, and update materials) may be found in the online DOI or iMeta Science http://www.imeta.science/.
REFERENCES
- 1. Liang, Long , Ridoutt Bradley G., Lal Rattan, Wang Dapeng, Wu Wenliang, Peng Peng, Hang Sheng, Wang Liyuan, and Zhao Guishen. 2019. “Nitrogen Footprint and Nitrogen Use Efficiency of Greenhouse Tomato Production in North China.” Journal of Cleaner Production 208: 285–96. 10.1016/j.jclepro.2018.10.149 [DOI] [Google Scholar]
- 2. Zhuang, Lubo , Li Yan, Wang Zhenshuo, Yu Yue, Zhang Nan, Yang Chang, Zeng Qingchao, and Wang Qi. 2021. “Synthetic Community With Six Pseudomonas Strains Screened From Garlic Rhizosphere Microbiome Promotes Plant Growth.” Microbial Biotechnology 14: 488–502. 10.1111/1751-7915.13640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dong, Chun Juan , Wang Ling Ling, Li Qian, and Shang QingMao. 2019. “Bacterial Communities in the Rhizosphere, Phyllosphere and Endosphere of Tomato Plants.” PLoS One 14: e0223847. 10.1371/journal.pone.0223847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cheng, Zhi Qiang , Lei Shaonan, Li Ye, Huang Wei, Ma Rong Qi, Xiong Juan, Zhang Ting, et al. 2020. “Revealing the Variation and Stability of Bacterial Communities in Tomato Rhizosphere Microbiota.” Microorganisms 8: 170. 10.3390/microorganisms8020170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bengtsson, Janne , AhnstrÖM Johan, and Weibull Ann‐Christin. 2005. “The Effects of Organic Agriculture on Biodiversity and Abundance: A Meta‐Analysis.” Journal of Applied Ecology 42: 261–9. 10.1111/j.1365-2664.2005.01005.x [DOI] [Google Scholar]
- 6. Bulgarelli, Davide , Schlaeppi Klaus, Spaepen Stijn, Ver Loren van Themaat Emiel, and Schulze‐Lefert Paul. 2013. “Structure and Functions of the Bacterial Microbiota of Plants” Annual Review of Plant Biology 64: 807–38. 10.1146/annurev-arplant-050312-120106 [DOI] [PubMed] [Google Scholar]
- 7. Genitsaris, Savvas , Stefanidou Natassa, Leontidou Kleopatra, Matsi Theodara, Karamanoli Katerina, and Mellidou Ifigeneia. 2020. “Bacterial Communities in the Rhizosphere and Phyllosphere of Halophytes and Drought‐Tolerant Plants in Mediterranean Ecosystems.” Microorganisms 8: 1708. 10.3390/microorganisms8111708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ghahremani, Zahra , Escudero Nuria, Saus Ester, Gabaldón Toni, and Sorribas F. Javier. 2019. “Pochonia Chlamydosporia Induces Plant‐Dependent Systemic Resistance to Meloidogyne Incognita” Frontiers in Plant Science 10: 945. 10.3389/fpls.2019.00945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de Lamo, Francisco J. , Constantin Maria E., Fresno David H., Boeren Sjef, Rep Martijn, and Takken Frank L. W.. 2018. “Xylem Sap Proteomics Reveals Distinct Differences Between R Gene‐ and Endophyte‐Mediated Resistance Against Fusarium Wilt Disease in Tomato.” Frontiers in Microbiology 9: 2977. 10.3389/fmicb.2018.02977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Toju, Hirokazu , Peay Kabir G., Yamamichi Masato, Narisawa Kazuhiko, Hiruma Kei, Naito Ken, Fukuda Shinji, et al. 2018. “Core Microbiomes for Sustainable Agroecosystems.” Nature Plants 4: 247–57. 10.1038/s41477-018-0139-4 [DOI] [PubMed] [Google Scholar]
- 11. Carrión, Víctor , Juan Perez‐Jaramillo J., Cordovez Viviane, Tracanna Vittorio, de Hollander Mattias, Ruiz‐Buck Daniel, Lucas W Mendes, et al. 2019. “Pathogen‐Induced Activation of Disease‐Suppressive Functions in the Endophytic Root Microbiome.” Science 366: 606–12. 10.1126/science.aaw9285 [DOI] [PubMed] [Google Scholar]
- 12. Zhao, Shuai , Li Li, Li Shan Hui, Wang Hong Fei, Hozzein Wael. N, Zhang Yong Guang, Wadaan Mohammed. A, Li Wen Jun, and Tian Chang Yan. 2015. “Actinotalea Suaedae Sp. Nov., Isolated From the Halophyte Suaeda Physophora in Xinjiang, Northwest China.” Antonie Van Leeuwenhoek 107: 1–7. 10.1007/s10482-014-0297-y [DOI] [PubMed] [Google Scholar]
- 13. Lee, Sang‐Moo , Kong Hyun Gi, Song Geun Cheol, and Ryu Choong‐Min. 2021. “Disruption of Firmicutes and Actinobacteria Abundance in Tomato Rhizosphere Causes the Incidence of Bacterial Wilt Disease.” ISME j 15: 330–47. 10.1038/s41396-020-00785-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Romero, F. M. , Marina M., and Pieckenstain F. L.. 2014. “The Communities of Tomato (Solanum lycopersicum L.) Leaf Endophytic Bacteria, Analyzed by 16S‐Ribosomal RNA Gene Pyrosequencing.” FEMS Microbiology Letters 351: 187–94. 10.1111/1574-6968.12377 [DOI] [PubMed] [Google Scholar]
- 15. Yang, Rui Heng , Bao Da Peng, Guo Ting, Li Yan, Ji Guang Yan, Ji Kai Ping, and Tan Qi. 2019. “Bacterial Profiling and Dynamic Succession Analysis of Phlebopus Portentosus Casing Soil Using MiSeq Sequencing.” Frontiers in Microbiology 10: 1927. 10.3389/fmicb.2019.01927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhechao, Zhang , Zimin Wei, Wei Guo, Yuquan Wei, Junqing Luo, Caihong Song, Qian Lu, and Yue Zhao. 2021. “Two Types Nitrogen Source Supply Adjusted Interaction Patterns Of Bacterial Community To Affect Humifaction Process Of Rice Straw Composting.” Bioresource Technology 332: 125129. 10.1016/j.biortech.2021.125129 [DOI] [PubMed] [Google Scholar]
- 17. Deng, Xuhui , Zhang Na, Shen Zongzhuan, Zhu Chengzhi, Liu Hongjun, Xu Zhihui, Li Rong, Shen Qirong, and Salles Joana Falcao. 2021. “Soil Microbiome Manipulation Triggers Direct and Possible Indirect Suppression Against Ralstonia Solanacearum and Fusarium Oxysporum.” NPJ Biofilms and Microbiomes 7: 33. 10.1038/s41522-021-00204-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xiong, Chao , Zhu Yongguan, Wang Juntao, Singh Brajesh Kumar, Han Li Li, Shen Ju Pei, Li Pei‐pei, et al. 2020. “Host Selection Shapes Crop Microbiome Assembly and Network Complexity.” New Phytologist Foundaion 229: 1091–104. 10.1111/nph.16890 [DOI] [PubMed] [Google Scholar]
- 19. Wongkiew, Sumeth , Chaikaew Pasicha, Takrattanasaran Natta, and Khamkajorn Thanachanok. 2022. “Evaluation of Nutrient Characteristics and Bacterial Community in Agricultural Soil Groups for Sustainable Land Management.” Scientific Reports 12: 7368. 10.1038/s41598-022-09818-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gu, Shaohua , Wei Zhong, Shao Zhengying, Friman Ville‐Petri, Cao Kehao, Yang Tianjie, Kramer Jos, et al. 2020. “Competition for Iron Drives Phytopathogen Control By Natural Rhizosphere Microbiomes.” Nature Microbiology 5: 1002–10. 10.1038/s41564-020-0719-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Han, Qin , Ma Qun, Chen Yong, Tian Bing, Xu Lanxi, Bai Yang, Chen Wenfeng, and Li Xia. 2020. “Variation in Rhizosphere Microbial Communities and its Association With the Symbiotic Efficiency of Rhizobia in Soybean.” ISME J 14: 1915–28. 10.1038/s41396-020-0648-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Malviya, Mukesh Kumar , Solanki Manoj Kumar, Li Chang Ning, Htun Reemon, Singh Rajesh Kumar, Singh Pratiksha, and Li Yang Rui. 2021. “7—Sugarcane Microbiome: Role in Sustainable Production.” Microbiomes and Plant Health edited by Solanki Manoj Kumar, Kashyap Prem Lal, Ansari Rizwan Ali and Kumari Baby, 225–42. Academic Press. 10.1016/B978-0-12-819715-8.00007-0 [DOI] [Google Scholar]
- 23. Lu, Yahai , Rosencrantz D., Liesack W., and Conrad R.. 2006. “Structure and Activity of Bacterial Community Inhabiting Rice Roots and the Rhizosphere.” Environmental Microbiology 8: 1351–60. 10.1111/j.1462-2920.2006.01028.x [DOI] [PubMed] [Google Scholar]
- 24. Ali, Shimaila , Saldias Soleda, Weerasuriya Nimalka, Delaney Kristen, Kandasamy Saveetha, and Lazarovits George. 2020. “Corn Microbial Diversity and its Relationship to Yield.” Canadian Journal of Microbiology 66: 457–73. 10.1139/cjm-2020-0002 [DOI] [PubMed] [Google Scholar]
- 25. Manzotti, Andrea , Bergna Alessandro, Burow Meike, Jørgensen Hans J. L., Cernava Tomislav, Berg Gabriele, Collinge David B., and Jensen Birgit. 2020. “Insights Into the Community Structure and Lifestyle of the Fungal Root Endophytes of Tomato by Combining Amplicon Sequencing and Isolation Approaches with Phytohormone Profiling.” FEMS Microbiology Ecology 96: fiaa052. 10.1093/femsec/fiaa052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xia, Ye , De Bolt Seth, Dreyer Jamin, Scott Delia, and Williams Mark A.. 2015. “Characterization of culturable bacterial endophytes and their capacity to promote plant growth from plants grown using organic or conventional practices.” Frontiers in Plant Science 6: 49. 10.3389/fpls.2015.00490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peiffer, Jason A. , Spor Ayme, Koren Omry, Jin Zhao, Tringe Susannah G., Dangl Jeffery L., Buckler Edward S., and Ley Ruth E.. 2013. “Diversity and Heritability of the Maize Rhizosphere Microbiome Under Field Conditions.” Proceedings of National Academy of Science of the United States of America 110: 6548–53. 10.1073/pnas.1302837110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gdanetz, Kristi , and Trail Frances. 2017. “The Wheat Microbiome Under Four Management Strategies, and Potential for Endophytes in Disease Protection.” Phytobiomes Journal 1: 158–68. 10.1094/PBIOMES-05-17-0023-R [DOI] [Google Scholar]
- 29. Bziuk, Nina , Maccario Lorrie, Douchkov Dimitar, Lueck Stefanie, Babin Doreen, Sørensen Søren J, Schikora Adam, and Smalla Kornelia. 2021. “Tillage Shapes the Soil and Rhizosphere Microbiome of Barley—But not its Susceptibility Towards Blumeria Graminis F. Sp. Hordei.” FEMS Microbiology Ecology 97: fiab018. 10.1093/femsec/fiab018 [DOI] [PubMed] [Google Scholar]
- 30. Zarraonaindia, Iratxe , Owens Sarah M., Weisenhorn Pamela, West Kristin, Hampton‐Marcell Jarrad, Lax Simon, Bokulich Nicholas A., et al. 2015. “The Soil Microbiome Influences Grapevine‐Associated Microbiota.” mBio 6: e02527‐14. 10.1128/mBio.02527-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ribeiro, Gabriel O. , Oss Daniela B., He Zhixiong, Gruninger Robert J., Elekwachi Chijioke, Forster Robert J., Yang Wen Zhu, Beauchemin Karen A., and McAllister Tim A.. 2017. “Repeated Inoculation of Cattle Rumen With Bison Rumen Contents Alters the Rumen Microbiome and Improves Nitrogen Digestibility In Cattle.” Scientific Reports 7: 1276. 10.1038/s41598-017-01269-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ryan, Robert P. , Germaine Kieran, Franks Ashley, Ryan David J., and Dowling David N.. 2008. “Bacterial Endophytes: Recent Developments and Applications.” FEMS Microbiology Letters 278: 1–9. 10.1111/j.1574-6968.2007.00918.x [DOI] [PubMed] [Google Scholar]
- 33. Seufert, Verena , Ramankutty Navin, and Foley Jonathan A.. 2012. “Comparing the Yields of Organic and Conventional Agriculture.” Nature 485: 229–32. 10.1038/nature11069 [DOI] [PubMed] [Google Scholar]
- 34. Zhao, Shuai , Zhou Na, Zhao Zheng Yong, Zhang Ke, and Tian Chang Yan. 2016. “High‐Throughput Sequencing Analysis of the Endophytic Bacterial Diversity and Dynamics in Roots of the Halophyte Salicornia Europaea.” Current Microbiology 72: 557–62. 10.1007/s00284-016-0990-3 [DOI] [PubMed] [Google Scholar]
- 35. St‐Pierre, Benoit , and Wright Andre D.. 2012. “Molecular Analysis of Methanogenic Archaea in the Forestomach of the Alpaca (Vicugna pacos).” BMC Microbiology 12: 1. 10.1186/1471-2180-12-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fierer, Noah . 2017. “Embracing the Unknown: Disentangling the Complexities of the Soil Microbiome.” Nature Reviews Microbiology 15: 579–90. 10.1038/nrmicro.2017.87 [DOI] [PubMed] [Google Scholar]
- 37. Wu, Lipeng , Wang Yidong, Zhang Shirong, Wei Wenliang, Kuzyakov Yakov, and Ding Xiaodong. 2021. “Fertilization Effects on Microbial Community Composition and Aggregate Formation in Saline‐Alkaline Soil.” Plant and Soil 463: 523–35. 10.1007/s11104-021-04909-w [DOI] [Google Scholar]
- 38. Zhang, Jingying , Liu Yong Xin, Zhang Na, Hu Bin, Jin Tao, Xu Haoran, Qin Yuan, et al. 2019. “NRT1.1B is Associated with Root Microbiota Composition and Nitrogen Use in Field‐Grown Rice.” Nature Biotechnology 37: 676–84. 10.1038/s41587-019-0104-4 [DOI] [PubMed] [Google Scholar]
- 39. Xue, Gang , Zhang Liangliang, Fan Xinyun, Luo Kaijie, Guo Shaopeng, Chen Hong, Li Xiang, and Jian Qiwei. 2022. “Responses of Soil Fertility and Microbiomes of Atrazine Contaminated Soil to Remediation by Hydrochar and Persulfate.” Journal of Hazardous Materials 435: 128944. 10.1016/j.jhazmat.2022.128944 [DOI] [PubMed] [Google Scholar]
- 40. Han, Hui , Teng Yanmin, Yang Hefa, and Li Ji. 2017. “Effects of Long‐Term Use of Compost on N2O and CO2 Fluxes in Greenhouse Vegetable Systems.” Compost Science & Utilization 25: S61–S9. 10.1080/1065657X.2016.1238786 [DOI] [Google Scholar]
- 41. Li, Huixiu , Cai Xiaoxu, Gong Jingyang, Xu Ting, Ding Guo Chun, and Li Ji. 2019. “Long‐Term Organic Farming Manipulated Rhizospheric Microbiome and Bacillus Antagonism Against Pepper Blight (Phytophthora Capsici).” Frontiers in Microbiology 10: 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ning, Wang , Huixin Li, Bo Wang, Jia Ding, Yingjie Liu, Yuquan Wei, Ji Li, and Guochun Ding. 2021. “Taxonomic and Functional Diversity of Rhizosphere Microbiome Recruited From Compost Synergistically Determined by Plant Species and Compost.” Frontiers of Microbiology 12: 798476. 10.3389/fmicb.2021.798476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhong, Wei , Friman Ville‐Petri, Pommier Thomas, Geisen Stefan, Jousset Alexandre, and Rong Shen Qi. 2020. “Rhizosphere immunity: targeting the underground for sustainable plant health management.” Frontiers of Agricultural Science and Engineering 7: 317–28. 10.15302/j-fase-2020346 [DOI] [Google Scholar]
- 44. McPherson, Morgan. R. , Wang Peng, Marsh Ellen L., Mitchell Robert B., and Schachtman Daniel P.. 2018. “Isolation and Analysis of Microbial Communities in Soil, Rhizosphere, and Roots in Perennial Grass Experiments.” Journal of Visualized Experiments 137: 57932. 10.3791/57932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sun, Lei , Qiu Fu Bin, Zhang Xia Xia, Dai Xin, Dong Xiu Zhu, and Song Wei. 2008. “Endophytic Bacterial Diversity in Rice (Oryza sativa L.) Roots Estimated By 16S rDNA Sequence Analysis.” Microbial Ecology 55: 415–24. 10.1007/s00248-007-9287-1 [DOI] [PubMed] [Google Scholar]
- 46. Vega‐Avila, A. D. , Gumiere Thiago, Andrade Pedro Avelino Maia, Lima‐Perim J. E., Durrer Ademir, Baigori Mario, Vazquez Fabio, and Andreote Fernando Dini. 2015. “Bacterial Communities In the Rhizosphere of Vitis vinifera L. Cultivated Under Distinct Agricultural Practices in Argentina.” Antonie van Leeuwenhoek 107: 575–88. 10.1007/s10482-014-0353-7 [DOI] [PubMed] [Google Scholar]
- 47. Constantin, Maria E. , de Lamo Francisco J., Vlieger Babette V., Rep Martijn, and Takken Frank L. W.. 2019. “Endophyte‐Mediated Resistance in Tomato to Fusarium Oxysporum Is Independent of ET, JA, and SA.” Front Plant Sci 10: 979. 10.3389/fpls.2019.00979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen, Tao , Nomura Kinya, Wang Xiaolin, Sohrabi Reza, Xu Jin, Yao Lingya, Paasch Bradley C., et al. 2020. “A Plant Genetic Network for Preventing Dysbiosis in the Phyllosphere.” Nature 580: 653–7. 10.1038/s41586-020-2185-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Klein, Eyal , Ofek Maya, Katan Jaacov, Minz Dror, and Gamliel Abraham. 2013. “Soil Suppressiveness to Fusarium Disease: Shifts in Root Microbiome Associated With Reduction of Pathogen Root Colonization.” Phytopathology 103: 23–33. 10.1094/phyto-12-11-0349 [DOI] [PubMed] [Google Scholar]
- 50. Edgar, Robert C. 2016. UNOISE2: Improved Error‐Correction for Illumina 16S and ITS Amplicon Sequencing. bioRxiv 081257. 10.1101/081257 [DOI]
- 51. Chen, Tingting , Chen Xu, Zhang Sisi, Zhu Junwei, Tang Bixia, Wang Anke, Dong Lili, et al. 2021. “The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types.” Genomics, Proteomics & Bioinformatics 19: 578–83. 10.1016/j.gpb.2021.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dixon, Philip . 2003. “VEGAN, a Package Of R Functions for Community Ecology.” Journal of Vegetation Science 14: 927–30. 10.1111/j.1654-1103.2003.tb02228.x [DOI] [Google Scholar]
- 53. Legendre, Pierre , Oksanen Jari, and ter Braak Cajo J. F.. 2011. “Testing the Significance of Canonical Axes In Redundancy Analysis.” Methods in Ecology and Evolution 2: 269–77. 10.1111/j.2041-210X.2010.00078.x [DOI] [Google Scholar]
- 54. Bastian, Mathieu , Heymann Sebastien, and Jacomy Mathieu. 2009. “Gephi: An Open Source Software for Exploring and Manipulating Networks.” Proceedings of the International AAAI Conference on Web and Social Media 3: 361–2. https://ojs.aaai.org/index.php/ICWSM/article/view/13937 [Google Scholar]
- 55. Liu, Yong Xin , Qin Yuan, and Bai Yang. 2019. “Reductionist Synthetic Community Approaches In Root Microbiome Research.” Current Opinion in Microbiology 49: 97–102. 10.1016/j.mib.2019.10.010 [DOI] [PubMed] [Google Scholar]
- 56. Valenzuela‐Aragon, Brenda , Parra‐Cota Fannie Isela, Santoyo Gustavo, Arellano‐Wattenbarger Guillermo Luis, and de los Santos‐Villalobos Sergio. 2019. “Plant‐Assisted Selection: a Promising Alternative for In Vivo Identification of Wheat (Triticum turgidum L. Subsp. Durum) Growth Promoting Bacteria.” Plant and Soil 435: 367–84. 10.1007/s11104-018-03901-1 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information.
Supplementary information.
Data Availability Statement
All data analyzed during this study are included in this published article and its Supporting Information files. The data sets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: PRJCA010522, and CRA007512 in the GSA. Supporting Information materials (figures, tables, scripts, graphical abstract, slides, videos, Chinese translated version, and update materials) may be found in the online DOI or iMeta Science http://www.imeta.science/.