

Abstract
Complex diseases such as cardiovascular disease (CVD), obesity, inflammatory bowel disease (IBD), kidney disease, type 2 diabetes (T2D), and cancer have become a major burden to public health and affect more than 20% of the population worldwide. The etiology of complex diseases is not yet clear, but they are traditionally thought to be caused by genetics and environmental factors (e.g., dietary habits), and by their interactions. Besides this, increasing pieces of evidence now highlight that the intestinal microbiota may contribute substantially to the health and disease of the human host via their metabolic molecules. Therefore, decoding the microbial genomes has been an important strategy to shed light on their functional potential. In this review, we summarize the roles of the gut microbiome in complex diseases from its functional perspective. We further introduce artificial tools in decoding microbial genomes to profile their functionalities. Finally, state‐of‐the‐art techniques have been highlighted which may contribute to a mechanistic understanding of the gut microbiome in human complex diseases and promote the development of the gut microbiome‐based personalized medicine.
Keywords: gut microbiome, metabolites, complex disease, host–microbe interactions
Functional roles of the gut microbiome in human health and disease. Commonly used tools in decoding microbial functionalities. State of the art techniques in validating microbial functionalities.
Highlights
Functional roles of the gut microbiome in human health and disease.
Commonly used tools in decoding microbial functionalities.
State of the art techniques in validating microbial functionalities.
INTRODUCTION
Genome‐wide association studies (GWAS) have dissected the genetic architecture of human complex diseases, which has advanced our understanding of disease etiology and promoted the development of genome‐based therapy [1]. However, genetics can only explain a limited proportion of an individual's risk of developing a complex disease [2]. For instance, GWAS can only explain the heritability of type 2 diabetes (T2D) and Crohn's disease with 6% and 20% [2] success, respectively. Recently, the contribution of the gut microbiome to the development of complex human diseases has increasingly been recognized ‐ and become a booming field of research [3, 4, 5, 6, 7, 8, 9, 10, 11].
The human intestines are colonized by a vast number of bacteria, archaea, microbial eukaryotes, and viruses, as abundant as our somatic cells, which are collectively known as the gut microbiome [12]. The gut microbiome has been involved in digesting food, training host immunity, regulating gut endocrine function and neurological signaling, modifying drug action and metabolism, eliminating toxins, and producing numerous compounds that influence the host [13]. In mice studies, gut microbiota has been shown to be essential for germ‐free animal models to develop inflammatory bowel disease (IBD) [14]. Human infants born from mothers with immune‐related diseases presented altered gut microbial compositions which was further proved to have the potential to trigger adaptive immune response [15, 16, 17]. All the evidence pinpoints to the critical roles of gut microbiota in developing complex diseases.
Rapid development of metagenomics sequencing technology and big cohort studies allow us to integrate gut microbiome profiles with host clinical phenotypes, to identify candidate disease‐related microbial features in a large scale. Numerous associations between the gut microbial composition and complex diseases have been reported, including but not limited to cardiovascular disease (CVD), diabetes, IBD, allergy, and cancer [3, 4, 5, 6, 7, 8, 9, 10]. Unlike the human genome, modification of gut microbial communities is feasible and ethical, the gut microbiome is thereby emerging as an attractive therapeutic target for disease prevention and treatment. However, there are still big gaps between research and clinical translation, including the lacking consistency of disease‐specific microbial taxa across studies, poor causal inference, and unsatisfactory efficiency of current microbiome‐based therapies in patients (e.g., fecal microbiome transplant and probiotic usage). These could be as a result of (1) many gut bacteria are opportunistic, and they could present adverse effects differently dependent on specific conditions, (2) most of studies focus on the microbial composition which is far from enough because different subspecies could behave differently. Functional analysis by decoding the microbial genomes found that microbial genes like cutC/D are responsible for the biosynthesis of phenylacetylglutamine and trimethylamine‐N‐oxide (TMAO), two metabolites that can induce CVD risk [18, 19]. Therefore, going beyond microbial composition and understanding the gut microbial functionalities could facilitate to shed light on the issues above.
Here, we summarize recent research advances in the intestinal microbiome related to human health and disease, with a particular focus on their functionalities, mainly including microbial virulence factors such as capsule and biofilm, microbiota‐derived small molecules, and drug metabolize. We further introduce artificial tools in decoding microbial genomes to characterize the functional potential. Finally, we highlight state‐of‐the‐art techniques that may help us gain a mechanistic understanding of the gut microbiome in human complex disease and to promote the development of gut microbiome‐based personalized medicine.
MICROBIAL FUNCTIONALITIES THAT AFFECT HUMAN HEALTH AND DISEASE
Via specific structures
The direct influence of gut microbes on the host can be attributed to the fundamental structures that lead to resistance and virulence such as flagella and fimbriae, capsule, spore, and biofilm, which facilitate the survival and activity of trillions of bacteria in the human intestine [20, 21] (Figure 1).
Figure 1.
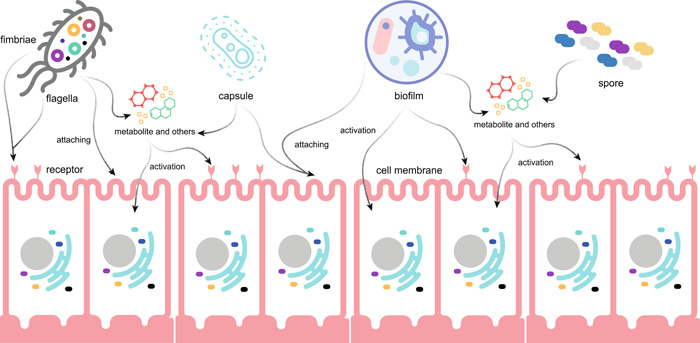
Microbial structures that contribute to resistance and virulence. The direct influence of gut microbes on the host can be attributed to the fundamental structures that lead to resistance and virulence such as flagella and fimbriae, capsule, spore, and biofilm, which facilitate the survival and activity of trillions of bacteria in the human intestine. Specific microbial structures can help microbes attach human cells or receptors which further activates various signaling pathways. Besides this, they may carry antibiotic resistance and virulence factors, as well as various metabolites and other disease‐relevant molecules
Fimbriae are straight filaments arising from the bacterial cell wall while flagella are much longer than fimbriae. Flagella spins the spirochete around and generates thrust, propelling bacteria moving forward. The formation of fimbriae and flagella always relies on gene clusters and varies substantially between species. For instance, the Salmonella fim cluster comprises 10 genes [22] while in Escherichia coli more than six gene clusters for fimbriae formation are identified [23]. Both can lead to host infection, but the mechanisms are different. Many bacterial pathogens require motility to infect, including E. coli, Salmonella enterica and others [24], thus flagella play key roles during this progress. Unlike flagella, fimbriae carry virulence factors and help in the adherence of bacteria to human cells. For instance, Bordetella pertussis uses its adhesin to bind to ciliated respiratory cells and cause whooping cough [25]. Fimbriae of Neisseria gonorrhea help it to bind to cervical cells and buccal cells to cause gonorrhea [26]. Without fimbriae to bind to the intestinal epithelium, E. coli and Campylobacter jejuni cannot cause diarrhea [27].
Capsule is a polysaccharide layer that lies outside the cell envelope and is considered a part of the outer envelope of a bacterial cell. The capsule is found in both Gram‐negative and positive bacteria. However, it is different from the second lipid membrane, which contains lipopolysaccharide (LPSs) and lipoproteins found only in Gram‐negative bacteria [20]. The capsule protects bacteria from mechanical injury and environmental changes (such as temperature, drying, bacteriophages, and eukaryotic cells) [28]. It also helps in the adherence of bacteria to smooth surfaces. For example, Streptococcus mutans, which causes dental caries, attaches to the surface of the teeth by its capsule [29]. The capsule is essential for pathogenic microorganisms to invade the host immune system and prevents them from being phagocytosed by macrophages and neutrophils [30]. A study revealed that the thickness of the capsule in Streptococcus pneumoniae was associated with the severity of meningitis [31]. Interacting with β‐glucans on the fungal cell wall during fungi infection leads to host Dectin‐1‐related CARD9 signaling pathways activation which can induce inflammation [32]. However, capsular materials have also been successfully used as vaccination against S. pneumoniae and Haemophilus influenza [33].
Unlike capsule, the spore is a very hardy cell and allows a bacterial cell to survive under even the worst conditions. Therefore, spore can protect the pathogenic bacteria from antibiotics and other injures to produce virulence factors [34]. Bacillus and Clostridium species are the most common bacteria to create spores and can induce various infection diseases [34]. For example, Bacillus cereus is well‐known for its ability to cause foodborne illness because of its spores surviving various temperatures [35]. Spores of B. anthracis cause cutaneous, gastrointestinal, inhalational, and injection anthrax via the production of anthrax toxins and the formation of a poly‐γ‐d‐glutamic acid capsule, which protects the bacteria from phagocytosis and immune surveillance [36].
Biofilm is defined as a bacterial colony with a self‐produced matrix of extracellular polymeric substances that protects the bacterial cells from unfavorable external influences, such as temperature changes, dehydration, and biocides [21]. Bacterial biofilms are usually pathogenic, and it has been estimated that up to 80% of microbial infections in humans, including endocarditis, cystic fibrosis, periodontitis, rhinosinusitis, osteomyelitis, nonhealing chronic wounds, meningitis, kidney infections, and prosthesis and implantable device‐related infections, are associated with biofilm formation [37]. Many bacteria can form biofilms, with the most common ones being E. coli, Enterococcus faecalis, Staphylococcus aureus, Staphylococcus epidermidis, Streptococcus viridans, Klebsiella pneumoniae, Proteus mirabilis, and Pseudomonas aeruginosa [38]. Notably, S. aureus and S. epidermidis are estimated to cause approximately 50% of prosthetic heart valve infections, 70% of catheter biofilm infections, and 80% of bloodstream infections [38]. Unfortunately, the use of antibiotics alone is ineffective in treating biofilm‐related infections. This is because biofilms can delay or prevent the penetration of antibiotics [39], acquire resistance via horizontal gene transfer [40], and use multidrug efflux pumps to pump antibiotic agents out of the maturing biofilms and into the extracellular matrix [41]. In addition, biofilms can activate the innate immune system via secretion of C‐di‐NMPs, which induce an immune response through STING and subsequently activate type 1 IFNs [42].
Via metabolic molecules
Gut microbes are involved in the biosynthesis and biotransformation of a series of bioactive metabolites that can act as substrates and signaling molecules, contributing to normal human physiological functions or eliciting complex diseases [13]. Specific classes of microbiota related metabolic molecules mainly include short‐chain fatty acids (SCFAs) [43], amino acids (AAs) [44], vitamins [45], bile acids (BAs) [46], toxins [47], anthocyanins [48], and phytoestrogens [49] (Figure 2).
Figure 2.
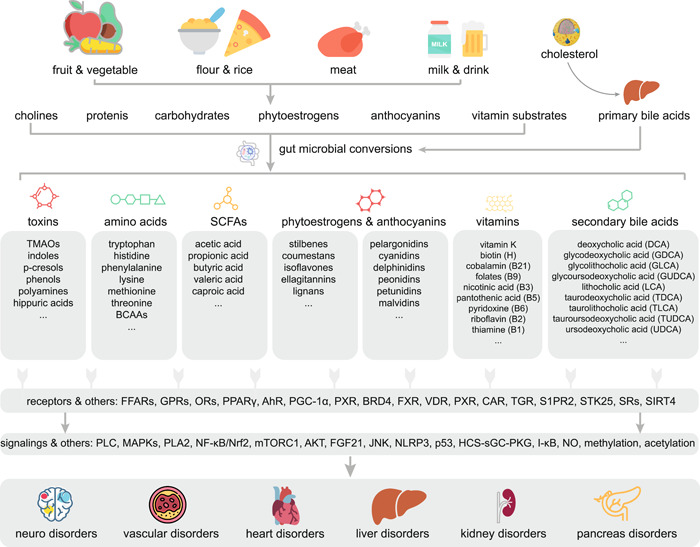
Gut microbial‐related metabolites that affect human health. Gut microbes are involved in the biosynthesis and biotransformation of a series of bioactive metabolites that can act as substrates and signaling molecules, contributing to normal human physiological functions or eliciting complex diseases. Specific classes of microbiota related metabolic molecules mainly include short‐chain fatty acids (SCFAs), amino acids, vitamins, bile acids, toxins, anthocyanins, and phytoestrogens
SCFAs can be biosynthesized by gut microbes from the colon via fermentation of carbohydrates (e.g., glucose, starch, and fiber) or AAs (e.g., lysine, arginine, glycine, leucine, valine, and isoleucine) [50]. Gut microbial‐derived SCFAs mainly include acetic, propionic, butyric, valeric, and caproic acids. Notably, acetic, propionic, and butyric acids account for more than 95% of the total SCFAs and are present at a molar ratio of approximately 60:20:20 in the human gut [51]. Biosynthesis of acetate mainly relies on microbial genes encode phosphotransacetylase or acetate kinase [52]. For propionate, genes involved in succinate pathway, acrylate pathway, and propanediol pathway are essential [53]. For butyrate biosynthesis, pyruvate pathway, 4‐aminobutyrate pathway, glutarate pathway, and lysine pathway have been characterized [54]. Well‐known butyrate producers include a wide range of species that mainly belong to the Firmicutes phylum, including Faecalibacterium prausnitzii, Eubacterium spp., Coprococcus spp., and Roseburia spp. mediated by butyrate kinase or butyryl CoA:acetate CoA transferase [55]. The genome of Bifidobacterium spp. harbors several carbohydrases, which allow them to participate in the production of acetate and lactate during nondigestible carbohydrate breakdown. However, laboratory studies have shown that the ability of Bifidobacterium spp. to produce SCFAs is highly strain‐dependent owing to the variety in gene content [56, 57]. In addition, Akkermansia muciniphila produces SCFAs by mucin degradation and is a beneficial bacterium that induces the secretion of anti‐inflammatory cytokines and enhances the intestinal mucosal barriers [58].
SCFAs are important fuels for the human host, and they control the luminal pH. In addition, SCFAs are closely related to human health and disease. The production of the SCFA butyrate by the gut is associated with improved insulin response after an oral glucose tolerance test, whereas abnormalities in the production or absorption of propionate are related to an increased risk of T2D [59]. The mechanisms underlying the important roles of SCFAs in human physiological processes may rely on their signaling capacities in activating the free fatty acid receptors (FFARs, FFAR2, and FFAR3), G protein‐coupled receptors (GPR109A and GPR42), olfactory receptors (OR51E1 and OR51E2), peroxisome proliferator‐activated receptor‐γ and the aryl hydrocarbon receptor (AhR) [60, 61, 62]. Activation of these receptors by acetic, propionic, and butyric acids can further result in the activation of signaling cascades, including phospholipase C, mitogen‐activated protein kinase (MAPK), phospholipase A2, and nuclear factor‐κB (NF‐κB) pathways. These pathways are known to be involved in the etiology of various complex diseases owing to their functional roles in regulating satiety, energy harvesting, fat storage, adipose inflammation, and neuro system [60, 61, 62]. In addition, intracellular SCFAs can influence acetylation and deacetylation of histones (mainly 3 and 4), which mainly occurs on the epsilon amino groups of lysine residues on the N‐terminal tails. This increases the accessibility of the transcriptional machinery to promote gene transcription. This process occurs by inhibiting the activity of histone deacetylases (HDACs), resulting in more transcriptionally active chromatin, or by increasing the activity of histone acetyltransferases, thereby stimulating acetylation. HDACs are involved in a range of complex diseases, including colorectal cancer and Alzheimer's disease [61]. Butyrate, propionate, and acetate inhibit HDACs, with butyrate being the most potent [63]. Therefore, SCFAs produced by gut microbes may act as modulators of complex diseases.
AAs can be produced by gut microbes via digestion of food proteins or through de novo biosynthesis. Importantly, all the nine human essential AAs, including histidine, lysine, methionine, phenylalanine, threonine, tryptophan, isoleucine, leucine, and valine can be biosynthesized by the gut microbiota [64], through a large group of oxaloacetate/aspartate AAs biosynthesis genes [65]. Studies have shown that manipulating microbial genomes, for example, fldC in Clostridium sporogenes, could change the human blood aromatic AAs [66]. In addition to the roles as substrate for protein assembly and fermentation of SCFAs, deficiency of AAs is related to human disorders. Among them, tryptophan is the most chemically complex AA, which is associated with both host‐ and microbiota‐dominated pathways. Tryptophan decarboxylases have been observed in several bacterial genomes, including Lactobacillus spp., Peptostreptococcus spp., Bacteroides spp., and Bifidobacterium spp., which play an important role in the conversion of tryptophan to tryptamine and indole derivatives [67, 68]. The downstream metabolites can be sensed by different host intestinal receptors and thereby participate in regulating a variety of molecular pathways. These receptors include GPR35, AhR, serotonin receptors (5‐HT4R and 5‐HT3R), peroxisome proliferator‐activated receptor‐γ coactivator 1α (PGC‐1α), and pregnane X receptor (PXR) that are associated with brain, skeletal muscle, pancreas, and kidney disorders [50]. Histidine may impair insulin signaling in T2D through activation of the p38γ–p62–mTORC1 pathway [69]. Phenylalanine can be derived from dopamine and is associated with nervous system disorders such as Parkinson's disease [70]. Lysine, methionine, and threonine are derived from the oxaloacetate/aspartate AA biosynthesis pathway that is involved in insulin secretion and glucose metabolism via mitochondrial sirtuin 4 (SIRT4) [71], fibroblast growth factor 21 [72], and serine/threonine‐protein kinase 25 [73], respectively. In addition, leucine, isoleucine, and valine are branched‐chain amino acids associated with insulin resistance and glucose intolerance; however, the mechanism is unclear [74].
Toxins can be generated by gut microbes from various substrates, including AAs and choline class compounds. Protein‐bound uraemic toxins such as TMAO, indole, p‐cresol, phenol, and their sulfates and glucuronides, polyamines, as well as hippuric acid are derived from AAs by gut microbes [47]. Microbial genes that encode choline‐TMA lyase (cutC/D), carnitine monooxygenase, betaine reductase, and TMAO reductase are responsible for TMAO and derivatives [75]. The production of AAs derived uremic toxins such as indole, p‐cresol, and phenol largely depends on the gene content across different taxa [75]. For example, the gene coding tryptophanases presents differently in Bacteroides species and therefore, only certain Bacteroides spp. produce indoxyl sulfate [76]. Such toxins can further induce chronic kidney disease and CVD through the NF‐κB, MAPK, and Jun N‐terminal kinase pathways, thereby initiating the transcription of proinflammatory cytokines and adhesion molecules leading to inflammation and oxidative stress [47, 77]. Toxins derived from choline class compounds chiefly include TMAO and its derivatives, which have several roles in CVD, and probably act via MAPK and NF‐κB signaling [78], as well as via NLRP3 inflammasome [79], leading to inflammation. In addition, Gram‐negative bacteria, primarily from the Bacteroidales order [80], can biosynthesize the toxin LPS, which plays a role in coronary artery disease through the NF‐κB pathway [81].
Vitamins are essential human nutrients that must be obtained from exogenous sources, including food and the gut microbiota. The gut microbiota mainly synthesizes vitamin K and most of the water‐soluble B vitamins, such as biotin (H), cobalamin (B12), folate (B9), nicotinic acid (B3), pantothenic acid (B5), pyridoxine (B6), riboflavin (B2), and thiamine (B1), which are produced by 40%–65% of human gut bacteria [82]. The potential microbial pathways that is responsible for B vitamins biosynthesis have been introduced recently [83]. It has been estimated that up to half of the daily vitamin K requirement is provided by the gut microbiota (e.g., Bacteroides, Bifidobacterium, and Enterococcus) [84]. Notably, the production of vitamin K and water‐soluble B vitamins isoforms vary across strains with different enzymes [85]. Vitamin K plays a key role in blood clotting and building bones, as both prothrombin and osteocalcin require this vitamin [86]. In addition, vitamin K can regulate the NF‐κB/Nrf2 pathway via activation of Gla proteins to influence vascular inflammation in T2D [87]. Furthermore, B vitamins have transcriptional regulatory roles. For example, biotin acts via the holocarboxylase synthetase‐soluble guanylate cyclase‐cGMP‐dependent protein kinase (PKG) pathway [88], pyridoxine, cobalamin, and pantothenic acid act via Nrf2 [89, 90, 91], folate acts via interaction with bromodomain‐containing protein 4 and the folate pathway enzyme methylenetetrahydrofolate dehydrogenase, cyclohydrolase, and formyltetrahydrofolate synthetase 1 [92], nicotinic acid acts via G protein‐coupled receptor 109 [93], riboflavin acts via DNA methylation [94] and thiamine via p53 [95].
BAs are amphipathic steroids that are synthesized from cholesterol in the liver, referred to as primary BAs. Primary BAs can be reabsorbed from the small intestine and further be structurally modified by colonic microbes to form secondary BAs [96]. This process is mediated by 7α/β‐dehydroxylation enzymes. A recent study has characterized hundreds of microbial genetic structural variation associations to the human plasma BAs, but the functionalities of majority of those structural variation were unknown [97]. Microbial structural variants (SVs) are highly variable segments of bacterial genomes, including presence/absence (deletion SVs) and copy number variations (variable SVs) that have been defined in recent years based on metagenomic sequencing data [98]. In addition to their roles in bile formation, facilitating the absorption of intestinal lipid and fat‐soluble vitamins, maintenance of cholesterol homeostasis, and antimicrobial actions in the small intestine [99], several other functions of BAs have been discovered in the past two decades [46, 100]. It has been established that BAs exert hormone‐like actions to control glucose, lipid, and energy metabolism modulate immune functions and cellular proliferation and control detoxification reactions [46, 100]. The actions of BAs are mediated through activation of nuclear receptors, that is, the established BA receptor farnesoid X receptor as well as vitamin D receptor, PXR, constitutive androstane receptor as well as membrane‐bound receptors, such as Takeda G protein‐coupled receptor 5 and sphingosine‐1‐phosphate receptor 2 [101]. Importantly, differently structured primary and secondary BAs that are present within a certain type and between different types show wide variability in their capacities to exert classical as well as signaling functions [102]. This appears to be of physiological relevance since remarkable interindividual variations in plasma BA concentration and composition have been reported in several human cohorts associated with liver fat content [103], fatty liver disease [104], T2D [105], as well as various plasma lipid parameters [103].
Anthocyanins are flavones containing a phenolic structure that are widely distributed in plant vacuoles and demonstrate pH‐dependent color. Anthocyanins are known for their possible health benefits in preventing various conditions, including CVD, cancer and neurodegenerative disorders, and improving visual and brain functions [48]. Pelargonidin, cyanidin, delphinidin, peonidin, petunidin, and malvidin are the common anthocyanins occurring naturally in food [75, 106]. Notably, the prebiotic effects of anthocyanins rely on microbial modulations. For instance, catabolism of the anthocyanin cyanidin‐3‐glucoside in the gut microbiome results in the production of phenolic compounds, including protocatechuic acid, vanillic acid, phloroglucinaldehyde, and ferulic acid, which have an effect on oxidative stress and inflammation in the gut via activation of the Nrf2, MAPK, and NF‐κB pathways [106]. The microbiota anthocyanin metabolite gallic acid (GA) has been shown to increase the levels of nitric oxide by increasing the phosphorylation of endothelial nitric oxide synthase [107]. GA also inhibits the angiotensin‐I converting enzyme, leading to a reduction in blood pressure [108].
Phytoestrogens are nonsteroidal secondary metabolites of plants with unique diphenolic structures that include different classes of chemical compounds such as stilbenes, coumestans, isoflavones, ellagitannins, and lignans [49]. Phytoestrogens can be found in our daily diet and exhibit various physicochemical and biological effects, including antioxidative, antibacterial, anti‐inflammatory, anticarcinogenic, and cardioprotective effects [109]. Similar to anthocyanins, phytoestrogens preferentially bind to estrogen receptors (ERs) with weak affinity [110]. However, the variants of phytoestrogens transformed by the gut microbiome through novel enzymatic reactions can substantially enhance their bioactivities. The gut microbiome can transform phytoestrogens into molecules, such as equol, enterolactone, and enterodiol [111]. Equol can bind to the nuclear ERs expressed in many regions of the brain to improve the development of the cerebellum [112]. Both enterolactone and enterodiol can alleviate the effect of peripheral blood lymphocytes activated by LPSs, which further leads to inhibitory‐κB degradation and NF‐κB activation, thereby resulting in the production of TNF‐α [113].
Via interactions with drugs
The gut microbiome can influence human health and disease through bidirectional interactions with drugs (Figure 3). On one hand, antibiotics can kill most of the gut bacterial species that play important roles in maintaining the metabolic health of the host via a series of mechanisms [114, 115]. For instance, penicillin works by attacking the cell wall of bacteria to prevent them from synthesizing peptidoglycan, which provides strength to the wall required for survival in the human body [116]. Quinolones target DNA gyrase, an important enzyme that helps unwind DNA for replication to prevent bacterial multiplication [117]. Tetracycline prevents key molecules from binding to selected sites on ribosomes to stop asexual reproduction [118]. The antituberculosis antibiotics belonging to the rifamycin group exert a similar effect by inhibiting the synthesis of RNA [119].
Figure 3.
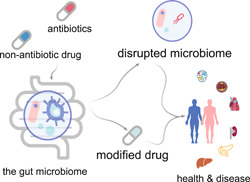
Microbial interactions with drugs. The gut microbiome can influence human health and disease through bidirectional interactions with drugs. On one hand, antibiotics can kill most of the gut bacteria that play important roles in maintaining the metabolic health of the host via a series of mechanisms. On the other hand, commonly used nonantibiotic drugs can be influenced by the gut microbiome via an enzymatic transformation that changes their bioavailability, bioactivity, or toxicity
In contrast, commonly used nonantibiotic drugs can be influenced by the gut microbiome via enzymatic transformation that changes their bioavailability, bioactivity, or toxicity [120]. A recent study conducted in vitro tests to assess the ability of 76 bacterial strains from the human gut, representing 68 species from the main bacterial taxonomic groupings, to metabolize 271 drugs. The drugs were chosen to include a diverse group based on factors, such as molecular structure or effect on the body. The study reported that 176 drugs demonstrated a substantial metabolic change caused by at least one bacterial strain, which resulted in reduced levels of the active drug molecule in the bacteria [121]. These results state the possibility that most drugs are modified by the microbiota, and such tests could prove useful during drug selection by isolating the agents that would probably be deactivated by specific gut microbes.
TOOLS FOR DECODING THE GUT MICROBIAL FUNCTIONALITIES
Based on high‐throughput next‐generation sequencing, which provides targeted or the whole microbial genomes, a series of bioinformatics tools have been developed to decode the microbial DNA sequence and predict their functionalities. In general, such tools can be divided into three categories based on their theories, namely, taxonomic marker gene‐based indirect prediction, gene homology‐based direct prediction, and sequence similarity‐based de novo prediction.
Taxonomic marker gene‐based indirect prediction
Amplicon‐based sequencing of marker genes like 16S ribosomal RNA is a powerful tool to assess and compare the structure of microbial communities within or between samples. However, insights into the functional capabilities of the gut microbiome are limited because the sequence information is only derived from specific genomic regions. Nevertheless, researchers often infer functions of uncultured organisms from their cultured counterparts, as a clade's core genome consists of genes, which its members can be expected to carry with a high probability. Thus, functions encoded in the genome of an organism may partially be predicted based on the functions encoded in closely related and well‐annotated genomes. Based on this theory, tools including PICRUSt2 [122], Tax4Fun2 [123], BugBase [124], and Piphillin [125] have been developed to profile functional components of the gut microbiome based on taxonomy information (Table 1).
Table 1.
Summary of taxonomic marker gene‐based functional prediction tools
| Tool | Description | Taxa database | Functional gene database | Functional pathway database | Link |
|---|---|---|---|---|---|
| PICRUSt2 | Ancestral‐state reconstruction based | ASVs derived from IMG | KEGG, EC number | MetaCyc | https://github.com/picrust/picrust2 |
| Tax4Fun2 | 16S rRNA gene BLAST based | Ref100NR derived from NCBI RefSeq | KEGG | https://github.com/bwemheu/Tax4Fun2 | |
| Bugbase | PICRUSt based but with clinical phenotypes | Greengenes | KEGG | https://bugbase.cs.umn.edu/index.html | |
| Piphillin | Nearest‐neighbor matching based | Manully created | KEGG | https://piphillin.secondgenome.com |
Abbreviation: KEGG, Kyoto Encyclopedia of Genes and Genomes.
PICRUSt2 [122] uses an extended ancestral‐state reconstruction algorithm based on IMG [126] to predict the gene families present and subsequently combines the gene families by a weighting method to estimate the composite metagenome. Tax4Fun2 [123] relies on the identification of the nearest neighbor with Ref100NR and generates Kyoto Encyclopedia of Genes and Genomes (KEGG) [127] outputs with normalizations and linear combinations. Piphillin [125] uses global nearest neighbor matching to generate operational taxonomic unit abundance tables that are independent of any proposed phylogenetic tree and further links to the most updated KEGG to profile the functional components. BugBase [124] utilizes a phylogenetic approach to predict genomic content based on 16S and biologically interpretable phenotypes such as oxygen tolerance, Gram staining, and pathogenic potential with existing knowledge. As the predictive power of the aforementioned tools chiefly relies on the functional information derived from the available genomes, recent progress in the construction of metagenomic‐assembled genomes [128] is likely to enhance the accuracy of functional inferences after incorporation.
Gene homology‐based direct prediction
Compared with the taxonomic marker gene‐based indirect prediction, massive sequencing reads generated by shotgun metagenomic/metatranscriptomic sequencing (MGS) that cover the entire genomes rather than marker genes can result in more accurate prediction of the gut microbial functionalities via directly mapping reads against well‐annotated gene databases. The commonly used tools for functional prediction of MGS include HUMAnN3 [129], MEGAN [130], ShotMAP [131], and gutSMASH [132] (Table 2). HUMAnN3 [129] generates species‐level gene abundances based on UniRef [133] and further assigns them to MetaCyc pathways [134]. MEGAN [130] profiles microbial functionalities based on SEED [135], eggNOG [136], and KEGG. ShotMAP [131] translates reads into predicted open reading frames and further searches the SFams [137] protein family database. GutSMASH [132] mines primary specialized metabolic gene clusters that are responsible for the biosynthesis of various metabolites in the human gut microbiome with the taxonomic resolution based on the KnownClusterBlast and ClusterBlast databases.
Table 2.
Summary of gene homology‐based functional prediction tools
| Tool | Description | Taxa database | Functional gene database | Functional pathway database | Link |
|---|---|---|---|---|---|
| HUMAnN3 | Profiling gene and pathway abundances with species resolution | UniRef | UniRef | MetaCyc | https://huttenhower.sph.harvard.edu/humann |
| MEGAN | Cross‐platform software for taxonomic level functionalities | NCBI taxonomy | SEED, eggNOG, KEGG, Pro2GO | https://github.com/danielhuson/megan-ce | |
| ShotMAP | ORF finding based homology inference and family classification | KEGG, FIGfams, SFams | https://github.com/sharpton/shotmap | ||
| gutSMASH | Profiling pathways for primary metabolites with strain resolution | 1621 Microbial genomes | MGCs | MGCs‐derived pathways | https://gutsmash.bioinformatics.nl/help.html#Validation |
Abbreviations: KEGG, Kyoto Encyclopedia of Genes and Genomes; MGC, metabolic gene clusters; NCBI, National Center for Biotechnology Information; ORF, open reading frame.
Sequence‐based de novo prediction
The current microbial genomic annotation pipelines are based on the principle of sequence similarity with existing databases, such as UniRef [133], MetaCyc [134], SEED [135], eggNOG [136], and KEGG [127]. Currently, only approximately 60% of the microbiome genomes can be annotated [128] with a homolog‐based approach, and nearly half of the microbial functionalities remain a mystery. In addition, genetic polymorphisms arise rapidly through de novo mutations (e.g., single‐nucleotide variations), which could have regulatory effects on gene expression and functions. Notably, a prevailing belief across modern molecular biology research is that a gene sequence defines the structure of the gene product and this structure, in turn, designates a unique function [138]. In other words, even with 99% similarity between the sequences of two genes, their functionalities may be completely different due to structural differences caused by variations in the remaining 1%. Thus, predicting the functionality of microbial genes based on the structure of the end product, for example, protein structure, can be a promising approach. Recently, a novel machine learning approach named AlphaFold has been developed to predict protein structures with atomic accuracy even in cases where the homologous protein structure is not known [139]. AlphaFold incorporates physical and biological knowledge regarding the protein structure and leverages multisequence alignments into the design of the deep learning algorithm. It does not impose known rules of protein biophysics or mimic the physical process of protein folding. Instead, AlphaFold performs purely geometric refinements learned from repeated attempts to predict protein structures. Thus, it may sweep the field of decoding microbial functionality in a novel manner.
However, before utilizing shotgun metagenomic sequencing data for decoding strain‐level microbial functionalities, the binning of short reads is considered a crucial step. There are two types of binning approaches, including reference‐dependent and independent binning. Reference‐dependent approach basically maps reads against a database of existing microbial reference genomes using tools such as bowtie2 [140]. But the main drawback is that it lacks the ability to characterize unknown microbial genomes. Reference independent approach is an unsupervised method to cluster contigs into individual genome bins without the assistance of any reference databases. The performance of various tools for metagenomic genome binning has been evaluated recently [141], and highlighted that most genome binning tools performed well for unique strains but reconstructing common strains still is a substantial challenge for all genome binning tools [141]. This may be due to the fact that common strains shared similar genomes that cannot be discriminated easily. Nevertheless, advances in the long‐read sequencing may facilitate de novo binning [142].
TECHNIQUES FOR VALIDATING MICROBIAL FUNCTIONALITIES
In silico approaches have identified hundreds of microbes through association‐based theory, which are likely to be important in human health and disease. However, the proposed putative functionalities of gut microbes of interest lack functional validation. Thus, taking advantage of state‐of‐the‐art techniques such as culturomics, genome editing, novel models, as well as multiomics may further strengthen our understanding of their functionalities and entirely develop the gut microbiome‐based personalized medicine (Figure 4).
Figure 4.
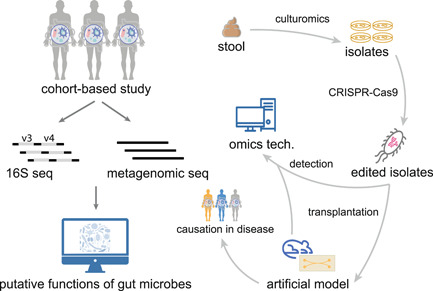
Verifying microbial functionalities with state‐of‐the‐art techniques. Many gut microbes have been found to be associated with various diseases in human participant‐based studies. Although the potential functional role of the gut microbes can be predicted via various bioinformatics tools, the proposed putative functionalities of gut microbes of interest lack functional validation. Thus, taking advantage of state‐of‐the‐art techniques such as culturomics, genome editing, novel models, as well as multiomics may further strengthen our understanding of their functionalities and entirely develop the gut microbiome‐based personalized medicine
Culturomics
Sequencing the gut microbiome highlighted that most bacteria in the gut remain uncultured and revealed the functional importance of specific gut microbes. However, the technique might be associated with bias in DNA extraction protocols, bioinformatics tools, as well as minority microbial populations. Consequently, culturomics was developed to culture and identify unknown bacteria that inhabit the human gut for direct functional validation and clinical application. Culturomics is a culturing approach that uses multiple culture conditions, mass spectrometry(, and a sequencing approach to identify bacterial species [143]. The first step in culturomics is to enable the provision of multiple culture conditions and promote the growth of fastidious bacteria from the human gut. This is achieved by improving the culture media to promote the growth of minority populations. Next, mass spectrometry is performed for the rapid identification of microbial species, which relies on the comparison of the protein mass spectra of the isolate with the most updated database. Following this, 16S and whole‐genome sequencing are applied to confirm the new taxa by comparing the existing microbial genomes recovered from humans. The application of culturomics has resulted in thousands of bacterial isolates, and a substantial proportion of them have been considered novel species/strains [144, 145, 146, 147]. Such resources allow us to test the association‐based functional hypotheses directly with isolates in mechanistic studies when coupled with in vitro and animal models or clinical applications such as microbiota transplantation and microbial editing with CRISPR‐Cas9.
Genome editing
It has been shown that structure variations widely exist in the gut microbial genomes [11]. Besides, single nucleotide polymorphism (SNP) level phylogenetic analysis of worldwide metagenomic samples showed remarkable within‐species genetic variability [128]. Variations observed in the genomes of microbial strains from the same species may vary in their functionalities. To understand the role of genomic variations, modifying genomes of microbial isolates with genome editing tools such as CRISPR‐Cas9 [148] is a promising approach to test genetic regulations of microbial functionalities.
Humanized animal and organ‐on‐chip models
Human participants cannot be directly subjected to verification of unpredictable functional roles of the gut microbiome in health and disease due to ethical issues. Consequently, novel in vivo and in vitro models such as humanized animal and organ‐on‐chip technology have emerged as the next‐generation disease and drug models [149, 150]. Humanized animal models are animal models with human‐like phenotypes obtained by editing the animal genome or inducing external perturbations. For instance, a mouse model with a human‐like BA pool has been generated by knocking out the Cyp2c70 gene with CRISPR/Cas9 [148], which might be a powerful tool to reveal the effects of the gut microbiota on BA metabolism and CVD. In the organ‐on‐a‐chip model, human induced pluripotent stem cells can be differentiated to obtain different tissue and cell types that can be used to construct organs‐on‐chips. In particular, organs‐on‐a‐chip such as gut‐on‐a‐chip and liver‐on‐a‐chip would be very interesting to investigate microbe‐intestine and host–microbe metabolic interactions.
Multiomics
Instead of accessing microbial functionalities via genomes, generating various types of omics datasets such as metabolomics, proteomics, and transcriptomics, and further linking them to the gut microbiome may enhance our understanding regarding the importance of gut microbes in complex diseases progressing from taxonomical association to potential functionality. However, challenges persist in both proteomics and metabolomics that prohibit further exploration of the functionality of gut microbes. Although the traditional targeted approach can result in accurate identification and quantification of individual metabolites or proteins. But its low throughput and relatively high cost make it less suitable for application in large cohort studies. Untargeted approaches by innovative tandem mass spectrometry approaches can profile thousands of molecules after a single injection; however, functional annotation and quantification remain a bottleneck in this approach. Although community guidelines for metabolite identification were published over a decade ago, adoption of the recommended standards has been limited [151]. Developing targeted extraction/identification protocols for specific metabolite and protein classes might be a promising approach to resolve these issues. In addition, with a better understanding of enzymatic functions, the gap in knowledge regarding unknown metabolites and proteins is reducing. Knowledge of metabolic reactions should promote the development of more powerful identification tools. For transcriptomics, a major challenge would be the isolation of microbial RNA, as the fecal samples are complex which makes it hard to get high‐quality microbial RNA. However, the development of a single bacterial sequencing technology might be a good solution.
FUTURE PERSPECTIVES
Nowadays, microbiome studies are mainly focused on the abundance of microbial taxa and functional genes. However, we have to bear in mind that the abundance of gut microbes generated by bioinformatic pipelines from sequencing data cannot really reflect the real density (absolute abundance) of microbial organisms in the human gut. If the microbial load varies substantially between samples, relative profiling will hamper attempts to link microbiome features to quantitative data such as metabolite concentrations [152]. As a cause, the quantitative microbiome profiling method that combines microbial cell count using flow cytometry with fecal microbiome sequencing data has been developed recently [153], which provides more power in assessing microbial variation within and between individuals. However, drawbacks still exist as the method is lab skill dependent in which a single measurement does not estimate the equilibrium abundance well. Besides this, it is expensive and time‐consuming and not suitable for big cohort‐based studies. Thus, future improvements in this technique are needed.
Apart from the importance of variations of the gut microbial genes in copy numbers, studying variations in microbial genomes is another essential direction to go. Like the human genome, SNPs, SVs (e.g., insertion and deletion), mobile genetic elements (e.g., bacteriophages and transposable elements) in the microbial DNA sequences may also be important for microbial functionalities and related to human diseases [11, 154]. In the past decades, genetic regulation of microbial functionalities mainly focused on the limited number of well‐known genes. Yet, systematic microbial genome‐wide association (e.g., SNPs and SVs) to physiology measurements or metabolite concentrations generated with omics techniques is still absent, which may reveal novel knowledge regarding the functional role of the human gut microbiome in host health and disease with a much higher resolution than microbial abundances. However, many challenges remain in both bioinformatics (e.g., problems in genome binning that have been discussed in the above sections) and statistics (e.g., millions of SNPs in the microbial genomes increase the number of statistical tests, which will have a negative effect on detection power). The employment of long‐read sequencing and common variants might be a potential direction to explore at the early stage.
CONCLUSIONS
The effects of gut microbiota on the host are mainly mediated by microbial virulence factors, metabolic molecules, and bidirectional interaction with drugs. We have highlighted the functional potential of gut microbes in human health and disease and summarized the associated molecular mechanisms. In addition, bioinformatics tools have been introduced that can be applied to decode the functionalities from microbial genomes, which might be helpful for researchers to prioritize them for a specific purpose. Finally, we highlighted the importance of culturomics and genome editing, multiomics, as well as novel models for functional verification and stated the possibilities of modulating the gut microbiome to improve human health.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Lianmin Chen contributed to conceptualization and writing. Yifeng Wang, Quanbin Dong, Shixian Hu, Huayiyang Zou, Tingting Wu, Jing Shi, Huayiyang Zou, Yanhui Sheng, Wei Sun, and Xiangqing Kong contributed to the discussion of the content. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
The authors thank the reviewers and editors for fruitful discussion and critical reading. Lianmin Chen is supported by the NJMU starting grant (303073572NC21) and the Foundation De Cock‐Hadders grant (20:20‐13). Xiangqing Kong is supported by the National Natural Science Foundation of China (NSFC, 82150002 and 82170425) and the foundation of Gusu School, Nanjing Medical University (GSKY20210105). Yifeng Wang is supported by the NSFC (82103925). Yanhui Sheng is supported by the foundation of Gusu School, Nanjing Medical University (GSRCKY20210203). Wei Sun is supported by the National Key R&D Program of China (2019YFA0210104 and 2019YFA0210100).
Wang, Yifeng , Dong Quanbin, Hu Shixian, Zou Huayiyang, Wu Tingting, Shi Jing, Zhang Haifeng, et al. 2022. “Decoding Microbial Genomes to Understand their Functional Roles in Human Complex Diseases.” iMeta 1, e14. 10.1002/imt2.14
Contributor Information
Xiangqing Kong, Email: kongxq@njmu.edu.cn.
Lianmin Chen, Email: lianminchen@njmu.edu.cn.
DATA AVAILABILITY STATEMENT
This manuscript does not generate any script and data. Supporting Information (graphical abstract, slides, videos, Chinese translated version, and update materials) are available online DOI or http://www.imeta.science
REFERENCES
- 1. Wijmenga, Cisca , and Zhernakova Alexandra. 2018. “The Importance of Cohort Studies in the Post‐GWAS Era.” Nature Genetics 50: 322–8. 10.1038/s41588-018-0066-3 [DOI] [PubMed] [Google Scholar]
- 2. Manolio, Teri A. , Collins Francis S., Cox Nancy J., Goldstein David B., Hindorff Lucia A., Hunter David J., McCarthy Mark I., et al. 2009. “Finding the Missing Heritability of Complex Diseases.” Nature 461: 747–753. 10.1038/nature08494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhernakova, Alexandra , Kurilshikov Alexander, Bonder Marc Jan, Tigchelaar Ettje F., Schirmer Melanie, Vatanen Tommi, Mujagic Zlatan, et al. 2016. “Population‐Based Metagenomics Analysis Reveals Markers for Gut Microbiome Composition and Diversity.” Science 352: 565–9. 10.1126/science.aad3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Falony, Gwen , Joossens Marie, Vieira‐Silva Sara, Wang Jun, Darzi Youssef, Faust Karoline, Kurilshikov Alexander, et al. 2016. “Population‐Level Analysis of Gut Microbiome Variation.” Science 352: 560–4. 10.1126/science.aad3503 [DOI] [PubMed] [Google Scholar]
- 5. Stewart, Christopher J. , Ajami Nadim J., O'Brien Jacqueline L., Hutchinson Diane S., Smith Daniel P., Wong Matthew C., Ross Matthew C., et al. 2018. “Temporal Development of the Gut Microbiome in Early Childhood from the TEDDY Study.” Nature 562: 583–8. 10.1038/s41586-018-0617-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vatanen, Tommi , Franzosa Eric A., Schwager Randall, Tripathi Surya, Arthur Timothy D., Vehik Kendra, Lernmark Åke, et al. 2018. “The Human Gut Microbiome in Early‐Onset Type 1 Diabetes from the TEDDY Study.” Nature 562: 589–94. 10.1038/s41586-018-0620-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Visconti, Alessia , Le Roy Caroline I., Rosa Fabio, Rossi Niccolò, Martin Tiphaine C., Mohney Robert P., Li Weizhong, et al. 2019. “Interplay Between the Human Gut Microbiome and Host Metabolism.” Nature Communications 10: 1–10. 10.1038/s41467-019-12476-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Manor, Ohad , Dai Chengzhen L., Kornilov Sergey A., Smith Brett, Price Nathan D., Lovejoy Jennifer C., Gibbons Sean M., and Magis Andrew T.. 2020. “Health and Disease Markers Correlate with Gut Microbiome Composition across Thousands of People.” Nature Communications 11: 5206. 10.1038/s41467-020-18871-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gacesa, Ranko , Kurilshikov Alexander, Vila Arnau Vich, Sinha Trishla, Klaassen Marjolein A. Y., Bolte Laura A., et al. 2020. “The Dutch Microbiome Project Defines Factors that Shape the Healthy Gut Microbiome.” bioRxiv, 401125. 10.1101/2020.11.27.401125 [DOI] [Google Scholar]
- 10. Salosensaari, Aaro , Laitinen Ville, Havulinna Ville S., Meric Guillaume, Cheng Susan, Perola Susan, Valsta Liisa, et al. 2021. “Taxonomic Signatures of Cause‐specific Mortality Risk in Human Gut Microbiome.” Nature Communications 12: 2671. 10.1038/s41467-021-22962-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen, Lianmin , Wang Daoming, Garmaeva Sanzhima, Kurilshikov Alexander, Vich Vila Arnau, Gacesa Ranko, Sinha Trishla, et al. 2021. “The Long‐Term Genetic Stability and Individual Specificity of the Human Gut Microbiome.” Cell 184: 2302–15. 10.1016/j.cell.2021.03.024 [DOI] [PubMed] [Google Scholar]
- 12. Sender, Ron , Fuchs Shai, and Milo Ron. 2016. “Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans.” Cell 164: 337–40. 10.1016/j.cell.2016.01.013 [DOI] [PubMed] [Google Scholar]
- 13. Fan, Yong , and Pedersen Oluf. 2021. “Gut Microbiota in Human Metabolic Health and Disease.” Nature Reviews Microbiology 19: 55–71. 10.1038/s41579-020-0433-9 [DOI] [PubMed] [Google Scholar]
- 14. Roy, Urmi , Gálvez Eric J. C., Iljazovic Aida, Lesker Till Robin, Błażejewski Adrian J., Pils Marina C., Heise Ulrike, et al. 2017. “Distinct Microbial Communities Trigger Colitis Development Upon Intestinal Barrier Damage via Innate or Adaptive Immune Cells.” Cell Reports 21: 994–1008. 10.1016/j.celrep.2017.09.097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Torres, Joana , Hu Jianzhong, Seki Akihiro, Eisele Caroline, Nair Nilendra, Huang Ruiqi, Tarassishin Leonid, et al. 2020. “Infants Born to Mothers with IBD Present with Altered Gut Microbiome that Transfers Abnormalities of the Adaptive Immune System to Germ‐Free Mice.” Gut 69: 42–51. 10.1136/gutjnl-2018-317855 [DOI] [PubMed] [Google Scholar]
- 16. Brand, Eelco C. , Klaassen Marjolein A. Y., Gacesa Ranko, Vich Vila Arnau, Ghosh Hiren, de Zoete Marcel R., Boomsma Dorret I., et al. 2021. “Healthy Cotwins Share Gut Microbiome Signatures with their Inflammatory Bowel Disease Twins and Unrelated Patients.” Gastroenterology 160: 1970–85. 10.1053/j.gastro.2021.01.030 [DOI] [PubMed] [Google Scholar]
- 17. Stokholm, Jakob , Blaser Martin J., Thorsen Jonathan, Rasmussen Morten A., Waage Johannes, Vinding Rebecca K., et al. 2018. “Maturation of the Gut Microbiome and Risk of Asthma in Childhood.” Nature Communications 9: 1–10. 10.1038/s41467-017-02573-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nemet, Ina , Saha Prasenjit Prasad, Gupta Nilaksh, Zhu Weifei, Romano Kymberleigh A., Skye Sarah M., Cajka Tomas, et al. 2020. “A Cardiovascular Disease‐Linked Gut Microbial Metabolite Acts via Adrenergic Receptors.” Cell 180: 862–77. 10.1016/j.cell.2020.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhu, Weifei , Romano Kymberleigh A., Li Lin, Buffa Jennifer A., Sangwan Naseer, Prakash Prem, Tittle Aaron N., et al. 2021. “Gut Microbes Impact Stroke Severity via the Trimethylamine N‐Oxide Pathway.” Cell Host & Microbe 29: 1199–208. 10.1016/j.chom.2021.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sutherland, Ian W. 1988. “Bacterial Surface Polysaccharides: Structure and Function.” International Review of Cytology 113: 187–231. 10.1016/s0074-7696(08)60849-9 [DOI] [PubMed] [Google Scholar]
- 21. Khatoon, Zohra , McTiernan Christopher D., Suuronen Erik J., Mah Thien‐Fah F., and Alarcon Emilio I.. 2018. “Bacterial Biofilm Formation on Implantable Devices and Approaches to Its Treatment and Prevention.” Heliyon 4: e01067. 10.1016/j.heliyon.2018.e01067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kolenda, Rafal , Ugorski Maciej, and Grzymajlo Krzysztof. 2019. Everything You Always Wanted to Know About Salmonella Type 1 Fimbriae, but were Afraid to Ask. Frontiers in Microbiology 10: 1017. 10.3389/fmicb.2019.01017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Elpers, Laura , and Hensel Michael. 2020. “Expression and Functional Characterization of Various Chaperon‐Usher Fimbriae, Curli Fimbriae, and Type 4 Pili of Enterohemorrhagic Escherichia coli O157:H7 Sakai.” Frontiers in Microbiology 11: 378. 10.3389/fmicb.2020.00378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chang, Yunjie , Moon Ki Hwan, Zhao Xiaowei, Norris Steven J., Motaleb Md A., and Liu Jun. 2019. “Structural Insights into Flagellar Stator‐Rotor Interactions.” eLife 8: e48979. 10.7554/eLife.48979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dorji, Dorji , Mooi Frits, Yantorno Osvaldo, Deora Rajendar, Graham Ross M., and Mukkur Trilochan K.. 2018. “ Bordetella Pertussis Virulence Factors in the Continuing Evolution of Whooping Cough Vaccines for Improved Performance.” Medical Microbiology and Immunology 207: 3–26. 10.1007/s00430-017-0524-z [DOI] [PubMed] [Google Scholar]
- 26. Semchenko, Evgeny A. , Everest‐Dass Arun V., Jen Freda E., Mubaiwa Tsitsi D., Day Christopher J., and Seib Kate L.. 2019. “Glycointeractome of Neisseria gonorrhoeae: Identification of Host Glycans Targeted by the Gonococcus to Facilitate Adherence to Cervical and Urethral Epithelial Cells.” mBio 10: e01339‐19. 10.1128/mBio.01339-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Laird, Renee M. , Ma Zuchao, Dorabawila Nelum, Pequegnat Brittany, Omari Eman, Liu Yang, Maue Alexander C., et al. 2018. “Evaluation of a Conjugate Vaccine Platform Against Enterotoxigenic Escherichia Coli (ETEC), Campylobacter jejuni and Shigella.” Vaccine 36: 6695–702. 10.1016/j.vaccine.2018.09.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schembri, Mark A. , Dalsgaard Dorte, and Klemm Per. 2004. “Capsule Shields the Function of Short Bacterial Adhesins.” Journal of Bacteriology 186: 1249–57. 10.1128/JB.186.5.1249-1257.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wen, Zezhang , and Burne Robert A.. 2002. “Functional Genomics Approach to Identifying Genes Required for Biofilm Development by Streptococcus mutans .” Applied and Environmental Microbiology 68: 1196–203. 10.1128/AEM.68.3.1196-1203.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wilson, John W. , Schurr Michael J., LeBlanc Carly L., Ramamurthy Racchana, Buchanan Kelly L., and Nickerson Cheryl A.. 2002. “Mechanisms of Bacterial Pathogenicity.” Postgraduate Medical Journal 78: 216–24. 10.1136/pmj.78.918.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hathaway, Lucy J. , Grandgirard Denis, Valente Luca G., Täuber Martin G., and Leib Stephen L.. 2016. “ Streptococcus pneumoniae Capsule Determines Disease Severity in Experimental Pneumococcal meningitis .” Open Biology 6: 150269. 10.1098/rsob.150269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van der Meer, Jos W. M. , van de Veerdonk Frank L., Joosten Leo A. B., Kullberg Bart‐Jan, and Netea Mihai G.. 2010. “Severe Candida spp. Infections: New Insights into Natural Immunity.” International Journal of Antimicrobial Agents 36: S58–62. 10.1016/j.ijantimicag.2010.11.013 [DOI] [PubMed] [Google Scholar]
- 33. Park, Yoon‐Dong , and Williamson Peter R.. 2015. “Masking the Pathogen: Evolutionary Strategies of Fungi and their Bacterial Counterparts.” Journal of Fungi 1: 397–421. 10.3390/jof1030397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mallozzi, Michael , Viswanathan V. K., and Vedantam Gayatri. 2010. “Spore‐Forming Bacilli and Clostridia in Human Disease.” Future Microbiology 5: 1109–23. 10.2217/fmb.10.60 [DOI] [PubMed] [Google Scholar]
- 35. Huang, Yiying , Flint Steve H., and Palmer Jon S.. 2020. “ Bacillus cereus Spores and Toxins—the Potential Role of Biofilms.” Food Microbiology 90: 103493. 10.1016/j.fm.2020.103493 [DOI] [PubMed] [Google Scholar]
- 36. Savransky, Vladimir , Ionin Boris, and Reece Joshua. 2020. “Current Status and Trends in Prophylaxis and Management of Anthrax Disease.” Pathogens 9: 370. 10.3390/pathogens9050370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jamal, Muhsin , Ahmad Wisal, Andleeb Saadia, Jalil Fazal, Imran Muhammad, Nawaz Muhammad A., Hussain Tahir, et al. 2018. “Bacterial Biofilm and Associated Infections.” Journal of the Chinese Medical Association 81: 7–11. 10.1016/j.jcma.2017.07.012 [DOI] [PubMed] [Google Scholar]
- 38. Chen, Meng , Yu Qingsong, and Sun Hongmin. 2013. “Novel Strategies for the Prevention and Treatment of Biofilm Related Infections.” International Journal of Molecular Science 14: 18488–501. 10.3390/ijms140918488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hall, Clayton W. , and Mah Thien‐Fah. 2017. “Molecular Mechanisms of Biofilm‐Based Antibiotic Resistance and Tolerance in Pathogenic Bacteria.” FEMS Microbiology Reviews 41: 276–301. 10.1093/femsre/fux010 [DOI] [PubMed] [Google Scholar]
- 40. Abe, Kimihiro , Nomura Nobuhiko, and Suzuki Satoru. 2020. “Biofilms: Hot Spots of Horizontal Gene Transfer (HGT) in Aquatic Environments, with a Focus on a New HGT Mechanism.” FEMS Microbiology Ecology 96: fiaa031. 10.1093/femsec/fiaa031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Miryala, Sravan Kumar , Anbarasu Anand, and Ramaiah Sudha. 2019. “Systems Biology Studies in Pseudomonas aeruginosa PA01 to Understand their Role in Biofilm Formation and Multidrug Efflux Pumps.” Microbial Pathogenesis 136: 103668. 10.1016/j.micpath.2019.103668 [DOI] [PubMed] [Google Scholar]
- 42. Venkiteswaran, Nityasri , Ellepola Kassapa, Seneviratne Chaminda Jayampath, Lee Yuan Kun, Puan Kia Joo, and Wong Siew Cheng. 2017. “Host–Biofilm Interactions at Mucosal Surfaces and Implications in Human Health." In Microbial Biofilms, pp. 203–36. CRC Press. [Google Scholar]
- 43. Martin‐Gallausiaux, Camille , Marinelli Ludovica, Blottiere Herve M., Larraufie Pierre, and Lapaque Nicolas. 2021. “SCFA: Mechanisms and Functional Importance in the Gut.” Proceedings of the Nutrition Society 80: 37–49. 10.1017/S0029665120006916 [DOI] [PubMed] [Google Scholar]
- 44. Liu, Yali , Hou Yuanlong, Wang Guangji, Zheng Xiao, and Hao Haiping. 2020. “Gut Microbial Metabolites of Aromatic Amino Acids as Signals in Host–Microbe Interplay.” Trends in Endocrinology & Metabolism 31: 818–34. 10.1016/j.tem.2020.02.012 [DOI] [PubMed] [Google Scholar]
- 45. Rudzki, Leszek , Trevor W. Stone, Maes Michael, Misiak Blazej, Samochowiec Jerzy, and Szulc Agata. 2021. “Gut Microbiota‐Derived Vitamins—Underrated Powers of a Multipotent Ally in Psychiatric Health and Disease.” Progress in Neuro‐Psychopharmacology & Biological Psychiatry 107: 110240. 10.1016/j.pnpbp.2020.110240 [DOI] [PubMed] [Google Scholar]
- 46. Li, Rumei , Andreu‐Sanchez Sergio, Kuipers Folkert, and Fu Jingyuan. 2021. “Gut Microbiome and Bile Acids in Obesity‐Related Diseases.” Best Practice & Research Clinical Endocrinology & Metabolism 35: 101493. 10.1016/j.beem.2021.101493 [DOI] [PubMed] [Google Scholar]
- 47. Graboski, Amanda L. , and Redinbo Matthew R.. 2020. “Gut‐Derived Protein‐Bound Uremic Toxins.” Toxins 12: 590. 10.3390/toxins12090590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hair, Rachel , Sakaki Junichi R., and Chun Ock K.. 2021. “Anthocyanins, Microbiome and Health Benefits in Aging.” Molecules 26: 537. 10.3390/molecules26030537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Seyed Hameed, Ahkam S. , Rawat Parkash S., Meng Xiangfeng, and Liu Weifeng. 2020. “Biotransformation of Dietary Phytoestrogens by Gut Microbes: A Review on Bidirectional Interaction Between Phytoestrogen Metabolism and Gut Microbiota.” Biotechnology Advances 43: 107576. 10.1016/j.biotechadv.2020.107576 [DOI] [PubMed] [Google Scholar]
- 50. Krautkramer, Kimberly A. , Fan Jing, and Backhed Fredrik. 2021. “Gut Microbial Metabolites as Multi‐Kingdom Intermediates.” Nature Reviews Microbiology 19: 77–94. 10.1038/s41579-020-0438-4 [DOI] [PubMed] [Google Scholar]
- 51. Cummings, John H. , Pomare E. W., Branch W. J., Naylor C. P., and Macfarlane G. T.. 1987. “Short Chain Fatty Acids in Human Large Intestine, Portal, Hepatic and Venous Blood.” Gut 28: 1221–7. 10.1136/gut.28.10.1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang, Bo , Lingga Christopher, Bowman Courtney, and Hackmann Timothy J.. 2021. “A New Pathway for Forming Acetate and Synthesizing ATP during Fermentation in Bacteria.” Applied and Environmential Microbiology 87: e0295920. 10.1128/AEM.02959-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Reichardt, Nicole , Duncan Sylvia H., Young Pauline, Belenguer Alvaro, McWilliam Leitch Carol, Scott Karen P., Flint Harry J., and Louis Petra. 2014. “Phylogenetic Distribution of Three Pathways for Propionate Production within the Human Gut Microbiota.” ISME Journal 8: 1323–35. 10.1038/ismej.2014.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Anand, Swadha , Kaur Harrisham, and Mande Sharmila S.. 2016. “Comparative In silico Analysis of Butyrate Production Pathways in Gut Commensals and Pathogens.” Frontiers in Microbiology 7: 1945. 10.3389/fmicb.2016.01945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Parada Venegas, Daniela , De la Fuente Marjorie K., Landskron Glauben, González María Julieta, Quera Rodrigo, Dijkstra Gerard, Harmsen Hermie, et al. 2019. “Short Chain Fatty Acids (SCFAs)‐Mediated Gut Epithelial and Immune Regulation and its Relevance for Inflammatory Bowel Diseases.” Frontiers in Immunology 10: 277. 10.3389/fimmu.2019.00277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Horiuchi, Hiroko , Kamikado Kohei, Aoki Ryo, Suganuma Natsuki, Nishijima Tomohiko, Nakatani Akiho, and Kimura Ikuo. 2020. “ Bifidobacterium animalis Subsp. Lactis GCL2505 Modulates Host Energy Metabolism via The Short‐Chain Fatty Acid Receptor GPR43.” Scientific Reports 10: 1–8. 10.1038/s41598-020-60984-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Solito, Arianna , Bozzi Cionci Nicole, Calgaro Matteo, Caputo Marina, Vannini Lucia, Hasballa Iderina, Archero Francesca, et al. 2021. “Supplementation with Bifidobacterium breve BR03 and B632 Strains Improved Insulin Sensitivity in Children and Adolescents with Obesity in a Cross‐Over, Randomized Double‐Blind Placebo‐Controlled Trial.” Clinical Nutrition 40(7): 4585–94. 10.1016/j.clnu.2021.06.002 [DOI] [PubMed] [Google Scholar]
- 58. Kim, Seungil , Shin Yun‐Chan, Kim Tae‐Young, Kim Yeji, Lee Yong‐Soo, Lee Su‐Hyun, and Kim Mi‐Na. 2021. “Mucin Degrader Akkermansia muciniphila Accelerates Intestinal Stem Cell‐Mediated Epithelial Development.” Gut Microbes 13: 1–20. 10.1080/19490976.2021.1892441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sanna, Serena , van Zuydam Natalie R., Mahajan Anubha, Kurilshikov Alexander, Vich Vila Arnau, Võsa Urmo, Mujagic Zlatan, et al. 2019. “Causal Relationships Among the Gut Microbiome, Short‐Chain Fatty Acids and Metabolic Diseases.” Nature Genetics 51: 600–605. 10.1038/s41588-019-0350-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Koh, Ara , De Vadder Filipe, Kovatcheva‐Datchary Petia, and Backhed Fredrik. 2016. “From Dietary Fiber to Host Physiology: Short‐Chain Fatty Acids as Key Bacterial Metabolites.” Cell 165: 1332–45. 10.1016/j.cell.2016.05.041 [DOI] [PubMed] [Google Scholar]
- 61. Dalile, Boushra , Van Oudenhove Lukas, Vervliet Bram, and Verbeke Kristin. 2019. “The Role of Short‐Chain Fatty Acids in Microbiota‐Gut‐Brain Communication.” Nature Reviews Gastroenterology and Hepatology 16: 461–78. 10.1038/s41575-019-0157-3 [DOI] [PubMed] [Google Scholar]
- 62. Marinelli, Ludovica , Martin‐Gallausiaux Camille, Bourhis Jean‐Marie, Beguet‐Crespel Fabienne, Blottiere Herve M., and Lapaque Nicolas. 2019. “Identification of the Novel Role of Butyrate as Ahr Ligand in Human Intestinal Epithelial Cells.” Scientific Reports 9: 643. 10.1038/s41598-018-37019-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sealy, Linda , and Chalkley Roger. 1978. “The Effect of Sodium Butyrate on Histone Modification.” Cell 14: 115–21. 10.1016/0092-8674(78)90306-9 [DOI] [PubMed] [Google Scholar]
- 64. Zhao, Jianfei , Zhang Xiaoya, Liu Hongbin, Brown Michael A., and Qiao Shiyan. 2019. “Dietary Protein and Gut Microbiota Composition and Function.” Current Protein and Peptide Science 20: 145–54. 10.2174/1389203719666180514145437 [DOI] [PubMed] [Google Scholar]
- 65. Chen, Lianmin , Collij Valerie, Jaeger Martin, van den Munckhof Inge C. L., Vich Vila Arnau, Kurilshikov Alexander, Gacesa Ranko, et al. 2020. “Gut Microbial Co‐Abundance Networks Show Specificity in Inflammatory Bowel Disease and Obesity.” Nature Communications 11: 4018. 10.1038/s41467-020-17840-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dodd, Dylan , Spitzer Matthew H., Van Treuren William, Merrill Bryan D., Hryckowian Andrew J., Higginbottom Steven K., Le Anthony, et al. 2017. “A Gut Bacterial Pathway Metabolizes Aromatic Amino Acids into Nine Circulating Metabolites.” Nature 551: 648–52. 10.1038/nature24661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bian, Shuang , Yu Zhiquan, Gong Junfeng, Yang Rong, Cheng Xiaogan, and Lin Xiubin, et al. 2021. “Spatiotemporal Distribution and Geodynamic Mechanism of the Nearly NS‐trending Rifts in the Tibetan Plateau.” Journal of Geomechanics 27: 178–94. 10.12090/j.issn.1006-6616.2021.27.02.018 [DOI] [Google Scholar]
- 68. Qi, Qibin , Li Jun, Yu Bing, Moon Jee‐Young, Chai Jin C., Merino Jordi, and Hu Jie, et al. 2021. “Host and Gut Microbial Tryptophan Metabolism and Type 2 Diabetes: An Integrative Analysis of Host Genetics, Diet, Gut Microbiome and Circulating Metabolites in Cohort Studies.” BMJ Journals, 324053. 10.1136/gutjnl-2021-324053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Koh, Ara , Molinaro Antonio, Ståhlman Marcus, Khan Muhammad T., Schmidt Caroline, Mannerås‐Holm Louise, Wu Hao, et al. 2018. “Microbially Produced Imidazole Propionate Impairs Insulin Signaling through MTORC1.” Cell 175: 947–61. 10.1016/j.cell.2018.09.055 [DOI] [PubMed] [Google Scholar]
- 70. Akram, Muhammad , Daniyal Muhammad, Ali Aatiqa, Zainab Rida, Muhammad Ali Shah Syed, Munir Naveed, and Mahmood Tahir Imtiaz. 2020.” Role of Phenylalanine and its Metabolites in Health and Neurological Disorders." In Synucleins‐Biochemistry and Role in Diseases, edited by Surguchov Andrei, London: IntechOpen. 10.5772/intechopen.83648 [DOI] [Google Scholar]
- 71. Anderson, Kristin A. , Huynh Frank K., Fisher‐Wellman Kelsey, Stuart J. Darren, Peterson Brett S., Douros Jonathan D., Wagner Gregory R., et al. 2017. “SIRT4 is a Lysine Deacylase that Controls Leucine Metabolism and Insulin Secretion.” Cell Metabolism 25: 838–55. 10.1016/j.cmet.2017.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wanders, Desiree , Forney Laura A., Stone Kirsten P., Burk David H., Pierse Alicia, and Gettys Thomas W.. 2017. “FGF21 Mediates the Thermogenic and Insulin‐Sensitizing Effects of Dietary Methionine Restriction but not its Effects on Hepatic Lipid Metabolism.” Diabetes 66: 858–67. 10.2337/db16-1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Nuñez‐Durán, Esther , Aghajan Mariam, Amrutkar Manoj, Sütt Silva, Cansby Emmelie, Booten Sheri L., Watt Andrew, et al. 2018. “Serine/Threonine Protein Kinase 25 Antisense Oligonucleotide Treatment Reverses Glucose Intolerance, Insulin Resistance, and Nonalcoholic Fatty Liver Disease in Mice.” Hepatology Communications 2: 69–83. 10.1002/hep4.1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhou, Meiyi , Shao Jing, Wu Cheng‐Yang, Shu Le, Dong Weibing, Liu Yunxia, Chen Mengping, et al. 2019. “Targeting BCAA Catabolism to Treat Obesity‐Associated Insulin Resistance.” Diabetes 68: 1730–46. 10.2337/db18-0927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang, Zeneng , and Zhao Yongzhong. 2018. “Gut Microbiota Derived Metabolites in Cardiovascular Health and Disease.” Protein & Cell 9: 416–31. 10.1007/s13238-018-0549-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Devlin, A. Sloan , Marcobal Angela, Dodd Dylan, Nayfach Stephen, Plummer Natalie, Meyer Tim, Pollard Katherine S., et al. 2016. “Modulation of a Circulating Uremic Solute via Rational Genetic Manipulation of the Gut Microbiota.” Cell Host & Microbe 20: 709–15. 10.1016/j.chom.2016.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Franko, Benoit , Brault Julie, Jouve Thomas, Beaumel Sylvain, Benhamou Pierre‐Yves, Zaoui Philippe, and Stasia Marie José. 2014. “Differential Impact of Glucose Levels and Advanced Glycation End‐products on Tubular Cell Viability and Pro‐Inflammatory/Profibrotic Functions.” Biochemical and Biophysical Research Communications 451: 627–31. 10.1016/j.bbrc.2014.08.042 [DOI] [PubMed] [Google Scholar]
- 78. Seldin, Marcus M. , Meng Yonghong, Qi Hongxiu, Zhu WeiFei, Wang Zeneng, Hazen Stanley L., Lusis Aldons J., and Shih Diana M.. 2016. “Trimethylamine N‐Oxide Promotes Vascular Inflammation through Signaling of Mitogen‐Activated Protein Kinase and Nuclear Factor‐κB.” Journal of the American Heart Association 5: e002767. 10.1161/JAHA.115.002767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chen, Ming‐Liang , Zhu Xiao‐Hui, Ran Li, Lang He‐Dong, Yi Long, and Mi Man‐Tian. 2017. “Trimethylamine‐N‐Oxide Induces Vascular Inflammation by Activating the NLRP3 Inflammasome through the SIRT3‐SOD2‐mtROS Signaling Pathway.” Journal of the American Heart Association 6: e006347. 10.1161/JAHA.117.006347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. d'Hennezel, Eva , Abubucker Sahar, Murphy Leon O., and Cullen Thomas W.. 2017. “Total Lipopolysaccharide from the Human Gut Microbiome Silences Toll‐Like Receptor Signaling.” mSystems 2: e00046‐00017. 10.1128/mSystems.00046-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lu, Yong‐Chen , Yeh Wen‐Chen, and Ohashi Pamela S.. 2008. “LPS/TLR4 Signal Transduction Pathway.” Cytokine 42: 145–151. 10.1016/j.cyto.2008.01.006 [DOI] [PubMed] [Google Scholar]
- 82. Rowland, Ian , Gibson Glenn, Heinken Almut, Scott Karen, Swann Jonathan, Thiele Ines, and Tuohy Kieran. 2018. “Gut Microbiota Functions: Metabolism of Nutrients and other Food Components.” European Journal of Nutrition 57: 1–24. 10.1007/s00394-017-1445-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Magnusdottir, Stefania , Ravcheev Dmitry, Crecy‐Lagard Valerie de, and Thiele Ines. 2015. “Systematic Genome Assessment of B‐vitamin Biosynthesis Suggests Co‐Operation among Gut Microbes.” Frontiers in Genetics 6: 148. 10.3389/fgene.2015.00148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hill, M. J. 1997. “Intestinal Flora and Endogenous Vitamin Synthesis.” European Journal of Cancer Prevention 6(Suppl 1): S43–5. 10.1097/00008469-199703001-00009 [DOI] [PubMed] [Google Scholar]
- 85. Zhang, Zimeng , Linxia Liu Chuan Liu, and Yumei Sun Dawei Zhang. 2021. “New Aspects of Microbial Vitamin K2 Production by Expanding the Product Spectrum.” Microbial Cell Factories 20: 1–12. 10.1186/s12934-021-01574-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. LeBlanc, Jean Guy , Milani Christian, Savoy De Giori Graciela, Sesma Fernando, Van Sinderen Douwe, and Ventura Marco. 2013. “Bacteria as Vitamin Suppliers to their Host: A Gut Microbiota Perspective.” Current Opinion in Biotechnology 24: 160–168. 10.1016/j.copbio.2012.08.005 [DOI] [PubMed] [Google Scholar]
- 87. Dihingia, Anjum , Ozah Dibyajyoti, Baruah Pranab K., Kalita Jatin, and Manna Prasenjit. 2018. “Prophylactic Role of Vitamin K Supplementation on Vascular Inflammation in Type 2 Diabetes by Regulating the NF‐Kappab/nrf2 Pathway va Activating Gla Proteins.” Food & Function 9: 450–62. 10.1039/c7fo01491k [DOI] [PubMed] [Google Scholar]
- 88. Leon‐Del‐Rio, Alfonso . 2019. “Biotin in Metabolism, Gene Expression, and Human Disease.” Journal of Inherited Metabolic Disease 42: 647–54. 10.1002/jimd.12073 [DOI] [PubMed] [Google Scholar]
- 89. Schrier, Matthew Scott , Zhang Yiting, Trivedi Malav Suchin, and Deth Richard Carlton. 2021. “Decreased Cortical Nrf2 Gene Expression in Autism and its Relationship to Thiol and Cobalamin Status.” Biochimie 192: 1–12. 10.1016/j.biochi.2021.09.006 [DOI] [PubMed] [Google Scholar]
- 90. Yokota, Mariko , Yahagi Shoichi, and Masaki Hitoshi. 2018. “Ethyl 2,4‐Dicarboethoxy Pantothenate, a Derivative of Pantothenic Acid, Prevents Cellular Damage Initiated by Environmental Pollutants through Nrf2 Activation.” Journal of Dermatological Science 92: 162–71. 10.1016/j.jdermsci.2018.08.012 [DOI] [PubMed] [Google Scholar]
- 91. Wei, Yao , Lu Yao, Mei Yao, Wang Haoran, Han Zhitao, Chen Miaomiao, Yao Hang, et al. 2020. “Pyridoxine Induces Glutathione Synthesis via Pkm2‐mediated Nrf2 Transactivation and Confers Neuroprotection.” Nature Communications 11: 941. 10.1038/s41467-020-14788-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sdelci, Sara , Rendeiro Andre F., Rathert Philipp, You Wanhui, Lin Jung‐Ming G., Ringler Anna, Hofstätter Gerald, et al. 2019. “MTHFD1 Interaction with BRD4 Links Folate Metabolism to Transcriptional Regulation.” Nature Genetics 51: 990–8. 10.1038/s41588-019-0413-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kang, Insug , Kim Sang‐Wook W., and Youn Jang H.. 2011. “Effects of Nicotinic Acid on Gene Expression: Potential Mechanisms and Implications for Wanted and Unwanted Effects of the Lipid‐Lowering Drug.” Journal of Clinical Endocrinology & Metabolism 96: 3048–55. 10.1210/jc.2011-1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Amenyah, Sophia D. , McMahon Amy, Ward Mary, Deane Jennifer, McNulty Helene, Hughes Catherine F., Strain J. J., et al. 2020. “Riboflavin Supplementation alters Global and Gene‐Specific DNA Methylation in Adults with the MTHFR 677 TT Genotype.” Biochimie 173: 17–26. 10.1016/j.biochi.2020.04.007 [DOI] [PubMed] [Google Scholar]
- 95. Chornyy, Sergiy , Parkhomenko Yulia, and Chorna Nataliya. 2017. “Thiamine Antagonists Trigger p53‐Dependent Apoptosis in Differentiated SH‐SY5Y Cells.” Scientific Reports 7: 10632. 10.1038/s41598-017-10878-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. de Boer, Jan Freark , Bloks Vincent W., Verkade Esther, Heiner‐Fokkema M. Rebecca, and Kuipers Folkert. 2018. “New Insights in the Multiple Roles of Bile Acids and their Signaling Pathways in Metabolic Control.” Current Opinion in Lipidology 29: 194–202. 10.1097/MOL.0000000000000508 [DOI] [PubMed] [Google Scholar]
- 97. Wang, Daoming , Doestzada Marwah, Chen Lianmin, Andreu‐Sánchez Sergio, van den Munckhof Inge C. L., Augustijn Hannah E., and Koehorst Martijn, et al. 2021. “Characterization of Gut Microbial Structural Variations as Determinants of Human Bile Acid Metabolism.” Cell Host & Microbe 29: 1802–14. 10.1016/j.chom.2021.11.003 [DOI] [PubMed] [Google Scholar]
- 98. Zeevi, David , Korem Tal, Godneva Anastasia, Bar Noam, Kurilshikov Alexander, Lotan‐Pompan Maya, Weinberger Adina, et al. 2019. “Structural Variation in the Gut Microbiome Associates with Host Health.” Nature 568: 43–8. 10.1038/s41586-019-1065-y [DOI] [PubMed] [Google Scholar]
- 99. Houten, Sander M. , Watanabe Mitsuhiro, and Auwerx Johan. 2006. “Endocrine Functions of Bile Acids.” EMBO Journal 25: 1419–25. 10.1038/sj.emboj.7601049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Yang, Jiufang , Palmiotti Anna, and Kuipers Folkert. 2021. “Emerging Roles of Bile Acids in Control of Intestinal Functions.” Current Opinion in Clinical Nutrition & Metabolic Care 24: 127–33. 10.1097/MCO.0000000000000709 [DOI] [PubMed] [Google Scholar]
- 101. Fiorucci, Stefano , Biagioli Michele, Zampella Angela, and Distrutti Eleonora. 2018. “Bile Acids Activated Receptors Regulate innate immunity.” Frontiers in Immunology 9: 1853. 10.3389/fimmu.2018.01853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hofmann, Alan F. , and Hagey Lee R.. 2014. “Key Discoveries in Bile Acid Chemistry and Biology and their Clinical Applications: History of the Last Eight Decades.” Journal of Lipid Research 55: 1553–95. 10.1194/jlr.R049437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Chen, Lianmin , van den Munckhof Inge C. L., Schraa Kiki, Ter Horst Rob, Koehorst Martijn, van Faassen Martijn, van der Ley Claude, et al. 2020. “Genetic and Microbial Associations to Plasma and Fecal Bile Acids in Obesity Relate to Plasma Lipids and Liver Fat Content.” Cell Reports 33: 108212. 10.1016/j.celrep.2020.108212 [DOI] [PubMed] [Google Scholar]
- 104. Jiao, Na , Baker Susan S., Chapa‐Rodriguez Adrian, Liu Wensheng, Nugent Colleen A., Tsompana Maria, Mastrandrea Lucy, et al. 2018. “Suppressed Hepatic Bile Acid Signalling Despite Elevated Production of Primary and Secondary Bile Acids in NAFLD.” Gut 67: 1881–91. 10.1136/gutjnl-2017-314307 [DOI] [PubMed] [Google Scholar]
- 105. Chavez‐Talavera, Oscar , Tailleux Anne, Lefebvre Philippe, and Staels Bart. 2017. “Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease.” Gastroenterology 152: P1679–94. 10.1053/j.gastro.2017.01.055 [DOI] [PubMed] [Google Scholar]
- 106. Tan, Jijun , Li Yanli, Hou De‐Xing, and Wu Shusong. 2019. “The Effects and Mechanisms of Cyanidin‐3‐Glucoside and its Phenolic Metabolites in Maintaining Intestinal Integrity.” Antioxidants (Basel) 8(10): 479. 10.3390/antiox8100479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Radtke, Oliver A. , Kiderlen Albrecht F., Kayser Oliver, and Kolodziej Herbert. 2004. “Gene Expression Profiles of Inducible Nitric Oxide Synthase and Cytokines in Leishmania Major‐Infected Macrophage‐Like RAW 264.7 Cells Treated with Gallic Acid.” Planta Medica 70: 924–8. 10.1055/s-2004-832618 [DOI] [PubMed] [Google Scholar]
- 108. Kang, Nalae , Lee Ji‐Hyeok, Lee WonWoo, Ko Ju‐Young, Kim Eun‐A., Kim Jin‐Soo, Heu Min‐Soo, et al. 2015. “Gallic Acid Isolated from Spirogyra sp. Improves Cardiovascular Disease through a Vasorelaxant and Antihypertensive Effect.” Environmental Toxicology and Pharmacology 39: 764–72. 10.1016/j.etap.2015.02.006 [DOI] [PubMed] [Google Scholar]
- 109. Boronat, Anna , Rodriguez‐Morató Jose, Serreli Gabriele, Fitó Montserrat, Tyndale Rachel F., Deiana Monica, and de la Torre Rafael. 2021. “Contribution of Biotransformations Carried out by the Microbiota, Drug‐Metabolizing Enzymes, and Transport Proteins to the Biological Activities of Phytochemicals Found in the Diet.” Advances in Nutrition 12: 2172–89. 10.1093/advances/nmab085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Rowe, Isobel J. , and Baber Rodney J.. 2021. “The Effects of Phytoestrogens on Postmenopausal Health.” Climacteric 24: 57–63. 10.1080/13697137.2020.1863356 [DOI] [PubMed] [Google Scholar]
- 111. Gaya, Pilar , Medina Margarita, Sanchez‐Jimenez Abel, and Landete Jose M.. 2016. “Phytoestrogen Metabolism by Adult Human Gut Microbiota.” Molecules 21: 1034. 10.3390/molecules21081034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ariyani, Winda , Miyazaki Wataru, and Koibuchi Noriyuki. 2019. “A Novel Mechanism of S‐equol Action in Neurons and Astrocytes: The Possible Involvement of GPR30/GPER1.” International Journal of Molecular Sciences 20: 5178. 10.3390/ijms20205178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Corsini, Emanuela , Dell'Agli Mario, Facchi Alessandra, De Fabiani Emma, Lucchi Laura, Boraso Maria S., Marinovich Marina, and Galli Corrado L.. 2010. “Enterodiol and eNterolactone Modulate the Immune Response by Acting on Nuclear Factor‐KappaB (NF‐kappaB) Signaling.” Journal of Agricultural and Food Chemistry 58: 6678–84. 10.1021/jf100471n [DOI] [PubMed] [Google Scholar]
- 114. Maier, Lisa , Goemans Camille V., Wirbel Jakob, Kuhn Michael, Eberl Claudia, Pruteanu Mihaela, Müller Patrick, et al. 2021. “Unravelling the Collateral Damage of Antibiotics on Gut Bacteria.” Nature 599: 120–4. 10.1038/s41586-021-03986-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Vázquez‐Laslop, Nora , and Mankin Alexander S. 2018. “How Macrolide Antibiotics Work.” Trends in Biochemical Sciences 43: 668–84. 10.1016/j.tibs.2018.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Sauvage, Eric , Kerff Frederic, Terrak Mohammed, Ayala Juan A., and Charlier Paulette. 2008. “The Penicillin‐Binding Proteins: Structure and Role in Peptidoglycan Biosynthesis.” FEMS Microbiology Reviews 32: 234–58. 10.1111/j.1574-6976.2008.00105.x [DOI] [PubMed] [Google Scholar]
- 117. Couturier, Martine , Bahassi EI Mustapha, and Van Melderen Laurence. 1998. “Bacterial Death by DNA Gyrase Poisoning.” Trends in Microbiology 6: 269–75. 10.1016/s0966-842x(98)01311-0 [DOI] [PubMed] [Google Scholar]
- 118. Dahl, Erica L. , Shock Jennifer L., Shenai Bhaskar R., Gut Jiri, DeRisi Joseph L., and Rosenthal Philip J.. 2006. “Tetracyclines Specifically Target the Apicoplast of the Malaria Parasite Plasmodium falciparum .” Antimicrob Agents Chemother 50: 3124–31. 10.1128/AAC.00394-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Campbell, Elizabeth A. , Korzheva Nataliya, Mustaev Arkady, Murakami Katsuhiko, Nair Satish, Goldfarb Alex, and Darst Seth A.. 2001. “Structural Mechanism for Rifampicin Inhibition of Bacterial RNA Polymerase.” Cell 104: 901–12. 10.1016/s0092-8674(01)00286-0 [DOI] [PubMed] [Google Scholar]
- 120. Weersma, Rinse K. , Zhernakova Alexandra, and Fu Jingyuan. 2020. “Interaction Between Drugs and the Gut Microbiome.” Gut 69: 1510–19. 10.1136/gutjnl-2019-320204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Zimmermann, Michael , Zimmermann‐Kogadeeva Maria, Wegmann Rebekka, and Andrew L. Goodman. 2019. “Mapping Human Microbiome Drug Metabolism by Gut Bacteria and their Genes.” Nature 570: 462–7. 10.1038/s41586-019-1291-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Douglas, Gavin M. , Maffei Vincent J., Zaneveld Jesse R., Yurgel Svetlana N., Brown James R., Taylor Christopher M., Huttenhower Curtis, and Langille Morgan. 2020. “PICRUSt2 for Prediction of Metagenome Functions.” Nature Biotechnology 38: 685–8. 10.1038/s41587-020-0548-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wemheuer, Franziska , Taylor Jessica A., Daniel Rolf, Johnston Emma, Meinicke Peter, Thomas Torsten, and Wemheuer Bernd. 2020. “Tax4Fun2: Prediction of Habitat‐Specific Functional Profiles and Functional Redundancy Based on 16S rRNA Gene Sequences.” Environmential Microbiome 15: 11. 10.1186/s40793-020-00358-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Ward, Tonya , Larson Jake, Meulemans Jeremy, Hillmann Ben, Lynch Joshua, and Sidiropoulos Dimitri, et al. 2017. “BugBase Predicts Organism‐Level Microbiome Phenotypes.” bioRxiv 133462. 10.1101/133462 [DOI] [Google Scholar]
- 125. Iwai, Shoko , Weinmaier Thomas, Schmidt Brian L., Albertson Donna G., Poloso Neil J., Dabbagh Karim, and DeSantis Todd Z.. 2016. “Piphillin: Improved Prediction of Metagenomic Content by Direct Inference from Human Microbiomes.” PLoS One 11: e0166104. 10.1371/journal.pone.0166104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Markowitz, Victor M. , Chen I‐Min, Palaniappan Krishna, Chu Ken, Szeto Ernest, Grechkin Yuri, Ratner Anna, et al. 2012. “IMG: The Integrated Microbial Genomes Database and Comparative Analysis System.” Nucleic Acids Research 40: D115–22. 10.1093/nar/gkr1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Kanehisa, Minoru , and Goto Susumu. 2000. “KEGG: Kyoto Encyclopedia of Genes and Genomes.” Nucleic Acids Research 28: 27–30. 10.1093/nar/28.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Almeida, Alexandre , Nayfach Stephen, Boland Miguel, Strozzi Francesco, Beracochea Martin, Shi Zhou Jason, Pollard Katherine S., et al. 2021. “A Unified Catalog of 204,938 Reference Genomes from the Human Gut Microbiome.” Nature Biotechnology 39: 105–14. 10.1038/s41587-020-0603-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Franzosa, Eric A. , McIver Lauren J., Rahnavard Gholamali, Thompson Luke R., Schirmer Melanie, Weingart George, Lipson Karen Schwarzberg, et al. 2018. “Species‐Level Functional Profiling of Metagenomes and Metatranscriptomes.” Nature Methods 15: 962–8. 10.1038/s41592-018-0176-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Huson, Daniel H. , Beier Sina, Flade Isabell, Górska Anna, El‐Hadidi Mohamed, Mitra Suparna, Ruscheweyh Hans‐Joachim, and Tappu Rewati. 2016. “MEGAN Community Edition—Interactive Exploration and Analysis of Large‐Scale Microbiome Sequencing Data.” PLoS Computional Biology 12: e1004957. 10.1371/journal.pcbi.1004957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Nayfach, Stephen , Bradley Patrick H., Wyman Stacia K., Laurent Timothy J., Williams Alex, Eisen Jonathan A., Pollard Katherine S., and Sharpton Thomas J.. 2015. “Automated and Accurate Estimation of Gene Family Abundance from Shotgun Metagenomes.” PLoS Computional Biology 11: e1004573. 10.1371/journal.pcbi.1004573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Pascal Andreu, Victoria , Roel‐Touris Jorge, Dodd Dylan, Fischbach Michael A., and Medema Marnix H.. 2021. “The gutSMASH wEb Server: Automated Identification of Primary Metabolic Gene Clusters from the Gut Microbiota.” Nucleic Acids Research 49: W263–70. 10.1093/nar/gkab353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Apweiler, Rolf , Bairoch Amos, Wu Cathy H., Barker Winona C., Boeckmann Brigitte, Ferro Serenella, Gasteiger Elisabeth, et al. 2004. “UniProt: The Universal Protein Knowledgebase.” Nucleic Acids Research 32: D115–9. 10.1093/nar/gkh131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Caspi, Ron , Billington Richard, Fulcher Carol A., Keseler Ingrid M., Kothari Anamika, Krummenacker Markus, Latendresse Mario, et al. 2018. “The MetaCyc Database of Metabolic Pathways and Enzymes.” Nucleic Acids Research 46: D633–9. 10.1093/nar/gkx935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Overbeek, Ross , Olson Robert, Pusch Gordon D., Olsen Gary J., Davis James J., Disz Terry, Edwards Robert A., et al. 2014. “The SEED and the Rapid Annotation of Microbial Genomes using Subsystems Technology (RAST).” Nucleic Acids Research 42: D206–14. 10.1093/nar/gkt1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Huerta‐Cepas, Jaime , Szklarczyk Damian, Heller Davide, Hernández‐Plaza Ana, Forslund Sofia K., Cook Helen, Mende Daniel R., et al. 2019. “eggNOG 5.0: A Hierarchical, Functionally and Phylogenetically Annotated Orthology Resource Based on 5090 Organisms and 2502 Viruses.” Nucleic Acids Research 47: D309–14. 10.1093/nar/gky1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Sharpton, Thomas J. , Jospin Guillaume, Wu Dongying, Langille Morgan G. I., Pollard Katherine S., and Eisen Jonathan A.. 2012. “Sifting through Genomes with Iterative‐Sequence Clustering Produces a Large, Phylogenetically Diverse Protein‐Family Resource.” BMC Bioinformatics 13: 264. 10.1186/1471-2105-13-264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Danchin, Antoine , Ouzounis Christos, Tokuyasu Taku, and Zucker Jean‐Daniel. 2018. “No Wisdom in the Crowd: Genome Annotation in the Era of Big Data—Current Status and Future Prospects.” Microbial Biotechnology 11: 588–605. 10.1111/1751-7915.13284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Jumper, John , Evans Richard, Pritzel Alexander, Green Tim, Figurnov Michael, Ronneberger Olaf, Tunyasuvunakool Kathryn, et al. 2021. “Highly Accurate Protein Structure Prediction with AlphaFold.” Nature 596: 583–9. 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Langmead, Ben , Wilks Christopher, Antonescu Valentin, and Charles Rone. 2019. “Scaling Read Aligners to Hundreds of Threads on General‐Purpose Processors.” Bioinformatics 35: 421–32. 10.1093/bioinformatics/bty648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Yue, Yi , Huang Hao, Qi Zhao, Dou Hui‐Min, Liu Xin‐Yi, Han Tian‐Fei, Chen Yue, et al. 2020. “Evaluating Metagenomics Tools for Genome Binning with Real Metagenomic Datasets and CAMI Datasets.” BMC Bioinformatics 21: 334. 10.1186/s12859-020-03667-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Karst, Soren M. , Ziels Ryan M., Kirkegaard Rasmus H., Sørensen Emil A., McDonald Daniel, Zhu Qiyun, Knight Rob, and Albertsen Mads. 2021. “High‐Accuracy Long‐Read Amplicon Sequences using Unique Molecular Identifiers with Nanopore or PacBio Sequencing.” Nature Methods 18: 165–9. 10.1038/s41592-020-01041-y [DOI] [PubMed] [Google Scholar]
- 143. Lagier, Jean‐Christophe , Dubourg Gregory, Million Matthieu, Cadoret Frederic, Bilen Melhem, Fenollar Florence, Levasseur Anthony, et al. 2018. “Culturing the Human Microbiota and Culturomics.” Nature Reviews Microbiology 16: 540–50. 10.1038/s41579-018-0041-0 [DOI] [PubMed] [Google Scholar]
- 144. Poyet, Mathilde , Groussin Mathieu, Gibbons Sean M., Avila‐Pacheco Sean, Jiang Xiaofang, Kearney Sean M., Perrotta A. R., et al. 2019. “A Library of Human Gut Bacterial Isolates Paired with Longitudinal Multiomics Data Enables Mechanistic Microbiome Research.” Nature Medicine 25: 1442–52. 10.1038/s41591-019-0559-3 [DOI] [PubMed] [Google Scholar]
- 145. Zou, Yuanqiang , Xue Wenbin, Luo Guangwen, Deng Ziqing, Qin Panpan, Guo Ruijin, Sun Haipeng, et al. 2019. “1,520 Reference Genomes from Cultivated Human Gut Bacteria Enable Functional Microbiome Analyses.” Nature Biotechnology 37: 179–85. 10.1038/s41587-018-0008-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Forster, Samuel C. , Kumar Nitin, Anonye Blessing O., Almeida Alexandre, Viciani Elisa, Stares Mark D., Dunn Matthew, et al. 2019. “A Human Gut Bacterial Genome and Culture Collection for Improved Metagenomic Analyses.” Nature Biotechnology 37: 186–92. 10.1038/s41587-018-0009-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Liu, Chang , Du Meng‐Xuan, Abuduaini Meng‐Xuan, Yu Hai‐Ying, Li Dan‐Hua, Wang Yu‐Jing, Zhou Nan, et al. 2021. “Enlightening the Taxonomy Darkness of Human Gut Microbiomes with a Cultured Biobank.” Microbiome 9: 119. 10.1186/s40168-021-01064-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. de Boer, Jan Freark , Verkade Esther, Mulder Niels L., de Vries Hilde D., Huijkman Nicolette, Koehorst Martijn, Boer Theo, et al. 2020. “A Human‐Like Bile Acid Pool Induced by Deletion of Hepatic Cyp2c70 Modulates Effects of FXR Activation in Mice.” Journal of Lipid Research 61: 291–305. 10.1194/jlr.RA119000243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Huh, Dongeun , Matthews Benjamin D., Mammoto Akiko, Montoya‐Zavala Martin, Yuan Hsin Hong, and Ingber Donald E.. 2010. “Reconstituting Organ‐Level Lung Functions on a Chip.” Science 328: 1662–8. 10.1126/science.1188302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Zhang, Boyang , Montgomery Miles, Chamberlain M. Miles, Ogawa Miles, Korolj Miles, Pahnke Miles, Wells Laura A., et al. 2016. “Biodegradable Scaffold with Built‐in Vasculature for Organ‐on‐a‐Chip Engineering and Direct Surgical Anastomosis.” Nature Materials 15: 669–78. 10.1038/nmat4570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Chaleckis, Romanas , Meister Isabel, Zhang Pei, and Wheelock Craig E.. 2019. “Challenges, Progress and Promises of Metabolite Annotation for LC‐MS‐Based Metabolomics.” Current Opinion in Biotechnology 55: 44–50. 10.1016/j.copbio.2018.07.010 [DOI] [PubMed] [Google Scholar]
- 152. Morton, James T. , Sanders Jon, Quinn Robert A., McDonald Robert, Gonzalez Antonio, Vázquez‐Baeza Yoshiki, Navas‐Molina Jose A., et al. 2017. “Balance Trees Reveal Microbial Niche Differentiation.” mSystems 2: e00162. 10.1128/mSystems.00162-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Vandeputte, Doris , Kathagen Gunter, D'Hoe Gunter, Vieira‐Silva Sara, Valles‐Colomer Mireia, Sabino Joao, Wang Jun, et al. 2017. “Quantitative Microbiome Profiling Links Gut Community Variation to Microbial Load.” Nature 551: 507–11. 10.1038/nature24460 [DOI] [PubMed] [Google Scholar]
- 154. Jiang, Xiaofang , Hall A. Brantley, Arthur Timothy D., Plichta Damian R., Covington Christian T., Poyet Mathilde, Crothers Jessica, et al. 2019. “Invertible Promoters Mediate Bacterial Phase Variation, Antibiotic Resistance, and Host Adaptation in the Gut.” Science 363: 181–7. 10.1126/science.aau5238 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This manuscript does not generate any script and data. Supporting Information (graphical abstract, slides, videos, Chinese translated version, and update materials) are available online DOI or http://www.imeta.science