

Abstract
Metagenomic evidence of great genetic diversity within the nonconserved regions of the human gut microbial genomes appeals for new methods to elucidate the species‐level variability at high resolution. However, current approaches cannot satisfy this methodologically challenge. In this study, we proposed an efficient binning‐first‐and‐assembly‐later strategy, named MetaTrass, to recover high‐quality species‐resolved genomes based on public reference genomes and the single‐tube long fragment read (stLFR) technology, which enables cobarcoding. MetaTrass can generate genomes with longer contiguity, higher completeness, and lower contamination than those produced by conventional assembly‐first‐and‐binning‐later strategies. From a simulation study on a mock microbial community, MetaTrass showed the potential to improve the contiguity of assembly from kb to Mb without accuracy loss, as compared to other methods based on the next‐generation sequencing technology. From four human fecal samples, MetaTrass successfully retrieved 178 high‐quality genomes, whereas only 58 ones were provided by the optimal performance of other conventional strategies. Most importantly, these high‐quality genomes confirmed the high level of genetic diversity among different samples and unveiled much more. MetaTrass was designed to work with metagenomic reads sequenced by stLFR technology, but is also applicable to other types of cobarcoding libraries. With the high capability of assembling high‐quality genomes of metagenomic data sets, MetaTrass seeks to facilitate the study of spatial characters and dynamics of complex microbial communities at enhanced resolution. The open‐source code of MetaTrass is available at https://github.com/BGI-Qingdao/MetaTrass.
Keywords: metagenome assembly, microbiome composition, synthetic long reads, taxonomic binning
We developed a tool to get high‐quality genomes with high taxonomic resolution by combining the cobarcoding information with public references. Compared with the conventional combination strategies, our pipeline generated a large number of high‐quality genomes for the human microbial cobarcoding data sets.
Highlights
MetaTrass is the first metagenome assembly tool implemented by the binning‐first‐assembly‐later strategy and it is freely available at https://github.com/BGI-Qingdao/MetaTrass.
MetaTrass integrates the cobarcoding and the reference genome information to achieve a high capability of generating high‐quality genomes at the species level for the human gut microbial communities.
MetaTrass uses the cobarcoding correlation between short‐length reads to reduce false‐negatives in conventional taxonomic binning and improve the contiguity of draft assemblies.
INTRODUCTION
Through sequencing and analyzing the DNA of microbial communities directly from the environment, metagenomics has shown significant promise in advancing the study of uncultured microbiomes [1, 2]. A high level of genetic diversity has been unveiled by the growing number of comprehensive metagenome‐assembled genomes (MAGs), particularly from human gut microbiomes [3, 4]. The progress in metagenomics has shed new light on the study of spatial distribution and dynamics of complex microbial communities from the human gut [5–7].
Functional mining of high‐quality strain‐resolved genomes revealed a strong correlation between genotypic differences among strains and their phenotypic differences [8, 9]. Intra‐species nonhomologous genes can serve as biomarkers to distinguish pathogenic strains from their commensal counterparts within a species [10–12]. The percentage of conserved intra‐species homologous genes shared among strains could be as low as 40% [13], and the remaining nonconserved genome sequences are considered to have a significant contribution to the phenotypic diversity of microorganisms. Therefore, complete genomes from a microbial sample at the species level will enable a more comprehensive view of intra‐species genome diversity. But it is still a challenge to generate sufficient high‐quality genomes from metagenomic data sets.
Most of the current approaches to analyzing microbial communities are designed to work with economical next‐generation sequencing (NGS or high‐throughput sequencing [HTS]) reads [14]. Many highly modularized computational tools have been developed, including genome assemblers, genome binners, taxonomic binners, and taxonomic profilers [15–17]. The assembly‐first‐and‐binning‐later strategies have been commonly used to generate MAGs. In these conventional strategies, short reads from a microbial community are first assembled into longer sequences by metagenomic assemblers with the consideration of uneven coverage depths of different microbial species [18–20] and further grouped into individual genomes by genome binners based on K‐mer composition and read coverage [21–23]. However, these conventional strategies often failed to resolve the long inter‐species repeats during contig reconstruction. Therefore, the contiguity of the draft genomes recovered from NGS reads remains not long enough to study large structural variations in microbial genomes.
Various sequencing technologies with long‐range information accompanied by specialized computational tools have been released to overcome the problem of long repeats. The third‐generation single‐molecule real‐time sequencing (TGS) technologies developed by Pacific Biosciences and Oxford Nanopore Technology (ONT) can produce contiguous reads with lengths up to hundreds of kilobases, and they show great potential to generate complete genomes from both cultured and uncultured microbial communities [24–26]. With the use of chromatin‐level contact probability information generated by high‐throughput chromosome conformation capture (Hi‐C) technology, more high‐quality MAGs with improved contiguity can be retrieved [27]. For NGS data sets, the coabundance of species in multiple samples with the common K‐mer composition is also used to improve the capability to achieve better assembly quality [28]. However, these approaches have several limitations. The high sequencing error rate in TGS long reads hampers the distinction between true biological variations and sequencing errors. An effective contact map with an Hi‐C library can only be established for a draft genome with long contiguity. Constructing coabundance in multiple samples ignores the genome characteristics of a single sample and increases the sequencing cost.
The cobarcoding sequencing library [29–33], an improved short‐read sequencing technology with long‐range genomic information, can provide an alternative way to improve metagenomic analysis. In cobarcoding library construction, long fragments of DNA molecules are first distributed into different isolated partitions and further sheared into shorter subfragments. Then these subfragments are indexed with a unique barcode (DNA cobarcoding). Finally, the cobarcoded subfragments are sequenced by standard short‐read sequencing platforms. For different cobarcoding libraries, such as BGI's single tube long fragment reads (stLFR) [32], 10X Genomics' linked‐reads [34], and Illumina's contiguity preserving transposase sequencing [30], technical differences in the number of barcodes and the short‐read coverage of the original long fragment have a great impact on the downstream analysis [35–38]. The cobarcoding correlation on the draft sequences or the assembled graph has been successfully applied to improve the contiguity of assembled genomes for both large eukaryotic genomes [39–41] and metagenomes [31, 42, 43]. However, none of these conventional strategies can conquer the inherent problem of long repeats among species with uneven abundance for complex microbial communities.
In this study, we introduce a pipeline named Metagenomic Taxonomy Read Assembly of Single Species (MetaTrass) based on cobarcoding sequencing data and reference genomes. Unlike conventional strategies, MetaTrass is featured with an integrative binning‐first‐and‐assembly‐later approach. The cobarcoding information is mainly used for two purposes. First, it facilitates the initial taxonomic binning procedure by clustering reads with the same barcode. Second, it improves the assemblies by providing long‐range information. We applied MetaTrass to stLFR data sets of a mock microbial community and four human gut microbial communities to evaluate its capability of producing high‐quality draft genomes with long contiguity and high taxonomic resolution. The microbiome composition and genetic diversity in the four human gut microbial communities were quantitatively analyzed using the high‐quality draft genomes assembled by MetaTrass. In addition, the high‐quality draft genomes assembled by MetaTrass have clear taxonomic information determined by the taxonomic binning, thus benefiting the microbial downstream analysis. All results were benchmarked by comparison with the existing mainstream tools.
RESULTS
MetaTrass pipeline
In this study, we developed a metagenome assembly pipeline named MetaTrass that integrates the information of reference genomes and DNA cobarcoding technology. As shown in the flowchart (Figure 1A), the taxonomic binning of stLFR reads was carried out before the genome assembling, unlike conventional assembly‐first‐and‐binning‐later strategies. In taxonomic binning, the metagenomic stLFR reads were classified into different taxonomic ranks by Kraken2 [44]. Due to the inherent limitations of Kraken2, only reads from conserved species‐specific regions can be successfully classified into the corresponding species. On the contrary, reads from inter‐species repeat and unique genome‐specific regions could not be effectively classified (Figure 1B). Here, the species‐specific regions represent similar or repeat sequences among genomes from the same species. The inter‐species repeat regions represent similar or repeat sequences among genomes from different species. The unique genome‐specific regions represent dissimilar sequences between genomes from the same species. The reads from inter‐species repeat regions are classified into the higher taxonomic ranks of the target species and those from unique genome‐specific regions are categorized as unclassified. In total, about 10% of the reads were classified into ranks higher than the bacterial species level for the four human fecal data sets and about 9% of the reads were unclassified (Supporting Information: Table S1). In the step of cobarcoded read refinement, the cobarcoding correlation between the reads from species‐specific regions and those from inter‐species repeat or unique genome‐specific regions was used to refine the final read set for a target species (Supporting Information: Figure 1B). The barcodes of the species‐specific reads were first extracted as the candidate barcodes. Then, the final barcodes were collected by a constraint of data size and the quality of cobarcoding information. Finally, reads of the target species were collected based on the final barcodes to be assembled in the following step. A data size threshold was set to reduce computational consumption for the species with extremely high abundance. The data size threshold was set to 300× by default according to the parameter sweep results of the sample P_Gut_Meta01 (Supporting Information: Table S2). The quality of cobarcoding information of a specific barcode was quantified by the number of reads with a confident species classification and the proportion of these reads among the total reads. In cobarcoded read assembly, the refined reads of each species were independently assembled by Supernova [39]. As aforementioned, there is a very small chance that long fragments from different species could be indexed with the same barcode in the stLFR libraries (Supporting Information: Figure S1). So, some false‐positive reads were introduced in the cobarcoded refinement procedure, and they were assembled to form contaminant sequences in draft assemblies. In the step of contamination removal, we eliminated these contaminant assemblies according to their dissimilarity to the reference genomes. The thresholds of average nucleotide identity (ANI) and alignment fraction (AF) were set to 90% and 50% by default according to the optimal results of the sample P_Meta_Gut01 (Supporting Information: Table S3). Overall, the comprehensive use of cobarcoding information and references in our approach could reduce the false‐negative effects of taxonomic binning and the false‐positive effects of cobarcoded read refinement.
Figure 1.
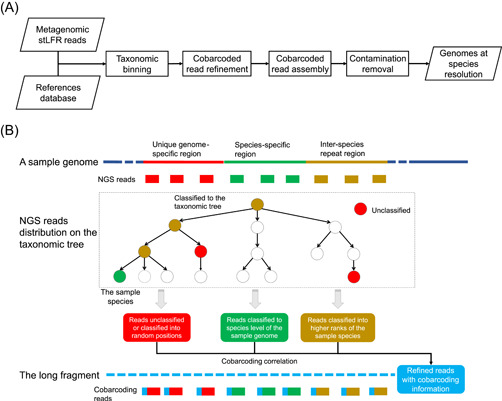
Flowchart and scheme of MetaTrass. (A) Flowchart of the MetaTrass assembling pipeline. (B) Scheme of cobarcoding correlation and taxonomic distribution of reads from different regions.
Assembly of the mock microbiome
The binning‐first‐and‐assembly‐later strategy has been widely adopted to assemble haplotype genomes for eukaryotes with large genome sizes [36, 45]. However, it has rarely been used to assemble metagenomes. We first applied MetaTrass to assemble stLFR read sets of the mock microbial community. In total, up to 99.4% of the reads were confidently assigned species‐level taxonomy given the simplicity of the mock microbial community with low inter‐species repeats and unique genome‐specific regions (Supporting Information: Table S4). To investigate the efficiency of our strategy, we compared it with the mainstream mixed assembly approaches (Figure 2A). Besides the MetaTrass analysis, the stLFR reads were also directly assembled by IDBA‐UD [20], MEGAHIT [18], Supernova [39], CloudSPAdes [42], and Athena [31]. Additionally, the optimal mixed assemblies of ONT and Illumina NGS reads in Nicholls's work [46] were also included for comparison, and assembled by WTDBG [47] and by SPAdes [48], respectively. The draft genome of each species in a mixed assembly was refined by our contamination removal module.
Figure 2.
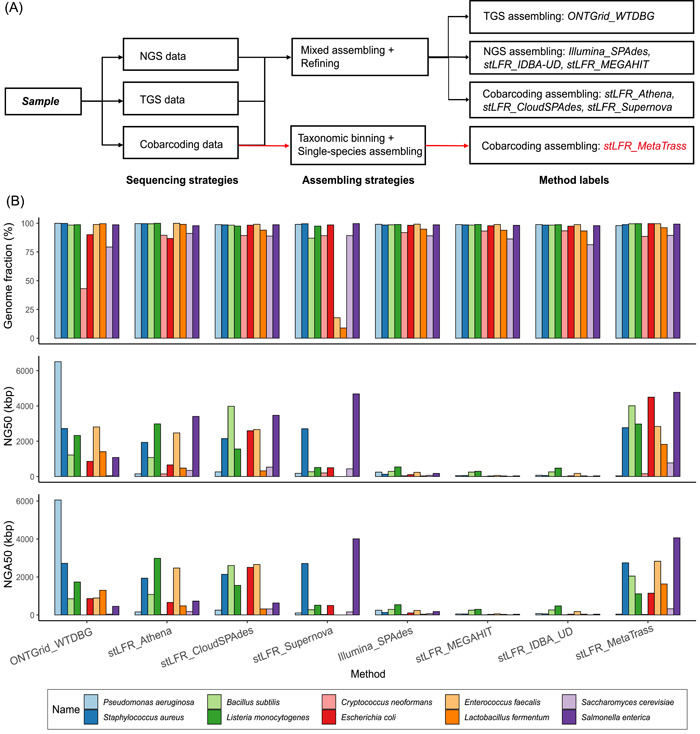
Scheme and evaluations for different strategies. (A) Difference labels of the assemblies based on different sequencing and assembling strategies. (B) Genome fraction, NG50, and NGA50 evaluated by QUAST for the assemblies.
Overall, our pipeline was superior in the production of draft genomes with high genome fractions and long contiguity (Figure 2). Two species, Enterococcus faecalis and Lactobacillus fermentum, were incompletely assembled by Supernova with genome fractions as low as 17.7% and 8.9%. On the contrary, both species were properly recovered by MetaTrass, indicating that the assembly complexity caused by uneven abundances was reduced by taxonomic binning. When compared to the results of TGS reads assembled by WTDBG, the assemblies by MetaTrass had high genome fractions, similar to those by NGS or cobarcoding assemblers designed for metagenomes. As compared with the NGS assemblers, the cobarcoding and TGS assemblers produced draft genomes with better contiguity, among which MetaTrass showed the best performance. MetaTrass produced seven draft assemblies with NG50 around 2 Mb and obtained the highest number of assemblies with NGA50 around 2 Mb. Further, MetaTrass yielded fewer assembly errors as compared to the TGS assembler (Supporting Information: Figure S2). The average numbers of mismatches and indels per 100 kb in assemblies of stLFR reads were 60 and 10, respectively, which were smaller than those of the ONT assemblies. It was worth noting that the improvement of the assembly of Pseudomonas aeruginosa genome by MetaTrass was not large, and this may come from the fact that this species had the least amount of valid cobarcoding information (Supporting Information: Table S5). The valid cobarcoding information about each species was evaluated by counting the number of valid long fragments from each species. A valid fragment was defined as a cluster with more than five paired‐end reads and a maximum distance between two paired‐end reads longer than 10 kb, by aligning paired‐end reads to reference.
Assembly of four human gut microbiomes
To evaluate the robustness of our approach, we applied MetaTrass to four human fecal samples to study human gut microbial communities. The comprehensive genome references of the Unified Human Gastrointestinal Genomes (UHGG V1.0) were used to classify NGS reads by Kraken2 [44], and the community compositions were estimated through the classified reads at different taxonomic ranks (Supporting Information: Figure S3–S6). The three healthy samples had a similar microbial community, in which the major microbiomes were from the bacterial phylum Firmicutes A. This microbial community was different from the patient microbial community dominated by the bacterial phylum Proteobacteria, which is demonstrated to be strongly correlated with enteric diseases caused by dysbiosis in gut microbiota [49]. The numbers of species with an abundance higher than 10× were 113, 108, 93, and 158 in samples H_Gut_Meta01, H_Gut_Meta02, H_Gut_Meta03, and P_Gut_Meta01, respectively.
The genome fraction of an assembly to the reference was used to evaluate the completeness of genome assembly. The genome fraction for all samples widely ranges from 0% to 90%, and the distributions of H_Gut_Meta01 and H_Gut_Meta02 were more concentrated than those of H_Gut_Meta03 and P_Gut_Meta01 (Figure 3A). More than half of the assemblies with confident species‐level taxonomy had a genome fraction higher than 50%. Considering the large genetic diversity between sample genomes and reference genomes [8], these results suggested that our pipeline was able to obtain complete genomic information for the species with an abundance higher than 10×. The genetic diversity was also confirmed by the significant differences in genome fraction and the ratio of assembled length to the reference length among the four samples (Supporting Information: Figure S7). The distribution of genome N50 values was generally dispersed ranging from a few kb to several Mb, and the medians of H_Gut_Meta02 and H_Gut_Meta03 were higher than those of H_Gut_Meta01 and P_Gut_Meta01 (Figure 3B). Nevertheless, the third quartiles in the box plots for all the samples were larger than 100 kb, demonstrating that our pipeline has a strong capability to generate draft genomes with long contiguity. Note that from these three healthy samples, plenty of draft genomes with ultra‐long contiguity (N50 > 1 Mb) were obtained, which provide possibilities to study the large genome difference in the microbiome.
Figure 3.
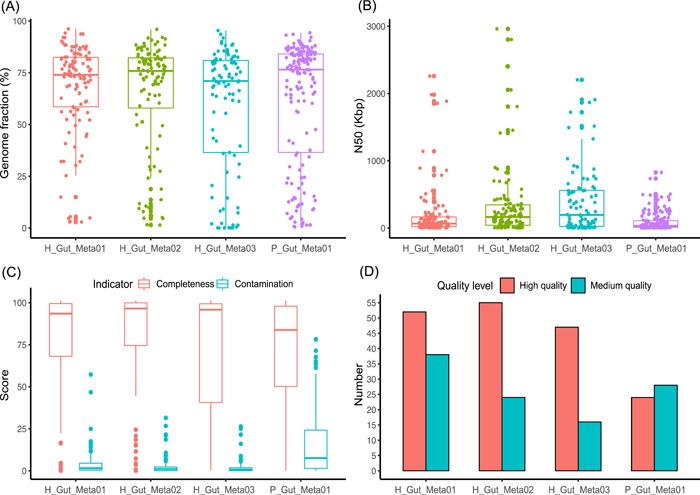
QUAST and CheckM evaluations of MetaTrass assemblies for the four human fecal samples. (A) Genome fraction. (B) Scaffold N50. (C) Box plot of completeness and contamination. (D) Number of high‐ and medium‐quality genomes
Considering the intra‐species genetic diversity, we also evaluated the quality of metagenomic assemblies by CheckM. The medians of the completeness of all assembled genomes in the three healthy samples were larger than 92%, while the medians of contamination were smaller than 2% (Figure 3C). The median completeness of the patient sample was about 83%, and the median contamination was about 7% (Supporting Information: Figure S8). A great number of high‐ and medium‐quality genomes were recovered by MetaTrass from the four samples (Figure 3D). Fifty‐two high‐quality and 37 medium‐quality genomes were produced for H_Gut_Meta01, 55 and 24 for H_Gut_Meta02, 47 and 16 for H_Gut_Meta03, and 24 and 28 for P_Gut_Meta01, respectively.
Comparison to the assembly‐first‐and‐binning‐later strategies
To further evaluate our approach's efficiency, we compared it with conventional assembly‐first‐and‐binning‐later approaches as listed in the Section “Methods.” It should be noted that currently, there are still no genome binning tools to directly exploit the cobarcoding information. With regard to the number of genomes with a completeness of >50% (Supporting Information: Table S6), MetaTrass outperformed all the other methods on all data sets. Especially for P_Gut_Meta01, MetaTrass yielded 117 draft genomes with completeness higher than 50%, much more than the 66 ones obtained by the optimal combination of Supernova [39] and Maxbin2.0 [22].
Through the comprehensive analysis of the completeness, contamination, and taxonomic rank of each genome, we assessed MetaTrass and conventional strategies on the ability to get high‐ and medium‐quality genomes and resolution of taxonomic rank (Figure 4). For different samples, the best combinations to produce optimal results were different. The combinations of MetaSPAdes [19] and Maxbin2.0, Supernova and MetaBAT2 [23], MetaSPAdes and MetaBAT2, and Athena [31] and MetaBAT2 were optimal for H_Gut_Meta01, H_Gut_Meta02, H_Gut_Meta03, and P_Gut_Meta01, respectively. For the four samples, the optimal results of the conventional strategies were still inferior to those of MetaTrass. For the example of H_Gut_Meta01, the combination of MetaSPAdes and Maxbin2.0 produced 41 high‐ and medium‐quality genomes, which was much less than the 90 obtained by MetaTrass. There were only 3 out of a total of 18 high‐quality genomes with a taxonomic rank lower than the bacterial order, but 15 out of 52 for MetaTrass. Compared with the strategies those only used NGS‐read information, MetaTrass outcompeted all these approaches by producing higher quality and finer resolution. These results demonstrated that the usage of cobarcoding information in MetaTrass was more efficient and accurate than those in the conventional approaches.
Figure 4.
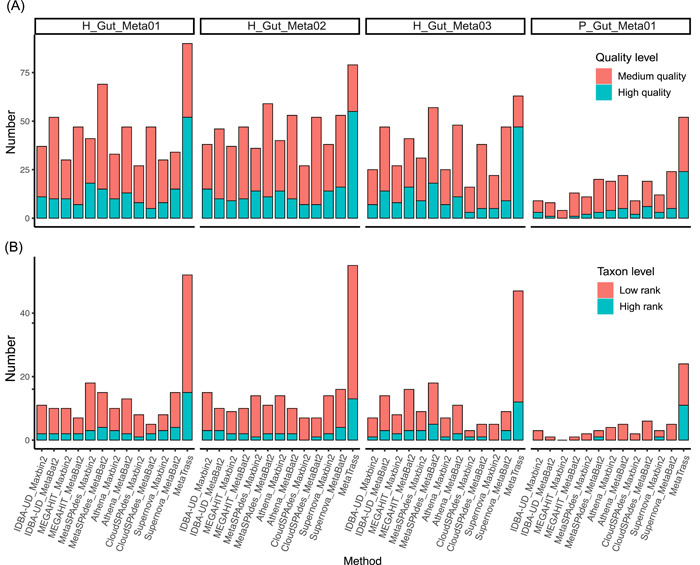
Comparison of metagenome assembly for different methods. (A) Number of high‐ and medium‐quality genomes assembled with different methods. (B) Number of high‐quality genomes with high‐ and low‐rank with different methods.
The human gut microbiome composition attracts much attention for many reasons, one of them being its strong correlation with personality traits [50]. To compare the microbiome composition of high‐quality genomes obtained with different methods, we uniformly used GTDB‐Tk [51] to annotate these genomes. The annotated taxonomic information was listed in Supporting Information: Table S7. Taxonomic trees of the high‐quality genomes obtained by MetaTrass were constructed based on the taxonomic information, and the corresponding N50 values were attached in the left histogram as shown in Figure 5. The high‐quality genomes obtained by the conventional strategies were marked in red in the middle of the heatmap (Figure 5) if the genome of the same species was also assembled by MetaTrass. The topology of the taxonomic tree of genomes assembled by MetaTrass gave comprehensive insights into the microbial composition structure. From the trees in Figure 5 and Supporting Information: Figures S9–S11, the numbers of the bacterial order with high‐quality genomes assembled by MetaTrass were 9, 11, 7, and 7 for H_Gut_Meta01, H_Gut_Meta02, H_Gut_Meta03, and P_Gut_Meta01, respectively. Notably, some bacterial orders contained more than five high‐quality genomes, and this could be used to study the microbiome structure at the genome‐wide scale in the same sample. For the sample H_Gut_Meta01 (Figure 5), there were 27 and 14 high‐quality genomes classified into the bacterial order Lachnospirales and Oscillospirales, respectively. These two were exactly the dominating bacterial orders according to the taxonomic abundance distribution. Similar results were obtained for the other two healthy samples (Supporting Information: Figures S9 and S10), indicating that the microbiome with higher sequencing coverage could be better assembled in MetaTrass. In contrast, the bacterial orders with more than five high‐quality genomes were Enterobacterales and Actinomycetales for P_Gut_Meta01 (Supporting Information: Figure S11). The obvious difference between the healthy and patient samples was consistent with the microbial composition differences observed in the taxonomic binning results. MetaTrass successfully assembled most of the high‐quality genomes of all the conventional strategies in our tests. Only 25 out of 137 genomes with high‐quality generated by all the conventional strategies were not assembled by MetaTrass (Figure 5). The heatmaps showed that most of the conventional strategies could assemble draft genomes for each bacterial order, but the numbers of genome in the same bacterial order were relatively small. The maximal number of genomes in one bacterial order was six and obtained by the combination of Supernova and MetaBAT2 for the bacterial order Lachnospirales. Moreover, 146 of 179 high‐quality genomes had N50 values larger than 100 kb, demonstrating that MetaTrass had a strong ability to improve the contiguity of assemblies.
Figure 5.
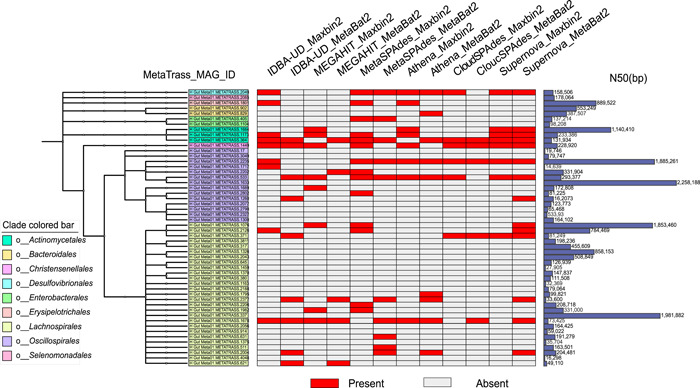
Taxonomic tree of the high‐quality genomes assembled by MetaTrass for H_Gut_Meta01. The taxonomic tree is on the left. The distribution of the high‐quality genomes assembled by other methods is colored red in the middle heatmap. N50 of each high‐quality genome is shown in the right histogram.
Genetic diversity in different samples
Different types of genomic variations in gut microbiomes are strongly associated with host health, and the genetic diversity among different microbiomes has been intensively studied to unravel the genetic origin of phenotypic differences among people of different geographical origins or health statuses [52, 53]. By aligning draft genomes to the references, we called variations for high‐quality genomes for each species in different samples, including single nucleotide variations (SNV) as well as small and large indels. For different variations, the number of SNVs was significantly larger than those of the small and large indels for the four samples (Supporting Information: Figure S12). Meanwhile, median values of variation numbers in the three healthy samples were similar but obviously larger than those in the patient sample. The smaller number of variations in the patient sample was due to the fewer alignments than those in the healthy samples according to the QUAST [54] evaluation. However, when we removed the effect of the total aligned length by calculating the SNV density, the patient sample showed denser SNV than the healthy samples (Supporting Information: Figure S12). The median was about 21 for the patient sample, but around nine for the healthy samples. This difference could be related to the individual's physiological state, which was related to the diseases, territory, and race [4].
Based on the taxonomic information of the high‐quality genomes, we found 15 species shared by three samples, among which 14 species appeared in the three healthy samples and one species Escherichia appeared in the patient and two healthy samples. By analyzing the SNV density and intersection of variations between different samples for each species in three healthy samples, we further investigated the genetic diversity between species from different samples. The SNV densities were different for distinct species even in the same sample but were similar for the same species in different samples (Figure 6A). From Figure 6B–D, the number of unique and shared variations in different types significantly fluctuated for different species, but their difference among samples showed great consistency. H_Gut_Meta01 and H_Gut_Meta02 shared the highest number of variations among all sample pairs. Furthermore, the ratio of large indels shared by all three healthy samples to the total was much smaller than those of SNVs and small indels. These results demonstrated that large variations were more sample‐specific than small variations, which is consistent with previous observations of the association between host health and structural variations in the human gut microbiome [52].
Figure 6.
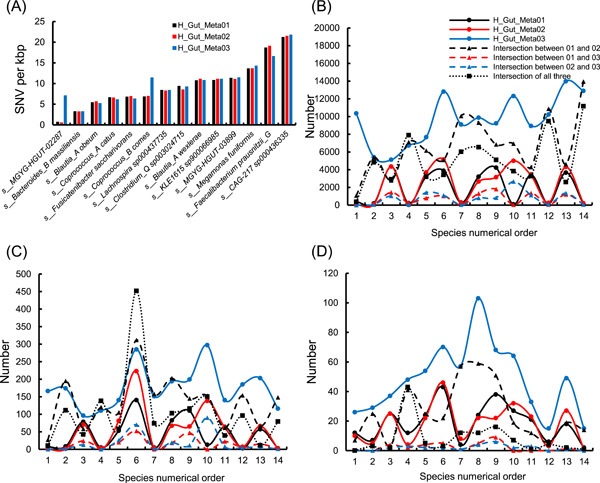
Single nucleotide variation (SNV) density and the number of unique and shared variations for each species appearing in all three healthy samples. (A) is the SNV density. (B), (C), and (D) are the number of SNVs, small, and large indels, respectively. The species numerical order in subfigures (B), (C), and (D) corresponds to the appearance order of species from left to right in subfigure A.
Computational performance
Runtime and used thread number of each assembler were recorded for all the human fecal data sets (Table 1). Most of the assemblers were tested on a small server of 24 Intel(R) Xeon(R) Silver 4116 CPU @ 2.10 GHz, except for Athena [31] and Supernova [39], which were tested on other high‐performance computing clusters due to their large memory requirements. The thread number set in each assembler was the same for different samples. The time consumption of the format conversion from stLFR reads to 10X linked‐reads was not included, which was about 500 min for a data set with 50 Gb in a single thread. We found that MetaTrass was less time‐consuming than Athena but more time‐consuming than other assemblers. This may be because both MetaTrass and Athena contained many subassembling, which took most of the time among all subprocesses in MetaTrass (Supporting Information: Table S8). Since the subassembling was independent, it could be run in parallel to further speed up by increasing the parallel number. Under the default parallel number of 8, the memory peaks used by MetaTrass were more than 50 Gb, but not more than 100 Gb for the four human fecal samples (Supporting Information: Table S9).
Table 1.
Runtimes and thread number of each assembler for all the human gut data sets
| Thread number | Runtime (min) | ||||
|---|---|---|---|---|---|
| Assembler | All samples | H_Gut_meta01 | H_Gut_Meta02 | H_Gut_Meta03 | P_Gut_Meta01 |
| IBDA‐UD | 6 | 863 | 884 | 911 | 2657 |
| MEGAHIT | 16 | 179 | 161 | 163 | 611 |
| MetaSPAdes | 16 | 1478 | 1289 | 1429 | 3459 |
| CloudSPAdes | 16 | 1024 | 1163 | 1039 | 2627 |
| Supernova | 8 | 1249 | 864 | 1098 | 6776 |
| Athena | 16 | 13,813 | 8689 | 6361 | – |
| MetaTrass | 16 | 5145 | 2631 | 3147 | 8363 |
Note: The exact runtime of assembling sample P_Gut_Meta01 by Athena was not collected correctly due to several uncontrolled interrupts on the high‐performance computing cluster.
DISCUSSION
High‐quality genomes at the species level are strongly demanded to investigate the genetic origins of diseases associated with the human gut microbiome. However, how to get a sufficient number of them in one sample is still a challenge due to the inter‐species repeats and uneven abundance in metagenome assembly. In this study, we developed a tool to get high‐quality genomes with fine taxonomic resolutions by combining the cobarcoding information with public references. Compared with conventional strategies, our pipeline generated a large number of high‐quality genomes for the human microbiome cobarcoding data sets. Meanwhile, plenty of draft genomes were also assembled with an NG50 value larger than 1 Mb, some of which were even longer than the references for both the mock and human fecal data sets. For all four human fecal samples, 178 draft genomes with high completeness and low contamination were generated by MetaTrass, but their genome fractions relative to the references were low. The differences between the sample genomes assembled by MetaTrass and the reference genomes demonstrated that the cobarcoding information could be used to reduce the false‐negative reads in taxonomic binning. These reads retrieved from inter‐species repeat and unique genome‐specific regions by cobarcoded read refinement could significantly improve the quality of assemblies. For the patient sample, the number of high‐quality genomes with long contiguity assembled by MetaTrass was significantly larger than that generated without cobarcoded read refinement (Supporting Information: Figure S13).
MetaTrass was originally designed for stLFR, but is also suitable for other kinds of cobarcoding sequencing reads. We analyzed the 10× Genomics linked‐reads data set of ATCC Mock‐20 used in the development of Athena [31], which contains two bacterial genera with multiple species. As a result, MetaTrass recovered all these species with high‐quality genomes, while the conventional strategies only gained several metagenomes with various degrees of missing, heterogeneity, and contamination (Supporting Information: Figure S14). This also demonstrated the capability of MetaTrass to recover metagenomes at species or strain resolution. Noted that Supernova [39] was replaced with CloudSPAdes [42] in the step of cobarcoded read assembly. Since the number of long fragments with the same barcode in linked‐reads is greater than that of stLFR reads [32], more false‐positive reads were introduced into the cobarcoded refined read sets, leading to the unsuccessful assembling of several species by Supernova.
The efficiency of our pipeline depends on the cobarcoding information quality and the cobarcoding assembler. The cobarcoding information quality has strong effects on the precision and sensitivity of cobarcoded read refinement. By comparing genome fractions of different read sets including the TRs defined in the Section “Methods,” the refined reads, and all reads for species with medium abundance in P_Gut_Meta01 (Supporting Information: Table S10), we evaluated the sensitivity of the cobarcoded read refinement. We observed that the fraction with high depths of the refined reads was higher than that of the TRs, but still lower than that of all aligned reads. These results indicated that there were still some false‐negative reads introduced by the low coverage or the short length of long fragments. The cobarcoded read assembly of Supernova consumed most of the computational time in the tests of the four human fecal samples and it cannot effectively assemble data sets with a high ratio of false‐positive reads in the test of linked‐reads. Thus, improvements on the cobarcoding library and algorithms of the cobarcoded read refinement and assembly would improve the performance of MetaTrass.
CONCLUSION
In summary, the application of MetaTrass in human fecal samples showed great promise in generating high‐quality genomes for a real complex microbial community at a fine resolution. With the increasing number of reference genomes from various microbial communities and the development of cobarcoding sequencing libraries, the binning‐first‐and‐assembly‐later strategy in MetaTrass will be strengthened and facilitate the investigation of the association between host phenotypes and microbial genotypes for different microbial communities.
METHODS
Data sets
A mock microbial and four human gut microbial communities were analyzed to evaluate the efficiency of MetaTrass. The mock microbial community (ZymoBIOMICS™ Microbial Community DNA Standard. Catalog D6305, Lot ZRC190812) consists of eight isolated bacteria and two fungi with an average abundance of about 12% and 2%, respectively (Supporting Information: Table S4). The four human gut microbial communities were sampled from the feces of three healthy volunteers and one patient with inflammatory bowel disease. The stLFR libraries were constructed according to the standard protocol [32]. The DNA samples were first sheared into long fragments, and then the long fragments were captured by magnetic microbeads with a unique barcode sequence. Finally, each long fragment was further fragmented and hybridized with a unique barcode by the Tn5 transposase on the surface of the microbead. The stLFR libraries of the mock microbial community and the patient sample were sequenced on the BGISEQ‐500 platform, and those of healthy samples were sequenced on the MGISEQ‐2000 platform. The length of paired‐end reads is 100 bp for all data sets. The three healthy sample libraries were individually allocated to a half lane and generated a total of about 50 Gb raw data on average. The mock and patient library was individually allocated to a full lane, resulting in about 85 and 100 Gb raw data, respectively. Barcode sequences were extracted from the end of the reverse read in a pair and then replaced with numerical symbols in the read names in the FASTQ file with our public tool stLFR_barcode_split [55]. SOAPfilter_v2.2 with parameters (‐y ‐F CTGTCTCTTATACACATCTTAGGAAGACAAGCACTGACGACATGA ‐R TCTGCTGAGTCGAGAACGTCTCTGTGAGCCAAGGAGTTGCTCTGG ‐p ‐M 2 ‐f ‐1 ‐Q 10) was used to remove low‐quality raw reads with adaptors, excessively confusing bases, and high duplications. Finally, 59.45 Gb of clean data were retained for the mock microbiome, 34.48 Gb for the first healthy sample (H_Gut_Meta01), 35.33 Gb for the second (H_Gut_Meta02), 37.88 Gb for the third (H_Gut_Meta03), and 97.20 Gb for the patient sample (P_Gut_Meta01). The clean data of all samples are available in the CNGB Sequence Archive (CNSA) [56] (https://db.cngb.org/cnsa/) of the China National GeneBank DataBase (CNGBdb) [57] with accession number CNP0002163.
Taxonomic binning
We adopted Kraken2 (version 2.0.9‐beta) [44] to classify stLFR reads into different species. First, customized Kraken databases were constructed using the reference genomes of a studied microbial community. Then, the stLFR reads were classified according to the database. Both processes of Kraken were run with default parameters. Specifically, references attached to the ZYMO product were used to construct the database for the mock sample. The Kraken2 database of the UHGG collection was downloaded for the human fecal samples, which included 4542 representative genomes at the species level [4].
Cobarcoded read refinement
A taxonomic tree of references was established to reduce the number of multiple hits of a K‐mer from inter‐species repeat sequences in Kraken2 [44]. The reads from these repetitive regions were classified into the lowest common ancient (LCA) rank higher than its corresponding species. Several previous studies tried to reallocate these reads to species by statistical inferences using the coverage depth or cobarcoding information [58, 59]. In the MetaTrass pipeline, the cobarcoding correlation between reads classified into a species and those classified into the LCA ranks higher than that species was used to reduce the false‐negative reads assigned to higher taxonomic ranks. Reads with a confident taxonomic assignment at the species level were treated as taxonomy reads (TRs) of the corresponding species. We collected and refined reads for each barcode according to the number of reads categorized as TRs (Num_T) and the ratio of these reads to the total reads (Ratio_T). Barcodes attached to TRs were first extracted as candidates. We ranked candidates according to Num_T in descending order first and then by Ratio_T also decreasingly. Finally, reads not more than 300× were collected to improve computational efficiency based on the rank of the barcodes. Since sufficient read coverage is required for assembling a complete genome, only the species with an abundance higher than 10× were retained in this step. Reads assigned to these species were further clustered and filtered with the cobarcoding correlation. The abundance of each species was estimated by the quotient of the total base number of TRs to that of the reference. The refined reads of each species were extracted using Seqtk (version 1.3‐r114‐dirty) [60]. Note that there were still some false‐positive reads, probably resulting from the collision of long fragments from different species captured by the same microbead, even though Ratio_T was set to minimize the number. Sequences assembled from these reads would be further filtered out in the way described in the Section, “Contamination removal.”
Cobarcoded read assembly
Reads of a single species with an abundance higher than 10× were assembled in Supernova (version 2.1.1) [39], a high‐performance cobarcoding de novo assembler for single large eukaryotic genomes. Supernova was designed for linked‐reads of 10× Genomics (http://www.10xgenomics.com/), which have different barcodes and formats from stLFR reads. Thus, we converted the stLFR reads into linked‐reads FASTQ format before running Supernova. Additionally, the parameter—accept‐extreme‐coverage was set to yes to allow for large coverage depth differences.
Contamination removal
As long fragments from different species could be captured by the same microbead in the stLFR library at low odds, reads from different species could putatively be labeled with the same barcode. As a result, a draft assembly of reads after cobarcoded read refinement might contain contaminated sequences from other species. We cleaned the assembly based on its similarity to the corresponding reference genome. The AF and ANI have been commonly adopted to circumscribe species [3, 4]. MetaTrass also adopted the metrics of AF and ANI. The ANI was independently calculated for each alignment. The AF was defined as the ratio of total alignment length with sufficiently high ANI to the total contig length. The alignments with ANI values higher than 90% were used in the AF calculation, and the contigs with AF values higher than 50% were retained to form the final assembly. The initial alignments between contigs and references were generated by QUAST (version 5.0.2) [54] with default parameters, except that the identity threshold to obtain valid alignment was set to 90%.
Assembly‐first‐and‐binning‐later approaches
Conventional standard analysis pipelines of the NGS metagenomes implement de novo metagenome assembly first and further bin the contigs into draft genomes. We compared different existing assembly‐first‐and‐binning‐later approaches to MetaTrass by analyzing a mock and four human gut microbial communities. In our tests, the stLFR cobarcoded reads were assembled by NGS assemblers, including IDBA‐UD (version 1.1.3) [20], MEGAHIT (version 1.1.3) [18], and MetaSPAdes (version 3.10.1) [19], or cobarcoding assemblers, including Supernova [39], Athena (version 1.3.0) [31], and CloudSPAdes (version 3.13.1) [42]. Then, all the draft assemblies were binned by two genome binners, namely, MetaBAT2 (version 2.12.1) [23] and Maxbin2.0 (version 2.2.5) [22]. Since Supernova, CloudSPAdes, and Athena were not designed for stLFR reads, we made an appropriate format conversion with the step_2_10X_fake.sh script in our public tool stlfr2supernova_pipeline [61]. Except for Supernova, all the assemblers were run with default parameters. To make Supernova able to assemble data sets with extremely high coverage depths, the parameter—accept‐extreme‐coverage was set to yes. All assembly results are available in the CNSA [56] (https://db.cngb.org/cnsa/) of the CNGBdb [57] with accession number CNP0002163. MetaBAT2 and Maxbin2.0 were implemented with default parameters.
Evaluations
Both reference‐based and reference‐free assessments were used to evaluate the quality of assemblies obtained using different strategies. For the mock microbial community, the reference‐based tool QUAST [54] was used to evaluate the contiguity and accuracy of metagenomic assemblies. Minimap2 (2.17‐r974‐dirty) [62] was used to map assemblies to references and get valid alignments with the identity threshold of 95%. Then, statistics such as genome fraction, NG50/NGA50, and the number of misassemblies were assessed from the alignments with default parameters. For the human gut microbial communities, the reference‐free tool CheckM (version 1.1.2) [63] was implemented with default parameters to evaluate the completeness and contamination of each genome. A definite number of marker genes conserved across almost all bacteria were used as the basis in CheckM. Following the guidance proposed in CheckM, we defined high‐quality assemblies (completeness > 90% and contamination < 5%) and medium‐quality assemblies (completeness > 50% and contamination < 10%, but not meet both completeness > 90% and contamination < 5%). In addition, the statistics of each genome notably N50, genome size, and taxonomic rank were also obtained by CheckM, and the taxonomic rank was used to show the resolution of a genome.
Genomic variation estimation and taxonomic classification
All the high‐quality genomes assembled by MetaTrass were used to call variations for the four human fecal samples. We aligned each genome to their corresponding reference using Minimap2 [62] with parameters (‐x asm5) to prevent an alignment extending to regions with diversity >5%. SAMtools (version 1.9) [64] and PAFtools (2.17‐r982‐dirty) [65] were used to convert the BAM file of initial unsorted alignments into a PAF file of sorted alignments. We identified variations using the “call” module in PAFtools with parameters (‐L 10000) to filter out the alignments shorter than 10,000 bp. We considered single‐nucleotide substitutions as SNVs but ignored single‐base insertions or deletions. Insertions or deletions with lengths shorter than 50 bp were defined as small indels, and the rest were defined as large indels. Those genomic variations with the same position and sequence information that belong to the same species in different samples were determined as shared variations.
We used the “classify_wf” function of GTDB‐Tk (version 0.3.1) [51] to conduct taxonomic annotation of the genomes obtained by both MetaTrass and the conventional strategies with default parameters. Considering the procedure of the UHGG database construction [4], genomes were assigned at the species level if the AF to the close species representative genomes was higher than 30% and ANI was higher than 95%. Interactive Tree of Life (iTOL version 4.4.2) [66] was used to visualize taxonomic trees constructed from the annotated species information.
AUTHOR CONTRIBUTIONS
Li Deng, Guangyi Fan, and Yanwei Qi contributed to the software design. Yanwei Qi, Shengqiang Gu, Yue Zhang, and Lidong Guo contributed to the software implementation. Li Deng, Yanwei Qi, Shengqiang Gu, Mengyang Xu, and Jianwei Chen contributed to the data analyses. Xiaofang Chen, Ou Wang, and Xiaodong Fang contributed to the data curation and collection. Guangyi Fan, Li Deng, and Xin Liu contributed to the benchmarking design. All authors contributed to the manuscript writing. Li Deng, Yanwei Qi, Lidong Guo, Mengyang Xu, and Ying Sun contributed to the manuscript revision. Li Deng and Guangyi Fan supervised the project. All authors read and approved the final manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supplementary information.
Supplementary information.
ACKNOWLEDGMENTS
We acknowledge the anonymous reviewers for their professional advice on this manuscript. We would like to thank Yufen Huang, Gaohong Xiao, and many other BGI‐Shenzhen employees for fruitful discussions during the development and performance test. We also thank Chingyee Moo for her help with language revision. This study was supported by the National Key Research and Development Program of China (2018YFD0900301‐05), the National Natural Science Foundation of China (32100514), and the Science Technology and Innovation Committee of Shenzhen Municipality of China (SGDX20190919142801722).
Qi, Yanwei , Gu Shengqiang, Zhang Yue, Guo Lidong, Xu Mengyang, Cheng Xiaofang, Wang Ou, et al. 2022. “MetaTrass: a high‐quality metagenome assembler of the human gut microbiome by cobarcoding sequencing reads.” iMeta 1, e46. 10.1002/imt2.46
Yanwei Qi and Shengqiang Gu contributed equally to this study.
Contributor Information
Li Deng, Email: dengli1@genomics.cn.
Guangyi Fan, Email: fanguangyi@genomics.cn.
DATA AVAILABILITY STATEMENT
MetaTrass is a command‐line application released to GitHub as open‐source software under General Public License v3.0 (https://github.com/BGI-Qingdao/MetaTrass). The assembling results and cleaned stLFR data sets of the mock and four human fecal samples were deposited into CNSA (https://db.cngb.org/cnsa/) of CNGBdb with accession number CNP0002163 and available from authors upon reasonable application through CNGBdb. The 10X Genomics linked‐reads data sets of Mock‐20 were downloaded from the website https://trace.ncbi.nlm.nih.gov/Traces/index.html?run=SRR6760785. Supplementary materials may be found in the online DOI or iMeta Science http://www.imeta.science/
REFERENCES
- 1. Schloss, Patrick D. , and Handelsman Jo. 2005. “Metagenomics for Studying Unculturable Microorganisms: Cutting the Gordian Knot.” Genome Biology 6: 1–4. 10.1186/gb-2005-6-8-229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Qin, Junjie , Li Ruiqiang, Raes Jeroen, Arumugam Manimozhiyan, Burgdorf Kristoffer Solvsten, Manichanh Chaysavanh, Nielsen Trine, et al. 2010. “A Human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing.” Nature 464: 59–65. 10.1038/nature08821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Parks, Donovan H. , Chuvochina Maria, Chaumeil Pierre‐Alain, Rinke Christian, Mussig Aaron J., and Hugenholtz Philip. 2020. “A Complete Domain‐to‐Species Taxonomy for Bacteria and Archaea.” Nature Biotechnology 38: 1079–86. 10.1038/s41587-020-0501-8 [DOI] [PubMed] [Google Scholar]
- 4. Almeida, Alexandre , Nayfach Stephen, Boland Miguel, Strozzi Francesco, Beracochea Martin, Shi Zhou Jason, Pollard Katherine S., et al. 2021. “A Unified Catalog of 204,938 Reference Genomes from the Human Gut Microbiome.” Nature Biotechnology 39: 105–14. 10.1038/s41587-020-0603-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sheth, Ravi U. , Li Mingqiang, Jiang Weiqian, Sims Peter A., Leong Kam W., and Wang Harris H.. 2019. “Spatial Metagenomic Characterization of Microbial Biogeography in the Gut.” Nature Biotechnology 37: 877–83. 10.1038/s41587-019-0183-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Martino, Cameron , Shenhav Liat, Marotz Clarisse A., Armstrong George, McDonald Daniel, Vázquez‐Baeza Yoshiki, Morton James T., et al. 2021. “Context‐Aware Dimensionality Reduction Deconvolutes Gut Microbial Community Dynamics.” Nature Biotechnology 39: 165–8. 10.1038/s41587-020-0660-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Qian, Xubo , Chen Tong, Xu Yiping, Chen Lei, Sun Fuxiang, Lu Meiping, and Liu Yongxin. 2020. “A Guide to Human Microbiome Research: Study Design, Sample Collection, and Bioinformatics Analysis.” Chinese Medical Journal 133: 1844–55. 10.1097/cm9.0000000000000871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Van Rossum, Thea , Ferretti Pamela, Maistrenko Oleksandr M., and Bork Peer. 2020. “Diversity within Species: Interpreting Strains in Microbiomes.” Nature Reviews: Microbiology 18: 491–506. 10.1038/s41579-020-0368-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Olm, Matthew R. , Crits‐Christoph Alexander, Bouma‐Gregson Keith, Firek Brian A., Morowitz Michael J., and Banfield Jillian F.. 2021. “Instrain Profiles Population Microdiversity from Metagenomic Data and Sensitively Detects Shared Microbial Strains.” Nature Biotechnology 39: 727–36. 10.1038/s41587-020-00797-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leimbach, Andreas , Hacker Jörg, and Dobrindt Ulrich. 2013. “ E. Coli as an All‐Rounder: The Thin Line between Commensalism and Pathogenicity.” Current Topics in Microbiology and Immunology 358: 3–32. 10.1007/82_2012_303 [DOI] [PubMed] [Google Scholar]
- 11. Pierce, Jessica V. , and Bernstein Harris D.. 2016. “Genomic Diversity of Enterotoxigenic Strains of Bacteroides Fragilis .” PLoS One 11: e0158171. 10.1371/journal.pone.0158171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yao, Guocai , Zhang Wenliang, Yang Minglei, Yang Huan, Wang Jianbo, Zhang Haiyue, Wei Lai, Xie Zhi, and Li Weizhong. 2020. “MicroPhenoDB Associates Metagenomic Data with Pathogenic Microbes, Microbial Core Genes, and Human Disease Phenotypes.” Genomics Proteomics Bioinformatics 18: 760–72. 10.1016/j.gpb.2020.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Welch, R. A. , Burland Valerie, Plunkett Guy, Redford Peter, Roesch Paula, Rasko David A., Buckles Eric. L., et al. 2002. “Extensive Mosaic Structure Revealed by the Complete Genome Sequence of Uropathogenic Escherichia Coli .” Proceedings of the National Academy of Sciences of the United States of America 99: 17020–4. 10.1073/pnas.252529799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu, Yongxin , Qin Yuan, Chen Tong, Lu Meiping, Qian Xubo, Guo Xiaoxuan, and Bai Yang. 2021. “A Practical Guide to Amplicon and Metagenomic Analysis of Microbiome Data.” Protein & Cell 12: 315–30. 10.1007/s13238-020-00724-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sczyrba, Alexander , Hofmann Peter, Belmann Peter, Koslicki David, Janssen Stefan, Dröge Johannes, Gregor Ivan, et al. 2017. “Critical Assessment of Metagenome Interpretation—A Benchmark of Metagenomics Software.” Nature Methods 14: 1063–71. 10.1038/nmeth.4458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Breitwieser, Florian P. , Lu Jennifer, and Salzberg Steven L.. 2019. “A Review of Methods and Databases for Metagenomic Classification and Assembly.” Briefings in Bioinformatics 20: 1125–36. 10.1093/bib/bbx120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eun Kang, Jee , Ciampi Antonio, and Hijri Mohamed. 2020. “SeSaMe: Metagenome Sequence Classification of Arbuscular Mycorrhizal Fungi‐Associated Microorganisms.” Genomics Proteomics Bioinformatics 18: 601–12. 10.1016/j.gpb.2018.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li, Dinghua , Liu Chi‐Man, Luo Ruibang, Sadakane Kunihiko, and Lam Tak‐Wah. 2015. “MEGAHIT: An Ultra‐Fast Single‐Node Solution for Large and Complex Metagenomics Assembly Via Succinct De Bruijn Graph.” Bioinformatics 31: 1674–6. 10.1093/bioinformatics/btv033 [DOI] [PubMed] [Google Scholar]
- 19. Nurk, Sergey , Meleshko Dmitry, Korobeynikov Anton, and Pevzner Pavel A.. 2017. “metaSPAdes: A New Versatile Metagenomic Assembler.” Genome Research 27: 824–34. 10.1101/gr.213959.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peng, Yu , Leung Henry C. M., Yiu Siu‐Ming, and Chin Francis Y. L.. 2012. “IDBA‐UD: A de novo Assembler for Single‐Cell and Metagenomic Sequencing Data with Highly Uneven Depth.” Bioinformatics 28: 1420–8. 10.1093/bioinformatics/bts174 [DOI] [PubMed] [Google Scholar]
- 21. Wu, Yu‐Wei , and Ye Yuzhen. 2011. “A Novel Abundance‐Based Algorithm for Binning Metagenomic Sequences Using l‐Tuples.” Journal of Computational Biology 18: 523–34. 10.1089/cmb.2010.0245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu, Yu‐Wei , Simmons Blake A., and Singer Steven W.. 2016. “MaxBin 2.0: An Automated Binning Algorithm to Recover Genomes from Multiple Metagenomic Datasets.” Bioinformatics 32: 605–7. 10.1093/bioinformatics/btv638 [DOI] [PubMed] [Google Scholar]
- 23. Kang, Dongwan D. , Froula Jeff, Egan Rob, and Wang Zhong. 2015. “MetaBAT, an Efficient Tool for Accurately Reconstructing Single Genomes from Complex Microbial Communities.” PeerJ 3: e1165. 10.7717/peerj.1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bertrand, Denis , Shaw Jim, Kalathiyappan Manesh, Ng Amanda Hui Qi, Kumar M. Senthil, Li Chenhao, Dvornicic Mirta, et al. 2019. “Hybrid Metagenomic Assembly Enables High‐Resolution Analysis of Resistance Determinants and Mobile Elements in Human Microbiomes” Nature Biotechnology 37: 937–44. 10.1038/s41587-019-0191-2 [DOI] [PubMed] [Google Scholar]
- 25. Chin, Chen‐Shan , Alexander David H., Marks Patrick, Klammer Aaron A., Drake James, Heiner Cheryl, Clum Alicia, et al. 2013. “Nonhybrid, Finished Microbial Genome Assemblies from Long‐Read SMRT Sequencing Data.” Nature Methods 10: 563–9. 10.1038/nmeth.2474 [DOI] [PubMed] [Google Scholar]
- 26. Kolmogorov, Mikhail , Bickhart Derek M., Behsaz Bahar, Gurevich Alexey, Rayko Mikhail, Shin Sung Bong, Kuhn Kristen, et al. 2020. “Metaflye: Scalable Long‐Read Metagenome Assembly Using Repeat Graphs.” Nature Methods 17: 1103–10. 10.1038/s41592-020-00971-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. DeMaere, Matthew Z. , and Darling Aaron E.. 2019. “bin3C: Exploiting Hi‐C Sequencing Data to Accurately Resolve Metagenome‐Assembled Genomes.” Genome Biology 20: 1–16. 10.1186/s13059-019-1643-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cleary, Brian , Brito Ilana Lauren, Huang Katherine, Gevers Dirk, Shea Terrance, Young Sarah, and Alm Eric J.. 2015. “Detection of Low‐Abundance Bacterial Strains in Metagenomic Datasets by Eigengenome Partitioning.” Nature Biotechnology 33: 1053–60. 10.1038/nbt.3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peters, Brock A. , Kermani Bahram G., Sparks Andrew B., Alferov Oleg, Hong Peter, Alexeev Andrei, Jiang Yuan, et al. 2012. “Accurate Whole‐Genome Sequencing and Haplotyping from 10 to 20 Human Cells.” Nature 487: 190–5. 10.1038/nature11236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adey, Andrew , Kitzman Jacob O., Burton Joshua N., Daza Riza, Kumar Akash, Christiansen Lena, Ronaghi Mostafa, et al. 2014. “In Vitro, Long‐Range Sequence Information for de novo Genome Assembly Via Transposase Contiguity.” Genome Research 24: 2041–9. 10.1101/gr.178319.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bishara, Alex , Moss Eli L., Kolmogorov Mikhail, Parada Alma E., Weng Ziming, Sidow Arend, Dekas Anne E., Batzoglou Serafim, and Bhatt Ami S.. 2018. “High‐Quality Genome Sequences of Uncultured Microbes by Assembly of Read Clouds” Nature Biotechnology 36: 1067–75. 10.1038/nbt.4266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang, Ou , Chin Robert, Cheng Xiaofang, Wu Michelle, Mao Qing, Tang Jingbo, Sun Yuhui, et al. 2019. “Efficient and Unique Cobarcoding of Second‐Generation Sequencing Reads from Long DNA Molecules Enabling Cost‐Effective and Accurate Sequencing, Haplotyping, and de novo Assembly.” Genome Research 29: 798–808. 10.1101/gr.245126.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen, Zhoutao , Pham Long, Wu Tsai‐Chin, Mo Guoya, Xia Yu, Chang Peter L., Porter Devin, et al. 2020. “Ultralow‐Input Single‐Tube Linked‐Read Library Method Enables Short‐Read Second‐Generation Sequencing Systems to Routinely Generate Highly Accurate and Economical Long‐Range Sequencing Information.” Genome Research 30: 898–909. 10.1101/gr.260380.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zheng, Grace X. Y. , Lau Billy T., Schnall‐Levin Michael, Jarosz Mirna, Bell John M., Hindson Christopher M., Kyriazopoulou‐Panagiotopoulou Sofia, et al. 2016. “Haplotyping Germline and Cancer Genomes with High‐Throughput Linked‐Read Sequencing.” Nature Biotechnology 34: 303–11. 10.1038/nbt.3432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Danko, David C. , Meleshko Dmitry, Bezdan Daniela, Mason Christopher, and Hajirasouliha Iman. 2019. “Minerva: An Alignment‐and Reference‐Free Approach to Deconvolve Linked‐Reads for Metagenomics.” Genome Research 29: 116–24. 10.1101/gr.235499.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu, Mengyang , Guo Lidong, Du Xiao, Li Lei, Peters Brock A., Deng Li, Wang Ou, et al. 2021. “Accurate Haplotype‐Resolved Assembly Reveals the Origin of Structural Variants for Human Trios.” Bioinformatics 37: 2095–102. 10.1093/bioinformatics/btab068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bishara, Alex , Liu Yuling, Weng Ziming, Kashef‐Haghighi Dorna, Newburger Daniel E., West Robert, Sidow Arend, and Batzoglou Serafim. 2015. “Read Clouds Uncover Variation in Complex Regions of the Human Genome.” Genome Research 25: 1570–80. 10.1101/gr.191189.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guo, Junfu , Shi Chang, Chen Xi, Wang Ou, Liu Ping, Yang Huanming, Xu Xun, Zhang Wenwei, and Zhu Hongmei. 2021. “stLFRsv: A Germline Structural Variant Analysis Pipeline Using Co‐Barcoded Reads.” Frontiers in Genetics 12: 222. 10.3389/fgene.2021.636239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weisenfeld, Neil I. , Kumar Vijay, Shah Preyas, Church Deanna M., and Jaffe David B.. 2017. “Direct Determination of Diploid Genome Sequences.” Genome Research 27: 757–67. 10.1101/gr.214874.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yeo, Sarah , Coombe Lauren, Warren René L, Chu Justin, and Birol Inanç. 2017. “ARCS: Scaffolding Genome Drafts with Linked Reads.” Bioinformatics 34: 725–31. 10.1093/bioinformatics/btx675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guo, Lidong , Xu Mengyang, Wang Wenchao, Gu Shengqiang, Zhao Xia, Chen Fang, Wang Ou, et al. 2021. “SLR‐superscaffolder: A de novo Scaffolding Tool for Synthetic Long Reads Using a Top‐to‐Bottom Scheme.” BMC Bioinformatics 22: 1–16. 10.1186/s12859-021-04081-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tolstoganov, Ivan , Bankevich Anton, Chen Zhoutao, and Pevzner Pavel A.. 2019. “cloudSPAdes: Assembly of Synthetic Long Reads Using De Bruijn Graphs.” Bioinformatics 35: i61–70. 10.1093/bioinformatics/btz349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kuleshov, Volodymyr , Snyder Michael P., and Batzoglou Serafim. 2016. “Genome Assembly From Synthetic Long Read Clouds.” Bioinformatics 32: i216–24. 10.1093/bioinformatics/btw267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wood, Derrick E. , Lu Jennifer, and Langmead Ben. 2019. “Improved Metagenomic Analysis with Kraken 2.” Genome Biology 20: 1–13. 10.1186/s13059-019-1891-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Koren, Sergey , Rhie Arang, Walenz Brian P., Dilthey Alexander T., Bickhart Derek M., Kingan Sarah B., Hiendleder Stefan, et al. 2018. “ de novo Assembly of Haplotype‐Resolved Genomes with Trio Binning.” Nature Biotechnology 36: 1174–82. 10.1038/nbt.4277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nicholls, Samuel M. , Quick Joshua C., Tang Shuiquan, and Loman Nicholas J.. 2019. “Ultra‐Deep, Long‐Read Nanopore Sequencing of Mock Microbial Community Standards.” GigaScience 8: giz043. 10.1093/gigascience/giz043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ruan, Jue , and Li Heng. 2020. “Fast and Accurate Long‐Read Assembly with wtdbg2.” Nature Methods 17: 155–8. 10.1038/s41592-019-0669-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bankevich, Anton , Nurk Sergey, Antipov Dmitry, Gurevich Alexey A., Dvorkin Mikhail, Kulikov Alexander S., Lesin Valery M., et al. 2012. “SPAdes: A New Genome Assembly Algorithm and its Applications to Single‐Cell Sequencing.” Journal of Computational Biology 19: 455–77. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shin, Na‐Ri , Whon Tae Woong, and Bae Jin‐Woo. 2015. “ Proteobacteria: Microbial Signature of Dysbiosis in Gut Microbiota.” Trends in Biotechnology 33: 496–503. 10.1016/j.tibtech.2015.06.011 [DOI] [PubMed] [Google Scholar]
- 50. Johnson, Katerina V‐A. 2020. “Gut Microbiome Composition and Diversity Are Related to Human Personality Traits.” Human Microbiome Journal 15: 100069. 10.1016/j.humic.2019.100069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chaumeil, Pierre‐Alain , Mussig Aaron J., Hugenholtz Philip, and Parks Donovan H.. 2019. “GTDB‐Tk: A Toolkit to Classify Genomes with the Genome Taxonomy Database.” Bioinformatics 36: 1925–7. 10.1093/bioinformatics/btz848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zeevi, David , Korem Tal, Godneva Anastasia, Bar Noam, Kurilshikov Alexander, Lotan‐Pompan Maya, Weinberger Adina, et al. 2019. “Structural Variation in the Gut Microbiome Associates with Host Health.” Nature 568: 43–8. 10.1038/s41586-019-1065-y [DOI] [PubMed] [Google Scholar]
- 53. Chen, Lianmin , Wang Daoming, Garmaeva Sanzhima, Kurilshikov Alexander, Vila Arnau Vich, Gacesa Ranko, Sinha Trishla, et al. 2021. “The Long‐Term Genetic Stability and Individual Specificity of the Human Gut Microbiome.” Cell 184: 2302–15. 10.1016/j.cell.2021.03.024 [DOI] [PubMed] [Google Scholar]
- 54. Gurevich, Alexey , Saveliev Vladislav, Vyahhi Nikolay, and Tesler Glenn. 2013. “QUAST: Quality Assessment Tool for Genome Assemblies.” Bioinformatics 29: 1072–5. 10.1093/bioinformatics/btt086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Guo, Lidong . stLFR_barcode_split. https://github.com/BGI-Qingdao/stLFR_barcode_split.
- 56. Guo, Xueqin , Chen Fengzhen, Gao Fei, Li Ling, Liu Ke, You Lijin, Hua Cong, et al. 2020. “CNSA: A Data Repository for Archiving Omics Data.” Database 2020: baaa055. 10.1093/database/baaa055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen, Fengzhen , You Lijin, Yang Fan, Wang Lina, Guo Xueqin, Gao Fei, Hua Cong, et al. 2020. “CNGBdb: China National Genebank Database.” Heredidas 42: 799–809. 10.16288/j.yczz.20-080 [DOI] [PubMed] [Google Scholar]
- 58. Lu, Jennifer , Breitwieser Florian P., Thielen Peter, and Salzberg Steven L.. 2017. “Bracken: Estimating Species Abundance in Metagenomics Data.” PeerJ Computer Science 3: e104. 10.7717/peerj-cs.104 [DOI] [Google Scholar]
- 59. Danko, David C. , Meleshko Dmitry, Bezdan Daniela, Mason Christopher, and Hajirasouliha Iman. 2019. Novel Algorithms for the Taxonomic Classification of Metagenomic Linked‐Reads. bioRxiv 549667. 10.1101/549667 [DOI]
- 60. Li, Heng . seqtk. https://github.com/lh3/seqtk
- 61. Guo, Lidong . stlfr2supernova_pipeline. https://github.com/BGI-Qingdao/stlfr2supernova_pipeline
- 62. Li, Heng . 2018. “Minimap2: Pairwise Alignment for Nucleotide Sequences.” Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Parks, Donovan H. , Imelfort Michael, Skennerton Connor T., Hugenholtz Philip, and Tyson Gene W.. 2015. “CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes.” Genome Research 25: 1043–1055. 10.1101/gr.186072.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li, Heng , Handsaker Bob, Wysoker Alec, Fennell Tim, Ruan Jue, Homer Nils, Marth Gabor, et al. 2009. “The Sequence Alignment/Map Format and SAMtools.” Bioinformatics 25: 2078–9. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li, Heng . paftools. https://github.com/lh3/minimap2/tree/master/misc
- 66. Letunic, Ivica , and Bork Peer. 2019. “Interactive Tree of Life (iTOL) v4: Recent Updates and New Developments” Nucleic Acids Research 47: W256–9. 10.1093/nar/gkz239 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information.
Supplementary information.
Data Availability Statement
MetaTrass is a command‐line application released to GitHub as open‐source software under General Public License v3.0 (https://github.com/BGI-Qingdao/MetaTrass). The assembling results and cleaned stLFR data sets of the mock and four human fecal samples were deposited into CNSA (https://db.cngb.org/cnsa/) of CNGBdb with accession number CNP0002163 and available from authors upon reasonable application through CNGBdb. The 10X Genomics linked‐reads data sets of Mock‐20 were downloaded from the website https://trace.ncbi.nlm.nih.gov/Traces/index.html?run=SRR6760785. Supplementary materials may be found in the online DOI or iMeta Science http://www.imeta.science/