

Significance
Computational protein structure prediction has been significantly improved by the application of coevolution information derived from multiple sequence alignment (MSA) and deep learning. Although very powerful, these methods do not perform well on small-sized protein families, flexible regions such as highly variable antibody Complementarity-determining region (CDR) regions, and single mutation effects. This study highlights the potential of integrating various protein language models and fine-tuning that may yield better performance than MSA-based methods and other single-sequence methods on antibody structure prediction (fine-tuning on antibody data), orphan protein structure prediction, and single mutation effect prediction.
Keywords: protein structure prediction, protein language model, single-sequence protein structure rediction, antibody structure prediction, single mutation effect
Abstract
Protein structure prediction has been greatly improved by deep learning in the past few years. However, the most successful methods rely on multiple sequence alignment (MSA) of the sequence homologs of the protein under prediction. In nature, a protein folds in the absence of its sequence homologs and thus, a MSA-free structure prediction method is desired. Here, we develop a single-sequence-based protein structure prediction method RaptorX-Single by integrating several protein language models and a structure generation module and then study its advantage over MSA-based methods. Our experimental results indicate that in addition to running much faster than MSA-based methods such as AlphaFold2, RaptorX-Single outperforms AlphaFold2 and other MSA-free methods in predicting the structure of antibodies (after fine-tuning on antibody data), proteins of very few sequence homologs, and single mutation effects. By comparing different protein language models, our results show that not only the scale but also the training data of protein language models will impact the performance. RaptorX-Single also compares favorably to MSA-based AlphaFold2 when the protein under prediction has a large number of sequence homologs.
In the past few years, computational protein structure prediction has been revolutionized by the application of coevolution information and deep learning, as evidenced first by RaptorX (1, 2) and then AlphaFold2 (3) and other similar methods. These successful methods make use of sequence homologs of the protein under prediction and coevolution information derived from their multiple sequence alignment (MSA). Although very powerful, these methods are not perfect. For example, in nature, a protein folds without knowledge of its sequence homologs, so predicting protein structure based upon some non-natural conditions such as MSAs (and coevolution information) does not reflect very well how a protein actually folds. It also takes much time to search for the sequence homologs of a protein, especially considering that the sequence databases are growing rapidly. Further, the MSA-based methods do not perform well on small-sized protein families. Experimental results show that MSA-based methods do not fare well on flexible regions such as loop regions (4) and highly variable antibody CDR regions. This can be attributed, in part, to the relatively weak presence of coevolutionary information within these regions. Further, MSA are not sensitive for sequence mutations and the MSA-based methods may not fare well on the prediction of single-point mutational effects (5).
To reduce the dependence of protein structure prediction on MSA, we have studied protein structure prediction without using coevolution information in Xu et al. (6) and demonstrated that in the absence of coevolution information, deep learning can still predict more than 50% of the most challenging CASP13 targets. This implies that deep learning indeed has captured sequence–structure relationships useful for tertiary structure prediction. Nevertheless, in this work, we still used sequence profiles derived from sequence homologs. In the past months, several groups (7–11) have studied deep learning methods for single-sequence-based protein structure prediction by making use of protein language models. Although running much faster, on average, these methods are less accurate than MSA-based AlphaFold2.
To further study the advantage of single-sequence-based methods, we have developed RaptorX-Single for single-sequence-based protein structure prediction. Our methods take an individual protein sequence as input and then feed it into protein language models to produce sequence embedding, which is then fed into a modified Evoformer module and a structure generation module to predict atom coordinates. Differing from other single-sequence methods, our methods use a combination of three well-developed protein language models (12–14) instead of only one. Experimental results show that RaptorX-Single not only runs much faster than MSA-based AlphaFold2 but also outperforms it on antibody structure prediction, orphan protein structure prediction, and single mutation effect prediction. RaptorX-Single also exceeds other single-sequence-based methods and compares favorably to AlphaFold2 when the protein under prediction has a large number of sequence homologs.
Results
Performance on the IgFold-Ab Dataset.
As shown in Table 1, our antibody-specific method RaptorX-Single-Ab consistently outperforms other methods for antibody structure prediction, especially in the CDR H3 region as shown in Fig. 1A. The MSA-based AlphaFold2 performs comparably with other single-sequence methods (HelixFold-Single, OmegaFold, and ESMFold), but is not as good as the antibody-specific methods DeepAb, IgFold, and EquiFold. AlphaFold2 (Single) performs much worse than AlphaFold2 (MSA), mainly because AlphaFold2 was trained on MSAs instead of single sequences. Antibodies are distinct from other proteins due to their constant structure in the framework region but large structure variability in the CDR regions. Consequently, the single-sequence methods trained on regular proteins do not perform well on antibody structure prediction, as evidenced by the performance of HelixFold-Single, OmegaFold, ESMFold, and RaptorX-Single in Table 1. After fine-tuning on antibody structures, our antibody-specific methods greatly outperform non-fine-tuned methods, suggesting the significance of the fine-tuning process on antibody data.
Table 1.
The average rmsd of predicted tertiary structures on the IgFold-Ab dataset
| rmsd (H) | rmsd (L) | |||||||
|---|---|---|---|---|---|---|---|---|
| Fr | CDR-1 | CDR-2 | CDR-3 | Fr | CDR-1 | CDR-2 | CDR-3 | |
| AlphaFold2 (MSA) | 0.48 | 0.77 | 0.76 | 3.55 | 0.43 | 0.96 | 0.45 | 1.26 |
| AlphaFold2 (Single) | 10.84 | 15.34 | 15.48 | 16.33 | 8.98 | 13.54 | 16.13 | 15.14 |
| HelixFold-Single | 0.56 | 0.85 | 0.95 | 5.01 | 0.51 | 1.10 | 0.57 | 1.60 |
| OmegaFold | 0.47 | 0.75 | 0.74 | 3.70 | 0.41 | 0.93 | 0.43 | 1.35 |
| ESMFold | 0.51 | 0.84 | 0.91 | 4.10 | 0.43 | 1.16 | 0.52 | 1.44 |
| DeepAb | 0.43 | 0.80 | 0.74 | 3.28 | 0.38 | 0.86 | 0.45 | 1.11 |
| IgFold | 0.45 | 0.80 | 0.75 | 2.99 | 0.45 | 0.83 | 0.51 | 1.07 |
| EquiFold | 0.44 | 0.74 | 0.69 | 2.86 | 0.40 | 0.78 | 0.40 | 1.02 |
| RaptorX-Single | 0.51 | 0.86 | 0.90 | 4.33 | 0.46 | 1.13 | 0.54 | 1.95 |
| RaptorX-Single-Ab | 0.38 | 0.63 | 0.60 | 2.65 | 0.35 | 0.69 | 0.39 | 0.88 |
Note: The performance of EquiFold was reported by its author.
Fig. 1.
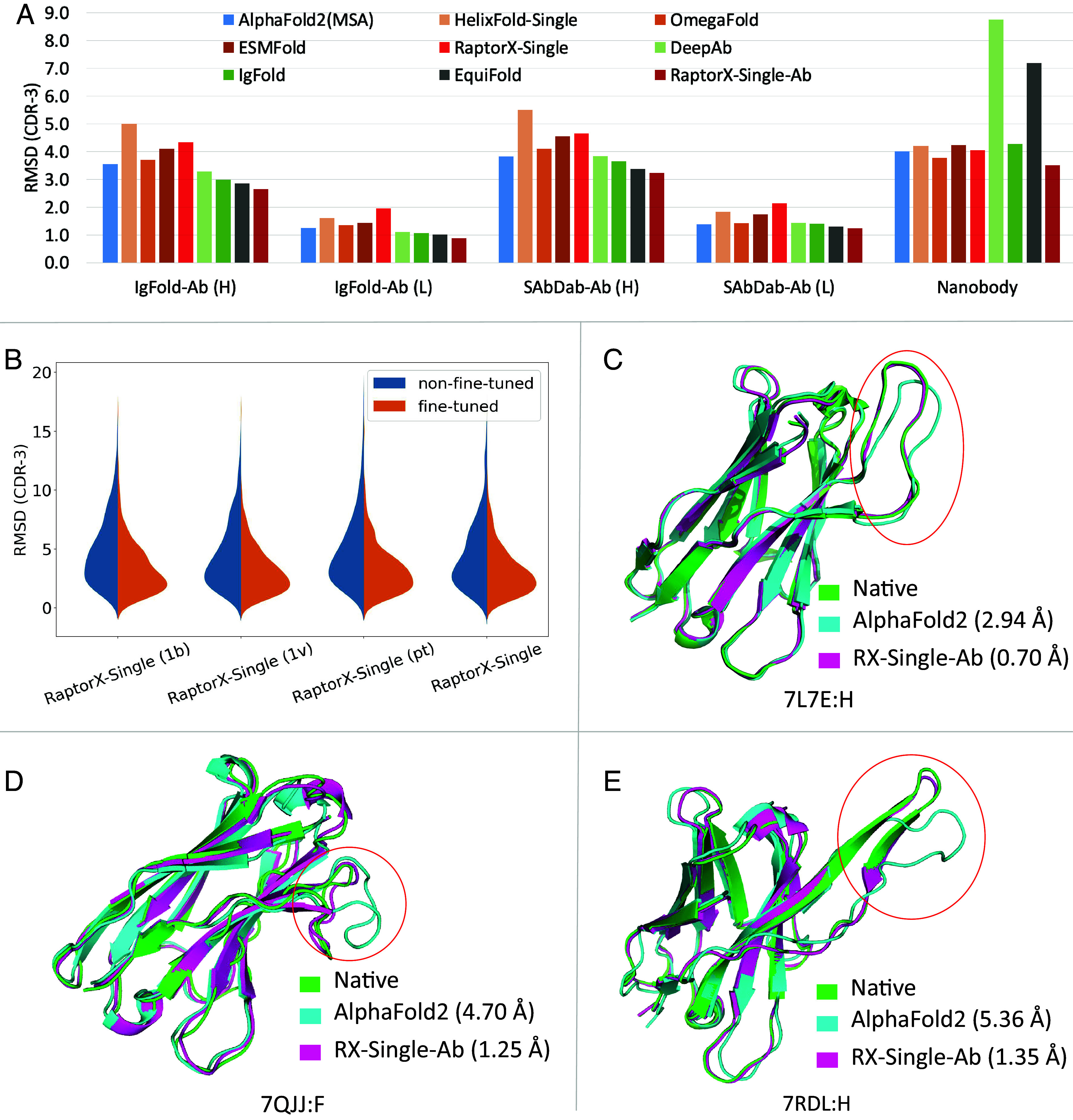
Performance comparison of various methods on antibody structure prediction. (A) The average rmsd of predicted CDR-3 structures on different datasets. (B) The CDR-3 rmsd distribution of our non-fine-tuned and antibody fine-tuned models by combining protein language models in different ways. The antibody fine-tuned methods consistently outperform the non-fine-tuned methods. (C–E) Comparison of RaptorX-Single-Ab and MSA-based AlphaFold2 on the CDR H3 structure prediction (marked by red circle) for targets 7L7E, 7QJJ, and 7RDL.
Performance on the SAbDab-Ab Dataset.
Our fine-tuned antibody-specific method greatly outperforms other methods regardless of metrics, as shown in Table 2. AlphaFold2 (MSA) performs slightly better than other single-sequence methods (HelixFold-Single, OmegaFold and ESMFold) and is comparable with the antibody-specific methods DeepAb and IgFold. Our fine-tuned antibody-specific methods outperform our non-fine-tuned methods, while there is no obvious difference between models using different protein language models as shown in SI Appendix, Table S5. As shown in Fig. 1 C–E, RaptorX-Single-Ab significantly outperforms AlphaFold2 (MSA) on some targets. High-accuracy structure prediction in antibody CDR regions holds great importance for antibody analysis, affinity maturation, and antibody design. As demonstrated in SI Appendix, Figs. S1–S5, in the case of certain targets’ CDR regions, RaptorX-Single-Ab has demonstrated the potential to achieve improvements of around 2 Å (rmsd) or more. Such improvements hold the promise of advancing antibody research by enabling more accurate structural insights. To explore the possibility of improving performance on CDR regions by predicting the VH and VL chains together, we investigated various strategies involving different linkers. Our results, as shown in SI Appendix, Tables S8 and S9, reveal that the utilization of different linkers did not yield a significant impact on performance.
Table 2.
The average rmsd of predicted tertiary structures on the SAbDab-Ab dataset
| rmsd (H) | rmsd (L) | |||||||
|---|---|---|---|---|---|---|---|---|
| Fr | CDR-1 | CDR-2 | CDR-3 | Fr | CDR-1 | CDR-2 | CDR-3 | |
| AlphaFold2 (MSA) | 0.63 | 1.08 | 0.89 | 3.82 | 0.59 | 0.89 | 0.69 | 1.39 |
| AlphaFold2 (Single) | 8.85 | 12.3 | 11.59 | 15.24 | 8.82 | 13.28 | 15.13 | 14.62 |
| HelixFold-Single | 0.71 | 1.15 | 1.1 | 5.5 | 0.66 | 1.1 | 0.79 | 1.84 |
| OmegaFold | 0.63 | 1.05 | 0.86 | 4.11 | 0.58 | 0.9 | 0.69 | 1.42 |
| ESMFold | 0.64 | 1.11 | 1.02 | 4.56 | 0.6 | 1.16 | 0.72 | 1.74 |
| DeepAb | 0.62 | 1.08 | 0.9 | 3.83 | 0.66 | 0.96 | 0.75 | 1.43 |
| IgFold | 0.66 | 1.15 | 0.95 | 3.65 | 0.65 | 0.96 | 0.8 | 1.4 |
| EquiFold | 0.6 | 1.05 | 0.89 | 3.37 | 0.57 | 0.87 | 0.72 | 1.31 |
| RaptorX-Single | 0.64 | 1.17 | 1.06 | 4.66 | 0.64 | 1.12 | 0.77 | 2.14 |
| RaptorX-Single-Ab | 0.57 | 1.01 | 0.82 | 3.24 | 0.53 | 0.79 | 0.66 | 1.24 |
Performance on the Nanobody Dataset.
Nanobody is an increasingly popular modality for therapeutic development (15). Compared with traditional antibodies, nanobodies lack a second immunoglobulin chain and have increased CDR3 loop length, which makes the nanobody structure prediction challenging. As shown in Table 3, no methods perform very well on the CDR-3 region, but our fine-tuned antibody-specific models still outperform other methods. AlphaFold2 (MSA) outperforms HelixFold-Single and ESMFold but underperforms OmegaFold. DeepAb performs poorly on nanobodies, possibly because it is mainly trained for traditional antibody structure prediction. EquiFold is a structure prediction method that relies solely on the primary sequence of proteins. While it demonstrates comparable performance to IgFold and RaptorX-Single-Ab, which are based on protein language models, for traditional antibody structure prediction, EquiFold’s performance significantly decays for nanobody structure prediction. This demonstrates the value of protein language models for the generalization ability of structure prediction methods across different contexts. Compared with the baseline methods, RaptorX-Single-Ab performs best, demonstrating the significance of combining three protein language models and fine-tuning on antibody data.
Table 3.
The average rmsd of the predicted structures on the Nanobody dataset
| rmsd | ||||
|---|---|---|---|---|
| Fr | CDR-1 | CDR-2 | CDR-3 | |
| AlphaFold2 (MSA) | 0.73 | 2.05 | 1.15 | 4.01 |
| AlphaFold2 (Single) | 9.34 | 12.67 | 12.39 | 17.87 |
| HelixFold-Single | 0.86 | 1.99 | 1.18 | 4.2 |
| OmegaFold | 0.71 | 2.02 | 1.12 | 3.77 |
| ESMFold | 0.80 | 2.06 | 1.12 | 4.23 |
| DeepAb | 0.92 | 2.38 | 1.34 | 8.76 |
| IgFold | 0.82 | 1.93 | 1.29 | 4.27 |
| EquiFold | 2.30 | 3.23 | 2.61 | 7.19 |
| RaptorX-Single | 0.83 | 2.19 | 1.14 | 4.06 |
| RaptorX-Single-Ab | 0.82 | 1.78 | 1.06 | 3.50 |
Performance of Various Single-Sequence Methods.
To provide a comprehensive evaluation of both single-sequence and MSA-based methods, we conducted additional assessments using the CASP14 and CAMEO datasets. The CASP14 dataset comprises 60 targets encompassing 88 domains, while the CAMEO dataset, consistent with the ESMFold benchmark, includes 194 targets released between April 2022 and June 2022. Our results, detailed in SI Appendix, Tables S10 and S11, emphasize the distinct advantage of MSA-based AlphaFold2 over single-sequence methods, particularly on the more challenging CASP14 dataset. Among the single-sequence methods, ESMFold outperforms others, highlighting the value of larger protein language models for improved performance.
Regarding our single-sequence methods, RaptorX-Single (1b), RaptorX-Single (1v), and RaptorX-Single (pt) employ ESM-1b, ESM-1v, and ProtTrans, respectively. Among three protein language models, ProtTrans contains 3 billion parameters, a significant increase over the 650 M parameters in ESM-1b and ESM-1v. On the general benchmark datasets (CASP14 and CAMEO), RaptorX-Single (pt) achieved the best performance compared to RaptorX-Single (1b) and RaptorX-Single (1v), which is consistent with the findings in ESMFold. However, it’s noteworthy that RaptorX-Single (pt) did not perform well on antibody datasets (SI Appendix, Tables S4–S6). Conversely, RaptorX-Single (1v) outperformed the others. The ESM-1v shares the same model with ESM-1b but differs in its training data, with ESM-1v trained on Uniref90 and ESM-1b trained on Uniref50. Thus, ESM-1v is more sensitive than ESM-1b to mutations. These results illustrate that not only the scale of protein language models but also the training strategy (like training data) will impact the performance. Furthermore, our results underscore the robust performance of RaptorX-Single and RaptorX-Single-Ab, which integrate three protein language models. By selecting models based on pLDDT scores from those predicted by four RaptorX-Single models, we can achieve further performance improvements.
Performance on the Orphan Protein Dataset.
Orphan proteins lack evolutionary homologs in structure and sequence databases and, thus, are very challenging for MSA-based methods. Here, we construct an orphan protein dataset, in which no protein has any sequence homologs in the 4 widely used sequence databases BFD, MGnify, Uniref90, and Uniclust30. As shown in Table 4, almost all single-sequence methods outperform AlphaFold2 on this dataset and our method RaptorX-Single achieved the best performance. SI Appendix, Fig. S6 shows the head-to-head performance comparison of our method with the competitive methods, and SI Appendix, Fig. S7 shows the comparison of predicted structures for two test targets 7W5Z_T3 and 7W5Z_T2, which demonstrates that RaptorX-Single exhibits superior performance in the loop and alpha-helix region over AlphaFold2. However, neither MSA nor single-sequence methods can predict the correct fold of most orphan proteins, mainly because current single-sequence methods are still implicitly making use of homologous information learned by protein language models.
Table 4.
The average model quality (measured by TMscore, GDT_TS and GHT_HA) of our method RaptorX-Single, AlphaFold2, HelixFold-Single, OmegaFold, and ESMFold on the Orphan dataset
| Method | TMscore | GDT_TS | GHT_HA |
|---|---|---|---|
| AlphaFold2 | 0.40 | 41.02 | 30.2 |
| HelixFold-Single | 0.42 | 44.19 | 30.95 |
| OmegaFold | 0.37 | 38.23 | 27.7 |
| ESMFold | 0.42 | 41.91 | 31.2 |
| RaptorX-Single | 0.43 | 43.4 | 32.14 |
Predicted Structure Changes of Single Mutations.
Previous works showed that the MSA-based methods do not work well on predicting mutational effects (4, 5, 16). To evaluate the advantage of our single-sequence methods over the MSA-based methods for mutational effect prediction, we analyze the correlation between predicted structure change and stability change of single mutations using data gathered from DMS datasets (17). The underlying hypothesis is that proteins tend to adopt the conformation with the lowest energy in their native state. Consequently, if a mutation reduces the protein's stability, the structure might change to preserve its lowest energy state. The structure change is measured by TMscore of predicted structures between the wildtype and mutated sequences. As shown in Fig. 2, our single-sequence method RaptorX-Single outperforms AlphaFold2 (MSA) on 9 of 14 targets, especially on targets EEHEE_rd3_1716, HEEH_rd3_0726, and HHH_0142. It also shows that performance varies with respect to protein language models. On average RaptorX-Single (1b) [PCC (Pearson correlation coefficient): 0.37] and RaptorX-Single (1v) (mean PCC: 0.37) perform better than RaptorX-Single (pt) (PCC: 0.34). AlphaFold2 (Single) outperforms AlphaFold2 (MSA), demonstrating the advantage of single-sequence methods for mutational effect prediction. It should be noted that the structure prediction methods were not specifically trained for stability prediction. Consequently, these methods may not exhibit the same level of competitiveness as state-of-the-art stability prediction methods.
Fig. 2.

Performance comparison of predicted structure change and stability change of single mutations. (A) The PCC between predicted structure change and stability change of all targets. (B) The scatter plot of predicted structure change by RaptorX-Single and stability change for target HHH_0142. (C) The scatter plot of predicted structure change by RaptorX-Single and stability change for target HHH_rd3_0138.
Impact of MSA Depth on Prediction Quality.
It is generally believed that current single-sequence methods are implicitly making use of sequence homologs of a protein under prediction. To study this, we compare our single-sequence method RaptorX-Single with the MSA-based AlphaFold2 on targets of various MSA depths. First, we used the 60 CASP14 targets and 194 CAMEO targets. Most of these targets have lots of sequence homologs, to balance the MSA depth distribution, we additionally collected 99 targets released from 01 January 2020, through 12 April 2022, that do not have sequence homologs in Uniclust30 but may have sequence homologs in BFD, MGnify or Uniref90. As shown in Fig. 3, when the test proteins have no sequence homologs (i.e., MSA depth = 1), RaptorX-Single (GDT_TS: 0.48) slightly outperforms the MSA-based AlphaFold2 (GDT_TS: 0.46). When the MSA is deep (depth>1e4), RaptorX-Single (GDT_TS: 0.79) slightly underperforms AlphaFold2 (GDT_TS: 0.86). When MSA is very deep (depth > 1e5), RaptorX-Single has a comparable performance (GDT_TS: 0.87) as AlphaFold2 (GDT_TS: 0.89). The average length of the proteins with deep MSAs (depth > 1e4) is 411. When their MSAs are not used, their structures predicted by AlphaFold2 have an average TMscore 0.31, indicating that the structures of these proteins cannot be easily predicted. This result implies that the protein language models implicitly learn coevolution information of large-sized protein families. RaptorX-Single significantly underperforms MSA-based AlphaFold2 when the MSA depth is between 100 and 1,000.
Fig. 3.

Performance comparison between our method RaptorX-Single and MSA-based AlphaFold2 with respect to MSA depth. The performance is measured by ΔGDT_TS (GDT_TS difference of the 3D models predicted by RaptorX-Single and that predicted by AlphaFold2). The MSA depth is the number of sequences in the MSA.
Discussion
We have presented deep learning methods that can predict the structure of a protein without explicitly making use of its sequence homologs and MSAs. Our experimental results show that our methods outperform MSA-based AlphaFold2 and other single-sequence methods on antibody structure prediction (after fine-tuning on antibody data), orphan protein structure prediction, and single mutation effect prediction, demonstrating the advantage of single-sequence methods on these specific prediction tasks. For antibody structure prediction, we are developing methods that may predict VH-VL complexes, which will further improve the structure prediction of traditional antibodies. However, although single-sequence methods do not explicitly make use of sequence homologs, the protein language models may implicitly encode some evolutionary and coevolution information by learning from a very large protein sequence database. Although outperforming AlphaFold2 on orphan proteins, our method and other similar ones still cannot predict the correct fold of most orphan proteins, possibly because all current single-sequence methods are still implicitly making use of sequence homologs of a protein under prediction. Our future plan is to develop a method that can indeed predict the structure of a protein directly from its primary sequence even without implicitly using any homologous information. Although EquiFold is developed to address this challenge, its performance is limited. Much effort is needed to achieve accurate structure prediction solely based on the primary sequence of proteins.
Materials and Methods
Protein Language Models.
Protein language models have been developed to model individual protein sequences. These models mainly use attention-based deep neural networks to capture long-range inter-residue relationships. Here, we make use of three pretrained protein language models including ESM-1b (12), ESM-1v (13), and ProtTrans (14). ESM-1b is a Transformer of ~650 M parameters that was trained on UniRef50 (18) of 27.1 million protein sequences. ESM-1v employs the same network architecture as ESM-1b, but was trained on Uniref90 with 98 million protein sequences. For ProtTrans, we use the ProtT5-XL model of 3 billion parameters that was trained on a newer UniRef50 of 45 million sequences.
Network Architecture.
The overall network architecture of our method is shown in Fig. 4, mainly consisting of three modules: the sequence embedding module, the modified Evoformer module, and the structure generation module. Given an individual protein sequence as input, the embedding module generates the sequence embedding of the input sequence and its pair representations, by making use of one or three protein language models. In the embedding module, the one-hot encoding of the input sequence passes through a linear layer to generate the initial sequence embedding, then it combines the sequence representation from protein language models to create a new sequential embedding. The initial pair embedding is generated by adding two primary sequences embedding (rowwise and columnwise), and it then combines the attention maps from the last two layers of protein language models to create a new pairwise embedding. We also add the relative positional encoding in the pairwise embedding.
Fig. 4.

Our deep network architecture for single-sequence-based protein structure prediction.
The sequence and pair embedding are updated iteratively in the Evoformer module consisting of 24 modified Evoformer layers. Our Evoformer differs from the original one in AlphaFold2 in that ours does not have the column self-attention layer, which is meaningless for an individual sequence.
The structure module is similar to that of AlphaFold2, which mainly consists of 8 IPA (invariant point attention) and transition layers with shared weights. Our structure module is different in that we use a linear layer to integrate the scalar, point, and pair attention values in the IPA model while AlphaFold2 uses only addition. The structure module outputs both the predicted atom 3D coordinates and confidence scores (i.e., pLDDT).
Training and Test Data.
Training data.
The training data consist of ~340 k proteins. Among them, there are 80,852 proteins of different sequences that have experimental structures released before January 2020 in PDB (denoted as BC100). We cluster the proteins in BC100 by 40% sequence identity and denote the clustering result as BC100By40. The remaining 264 k proteins have tertiary structures predicted by AlphaFold2 (denoted as distillation data). The proteins in the distillation dataset are extracted from Uniclust30_2018_08 (19) and share no more than 30% sequence identity. Given that the current experimental structures in the PDB database cover only a fraction of the vast sequence space, incorporating well-predicted structures as training data for distillation can enhance the generalization performance of the structure prediction model, which has been demonstrated by AlphaFold2 (3). During each training epoch, one protein is randomly sampled from each BC100By40 cluster to form a set of training proteins, with the acceptance rate determined by the sequence length (0.5 for lengths under 256, 0.5 to 1 for lengths between 256 and 512, and 1.0 for lengths over 512). In each epoch, proteins are also sampled from the distillation data by the ratio of 1:3 between BC100By40 and the distillation data.
Antibody data for fine-tuning.
In order to improve the performance on antibody targets, we construct an antibody training set for fine-tuning. Specifically, we use experimental structures from SAbDab (20) released before 2021/03/31 as training samples and split each target into chains. In total, the training set contains 5,033 heavy and light chains. The validation set consists of 178 antibody structures released between 2021/04/01 and 2021/06/30.
Training procedure.
The model was implemented using PyTorch (21), and its distributed training on multi-GPUs was based on PyTorch Lightning (22). We used the AdamW (23) optimizer with β1 = 0.9, β2 = 0.999, ε = 10 to 8, and weight decay of 0.0001 for all models. Over the first ~1,000 steps, the learning rate warmed up linearly from 1e-6 to 5e-4, remained at 5e-4 for the first 1/3 training steps, and then linearly decreased to 1e-6 for the remaining 2/3 training steps. The model is initially trained on protein crops of size 256 for the first 2/3 of the training steps and then trained on crop sizes of 384 for the remaining 1/3 of the training steps. The training losses include pairwise losses and structure losses. The pairwise losses are distogram distance and orientation loss, as employed in trRosetta (24). The structure losses include the Frame Aligned Point Error (3) loss with a clamp of 20 Å and the pLDDT loss. To improve the performance of the model, we also implemented the recycling strategy during training. The number of recycling iterations is randomly sampled from 0 to 3. Each model is trained on 32 GPUs with accumulated gradients 4, thus the batch size is 128.
In total, we have trained four models with 150 epochs by combining the protein language models in different ways. RaptorX-Single (1b), RaptorX-Single (1v), and RaptorX-Single (pt) make use of ESM-1b, ESM-1v, and ProtTrans, respectively, while RaptorX-Single makes use of the three protein language models together. In the fine-tuning stage for antibody, we fine-tuned all four models 50 epochs with the learning rate linearly decreasing from 2e-4 to 1e-5 and obtained the corresponding four models for antibody structure prediction: RaptorX-Single-Ab (1b), RaptorX-Single-Ab (1v), RaptorX-Single-Ab (pt), and RaptorX-Single-Ab.
Evaluation.
Benchmark datasets.
We evaluated our methods on three antibody datasets (SAbDab-Ab, IgFold-Ab, and Nanobody), one orphan protein dataset, and one single mutation effects dataset. It should be noted that there is no benchmark sequence present in the training dataset.
-
•
IgFold-Ab dataset: It includes 67 non-redundant antibodies from the IgFold work (25) that were released after 1 July 2021.
-
•
SAbDab-Ab dataset: It is a non-redundant dataset (sequence identity lower than 95%) derived from SAbDab (20), which has 202 antibodies released in the first 6 mo of 2022.
-
•
Nanobody dataset: It includes 60 nanobodies of different sequences derived from SAbDab released in the first 6 mo of 2022.
-
•
Orphan dataset: It consists of 11 proteins released between 01 January 2020, and 12 April 2022. The proteins in this set do not have any sequence homologs in BFD (3), MGnify (26), Uniref90 (18), and Uniclust30 (19). That is, they are not similar to any proteins used for training the protein language models and our deep learning models.
-
•
Rocklin dataset: The Rocklin dataset has 14 native and de novo designed proteins and their stability measures of 10,674 single mutations. The stability was evaluated using thermal and chemical denaturation. The source data is from Rocklin et al. (17) and here we use the data collected by Strokach et al. (27).
Performance metrics.
For antibodies, we evaluate the backbone rmsd of the framework (Fr) and CDR (CDR-1, CDR-2, and CDR-3) regions of the heavy and light chains separately. The CDR regions are classified using ANARCI (28), and the backbone rmsd is calculated by PyRosetta (29). For orphan targets, we evaluate the quality of the predicted structures by TMscore (30), global distance test–total score (GDT_TS), and global distance test–high accuracy (GHT_HA) (31). For the single mutation effect prediction, we calculate the PCC between the predicted structure changes and the stability data. The structure change is measured by the TMscore of predicted structures between the wild and mutant sequences.
Baseline methods.
We compared our methods with MSA-based AlphaFold2 (without templates) and three single-sequence methods [ESMFold (7), OmegaFold (8), and HelixFold-Single (11)]. For AlphaFold2, we search the Uniclust30 (19) as of August 2018, Uniref90 (18) as of January 2020, and Metaclust (26) as of December 2018 and BFD (3) using HHblits (32) and Jackhmmer (33) to build its MSA. Additionally, we also report the performance of AlphaFold2 using a single-sequence as input. While testing on the antibody datasets, we compare our method with antibody-specific methods DeepAb (34), IgFold (25), and EquiFold (35). EquiFold is a structure prediction method relying solely on protein primary sequence, which can be used to evaluate the effectiveness of protein language models in the context of structure prediction.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We are grateful to Dr. Lupeng Kong for collecting DMS data and valuable discussion.
Author contributions
X.J. and J.X. designed research; X.J., F.W., and X.L. performed research; X.J. and F.W. analyzed data; and X.J., F.W., and J.X. wrote the paper.
Competing interests
J.X. is on sabbatical and with MoleculeMind while working on this project. F.W. was an intern in MoleculeMind Ltd working on this project.
Footnotes
This article is a PNAS Direct Submission. S.O. is a guest editor invited by the Editorial Board.
Data, Materials, and Software Availability
The training and fine-tuning data were derived from PDB (https://www.rcsb.org/) (36) and SAbDab (https://opig.stats.ox.ac.uk/webapps/newsabdab/sabdab/) (37) as mentioned in main text. The source code and benchmark target lists are available at https://doi.org/10.5281/zenodo.10689900 (38).
Supporting Information
References
- 1.Wang S., Sun S., Li Z., Zhang R., Xu J., Accurate de novo prediction of protein contact map by ultra-deep learning model. PLOS Comput. Biol. 13, e1005324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu J., Distance-based protein folding powered by deep learning. Proc. Natl. Acad. Sci. U.S.A. 116, 16856–16865 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jumper J., et al. , Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akdel M., et al. , A structural biology community assessment of AlphaFold2 applications. Nat. Struct. Mol. Biol. 29, 1056–1067 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buel G. R., Walters K. J., Can AlphaFold2 predict the impact of missense mutations on structure? Nat. Struct. Mol. Biol. 29, 1–2 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu J., McPartlon M., Li J., Improved protein structure prediction by deep learning irrespective of co-evolution information. Nat. Mach. Intell. 3, 601–609 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin Z., et al. , Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023). [DOI] [PubMed] [Google Scholar]
- 8.Wu R., et al. , High-resolution de novo structure prediction from primary sequence. bioRxiv [Preprint] (2022). 10.1101/2022.07.21.500999 (Accessed 1 January 2024). [DOI]
- 9.Wang W., Peng Z., Yang J., Single-sequence protein structure prediction using supervised transformer protein language models. Nat. Comput. Sci. 2, 804–814 (2022). [DOI] [PubMed] [Google Scholar]
- 10.Chowdhury R., et al. , Single-sequence protein structure prediction using a language model and deep learning. Nat. Biotechnol. 40, 1617–1623 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fang X., et al. , A method for multiple-sequence-alignment-free protein structure prediction using a protein language model. Nat. Mach. Intell. 5, 1087–1096 (2023). [Google Scholar]
- 12.Rives A., et al. , Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. U.S.A. 118, e2016239118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meier J., et al. , Language models enable zero-shot prediction of the effects of mutations on protein function. bioRxiv [Preprint] (2021). 10.1101/2021.07.09.450648 (Accessed 1 January 2024). [DOI]
- 14.Elnaggar A., et al. , “ProtTrans: Towards cracking the language of life’s code through self-supervised deep learning and high performance computing” in IEEE Transactions on Pattern Analysis and Machine Intelligence (IEEE Computer Society, Washington DC, 2021). [Google Scholar]
- 15.Van Bockstaele F., Holz J.-B., Revets H., The development of nanobodies for therapeutic applications. Curr. Opin. Invest. Drugs 10, 1212–1224 (2009). [PubMed] [Google Scholar]
- 16.Pak M. A., et al. , Using AlphaFold to predict the impact of single mutations on protein stability and function. bioRxiv [Preprint] (2021). 10.1101/2021.09.19.460937 (Accessed 1 January 2024). [DOI] [PMC free article] [PubMed]
- 17.Rocklin G. J., et al. , Global analysis of protein folding using massively parallel design, synthesis, and testing. Science 357, 168–175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzek B. E., Wang Y., Huang H., McGarvey P. B., Wu C. H., UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mirdita M., et al. , Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 45, D170–D176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunbar J., et al. , SAbDab: The structural antibody database. Nucleic Acids Res. 42, D1140–D1146 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paszke A., et al. , PyTorch: An imperative style, high-performance deep learning library. arXiv [Preprint] (2019). 10.48550/arXiv.1912.01703 (Accessed 27 July 2022). [DOI]
- 22.Falcon W., The PyTorch lightning team (PyTorch Lightning, 2019). [Google Scholar]
- 23.Loshchilov I., Hutter F., Decoupled weight decay regularization. arXiv [Preprint] (2019). 10.48550/arXiv.1711.05101 (Accessed 27 July 2022). [DOI]
- 24.Yang J., et al. , Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. U.S.A. 117, 1496–1503 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruffolo J. A., Chu L.-S., Mahajan S. P., Gray J. J., Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies. bioRxiv [Preprint] (2022). 10.1101/2022.04.20.488972 (Accessed 1 January 2024). [DOI] [PMC free article] [PubMed]
- 26.Mitchell A. L., et al. , MGnify: The microbiome analysis resource in 2020. Nucleic Acids Res. 48, D570–D578 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strokach A., Lu T. Y., Kim P. M., ELASPIC2 (EL2): Combining contextualized language models and graph neural networks to predict effects of mutations. J. Mol. Biol. 433, 166810 (2021). [DOI] [PubMed] [Google Scholar]
- 28.Dunbar J., Deane C. M., ANARCI: Antigen receptor numbering and receptor classification. Bioinformatics 32, 298–300 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chaudhury S., Lyskov S., Gray J. J., PyRosetta: A script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics 26, 689–691 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y., Skolnick J., Scoring function for automated assessment of protein structure template quality. Proteins 57, 702–710 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Cozzetto D., Kryshtafovych A., Ceriani M., Tramontano A., Assessment of predictions in the model quality assessment category. Proteins 69, 175–183 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Remmert M., Biegert A., Hauser A., Söding J., HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 9, 173–175 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Johnson L. S., Eddy S. R., Portugaly E., Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 11, 431 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruffolo J. A., Sulam J., Gray J. J., Antibody structure prediction using interpretable deep learning. Patterns 3, 100406 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee J. H., et al. , EquiFold: Protein structure prediction with a novel coarse-grained structure representation. bioRxiv [Preprint] (2023). 10.1101/2022.10.07.511322 (Accessed 1 January 2024). [DOI]
- 36.wwPDB, Consortium, “Protein Data Bank: the single global archive for 3D macromolecular structure data”. Nucleic Acids Res. 47, 520–528 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneider C., Raybould M. I. J., Deane C. M., SAbDab in the Age of Biotherapeutics: Updates including SAbDab-Nano, the Nanobody Structure Tracker. Nucleic Acids Res. 50, D1368–D1372 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jing X., Wu F., Luo X., Xu J., Data for RaptorX-Single [Data set]. Zenodo. 10.5281/zenodo.10689900. Deposited 22 February 2024. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
The training and fine-tuning data were derived from PDB (https://www.rcsb.org/) (36) and SAbDab (https://opig.stats.ox.ac.uk/webapps/newsabdab/sabdab/) (37) as mentioned in main text. The source code and benchmark target lists are available at https://doi.org/10.5281/zenodo.10689900 (38).