

Summary
Background
Sarcomas represent an extensive group of malignant diseases affecting mesodermal tissues. Among sarcomas, the clinical management of chondrosarcomas remains a complex challenge, as high-grade tumours do not respond to current therapies. Mutations in the isocitrate dehydrogenase (IDH) 1 and 2 genes are among the most common mutations detected in chondrosarcomas and may represent a therapeutic opportunity. The presence of mutated IDH (mIDH) enzymes results in the accumulation of the oncometabolite 2-HG leading to molecular alterations that contribute to drive tumour growth.
Methods
We developed a personalized medicine strategy based on the targeted NGS/Sanger sequencing of sarcoma samples (n = 6) and the use of matched patient-derived cell lines as a drug-testing platform. The anti-tumour potential of IDH mutations found in two chondrosarcoma cases was analysed in vitro, in vivo and molecularly (transcriptomic and DNA methylation analyses).
Findings
We treated several chondrosarcoma models with specific mIDH1/2 inhibitors. Among these treatments, only the mIDH2 inhibitor enasidenib was able to decrease 2-HG levels and efficiently reduce the viability of mIDH2 chondrosarcoma cells. Importantly, oral administration of enasidenib in xenografted mice resulted in a complete abrogation of tumour growth. Enasidenib induced a profound remodelling of the transcriptomic landscape not associated to changes in the 5 mC methylation levels and its anti-tumour effects were associated with the repression of proliferative pathways such as those controlled by E2F factors.
Interpretation
Overall, this work provides preclinical evidence for the use of enasidenib to treat mIDH2 chondrosarcomas.
Funding
Supported by the Spanish Research Agency/FEDER (grants PID2022-142020OB-I00; PID2019-106666RB-I00), the ISC III/FEDER (PI20CIII/00020; DTS18CIII/00005; CB16/12/00390; CB06/07/1009; CB19/07/00057); the GEIS group (GEIS-62); and the PCTI (Asturias)/FEDER (IDI/2021/000027).
Keywords: Chondrosarcoma, Enasidenib, IDH2, Patient-derived models, Personalized medicine
Research in context.
Evidence before this study
Sarcomas represent an extensive group of malignant diseases affecting mesodermal tissues. The genomic nature of most sarcoma subtypes, displaying high inter- and intra-tumour heterogeneity with few recurrent driver mutations in a small portion of patients, makes these tumours especially indicated for personalized treatment approaches. For optimal development of these personalized protocols and a more efficient translation to the clinic, it is necessary to create patient-derived models suitable for testing the efficiency of candidate therapies. These strategies might be especially indicated for chondrosarcomas, a subtype of bone sarcoma that is inherently resistant to current therapies.
Added value of this study
To develop a personalized medicine strategy for sarcomas we have applied targeted sequencing protocols to detect druggable mutations in a collection of sarcomas cases with available patient-derived models. Among those potential druggable alterations detected in patient samples and avatar cell lines, we found IDH mutations in two chondrosarcomas. The presence of mutated IDH enzymes results in the accumulation of the oncometabolite 2-HG which contributes to driving tumour growth. In vitro and in vivo experiments evidenced the anti-tumour potential of the IDH mutant inhibitor enasidenib for the treatment of IDH2 mutant chondrosarcomas. Our transcriptomic and epigenomic analyses show that the mechanism of action of this drug is associated with the repression of proliferative pathways rather than with the promotion of tumour differentiation.
Implications of all the available evidence
This study suggests that enasidenib may represent an efficient therapeutic alternative for mutant IDH2 chondrosarcomas. This anti-proliferative mechanism of action of this drug may be especially relevant in dedifferentiated chondrosarcomas where reversal of this phenotype is not possible. In addition, this work provides support for the use of sarcoma patient-derived lines as avatar models capable of predicting (pre)-clinical responses in personalized medicine strategies.
Introduction
The rapid development of Next-Generation Sequencing (NSG) and its implementation in the analysis of molecular alterations in human disease foresee a paradigm shift in the way that patients with cancer will be treated in the near future.1 It is expected that oncological treatments will be based on the use of personalized strategies for each patient that combine the molecular information gained through NGS with functional drug screenings using patient-derived models to find the most efficient therapy for each case.2,3 These precision medicine approaches will especially benefit those types of tumours that show a high rate of inter- and intra-tumour heterogeneity. This is the case for most types of sarcomas, which are characterized by the presence of few recurrent driver mutations in a small portion of patients.4,5 In sarcomas, cytotoxic drugs (doxorubicin, ifosfamide, cisplatin, etc) and/or radiotherapy remain the cornerstone of systemic treatment and targeted therapies are restricted to the use of specific tyrosine kinase inhibitors in tumours harbouring mutations in KIT and/or PDGFRA or NTKR fusion genes.6
Among bone sarcomas, chondrosarcomas stand out as the subtype with fewer therapeutic options available. These types of cartilage-forming bone sarcomas are inherently resistant to conventional chemo and radiotherapy and nowadays there are no effective treatments available for metastatic or inoperable tumours.7, 8, 9 Most common genomic alterations described for chondrosarcoma include mutations in COL2A1 and TP53 in all subtypes, HEY1 rearrangement in mesenchymal chondrosarcoma, and specially isocitrate dehydrogenase 1 (IDH1) or IDH2 genes, which has been found in more than 50% of cases of central and dedifferentiated subtypes.10, 11, 12 Mutations in IDH1/2 genes are also common and play key roles in enchondromas, Ollier disease and Maffucci syndrome, which are benign lesions that may progress to chondrosarcoma.13,14 Also in support of a driver role of mutant IDH1/2 genes in the origin of chondrosarcomas, mutant IDH enzymes were reported to induce an epigenetic dysregulation of genes involved in stem cell-driven differentiation and lineage specification and the expression of mutant IDH2 in mesenchymal stem cells induced the formation of sarcomas in vivo.15,16 Besides chondrosarcomas, mutations in IDH genes are also common and play pro-tumour roles in gliomas (up to 80% of the cases),17 acute myeloid leukaemia (AML) (∼20%),18 and cholangiocarcinomas (∼20%).19
IDH enzymes catalyse the conversion of isocitrate to α-ketoglutarate (α-KG) in the tricarboxylic acid cycle. This enzymatic function is altered due to non-synonymous substitutions of arginine residues R132 in IDH1 and R172 or R140 in IDH2. The mutation of these critical amino acids of the catalytic domain results in the acquisition of a new enzymatic activity in which IDH mutant enzymes catalyse the conversion of α-KG into the oncometabolite d-2-hydroxyglutarate (D2HG).20 The pathological accumulation of D2HG results in the inhibition of α-KG-dependent dioxygenases such as methyl cytosine hydroxylases, and the histone demethylases of the TET family. Inhibition of these enzymes promotes DNA and histone hypermethylation, leading to a transcriptional dysregulation that results in the blockage of different cell differentiation pathways and the onset of oncogenic processes.9,21
In recent years, several inhibitors targeting IDH1/2 mutants have been developed.22,23 Some of these compounds have been demonstrated to be effective in pre-clinical studies, mainly in AML, gliomas and cholangiocarcinoma and are being tested in clinical trials.22, 23, 24 Among them, AG-120 (ivosidenib) and AG-221 (enasidenib), which are specific inhibitors against IDH1 and IDH2 mutants respectively, are first-in-class compounds and has been approved by FDA for the treatment of relapsed or refractory AML.25,26 IDH anti-mutant treatments may also represent a therapeutic alternative for chondrosarcomas, however, the effect of these compounds in this type of tumours has been barely explored. On one hand, different studies using IDH1 mutant inhibitors reached contradictory conclusions about the ability of this drug to suppress tumorigenic properties in chondrosarcoma cells.27, 28, 29 In the clinical setting, ivosidenib showed minimal toxicity and durable disease control in a small cohort of patients with chondrosarcoma enrolled in a phase I study.30 On the other hand, the effect of IDH2 mutant inhibitors in chondrosarcoma have not been preclinically tested so far and only a few patients with chondrosarcoma (n = 3) have been enrolled in a clinical trial (NCT02273739).
Here we aimed to assay a personalized medicine strategy for sarcomas based on the use of patient-derived cell lines as a drug-testing platform. Among those potential druggable alterations found in six sarcoma cases, we found IDH mutations in two chondrosarcomas. In vitro and in vivo experiments evidenced the anti-tumour potential of enasidenib for the treatment of IDH2 mutant chondrosarcomas.
Methods
Cell lines
CDS11 and CDS17 patient-derived lines and T-CDS17 xenograft lines were previously established and described.31 CDS23, SARC06, SARC20 and SYN01 lines were also established following the same methodology. L2975 cells (CVCL_D706) were described elsewhere.32 The SW1353 cell line (CVCL_0543) was purchased from the ATCC. The range of passes in which the different lines were used was: CDS11: 5–10; CDS17: 10–15; T-CDS17#1: 4–10; T-CDS17#3: 2–4; T-CDS17#4: 3–5; CDS23: 2–4; SARC06: 4–6; SARC20: 4–6; SYN01: 6–10; L2975: 46–49; SW1353: 28–32. An overview of patient and tumour characteristics associated with each cell line is given in Table S1. All the cell types were cultured as previously described.31,33 The identity of cell lines has been authenticated by Short Tandem Repeats analysis (Supplemental data). All cultures were regularly tested and found to be negative for mycoplasma contamination using the Biotools Mycoplasma Gel Detection kit (Cat#90.021-4542, B&M LABS, Madrid, Spain).
Drugs
Enasidenib (AG-221) was purchased from MedChemExpress (Cat#HY-18690, Monmouth Junction, NJ, USA) and ivosidenib (AG-120), vorasidenib (AG-881), AGI-5198, DS-1001b, metformin and CB-839 were acquired from Selleckchem (Cat#S8206, #S8611, #S7185, #S8881, #S1950 and #S7655, respectively, Houston, TX, USA). Stocks for in vitro experiments were prepared as 10 mM solutions in sterile DMSO, stored at −80 °C and diluted in culture medium to the final concentration just before use. For in vivo treatments, enasidenib was solubilized in distilled water containing 0.5% methylcellulose (Cat#M7027, Sigma–Aldrich, St Louis, MO, USA) and 0.2% Tween-80 (Cat#P8074, Sigma–Aldrich) before administration.
Next generation sequencing
Genomic DNA from matched samples of healthy tissue (blood), tumour tissue and tumour-derived cell lines was extracted using the QIAmp DNA Mini Kit (Cat#51306, Qiagen, Hilden, Germany). Subsequent targeted sequencing of 160 cancer-related genes was performed using a QIAseq Human Comprehensive Cancer Panel v2 (Cat#DHS-3501Z, Qiagen Inc, Valencia, CA) (Table S2). In addition, the panel includes more than 21 genes in which clinically actionable alterations have been described, as well as 29 genes known to predispose to cancer. Multiplex PCR was performed by amplifying 10 ng of DNA in each of 4 separate PCR reactions to generate the corresponding libraries for next-generation sequencing. All steps of library preparation were performed according to the manufacturer's protocol. Briefly, the amplicons were pooled, purified, labelled with a compatible sequencing adapter/index using NEBNext kit (Cat#E7370L, New England BioLabs, Evry, France) and finally purified again. The quality of the libraries was confirmed using the Agilent high-sensitivity DNA kit (Cat#5067-4626, Agilent Technologies, Santa Clara, CA, USA) to confirm the successful generation of 300-bp products for the GeneRead Human Comprehensive Cancer Panel. The individual libraries for each patient were quantified with Kapa Library Quantification Kit (Cat# KK4824, Kapa Biosystems, Manufacturing, R&D, Cape Town, South Africa).
For the Human Comprehensive Cancer Panel, up to 12 samples were pooled equimolar and paired-end reads 2 × 150bp sequencing on an Illumina NextSeq 500 instrument using the 300 cycle Mid Output v2.5 Reagent Kit (Cat#20024905, Illumina, San Diego, CA, USA) according to Illumina guidelines. Read mapping, variant discovery, and functional annotation were performed with the Qiagen GeneRead Variant Calling Pipeline. For the identification of somatic mutations, tumour and matched reference were compared between them using the Analyze GeneGlobe (https://geneglobe.qiagen.com/es/) software to detect single nucleotide variants (SNVs), short insertions and deletions (indels) and copy-number variants (CNVs). Variants categorized as non-synonymous, frameshift, stop gain or affect a conserved splice-site in any of the annotated transcripts associated with the cancer gene. The variants were classified as pathogenic or probably pathogenic. The resulting somatic variants with more than 100 reads coverage and a VAF above 5% were manually reviewed with the IGV browser (Integrative Genomics Viewer, Broad Institute, CA, USA) to detect possible sequencing artefacts, for example in poorly mapped regions, last base of amplicons or polynucleotide tracks. Variants that were observed in the control sample and suspected of being sequencing errors by visual inspection on the IGV browser were filtered-out.
The mutational status of IDH1, IDH2, SETD2, TSC1 and TP53 was confirmed by Sanger sequencing as described before.31 PCR reactions for Sanger sequencing were carried out using the primers detailed in Table S3.
RNA sequencing
RNA samples were extracted from triplicated cultures of control and enasidenib treated cell lines as previously described.34 cDNA libraries were prepared using the TruSeq RNA Library Preparation Kit (Cat# RS-122-2001, Illumina) and checked for quality and quantified using a TapeStation (Agilent Tecnologies). Paired-end sequencing was performed on an Illumina Novaseq 6000 (Illumina) using 100-base reads.
RNA-seq data preprocessing
All sequencing data files were evaluated using the fastQC software (v.0.11.9) for quality control. Adapters and poor-quality paired-end reads were removed using fastp (v.0.20.1) with the following options: --detect_adapter_for_pe --trim_poly_x --correction -r -M 10 -l 20.
Transcript-level quantification of RNA-seq data
Preprocessed reads were pseudo-aligned to the GRCh38 human genome and quantified at the transcript-level using the mapping-based mode of the Salmon package (v.1.3.0) with the following options: --validateMappings --gcBias. A Salmon index was previously generated from a gentrome file which was constructed by the gencode.v36 transcriptome and the GRCh38.p13 DNA primary assembly.
Differential gene expression analyses
Analyses of gene expression data were performed using R statistical software (v.4.0.2). Transcript-level information was summarized at the gene-level for both exploratory and differential analyses using the tximport package (v.1.16.1). For visualization purposes, the matrix of raw counts was rlog (regularized logarithm) transformed according to the DESeq2 package (v.1.28.1). Non-expressed genes (<2 counts across all conditions) were discarded to reduce the multiple testing penalty. Differential gene expression analyses between control and enasidenib treatment conditions were then performed for each chondrosarcoma cell line using the negative binomial generalized linear model fitting implemented in DESeq2. After adjusting p-values for multiple testing using the Benjamini-Hochberg method, differentially expressed genes (DEGs) were defined as those with FDR <0.05 and |log2 (FC)|>1 (FDR: false discovery rate, FC: fold change).
Gene annotation
The org.Hs.eg.db package (v.3.11.4) was used to annotate each Ensembl Stable Gene ID with their corresponding Entrez Gene ID and Gene Symbol categories.
Gene set enrichment analyses
Pathway enrichment analyses between control and enasidenib conditions were performed for each cell type using the fgsea package (v.1.14.0) on the gene sets from the Molecular Signatures Database (MSigDB), accessed via the msigdbr package (v.7.2.1). Gene rankings were provided to the fgsea algorithm using the log transformation of p-values: −log10 (p-values) x sign (log2FC).
Microarray analysis of chondrosarcoma samples
Microarray expression data (Affymetrix Human Gene 2.0 ST arrays) from Nicolle et al. (2019) were analysed to determine whether enasidenib treatment can alter transcriptome profiles associated with chondrosarcoma expression subtypes (E1/E2). To this end, E1/E2 signature genes shared between our RNA-seq experiments and microarray data were selected for further analyses. Next, their log expression values were normalized to the range [0, 1] independently for the microarray data and for each control–treatment pair of each cell line to reduce biases due to differences between biological systems and expression technologies. Finally, all samples were hierarchically clustered using Euclidean distances.
Over-representation analyses
For each chondrosarcoma cell line, enriched pathways in up-regulated (FDR <0.05, log2FC > 0) and down-regulated (FDR <0.05, log2FC <0) genes belonging to the E1/E2 expression signature were identified using the fora function of the fgsea package on the MSigDB GO BP gene sets.
Genome-wide DNA methylation analysis
Genomic DNA was extracted from triplicated cultures of control and enasidenib treated cell lines as described above. The EZ-96 DNA Methylation Kit (Cat#D5003, Zymo Research) was used to perform the bisulphite conversion. The processed DNA samples were then hybridised to Infinium MethylationEPIC BeadChips (Cat#20087709, Illumina, San Diego, CA, USA) following the Illumina Infinium HD Methylation Assay protocol.
Infinium MethylationEPIC data preprocessing
MethylationEPIC BeadChip data analyses were performed using the minfi package (v.1.32.0) in statistical software R (v.4.0.2). All samples validated the expected sex identity of the cell line, preserved the methylation landscape of SNP probes and passed the specific quality control for intensity signals in both methylated and unmethylated channels. After checking the quality of the samples, the ssNoob method was applied to correct background noise signals affecting methylation intensity values. β-values were calculated for each interrogated CpG site using the following formula: . Next, the beta-mixture quartile normalization (BMIQ) implemented in ChAMP (v.2.16.2) was selected to overcome the probe design bias of Illumina Infinium arrays. Finally, to avoid spurious methylation signals due to technical issues or unwanted biological sources of variation, probes were filtered according to the following exclusion criteria: (a) detection p-value >0.01 in any sample; (b) cross-reactive or multi-mapping nature, (c) sex chromosome location and (d) inclusion of SNPs with MAF ≥0.01 at their CpG or SBE sites (dbSNP v.147).
Differential methylation analyses
Linear regression models were built to detect differentially methylated probes (DMPs) using the limma package (v.3.44.3), with the methylation value as the dependent variable. To increase homoscedasticity in the models, β-values were logit-transformed to M-values using the beta2m function of the lumi package (v.2.40.0). Sex and study accession code were included as covariates in the linear modelling. Empirical Bayes-moderated t-tests were employed to define contrasts and the p-values were adjusted for multiple comparisons using the Benjamini-Hochberg method (FDR <0.05). In addition, filtering based on the absolute difference in beta values (|Δβ|) for each pairwise comparison (>20%) was applied to define functionally relevant DMPs. To detect differentially methylated regions (DMRs), the limma p-values from DMPs were inputted in the comb-p function of the Enmix package (v.1.28.2) using default parameters, discovering CpG sites that were spatially related at the level of statistical significance. DMRs were then selected under the following thresholds: (a) FDR <0.05, (b) Sidak-corrected p-value <0.05, (c) |Δβ|>20%, β: mean methylation value (region). In addition, DMRs displaying less than 66% of CpG sites with changes in the same direction (hyper: >5% gain; hypo: >5% loss; equal: ≤5% change) were filtered out, as were DMRs with less than 5 CpGs and those regions that were not annotated to any gene. Finally, only those genes whose DMRs changed in the same direction were retained as differentially methylated genes.
Probe annotation
The IlluminaHumanMethylationEPICanno.ilm10b4.hg19 package (v.0.6.0) was used to assign each probe to its CGI (CpG Island) and gene location status. For the annotation of regions, the probes belonging to each region were first individually annotated as described above. A single annotation was then assigned to each region according to the following criteria: (1) for CGI status, “Island”>“N_Shore”>“S_Shore”>“N_Shelf”>“S_Shelf”>“OpenSea”; and (2) for gene locations, “TSS1500”>“TSS200”>“5'UTR”>“1stExon”>“Body”>“ExonBnd”>“3'UTR”>“Intergenic”.
Pathway enrichment analyses
To interrogate the functionality of DMPs and DMRs, pathway enrichment analyses were performed using the gsameth and gsaregion functions of missMethyl package (v.1.22.0) [11] on the gene sets from the Molecular Signatures Database (MSigDB) [12], accessed via the msigdbr package (v.7.2.1). For both methods the number of probes mapping to each gene was taken into account as a bias factor for the enrichment analyses.
Integration of methylation array data from cartilage samples
Appropriate publicly available methylation datasets were considered to characterize the methylation landscape of healthy human cartilage samples (ArrayExpress accession codes: E-GEOD-63106, E-GEOD-73626). HumanMethylation450 BeadChip arrays from cartilage samples were integrated with MethylationEPIC BeadChip arrays to extract common CpGs between the two technologies (399,453 probes). All cartilage samples passed the quality controls applied to cell line samples, including validation of self-reported sex.
Cell viability assays
In cell survival assays, cells were plated in 24-well plates at cell densities of 1 × 104 or 5 × 103 cells per well, incubated overnight and treated for 72 or 144 h respectively with increasing concentration of different drugs. At the experimental end point, 18 μl/ml of WST-1 reagent was added to each well and the cells were incubated for 1 h at 37 °C. The colour change produced by the cleavage of the WST-1 reagent (Cat#11644807001, Roche, Mannheim, Germany) by mitochondrial dehydrogenases was measured by reading the absorbance at 440 nm with the use of a Synergy HT plate reader (BioTek, Winooski, VT, USA).35,36 IC50 for each treatment was determined by non-linear regression using GraphPad Prism 9.0.1 software (Graphpad Software Inc., San Diego, CA, USA). Alternatively, the cytotoxic potential of the assayed treatments was assayed in colony formation unit (CFU) assays as previously described.37 In these assays, cells were treated for 24 h and left to form colonies in drug-free medium for 10 days. The surviving fraction was determined by dividing the average number of colonies for each treatment by the average number of colonies in the control. The induction of apoptosis was assayed using the Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit (Cat#V13241, Life Technologies, San Francisco, CA, USA) following the manufacturer's instructions. All cell viability assays were performed in the presence of foetal bovine serum.
D-2-Hydroxygluturate measurement
D2HG levels were measured in adherent cultures of 1 × 107 chondrosarcoma cells treated with increasing concentrations of enasidenib for 48 h using the D2HG colorimetric Assay Kit (Cat#ab211070, Abcam, Cambridge, UK) following the manufacturer's recommendations.
Tumoursphere formation assay
Cells were plated at a density of 5000 cells per well in 6-well low-attachment plates (Cat#3736, Corning, NY, USA) and cultured in serum-free sphere medium containing Ham's-F12 (Cat# 10-080-CV, Gibco, CA, USA) supplemented with Glutamax (Cat#35050038, Gibco), B-27 Supplement (1:50; Cat#12587010, Gibco), Heparin (1:1000; Cat#H3149-10KU, Sigma–Aldrich, MO, USA), the growth factors human EGF (20 ng/ml; Cat# AF-100-15, PeproTech, London, UK) and human bFGF (10 ng/ml; Cat#AF-100-18B, PeproTech). Fresh aliquots of EGF and bFGF were added every three days. To analyse the effects of drugs, the tumourspheres formed after 10 days of culture were incubated in sphere medium containing different concentrations of the drugs for 96 h. After the treatments, tumourspheres were scored and cell viability was determined using the Cell Proliferation reagent WST-1 as described above.38,39
Immunofluorescence staining
The formation of DNA damage-induced γ-H2AX foci was analysed by Immunofluorescence staining using a mouse monoclonal anti-phospho-Histone H2A.X (Ser139, Sigma) (AB_477058) as described before.40 Quantification of dots per nuclei was performed in 100–120 randomly selected cells using Image J software (National Institute of Health, Bethesda, MD, USA).
Western blotting
Western blotting analyses were performed as previously described.41 Primary antibodies used in these analyses were: anti-H3K9me3 [Millipore (17–625), 1:1000 dilution] (AB_916348), H3K4me3 [abcam (ab8580), 1:1000] (AB_306649), H3K27me3 [abcam (ab6002), 1:1000] (AB_305237) and anti-β-Actin [Sigma Aldrich (A5441), 1:10,000] (AB_476744). The IRDye Infrared Fluorescent secondary antibodies anti-Rabbit and anti-Mouse IRDye 800CW and IRDye 680RD (AB_621843, AB_2721181, AB_2687825, AB_10956588, respectively, LI-COR Biosciences, Lincoln, NE, USA) were used for detection. Membranes were scanned with the Odyssey Fc Dual-Mode Imaging System (LI-COR Biosciences) using the red (700 nm) and green (800 nm) channels, and signal analysis was performed using Image Studio Lite software (LI-COR Biosciences).
Chondrogenic differentiation assay
To induce chondrogenic differentiation, 2.5 × 105 cells were placed in a 15 ml polypropylene tube (Corning, TX, USA) and centrifuged at 1.000 rpm for 3 min. The pellet was cultured in 500 μl of either standard culture medium or ready-to-use complete chondrogenic differentiation medium StemPro chondrogenesis differentiation kit (Cat#A1007101, Gibco-Thermo Fisher, Waltham, MA, USA) following the manufacturer's recommendations. The medium was carefully removed to avoid pellet resuspension and replaced every 3 days with or without drugs for 21 days. Then, pellets were washed in PBS, fixed at 4 °C with 4% formaldehyde and embedded in HistoGel (Cat#HG-4000-012, Epredia, Kalamazoo, MI, USA) firstly and in paraffin afterwards processed for histological analyses (H&E and PAS-alcian blue stainings) as described below. Slides were scanned with the digital scanner Motic EasyScan One (Motic, Xiamen, China) and the percentage of PAS-alcian positive areas relative to the total area was quantified using the ImageJ 2.1.0 software (National Institutes of Health, Bethesda, MD, USA).
Treatment of tumour xenografts
Athymic nude-Foxn1nu mice (Envigo, Barcelona, Spain) of 6 weeks old were inoculated subcutaneously (s.c.) with 5 × 105 cells mixed 1:1 with BD Matrigel Matrix High Concentration (Cat#354248, BD Biosciences, Erembodegem, Belgium) previously diluted 1:1 in culture medium. Once tumours reached approximately 100 mm3, the mice were randomly assigned by the researcher (n = 6 per group; n = 12 in total) to receive oral gavage doses of enasidenib (35 mg/kg per dose) or vehicle (0.5% methylcellulose and 0.2% Tween-80) twice a day (b.i.d.) for 21 days. Tumour size was measured using a calliper 3 times a week and tumour volume was determined using the equation (D × d2)/6 × 3.14, where D is the maximum diameter, and d is the minimum diameter. Primary outcome measure was the analysis of the effect of enasidenib on tumour growth. Relative tumour volume (RTV) for every xenograft was calculated as follows: RTV = tumour volume at the day of measurement (Vt)−tumour volume at the beginning of the treatment (Vo). End points was reached if tumour volumes exceeded 1000 mm3, severe weight loss were observed or when the animals reached the end of the study. Mice were sacrificed by cervical dislocation and tumours were extracted, fixed in 4% formaldehyde, and processed for histological analysis. This was a nonblinded study. No criteria were set for including or excluding animals and no animal were excluded from the study. Mice were treated and tumours were measured at the same time by one person. Mice cages were placed next to each other. Animal experimentation was performed in the Vivarium Unit of the University of Oviedo (Oviedo, Spain; https://www.sct.uniovi.es/unidades/experimentacion-animal/bioterio/presentacion).
Histological analysis
Xenograft tumour samples were fixed in 4% formaldehyde, embedded in paraffin and cut into 4-μm sections which were deparaffinized and rehydrated before further histological analyses.42,43 For PAS-Alcian Blue staining, slides were immersed in 1% alcian blue solution (Cat#1016470500, Sigma–Aldrich, MO, USA) (pH 2.5) for 30 min, sections were oxidized in 0.5% periodic acid (Cat#132320, ITW Reagents, MB, IT) solution for 5 min, washed in distilled water and placed in Schiff's reagent (Cat#109033, Sigma–Aldrich) for 15 min. After washing, sections were counterstained with Mayer's Hematoxylin (Cat#254766, ITW Reagents) for 1 min for nuclear visualization. For hematoxylin and eosin (H&E) staining, slides were immersed in Harris's hematoxylin (Cat#253949, ITW Reagents) solution for nuclear staining, followed by differentiation in acid alcohol and bluing in an alkaline solution. Afterward, counterstaining with eosin (Cat#256879, ITW Reagents) was conducted to visualize cytoplasmic components. Finally, slides were washed, dehydrated, and mounted using Entellan (Cat#107960, Sigma–Aldrich). Immunohistochemical analyses were performed in an automatic workstation (Dako Autostainer Plus) with anti-Ki67 (Clone MIB-1, Dako # JR626, Prediluted), using the Dako EnVision Flex + Visualization System (Cat# K8023, Dako Autostainer).41 Quantification of Ki67 staining was performed by counting the number of positive cells per 10 high-power fields (40×). To quantify PAS-alcian staining, slides were scanned with the digital scanner Motic EasyScan One (Motic, Xiamen, China) and the percentage of PAS-alcian positive areas relative to the total area was quantified using the ImageJ 2.1.0 software (National Institutes of Health, Bethesda, USA) in three random images (x200) per condition.
Prediction of protein conformation
Wild-type IDH1 and 2 structures were obtained in the AlphaFold Protein Structure Database (https://www.alphafold.ebi.ac.uk/). The in-silico mutation impact on the protein structure was determined via the Dynamut server (https://www.biosig.lab.uq.edu.au/dynamut/prediction).44 PDB files were visualized using UCSF ChimeraX program (https://www.cgl.ucsf.edu/chimerax/).
Statistics
Statistical analysis was performed using GraphPad Prism 9.0.1 software. Unless otherwise indicated, all data are presented as the mean (±standard deviation or SEM as indicated) of at least three independent experiments. A two-sided Student's t-test was performed to determine the statistical significance between groups. Multiple comparisons of the data were performed using one-way ANOVA and Tukey's test, p ≤ 0.05 values were considered statistically significant. Specific statistical tests and sample sizes for each experimental protocol are indicated in the relevant methods section and/or in the figure legends. These sections also indicate randomization and/or blinding procedures where applicable.
Ethics
All experimental protocols involving human samples were conducted following the institutional review board guidelines and in compliance with the WMA Declaration of Helsinki and were approved by the Institutional Ethics Committee of the Principado de Asturias (ref. 255/19). We collected samples from both male and female patients with sarcomas and the data were self-reported by study participants. All animal research protocols were prepared before the study, were approved by the Animal Research Ethical Committee of the University of Oviedo (ref. PROAE 34–2019) and were carried out in accordance with the institutional guidelines of the University of Oviedo.
Role of funders
The funders were not involved in the study design, data collection, data analysis, interpretation, writing of the manuscript or any other aspect pertinent to the study.
Results
Identification of targetable mutations in patients with sarcoma
We aimed to find actionable mutations in sarcoma cases with available matched patient-derived cell lines. In this analysis, we included tumours and patient-derived cell lines (CDS11 and CDS17) conventional chondrosarcoma (CDS11) and a dedifferentiated chondrosarcoma (CDS17), as well as several cells lines established from CDS17-developed xenografts (T-CDS17#1, #3, and #4), which have been previously characterized.31 In addition, we analysed two cases of undifferentiated pleomorphic sarcomas (SARC06 and SARC20), one synovial sarcoma (SYN01) and a secondary peripheral chondrosarcoma (CDS23) with newly developed cell lines (Table S1). First, we performed NGS targeted sequencing of 160 cancer-related genes (Table S2) in normal (non-tumoral) tissue, tumour samples and patient-derived models of SARC06, SAR20, SYN01 and CDS17 patients. The average nucleotide coverage in DNAseq studies was >2500 reads in all samples analysed (range 2488–5340 reads), and only the variants presenting more than 100 reads were selected for further analyses. Data from NGS analysis of the tumour and cell line samples were compared to that of normal tissue DNA to exclude germline alterations. To select those somatic mutations with potential driver roles in each case, we filter the results to select variants with non-synonymous effects on coded proteins which presented variant allele frequencies >0.30 in tumour and cell line samples and maximum allele frequencies <0.01 in population databases (dbSNP, ExAC, ESP, and 1000 Genomes) (Fig. 1a). Using these criteria, we found 3 somatic mutations that were shared by both tumour and cell line samples: SETD2 (p.E2089∗) and TP53 (p.V173L) in SARC06 and IDH2 (p.R172G) in CDS17/T-CDS17#1. In addition, we found mutations in TSC1 (p.SS331R) and TP53 (p.S215R) that appears de novo in the cell lines SARC20 and CDS17/T-CDS17#1 respectively, thus reflecting a certain level of genetic drift of tumour cells during their adaptation to in vitro growth conditions. Finally, we did not find relevant mutations in the SYN01 case. All detected mutations in NGS panels were confirmed by Sanger sequencing (Fig. 1b and Figure S1). In addition, Sanger sequencing analysis also confirmed the presence of the IDH2 (p.R172G) mutation in other cell lines (T-CDS17#3 and T-CDS17#4) derived from CDS17-generated xenografts (Fig. 1a and Figure S2). As previously seen, we also detected a heterozygous mutation in IDH1 (p.R132L) in CDS11 samples (Fig. 1a and b).31 Finally, we did not find IDH mutations in the CDS23 chondrosarcoma (Fig. 1a and Figure S2).
Fig. 1.

Mutations found in patient-derived sarcoma models. (a) Summary of somatic mutations found by NSG-panel and/or Sanger sequencing in tumour samples and/or cell lines derived from six patients with sarcoma. Variant allele frequency for each mutation detected by NSG sequencing is indicated. (b) Sanger sequencing validation of IDH mutations found in CDS11 and CDS17 models (Sanger validation of the rest of mutations is presented in Figs. S1 and S2). (c and d) In silico docking modelling of the conformational changes caused by the mutation in IDH1 (c) and IDH2 (d) in the substrate-binding pocket. Residues 132 in IDH1 and 172 in IDH2 are highlighted. [(nd): not determined; (−): not assayed].
Most common mutations reported in the R172 residue of IDH2 in chondrosarcoma include variants p.R172S (50%), p.R172G (30%), p.R172W (10%) and p.R172T (10%).45 On the other hand, reported variants in the R132 residue of IDH1 in chondrosarcomas include p.R132C (44%), p.R132G (17%) and p.R132L (11%) among others.45 As depicted in Fig. 1c and d, the R172 arginine in IDH2 and R132 in IDH1 are located in the isocitrate-binding pocket where these and other residues activated the catalytic conversion of isocitrate to α-KG.46 The presence of R172S/G/or W variants in IDH2 or R132C/G/or L variants in IDH1 induces conformational changes in isocitrate-binding pocket of these enzymes that may lead to the formation of D2HG (Fig. 1c and d).
Targeting IDH mutations in chondrosarcoma
Given the prominent role of IDH mutations in chondrosarcomas and the availability of drugs specifically directed against these mutations, we focused on studying the effect of targeting mutant IDH enzymes in the chondrosarcoma models. In these analyses, we also included two previously reported chondrosarcoma cell lines with IDH2 mutations: SW1353 (IDH2-R172S) and L2975 (IDH2-R172W)32 (Table S1). In 72-h dose–response cell survival assays, we found that the CDS17 cells and their associated xenograft-derived cell lines T-CDS17#1, #3 and #4, carrying the IDH2-R172G variant, were sensitive to low μM concentrations of IDH2mut inhibitor enasidenib (IC50 between 16.65 and 22.65 μM) (Fig. 2a and b). In these experiments SW1353 cells (IDH2-R172S; IC50 = 49.47 μM) displayed an intermediate sensitivity, whereas CDS11 (IDH1-R132L; IC50 = 63.53) and L2975 cells (IDH2-R172W; IC50 = 73.53 μM) were partially resistant to enasidenib. Finally, CDS23 cells (IDH-wt; IC50 > 100 μM) were fully resistant to this drug (Fig. 2a and b). A longer treatment period of 144 h confirmed that CDS17 models and SW1353 cells are sensitive to enasidenib, while L2975 is resistant to the anti-tumour activity of this inhibitor (Figure S3a and b). On the other hand, when treated with the IDH1mut specific inhibitors ivosidenib (Fig. 2c, Figure S3c), AGI-5198 (Figure S4a) or DS-1001b (Figure S4b) or with the dual inhibitor of both IDH1 and IDH2 mutants vorasidenib (AG-881) (Fig. 2d, Figure S3d), none of the assayed lines showed a significant response to these drugs.
Fig. 2.

Dose-dependent effect of several inhibitory compounds on chondrosarcoma cell lines. (a–f) Cell viability (WST-1 assays) measured after the treatment of wild-type and mutant IDH chondrosarcoma cell lines with increasing concentrations of enasidenib (a), ivosidenib (c), vorasidenib (d), CB-839 (e) and metformin (f) for 72 h. IDH mutation status of chondrosarcoma cell lines and IC550 values for enasidenib treatments are shown (b). Error bars represent the standard deviation of three independent biological replicates.
Further exploring the antiproliferative effects of enasidenib in IDH mutant models, we performed colony-forming assays with T-CDS17#1, SW1353 and L2975 IDH2 mutant cells following the treatment with increasing concentrations of enasidenib. Similar to our results in cell viability assays, we found that T-CDS17#1 and SW1353 lines were the most sensitive lines, whereas L2975 cells displayed a resistant phenotype (Fig. 3a and b). The cytotoxic effect of enasidenib was partially mediated by the induction of apoptosis, as evidenced by the time-dependent accumulation of the T-CDS17#4 cells staining positive for annexin V/propidium iodide after drug treatment (Figure S5a and b). In any case, the induction of apoptosis was not associated with the induction of DNA damage (Figure S5c and d).
Fig. 3.

Effect of enasidenib in IDH2 mutated chondrosarcoma cell lines. (a and b) Colony formation unit (CFU) assay of T-CDS17#1, SW1353 and L2975 cells treated with increasing concentrations of enasidenib for 24 h and left to form CFUs for 10 days. Representative pictures (a) and quantification (b) of CFU assays for each cell line are shown. (c–e) CSC-enriched tumourspheres of T-CDS17#4 cells were treated with increasing concentrations of enasidenib for 96 h. Representative pictures of the spheres cultures (c), quantification of the number of spheres (d) and cell viability (WST-1 assay) (e) of spheres at the end of the treatment are shown. Scale bar = 200 μm. (f) Intracellular D2HG levels were determined in T-CDS17#1 cells treated or not with the indicated concentrations of enasidenib for 48 h. Error bars represent the standard deviation of two biological replicates with technical triplicates for panel F and three biological replicates for the rest of the panels. Asterisks indicate statistically significant differences (∗∗: p < 0.01 one-way ANOVA).
Cell lines of the CDS17 model were able to grow as 3D tumourspheres with enhanced cancer stem cell (CSC)-related properties, as previously shown.31 To test whether enasidenib may also target these 3D models, we treated tumoursphere cultures of T-CDS17#1 cells with increasing concentrations of the inhibitor for 96 h. Following this treatment, tumourspheres displayed a disrupted and irregular morphology and their number and viability dramatically decreased in a dose-dependent fashion (Fig. 3c–e).
The anti-tumour activity of enasidenib in AML has been related to its ability to inhibit the production of 2DHG.26 In chondrosarcoma we found that enasidenib was able to significantly reduce the intracellular levels of this oncometabolite both in sensitive, T-CDS17#1 (Fig. 3f) and SW1353 (Figure S6a), and resistant, L2975 (Figure S6b), cell lines.
Another type of anti-IDHmut therapy tested in chondrosarcomas is based on the inhibition of pathways that lead to the synthesis of α-KG, the substrate that IDH1 and IDH2 mutant enzymes use to produce D2HG. Therefore, inhibitors of glutaminolysis such as metformin and CB-839 have been tested in different chondrosarcoma cell lines.47 In these experiments, only CB-839 was able to partially reduce cell viability in the IDH1 mutant cell line CDS11 but not in the CDS17/T-CDS17 IDH2 mutant models (Fig. 2e), while metformin treatment did not affect cell viability in any of these cell lines (Fig. 2f). CB-839, but not metformin, was able to reduce D2HG levels in T-CDS17#1 (IDH2mut) and CDS11 (IDH1mut) cells (Figure S6c and d). Therefore, the results showed that these inhibitors of glutaminolysis were ineffective to reduce the viability of IDHmut chondrosarcoma cell lines regardless of their ability to reduce D2HG levels.
DNA methylation and gene expression profiling of enasidenib-treated chondrosarcoma cells
The therapeutic activity of enasidenib and other IDH mutant inhibitors in AML and glioma was associated with the recovery of the D2HG-inhibited activity of αKG-dependent dioxygenases and the subsequent restoration of normal DNA methylation levels.24 Therefore, we analysed the effect of enasidenib in the methylome of chondrosarcoma cells. These analyses showed that T-CDS17#1 cells treated with enasidenib for either 48 h (Fig. 4a and b) or 120 h (Figure S7) did not show any noticeable differences in methylation values compared to control cells. Given this lack of effect, we performed an exploratory analysis to describe the global landscape of DNA methylation alterations in T-CDS17#1 cells treated or not with enasidenib for 48 h, along with healthy cartilage samples. As expected, T-CDS17#1 cells underwent a total remodelling of DNA methylation profiles in comparison with healthy cartilage (Fig. 4c). We performed differential analyses to identify genome-wide differentially methylated positions (DMPs, FDR <0.05, |Δβ|>20%) across the different groups (Fig. 4d). Using empirical Bayes moderated t-tests, we discovered abundant DNA methylation alterations between control T-CDS17#1 cells and cartilage samples (128,826 CON vs CART DMPs), and also when compared to enasidenib-treated cells (129,083 ENA vs CART DMPs). Despite this profound remodelling, we detected only 1 hypomethylated DMP between enasidenib-treated cells and their control counterparts. To verify the low impact of enasidenib treatment on DNA methylation, we evaluated the consistency between the DMPs identified in the CON vs CART and ENA vs CART comparisons, showing a high concordance (126,762 COMMON DMPs). Furthermore, the direction of DNA methylation alterations was well balanced in the comparisons CON vs CART (69,765 hypermethylated DMPs, 59,061 hypomethylated DMPs), ENA vs CART (69,419 hypermethylated DMPs, 59,664 hypomethylated DMPs) and COMMON (68,531 hypermethylated DMPs, 58,231 hypomethylated DMPs).
Fig. 4.

Effect of enasidenib on the methylome of IDH2-mutant chondrosarcoma cells. T-CDS-17#1 cells were treated in biological triplicates with DMSO (control; CON) or 20 μM enasidenib (ENA) for 48 h prior to be processed for DNA methylation analysis. (a) Scatter plot showing the PCA of CON and ENA samples according to the beta methylation values at the top 50,000 most variable CpG sites. (b) Heatmap diagram depicting the beta methylation values of CON and ENA samples at the top 5000 most variable CpG sites. No differentially methylated positions (FDR <0.05, |Δβ|>20%) between ENA and CON samples were found. (c–h) analysis of the methylation data of CON and ENA samples along with a public-available methylation datasets of healthy human cartilage samples (CART) (n = 39). (c) PCA of CON, ENA and cartilage samples according to the beta methylation values at the top 50,000 most variable CpG sites belonging to the HumanMethylation450K array. (d) Barplots depicting the number of hyper- and hypomethylated DMPs (FDR <0.05, |Δβ|>20%) found in CON vs CART, ENA vs CART and ENA vs CON comparisons. COMMON bar includes those hyper- and hypo- DMPs that are in both CON vs CART and ENA vs CART comparisons. (e) Barplots showing the number of significantly enriched pathways found using hyper- and hypomethylated common DMPs for different MSigDB gene sets. (f) Barplot depicting the number of hyper- and hypomethylated DMRs (FDR <0.05, |Δβ|>20%) found in gene regions using common DMPs. (g) On the left, a barplot showing the number of hyper- and hypomethylated genes. On the right, a heatmap plot showing the beta methylation values of the aforementioned differentially methylated genes. (h) Heatmap plot depicting the beta methylation values of those differentially methylated genes that belong to the GO BP chondrocyte differentiation pathway.
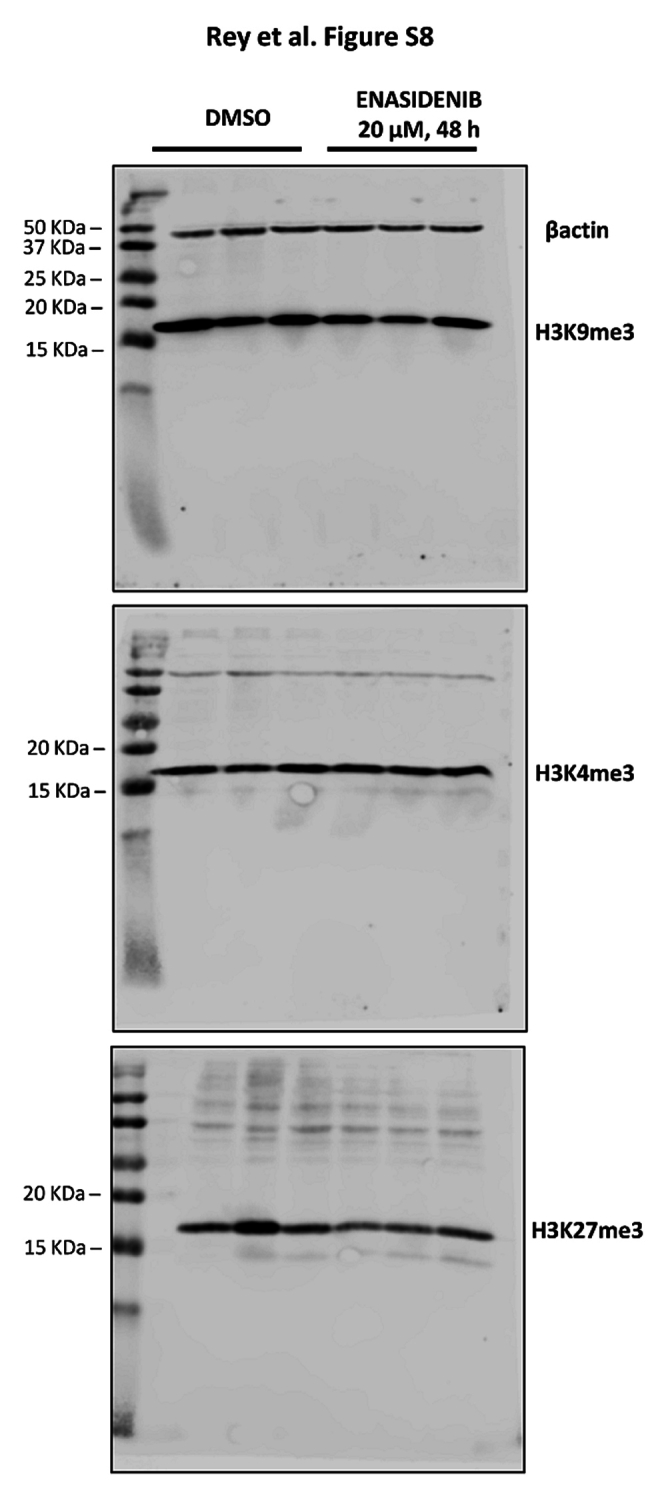
When we studied the functionality of the COMMON DMPs identified, we found that the majority of the enriched pathways were associated with hypermethylation alterations across the different molecular databases from the MSigDB (Fig. 4e). Consequently, although the number of methylation alterations is directionally balanced, only the methylation gain is functionally relevant to explain the epigenetic differences between healthy cartilage and T-CDS17#1 cells. Next, to further increase our power to detect functional DNA methylation alterations, we performed analysis at the regional level to define differentially methylated regions (DMRs, FDR <0.05, |Δβ|>20%). We then detected 2789 hypermethylated DMRs and 1009 hypomethylated DMRs (Fig. 4f), involving a total of 2367 hypermethylated and 741 hypomethylated genes (Fig. 4g). Therefore, in comparison with healthy cartilage, the IDH2 mutant T-CDS17#1 cell line showed a high remodelling of their DNA methylation landscape, with hypermethylated DMPs being more functionally relevant. Among the most significantly altered pathways between healthy cartilage and the T-CDS17#1 cell line, intense hypermethylation of genes involved in chondrocyte differentiation stands out (Fig. 4h). In any case, this hypermethylated status of genes was not affected by the enasidenib treatment of tumour cells (Fig. 4g and h). Furthermore, we have not detected either significant change in the levels of the tri-methylated forms of H3K4, H3K9 and H3K27 histones following the treatment of T-CDS17#1 cells with enasidenib (Figure S8).
In sharp contrast with this finding, RNAseq analyses showed that enasidenib induced a profound remodelling of the transcriptomic landscape in T-CDS17#1, SW1353 and L2975 IDH2 mutant chondrosarcoma cell lines (Fig. 5a). Therefore, we selected differentially expressed genes (DEG) (log2 fold change ≤−1 or ≥1 and FDR <0.01) in treated vs control samples. We found a higher proportion of downregulated vs. upregulated genes in all cell lines (Fig. 5b and Table S4). Among commonly regulated targets, there were 6 and 99 genes respectively upregulated and repressed by enasidenib in all cell lines (Fig. 5c, Table S5 and Table S6).
Fig. 5.

Effect of enasidenib on the transcriptome of IDH2-mutant chondrosarcoma cells. T-CDS-17#1, SW1353 and L2975 cells were treated in biological triplicates with DMSO (control) or 20 μM enasidenib for 48 h prior to be processed for RNA sequencing. (a) Principal component analysis of all samples according to rlog expression values in the top 1000 most variable genes between control and treatment conditions for each cell type. (b) Bar plot depicting the number of differentially expressed genes (DEGs, FDR <0.05 and |log2 (FC)|>1) that were up- or down-regulated in each cell type by enasidenib treatment. (c) Venn diagrams showing the intersections between DEGS upregulated (left panel) and downregulated (right panel) by enasidenib in T-CDS-17#1, SW1353 and L2975 cells. The list of genes commonly upregulated and downregulated in the different cell lines is presented in Tables S5 and S6 respectively. (d) Bubble plots (left panels) showing significantly enriched pathways (GSEA, FDR <0.05) from the MSigDB Hallmark collection in each enasidenib-treated cell type. Upset plot (right panel) depicting intersections of significantly enriched pathways (FDR <0.05) in T-CDS17, L2975 and SW1353 cell lines. Set size of pathways up downregulated in each cell line is showed as an inset. (e–h) Top panels: heatmap plots showing the expression values of those DEGs (FDR <0.05) of the Hallmarks TGFβ (e), mTORC1 (f), E2F targets (g) and G/M checkpoint (h) signalling pathways. Bottom panels: GSEA analysis of these signalling pathways in the indicated cell lines. Enrichment score (ES) and False discovery rate (FDR) values are indicated.
GSEA analysis of DEGs revealed that enasidenib induced a common pattern of regulation in those IDH2 mutant cell lines sensitive to the effect of enasidenib like T-CDS17#1 and SW1353 cells (Table S7). These cell lines shared 4 out of 6 pathways altered by enasidenib (Fig. 5d), including the downregulation of TGFβ signalling (nearly significant in SW1353, FDR = 0.055) (Fig. 5e), the upregulation of mTORC1 signalling (Fig. 5f) or the activation of unfold protein response signalling. On the other hand, the enasidenib-insensitive L2975 cell line responded very differently to the treatment with this inhibitor (Table S7). Thus, some of the pathways involved in the progression through the cell cycle and proliferation that were heavily downregulated in T-CDS17#1 cells, such as the activation of E2F downstream targets and G/2M checkpoint signalling, showed a significant upregulation in L2975 cells (Fig. 5d, g and h). Furthermore, unlike in enasidenib-sensitive lines, mTORC1 signalling and TGFβ signalling were not significantly modulated in L2975 cells.
These results indicate that enasidenib induced a different transcriptomic regulation in enasidenib-sensitive and -resistant IDH2 mutant chondrosarcoma models. Thus, the level of response to enasidenib seems to be associated to the different modulation of pathways involved in the regulation tumour growth, such as those controlled by mTORC1, TGFβ, and/or E2F.
Enasidenib treatment also induced profound deregulation of genes involved in the chondrogenic differentiation process. In this case all IDH2 mutant cell lines showed a similar trend of modulation (Fig. 6a). Some of these genes related with osteochondral differentiation, such as COL2A1, RUNX3, ACAN and/or MMP13, were downregulated, while other key factors involved in the terminal differentiation of chondrocytes such as SPP148 increased their expression following enasidenib treatment.
Fig. 6.

Effect of enasidenib treatment in the chondrogenic differentiation pathway and proliferative status. (a–c) Heatmap plots depicting the expression values of genes belonging to the GO BP chondrocyte differentiation pathway according to enasidenib treatment for each cell line. (b–c) Histological analysis of formalin-fixed paraffin-embedded T-CDS17#1 cell spheroids growth in chondrocyte differentiation medium with or without (control) 10 μM enasidenib for 21 days. Representative images of H&E and PAS-alcian staining (b) and quantification of PAS-alcian stain (c) are displayed. Scale bars = 100 μm. Error bars represent the standard deviation of three independent experiments. (d) Heatmap plot showing the expression values of those genes belonging to the mRNA expression signature developed by Nicolle et al. (2019), including their microarray experiments and the classification of chondrosarcoma samples into E1/E2 subtypes (n = 102). (e) Bubble plots showing the top 5 most significant pathways (ORA, FDR <0.05) from the MSigDB GO BP collection in each cell type for up-regulated (left) and down-regulated (right) E1/E2 genes.
To check whether these transcriptional enasidenib-induced changes resulted in a functional alteration of the chondrogenic differentiation potential of T-CDS17#1 cells, we performed a chondrogenic differentiation assay in which cell spheroids cultured in differentiation medium were exposed or not to a sub-lethal concentration of enasidenib for 21 days. Despite the transcriptional regulation induced by enasidenib, these experiments showed no differences in the chondrogenic differentiation capability of control and enasidenib-treated T-CDS17#1 cells (Fig. 6b and c).
A recent multi-omic analysis of a collection of chondrosarcomas has established a transcriptomic classification that identifies two tumour subtypes (E1 and E2) defined by a balance between chondrogenic tumour differentiation and cell cycle activation.49 The E1 phenotype is characterized by the overexpression of chondrogenic differentiation markers, while the E2 phenotype is characterized by the activation of proliferative pathways and greater chromosomal instability and is associated with a worse prognosis.49 The comparison of the transcriptome of the IDH2 mutant cell lines, treated or not with enasidenib, with those of the collection of chondrosarcomas described in this work, suggests that the treatment with enasidenib can reprogram the transcriptome of the enasidenib-sensitive models (T-CDS17#1 and SW1353) from a proliferative E2-like phenotype to a less aggressive E1-like one. On the other hand, the enasidenib-resistant cell line L2975 followed the opposite way (Fig. 6d). These results suggest that the different modulation of pathways associated to control of proliferation, such as E2F-mediated signalling, G2/M checkpoint signalling, TFGβ pathway and/or mTORC1 signalling may underlie the anti-tumour effect of enasidenib.
We performed over-representation analysis (ORA) to visualize the intensity of the reprograming of the different chondrosarcoma cell lines between subset of genes defining the E1 and E2 phenotypes (Fig. 6e, Tables S8 and S9). These analyses showed that a high percentage of genes downregulated by enasidenib in T-CDS17#1 and SW1353 cells belong to gene sets associated to cell cycle progression, such as chromosome segregation and mitotic nuclear division, while these gene sets were upregulated in the enasidenib-resistant L2975 cell line (Fig. 6e).
In vivo antitumour activity of enasidenib
Finally, we aimed to test the effectiveness of enasidenib in the in vivo setting. Therefore, we treated immunodeficient mice carrying T-CDS17#1 xenografts with vehicle or 35 mg/kg enasidenib b.i.d. for 21 days. A similar protocol was successfully used for treating in vivo models of AML.26 Enasidenib treatment completely inhibited tumour growth and even caused tumour regressions at the experimental end point (Fig. 7a). Likewise, tumour weights in the control series doubled those of the enasidenib-treated series at the end of the experiment (Fig. 7b, Figure S9). Notably, enasidenib treatment did not cause loss of weight (Fig. 7c) or other adverse effects. Histological examination showed that, in agreement with the in vitro assays, enasidenib treatment does not have a relevant impact on the differentiation status of tumours. Thus, both control and enasidenib-treated tumours displayed areas of chondrogenic differentiated (PAS-Alcian positive) and undifferentiated cells (Fig. 7d) with not significant changes induced by enasidenib (Fig. 7e, Figure S9). On the other hand, enasidenib-treated tumours exhibited a significantly lower proportion of cells staining positive for the proliferation marker ki67 (Fig. 7f and g, Figure S9).
Fig. 7.

In vivo effect of enasidenib. Established T-CDS17 xenografts were randomly assigned to two different groups (n = 6 per group) and treated b.i.d. with vehicle solvent (control) or 35 mg/kg enasidenib for 21 days. (a) Curves representing the mean relative tumour volume of T-CDS17 xenografts during the treatments. (b) Average tumour weight at the end of the experiment. (c) Change in the body weights of mice during the treatments. (d–g) Histological analysis of formalin-fixed paraffin-embedded tumours extracted at the end of the treatment. (d) Representative images of PAS-alcian staining of control and treated tumours. Two different areas of the tissue are magnified. Scale bars = 300 μm for left panels and 30 μm for magnified right panels. (e) Quantification of the percentage of PAS-alcian stained areas. (f) immuno-staining detection of Ki67. Scale bars = 100 μm (g) Quantification of Ki67 positive cells. A summary of the experimental variables associated to each mouse and the correlation between them is showed in Figure S9. Error bars represent the SD and asterisks indicate statistically significant differences between groups in a two-tailed unpaired t-test (∗: p < 0.05; ns: not significant).
Altogether, these results suggest that enasidenib may represent an efficient therapeutic alternative for the case CDS17 and possibly for other chondrosarcomas carrying R172G and R172S variants in the IDH2 gene.
Discussion
In this work, we have applied NGS protocols to detect targetable mutations in a collection of sarcomas with available patient-derived models. In these analyses, we have detected the oncogenic IDH2-R172G and IDH1-R132L variants in the CDS17 and CDS11 chondrosarcoma models, respectively. These patient-derived cell lines constitute the only models reported to date with these pro-tumour variants in chondrosarcoma. Following this personalized medicine approach, we focus on the specific targeting of IDH mutations in the corresponding patient-derived avatars. First, we analysed the effect of treating chondrosarcoma preclinical models with IDH2 mutant inhibitors. We found that the CDS17/T-CDS17 model carrying the R172G-IDH2 variant and the SW1353 cell line mutated in R172S-IDH2 were much more sensitive to antiproliferative effects of enasidenib than the rest of the cell lines with other mutations in IDH1/2 or without mutation in these genes. Besides, in vivo treatment with enasidenib of T-CDS17 xenograft models efficiently inhibited tumour growth at non-toxic concentrations. In accordance with the anti-tumour effects produced by enasidenib, we observed that this inhibitor drastically decreased the intracellular levels of D2HG, as previously reported in patients with AML treated with enasidenib.26,50 In any case, the reduction of D2HG levels was also observed in IDH2 mutant L2975 cells which did not respond to enasidenib treatment, suggesting that the inhibition of D2HG production is not sufficient to trigger an efficient anti-tumour activity in IDH2 mutant chondrosarcoma.
In contrast to the positive effects of enasidenib, we did not observe any relevant antiproliferative effect when treating our models either with the dual mIDH1/IDH2 inhibitor vorasidenib or with inhibitors of glutaminolysis. A previous study showed that SW1353 and L2975 mIDH2 cell lines were respectively highly and moderately sensitive to the in vitro treatment with the glutaminolysis inhibitor CB-83947. The fact that, unlike the study by Pertese et al., we performed the CB-839 treatments in the presence of serum and using different models of chondrosarcoma may contribute to the different results observed in both studies.
Regarding the specific targeting of mIDH1, previous studies have yielded contradictory results about the anti-tumour potential of mIDH1 inhibitors in chondrosarcomas. On the one hand, one study reported that the mIDH1 inhibitor DS-1001b was capable of inhibiting the in vitro and in vivo proliferation of chondrosarcomas with these mutations.28 Likewise, other study conducted with the AGI-5198 inhibitor reported in vitro anti-tumour activity of this drug in IDH1 mutant chondrosarcoma cell lines.27 On the other hand, a third study performed in a collection of wild-type and IDH mutant chondrosarcoma cell lines concluded that, despite being able to efficiently decrease D2HG levels, the AGI-5198 inhibitor did not affect the viability, proliferation, migration or methylation status of chondrosarcoma cells.29 in any case, Li et al. only observed a cytotoxic effect at the highest concentration assessed which has not been tested by Suijker et al. In line with this last study, we did not observe any effect of ivosidenib on the viability of our panel of chondrosarcoma cell lines, independently of the IDH1 mutation status.
The most described mechanism of action for inhibitors of mutant IDH enzymes in AML and glioma involves the recovery of the D2HG-inhibited activities of α-KG-dependent dioxygenases and the consequent restoration of normal levels of DNA and histones methylation. This reversal of the hypermethylated phenotype would lead to an increase in differentiation and therefore a decrease in tumour aggressiveness.23,24 However, it has also been reported that not all epigenetic effects caused by IDH mutants are reversible.24,51 In addition, clinical responses of patients with AML were not always associated with a complete reversal of mutant IDH-induced DNA hypermethylation.24,52 In line with these non-canonical cases, we have not found changes in the methylation levels of the CDS17 chondrosarcoma models after treatment with enasidenib. The relevance of IDH mutant-induced epigenetic changes in chondrosarcoma has been revealed in previous studies showing a global hypermethylated status of IDH mutant cases.49,53 Although there could be technical biases arising from the comparison of epigenetic features in tissue samples and cultured cell lines, our methylome analysis of normal cartilage samples and chondrosarcoma cell lines also showed a high rate of hypermethylated genes in IDH2 mutated cells. Moreover, recent work with IDH mutant enchondroma and chondrosarcoma samples indicates that epigenetic remodelling plays an important role in chondrosarcoma progression, although no vulnerabilities specifically associated with IDH mutations could be identified in an epigenetic compound screen.54 Altogether, it can be speculated that the intense epigenetic modulation induced by the mutation in our model of dedifferentiated chondrosarcoma has reached a state that cannot be reversed by lowering D2HG levels. In line with this hypothesis, our in vitro and in vivo functional studies showed that enasidenib treatment did not result in increased chondrogenic differentiation, and therefore the observed anti-tumour effects must be mediated by alternative mechanisms. In this sense, the mIDH1 inhibitor DS-1001b was able to induce chondrogenic differentiation in a model of conventional chondrosarcoma but not in a dedifferentiated chondrosarcoma line. Instead, the antitumour mechanism in this second model was associated with the induction of cell cycle arrest.28
Our transcriptomic studies support the involvement of pathways related to the control of cell differentiation and cell fate within the mechanism of action of enasidenib. Among them, signalling mediated by TGFβ and BMPs are among the most strongly repressed by the inhibitor in sensitive cell lines. In chondrosarcoma, these signalling pathways have been reported to be functionally active and are involved in tumour progression and regulation of the dedifferentiated phenotype in high-grade tumours.55 We can speculate that the TGFβ/BMP pathway was overactivated in IDH mutant chondrosarcoma cell lines and the treatment with enasidenib inhibited this signalling as part of its anti-tumour mechanisms. In addition, our transcriptome analysis provides clues indicating that the anti-tumour effect of enasidenib highly relies on the block of proliferation-related pathways, such as E2F-mediated signalling and G2/M checkpoint signalling. On the other hand, the resistant phenotype could be associated to the activation, rather than inhibition, of these proliferative pathways. In sum, we found a different transcriptomic regulation in enasidenib-sensitive (T-CDS17#1 and SW1353) and -resistant (L2975) IDH2 mutant chondrosarcoma models. Given that these three models carry different point mutations in the IDH2 gene, we may speculate that the type of mutation may influence the molecular changes triggered by enasidenib, with the chondrosarcoma models carrying the R172G or R172S variants those presenting a better anti-tumour response.
The comparison of the transcriptomic profiles of our IDH2 mutant chondrosarcoma cells lines with those from a multiomic study of a collection of chondrosarcomas49 supported the role of the inhibition of proliferative pathways as a relevant mechanism of action of enasidenib. Thus, the transcriptome of the enasidenib-sensitive T-CDS-17#1 and SW1353 cells corresponded to a proliferative profile associated with a worse prognosis (E2 profile), and the treatment with enasidenib was able to reprogram their transcriptomic landscape towards a phenotype with less aggressive characteristics and better prognosis (E1 profile). In contrast, the enasidenib-resistant cell line L2975 followed the opposite way. Considering the genes of the E1/E2 signatures, we can hypothesize that the inhibitor produces a negative regulation of those genes linked to the cell cycle, proliferation and division, which would explain the general repression of E2F-controlled targets in enasidenib-sensitive models and the opposite trend in enasidenib-resistant cells. Relevant to this finding, a recent study found that mutant IDH-induced methylation profiles in dedifferentiated chondrosarcomas were associated with upregulated expression of genes involved in G2/M checkpoints and E2F targets.53 Therefore, we can speculate that this blockade of cell growth pathways induced by enasidenib is at the base of its anti-tumour effect. This hypothesis is supported by our in vivo experiments, in which we did not observe cell differentiation in tumours, but we did observe tumour regression associated with a decrease in cell proliferation. Finally, for in vivo experiments we have tested a dose that has been successfully used in preclinical models of AML to support the initiation of clinical trials and the subsequent approval of the drug for the treatment of this disease.26 Therefore, the fact that this concentration was also effective and safe in the chondrosarcoma models assayed in this work, supports future clinical testing in patients with chondrosarcoma.
In summary, this study provides preclinical evidence of the effectiveness of targeted inhibition of IDH2 mutants in chondrosarcoma by enasidenib treatment. Our data suggest that the mechanism of action of this drug is associated with the repression of proliferative pathways and not with the promotion of tumour differentiation. This anti-proliferative mechanism of action may be especially relevant in dedifferentiated chondrosarcomas where reversal of this phenotype is not possible. In any case, future research should be designed to define reliable biomarkers capable of identifying those patients who are suitable to benefit from this treatment. Finally, this work provides support for the use of bone sarcoma patient-derived lines as avatar models capable of predicting (pre)-clinical responses in personalized medicine strategies.
Contributors
Verónica Rey: investigation; methodology; data curation; formal analysis; writing—original draft. Juan Tornín: investigation; methodology; data curation; formal analysis. Juan Jose Alba-Linares: data curation; software; formal analysis; methodology; writing—original draft. Cristina Robledo: investigation; methodology; formal analysis. Dzohara Murillo, Aida Rodríguez, Borja Gallego, Carmen Huergo and Cristinta Viera: investigation; methodology. Alejandro Braña: resources; methodology. Aurora Astudillo: pathological analysis, methodology. Dominique Heymann, Judith Bovée and Karoly Szuhai: resources; writing—review and editing. Agustín Fernández and Mario F. Fraga: data curation; software; formal analysis; writing—review and editing. Javier Alonso: methodology; formal analysis, funding acquisition, writing—review and editing. René Rodríguez: Conceptualization; resources; data curation; formal analysis; supervision; funding acquisition; project administration; writing—original draft; writing—review and editing. Verónica Rey and René Rodríguez verified the underlying data. All authors read and approved the submission of the final manuscript.
Data sharing statement
All relevant aggregate data supporting the findings of this study are available within the article and its supplementary information files. Raw data files obtained from RNA seq and DNA methylation analysis have been deposited at the GEO-NCBI repository with SuperSeries reference GSE235701 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE235701), which includes subseries GSE235605, GSE235697, GSE261421 and GSE261434. Other unprocessed data are available from the corresponding author on reasonable request.
Declaration of interests
The authors declare that they have no conflict of interest.
Acknowledgements
We acknowledge Daniela Corte-Torres from the Principado de Asturias BioBank (PT20/00161 and PT23/00077) for her support with histological analysis. We also thank Isaac Tamargo from the Fundación para la Investigación y la Innovación Biosanitaria del Principado de Asturias (FINBA) for his assistance with the protein conformation analyses. This work was supported by the Agencia Estatal de Investigación [AEI-MICINN/Fondo Europeo de Desarrollo Regional (FEDER) (grants PID2022-142020OB-I00 and PID2019-106666RB-I00 to R.R.)], the ISC III/FEDER (grants PI20CIII/00020, DTS18CIII/00005 to J.A.; Consorcio CIBERONC - CB16/12/00390 and Consorcio CIBERER–CB06/07/1009 and CB19/07/00057)]; the Spanish Group for Research on Sarcomas (GEIS) (grant GEIS-62 to R.R.); and the Plan de Ciencia Tecnología e Innovación del Principado de Asturias (PCTI)/FEDER (grant IDI/2021/000027 to R.R.). BG and DM were supported by Severo Ochoa predoctoral fellowships (Plan de Ciencia Tecnología e Innovación del Principado de Asturias/FEDER; grants BP- 20-046 and BP-21–084 respectively). CH was supported by a predoctoral fellowship from the Fundación Científca de la Asociación Española Contra el Cáncer (AECC). Finally, we would like to acknowledge the support from the following Patient Associations: Asociación GALBAN-Niños con Cancer (grant 2022 to J.T. and R.R.), Asociación Pablo Ugarte (grants TRPV205/18 and DGDO 195/22 to J.A.), Asociación Candela Riera, Asociación Todos Somos Iván, and Fundación Sonrisa de Alex (grants TVP333-19, TVP-1324/15 to J.A).
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.ebiom.2024.105090.
Appendix A. Supplementary data
Figure S1.

Figure S2.

Figure S3.

Figure S4.

Figure S5.

Figure S6.

Figure S7.

Figure S8.

Figure S9.

Western blots - Fig S8.
References
- 1.Salgado R., Moore H., Martens J.W.M., et al. Steps forward for cancer precision medicine. Nat Rev Drug Discov. 2018;17(1):1–2. doi: 10.1038/nrd.2017.218. [DOI] [PubMed] [Google Scholar]
- 2.Lee J.K., Liu Z., Sa J.K., et al. Pharmacogenomic landscape of patient-derived tumor cells informs precision oncology therapy. Nat Genet. 2018;50(10):1399–1411. doi: 10.1038/s41588-018-0209-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sayles L.C., Breese M.R., Koehne A.L., et al. Genome-informed targeted therapy for osteosarcoma. Cancer Discov. 2019;9(1):46–63. doi: 10.1158/2159-8290.CD-17-1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network Electronic address edsc, cancer genome atlas research N. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell. 2017;171(4):950–965.e928. doi: 10.1016/j.cell.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kovac M., Blattmann C., Ribi S., et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat Commun. 2015;6:8940. doi: 10.1038/ncomms9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grunewald T.G., Alonso M., Avnet S., et al. Sarcoma treatment in the era of molecular medicine. EMBO Mol Med. 2020;12(11) doi: 10.15252/emmm.201911131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boehme K.A., Schleicher S.B., Traub F., Rolauffs B. Chondrosarcoma: a rare misfortune in aging human cartilage? The role of stem and progenitor cells in proliferation, malignant degeneration and therapeutic resistance. Int J Mol Sci. 2018;19(1) doi: 10.3390/ijms19010311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Menendez S.T., Gallego B., Murillo D., Rodriguez A., Rodriguez R. Cancer stem cells as a source of drug resistance in bone sarcomas. J Clin Med. 2021;10(12) doi: 10.3390/jcm10122621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Speetjens F.M., de Jong Y., Gelderblom H., Bovee J.V. Molecular oncogenesis of chondrosarcoma: impact for targeted treatment. Curr Opin Oncol. 2016;28(4):314–322. doi: 10.1097/CCO.0000000000000300. [DOI] [PubMed] [Google Scholar]
- 10.Amary M.F., Bacsi K., Maggiani F., et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011;224(3):334–343. doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- 11.Amary M.F., Ye H., Forbes G., et al. Isocitrate dehydrogenase 1 mutations (IDH1) and p16/CDKN2A copy number change in conventional chondrosarcomas. Virchows Arch. 2015;466(2):217–222. doi: 10.1007/s00428-014-1685-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tarpey P.S., Behjati S., Cooke S.L., et al. Frequent mutation of the major cartilage collagen gene COL2A1 in chondrosarcoma. Nat Genet. 2013;45(8):923–926. doi: 10.1038/ng.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amary M.F., Damato S., Halai D., et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat Genet. 2011;43(12):1262–1265. doi: 10.1038/ng.994. [DOI] [PubMed] [Google Scholar]
- 14.Pansuriya T.C., van Eijk R., d'Adamo P., et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat Genet. 2011;43(12):1256–1261. doi: 10.1038/ng.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin Y., Elalaf H., Watanabe M., et al. Mutant IDH1 dysregulates the differentiation of mesenchymal stem cells in association with gene-specific histone modifications to cartilage- and bone-related genes. PLoS One. 2015;10(7) doi: 10.1371/journal.pone.0131998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu C., Venneti S., Akalin A., et al. Induction of sarcomas by mutant IDH2. Genes Dev. 2013;27(18):1986–1998. doi: 10.1101/gad.226753.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan H., Parsons D.W., Jin G., et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andersson A.K., Miller D.W., Lynch J.A., et al. IDH1 and IDH2 mutations in pediatric acute leukemia. Leukemia. 2011;25(10):1570–1577. doi: 10.1038/leu.2011.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kipp B.R., Voss J.S., Kerr S.E., et al. Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum Pathol. 2012;43(10):1552–1558. doi: 10.1016/j.humpath.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 20.Ward P.S., Patel J., Wise D.R., et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;173:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu W., Yang H., Liu Y., et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dang L., Yen K., Attar E.C. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann Oncol. 2016;27(4):599–608. doi: 10.1093/annonc/mdw013. [DOI] [PubMed] [Google Scholar]
- 23.Golub D., Iyengar N., Dogra S., et al. Mutant isocitrate dehydrogenase inhibitors as targeted cancer therapeutics. Front Oncol. 2019;9:417. doi: 10.3389/fonc.2019.00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Molenaar R.J., Wilmink J.W. IDH1/2 mutations in cancer stem cells and their implications for differentiation therapy. J Histochem Cytochem. 2022;70(1):83–97. doi: 10.1369/00221554211062499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DiNardo C.D., Stein E.M., de Botton S., et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386–2398. doi: 10.1056/NEJMoa1716984. [DOI] [PubMed] [Google Scholar]
- 26.Yen K., Travins J., Wang F., et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 2017;7(5):478–493. doi: 10.1158/2159-8290.CD-16-1034. [DOI] [PubMed] [Google Scholar]
- 27.Li L., Paz A.C., Wilky B.A., et al. Treatment with a small molecule mutant IDH1 inhibitor suppresses tumorigenic activity and decreases production of the oncometabolite 2-hydroxyglutarate in human chondrosarcoma cells. PLoS One. 2015;10(9) doi: 10.1371/journal.pone.0133813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakagawa M., Nakatani F., Matsunaga H., et al. Selective inhibition of mutant IDH1 by DS-1001b ameliorates aberrant histone modifications and impairs tumor activity in chondrosarcoma. Oncogene. 2019;38(42):6835–6849. doi: 10.1038/s41388-019-0929-9. [DOI] [PubMed] [Google Scholar]
- 29.Suijker J., Oosting J., Koornneef A., et al. Inhibition of mutant IDH1 decreases D-2-HG levels without affecting tumorigenic properties of chondrosarcoma cell lines. Oncotarget. 2015;6(14):12505–12519. doi: 10.18632/oncotarget.3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tap W.D., Villalobos V.M., Cote G.M., et al. Phase I study of the mutant IDH1 inhibitor ivosidenib: safety and clinical activity in patients with advanced chondrosarcoma. J Clin Oncol. 2020;38(15):1693–1701. doi: 10.1200/JCO.19.02492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rey V., Menendez S.T., Estupinan O., et al. New chondrosarcoma cell lines with preserved stem cell properties to study the genomic drift during in vitro/in vivo growth. J Clin Med. 2019;8(4) doi: 10.3390/jcm8040455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Oosterwijk J.G., de Jong D., van Ruler M.A., et al. Three new chondrosarcoma cell lines: one grade III conventional central chondrosarcoma and two dedifferentiated chondrosarcomas of bone. BMC Cancer. 2012;12:375. doi: 10.1186/1471-2407-12-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Estupinan O., Santos L., Rodriguez A., et al. The multikinase inhibitor EC-70124 synergistically increased the antitumor activity of doxorubicin in sarcomas. Int J Cancer. 2019;145(1):254–266. doi: 10.1002/ijc.32081. [DOI] [PubMed] [Google Scholar]
- 34.Menendez S.T., Rey V., Martinez-Cruzado L., et al. SOX2 expression and transcriptional activity identifies a subpopulation of cancer stem cells in sarcoma with prognostic implications. Cancers (Basel) 2020;12(4) doi: 10.3390/cancers12040964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Estupiñán Ó., Niza E., Bravo I., et al. Mithramycin delivery systems to develop effective therapies in sarcomas. J Nanobiotechnol. 2021;19(1):267. doi: 10.1186/s12951-021-01008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Estupiñán Ó., Rendueles C., Suárez P., et al. Nano-encapsulation of mithramycin in transfersomes and polymeric micelles for the treatment of sarcomas. J Clin Med. 2021;10(7):1358. doi: 10.3390/jcm10071358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Estupinan O., Rey V., Tornin J., et al. Abrogation of stemness in osteosarcoma by the mithramycin analog EC-8042 is mediated by its ability to inhibit NOTCH-1 signaling. Biomed Pharmacother. 2023;162 doi: 10.1016/j.biopha.2023.114627. [DOI] [PubMed] [Google Scholar]
- 38.Martinez-Cruzado L., Tornin J., Rodriguez A., et al. Trabectedin and campthotecin synergistically eliminate cancer stem cells in cell-of-origin sarcoma models. Neoplasia. 2017;19(6):460–470. doi: 10.1016/j.neo.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tornin J., Martinez-Cruzado L., Santos L., et al. Inhibition of SP1 by the mithramycin analog EC-8042 efficiently targets tumor initiating cells in sarcoma. Oncotarget. 2016;7(21):30935–30950. doi: 10.18632/oncotarget.8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gallego B., Murillo D., Rey V., et al. Addressing doxorubicin resistance in bone sarcomas using novel drug-resistant models. Int J Mol Sci. 2022;23(12) doi: 10.3390/ijms23126425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hermida-Prado F., Villaronga M.Á., Granda-Díaz R., et al. The SRC inhibitor dasatinib induces stem cell-like properties in head and neck cancer cells that are effectively counteracted by the mithralog EC-8042. J Clin Med. 2019;8(8):1157. doi: 10.3390/jcm8081157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rubio R., Abarrategi A., Garcia-Castro J., et al. Bone environment is essential for osteosarcoma development from transformed mesenchymal stem cells. Stem Cell. 2014;32(5):1136–1148. doi: 10.1002/stem.1647. [DOI] [PubMed] [Google Scholar]
- 43.Tornin J., Hermida-Prado F., Padda R.S., et al. FUS-CHOP promotes invasion in myxoid liposarcoma through a SRC/FAK/RHO/ROCK-Dependent pathway. Neoplasia. 2018;20(1):44–56. doi: 10.1016/j.neo.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodrigues C.H., Pires D.E., Ascher D.B. DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018;46(W1):W350–W355. doi: 10.1093/nar/gky300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dogan S., Frosina D., Geronimo J.A., et al. Molecular epidemiology of IDH2 hotspot mutations in cancer and immunohistochemical detection of R172K, R172G, and R172M variants. Hum Pathol. 2020;106:45–53. doi: 10.1016/j.humpath.2020.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Waitkus M.S., Diplas B.H., Yan H. Isocitrate dehydrogenase mutations in gliomas. Neuro Oncol. 2016;18(1):16–26. doi: 10.1093/neuonc/nov136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peterse E.F.P., Niessen B., Addie R.D., et al. Targeting glutaminolysis in chondrosarcoma in context of the IDH1/2 mutation. Br J Cancer. 2018;118(8):1074–1083. doi: 10.1038/s41416-018-0050-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Komori T. Runx2, an inducer of osteoblast and chondrocyte differentiation. Histochem Cell Biol. 2018;149(4):313–323. doi: 10.1007/s00418-018-1640-6. [DOI] [PubMed] [Google Scholar]
- 49.Nicolle R., Ayadi M., Gomez-Brouchet A., et al. Integrated molecular characterization of chondrosarcoma reveals critical determinants of disease progression. Nat Commun. 2019;10(1):4622. doi: 10.1038/s41467-019-12525-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stein E.M., DiNardo C.D., Pollyea D.A., et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722–731. doi: 10.1182/blood-2017-04-779405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rohle D., Popovici-Muller J., Palaskas N., et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340(6132):626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang F., Morita K., DiNardo C.D., et al. Leukemia stemness and co-occurring mutations drive resistance to IDH inhibitors in acute myeloid leukemia. Nat Commun. 2021;12(1):2607. doi: 10.1038/s41467-021-22874-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dermawan J.K.T., Nafa K., Mohanty A., et al. Distinct IDH1/2-associated methylation profile and enrichment of TP53 and TERT mutations distinguish dedifferentiated chondrosarcoma from conventional chondrosarcoma. Cancer Res Commun. 2023;33:431–443. doi: 10.1158/2767-9764.CRC-22-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Venneker S., Kruisselbrink A.B., Baranski Z., et al. Beyond the influence of IDH mutations: exploring epigenetic vulnerabilities in chondrosarcoma. Cancers (Basel) 2020;12(12):3589. doi: 10.3390/cancers12123589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boeuf S., Bovee J.V., Lehner B., et al. BMP and TGFbeta pathways in human central chondrosarcoma: enhanced endoglin and Smad 1 signaling in high grade tumors. BMC Cancer. 2012;12:488. doi: 10.1186/1471-2407-12-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.