

Abstract
Primary effusion lymphoma (PEL) is a rare, aggressive B cell non-Hodgkin’s lymphoma of the body cavities with malignant effusions. The prognosis is poor, and no optimal treatment has been established. CD38 is a type II transmembrane glycoprotein known to overexpress in multiple myeloma (MM). Daratumumab (DARA), a human CD38-targeting monoclonal antibody (mAb), is approved for MM treatment. In this study, we found expression of CD38 on PEL cells and assessed the anti-PEL activity of DARA. We found that both KHYG-1 and N6 (CD16-transfected KHYG-1) NK cell lines showed direct killing activity against PEL cells with induction of CD107a, and NK-mediated cytotoxicity by N6NK (CD16+) cells increased with DARA treatment. We confirmed direct NK activity and antibody-dependent cell cytotoxicity (ADCC) by expanded NK cells, indicating that DARA has high ADCC activity. We elucidated the antibody-dependent cell phagocytosis (ADCP) by using human monocyte-derived macrophages (MDMs) and mouse peritoneal macrophages. DARA also showed potent complement-dependent cytolysis (CDC) toward PEL. DARA also induced PEL cell death in the presence of a cross-linking antibody. Moreover, treatment with DARA inhibited tumor growth in a PEL xenograft mouse model. These results provide preclinical evidence that Ab targeting of CD38 could be an effective therapeutic strategy for the treatment of PEL.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00262-021-03054-8.
Keywords: Primary effusion lymphoma, CD38, Daratumumab, ADCC, CDC, ADCP
Introduction
Primary effusion lymphoma (PEL) is a rare and aggressive B cell lymphoma. This HIV-related non-Hodgkin’s lymphoma (NHL) accounts for approximately 4% of all HIV-associated NHL [1–4]. PEL has a very poor prognosis [5]. Treatment with cyclophosphamide, doxorubicin, vincristine, and prednisolone (CHOP) chemotherapy is the classical regimen for PEL. However, PEL is resistant to standard chemotherapy, and the median survival time is less than six months [1, 2, 4, 6]. PEL patients with more than one affected body cavity have a median overall survival of four months, compared with 18 months in patients with only one affected body cavity [7]. A large multicenter study of 28 patients reported a survival time of 6.2 months and a one-year overall survival rate of 39.3% [8]. Moreover, clinical trials to evaluate treatments for PEL are lacking because of its rarity [4].
Monoclonal antibody immunotherapy is a promising treatment in the area of B cell non-Hodgkin lymphoma, specifically employing anti-CD20 Ab such as rituximab [9–11], and obinutuzumab has become a revolutionary therapeutic mAb for B cell lymphomas [12]. However, most PEL cases are negative for CD20 expression [4]. One study found rituximab to be an effective treatment in rare cases of CD20- expressing PEL [13]. Brentuximab vedotin (SGN-35), an anti-CD30-drug conjugated agent, has been shown to prolong survival in a PEL xenograft mouse model [14]. Nevertheless, there is currently no effective immunotherapeutic option available for PEL.
The human anti-CD38 IgG1 kappa monoclonal antibody daratumumab (DARA) has demonstrated therapeutic activity in multiple myeloma (MM) [15–17]. The US Food and Drug Administration (FDA) approved DARA for use in combination treatments for MM in November 2015 [18]. DARA demonstrated strong anti-tumor activities in preclinical models. DARA exhibits diverse mechanisms of action, including an increase of cytolytic function of NK cells via antibody-dependent cell cytotoxicity (ADCC), complement-dependent cytolysis (CDC) [15], antibody-dependent phagocytosis (ADCP), and induction of cell death. At least three anti-CD38 antibodies have been designed to achieve therapeutic efficacy against MM. The other two, isatuximab [19] and MOR202 [20], have also been tested for the treatment of relapsed/refractory MM. While DARA was the first anti-CD38 Ab to be approved by the FDA for MM treatment, FDA also approved isatuximab in combination with pomalidomide and dexamethasone for the treatment of MM in March 2020 [21].
CD38 is a 46 kDa type II multifunctional transmembrane glycoprotein that has receptor as well as enzymatic functions. CD38 comprises a long 256 amino acid extracellular and transmembrane domain, and a short 20 amino acid intracellular domain [22–24]. CD38 serves as a receptor for its ligand, CD31 [25]. Engagement of ligand-receptor triggers activation of the intracellular signaling pathway and leads to cellular responses such as cell activation [26] proliferation and migration [27, 28]. The ectoenzymatic activities of CD38 contribute to intracellular calcium mobilization [29]. The expression of CD38 is broadly positive on hematopoietic cells, including plasma cells (PCs), natural killer (NK) cells, lymphoid cells, and myeloid cells [30]. CD38 shows especially broad and high expression levels in plasma cell tumors such as multiple myeloma (MM) [31–33]. CD38 targeting of antibodies is a significant activity in MM. Studies using other CD38-positive hematological malignancies such as non-Hodgkin’s lymphoma (NHL) [34], acute myeloid leukemia (AML) [35, 36], T cell-acute lymphoblastic leukemia (T-ALL) [37], and chronic lymphocytic leukemia (CLL) [38] demonstrate positive outcomes.
In this study, we found that CD38 was highly expressed on PEL cell lines. We aimed to test the effect of DARA in terms of ADCC, ADCP, and CDC toward PEL. Our results reveal that DARA has potential as a therapeutic mAb against CD38-expressing PEL cells, mediating ADCC, CDC, ADCP and cell death induction by cross-linking. Our results provide significant and noteworthy preclinical data supporting DARA for the treatment of PEL.
Materials and methods
Cells cultures and media
Primary effusion lymphoma (PEL) cell lines, BCBL-1 (AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, Bethesda, MD), BC-1, BC-2, BC-3, BCP-1 (ATCC, Rockville, MD), RMP-1 (JRCB cell bank, Osaka, Japan), GTO [39] and TY-1 (kindly provided by Dr. H Katano, NIID, Tokyo, Japan), and K562 (RIKEN cell bank, Tsukuba, Japan) were cultured in RPMI-1640 (Fujifilm Wako, Osaka, Japan) supplemented with 10% FBS, penicillin (100 IU/ml), and streptomycin (100 U/ml) at 37 °C in a humidified atmosphere containing 5% CO2 and 95% air. The CD16-negative human NK cell line, KHYG-1, was obtained from JCRB cell bank (Osaka, Japan). N6 cell line, KHYG-1 transduced human CD16 (V158 variant of FCGR3A), was kindly provided by Prof. Shuzo Matsushita (Kumamoto University, Japan). Recombinant Jurkat T cells expressing firefly luciferase gene under the control of NFAT response elements with constitutive expression of human FcγRIIIa, high affinity (V158) variant and Fcγ chain (ADCC-Jurkat) (#60,541, BPS Bioscience, San Diego, CA) were used to measure ADCC activity by reporter assay.
Peripheral blood mononuclear cells (PBMC) were separated using Pancoll Human (PAN Biotech, Geschäftszeiten, Germany) from blood donated by healthy donors with written informed consent. Study protocols were approved by Kumamoto University ethics committee in accordance with the Declaration of Helsinki (RINRI 548).
Antibodies and reagents
Daratumumab (DARA, humanized anti-human CD38 antibody: Darzalex) was purchased from Janssen Pharmaceutical KK Japan (Tokyo, Japan). Anti-human CD38 (HIT2)-APC, CD56 (HCD56)-APC and BV421, CD107a (H4A3)-APC, CD16 (3G8)-BV421, CD11b-APC, CD3 (UCHT1)-FITC were from BioLegend (San Diego, CA). FcR Blocking reagent was from Miltenyi Biotec (Bergisch Gladbach, Germany). ChromPure Human IgG, whole molecule (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) was used as the IgG control.
Determination of CD38 expression
PEL cells were stained with anti-human CD38-APC and analyzed with FACS Celesta (BD Bioscience, San Jose, CA). The collected data were analyzed with FlowJo Version 10 software (Tree Star, San Jose, CA).
NK cells isolation and expansion
The method of NK cell isolation and expansion was modified from our previous report [40]. Briefly, PBMC were isolated using Pancoll, and negative selection was performed using the NK Cell Isolation Kit (Miltenyi Biotec) by Magnetics Activated Cells Sorting (MACS) technique according to the manufacturer's instructions. The purity of NK cells (CD3−CD56+) was higher than 95%. NK cells were then cultured in 10%FCS RPMI1640 media with IL-2 (200 U/ml) and IL-15 (100 U/ml) at 37 °C, 5% CO2. The MHC class I negative K562 cells were added into the culture on days 3 and 7 at an E:T ratio of 1:10 [41]. Fresh growth medium was added to the NK cell co-culturing dish during NK cell expansion.
Antibody-dependent cell cytotoxicity (ADCC)
The ADCC reporter bioassay was performed according to the manufacturer’s instructions [42]. Briefly, Target PEL cells (5 × 104 cells) were incubated with either DARA or human IgG1 for 15 min at room temperature as described [38, 43]. ADCC-Jurkat cells (1 × 104 cells) were then added to each well and incubated at 37 °C, 5% CO2 for 18 h following by cell lysis and the addition of luciferin substrate. Finally, the luciferase signal was detected by a luminometer (Lumat3, Berthold Technologies, Bad Wildbad, Germany).
ADCC by calcein-AM releasing assay was conducted [15, 38]. PEL cells were labeled with 1 μM calcein-AM (Dojindo, Japan) for 15 min at 37 °C. Next, calcein-AM-labeling PEL was washed twice with growth media and plated at 1 × 104 cells per well in 96 well round bottom plates. The calcein-AM-labeling PEL was pre-incubated with DARA or isotype IgG1 control for 15 min. 1% Triton X-100 was used as a determination of the maximal calcein release. Furthermore, NK cells were added as indicated E:T ratio in each experimental design. The reaction plate was then incubated for 4 h at 37 °C. Afterward, the plate was centrifuged, supernatant was transferred into black plate (Costar, USA). The fluorescence signal was measured by the plate reader (excitation filter:485 ± 20 nm; band-pass filter 530 ± 20 nm). The percentage of cellular cytotoxicity was calculated using the following formula:
Cytotoxicity of PEL cells by ADCC was measured using flow cytometry [44]. Briefly, target cells were labeled with a carboxyfluorescein succinimidyl ester (CFSE) (Invitrogen, Carlsbad, CA) and incubated with either DARA or human IgG1 for 15 min at room temperature. CFSE-labeled target cells were co-cultured with effector NK cells for 4 h at 37 °C, 5% CO2. The cells were then stained with Ghost Dye 780 (Tonbo Biosciences, San Diego, CA) to determine dead cells. The percentage of target cell death was determined by the amount of double positive staining of both CFSE and Ghost Dye 780 with FACS Celesta and analyzed on FlowJo software version 10.4 (Tree Star, San Carlos, CA).
Detection of CD107a, a functional marker of NK cells
Target cells were incubated with either DARA or human IgG1 for 15 min at room temperature. Target cells were co-cultured with effector NK cells for 4 h at 37 °C, 5% CO2. The cells were then stained with BV421 conjugated anti-CD56 antibody (BioLegend) and APC conjugated anti-CD107a antibody (BioLegend). The percentages of CD56+CD107a+ cells were detected with FACS Celesta. To enhance the signal strength of CD107a, we counter stained cells with APC conjugated anti-CD107a antibody (BioLegend) during co-culturing in some experiments as indicated in figure legends.
Complement-dependent cytotoxicity (CDC)
CDC against PEL cells was assessed with flow cytometry by measuring the percentage of propidium iodide (PI)-positive cells, as previously described [45]. Standard rabbit complement (Pooled normal rabbit serum, Cedarlane corp., Burlington, Ontario, Canada) was used as a source of complement. Heat-inactivated rabbit complement (56 °C for 30 min) was used as a negative control. PEL cells were incubated with DARA for 15 min at room temperature. The complement was then added to the reaction and incubated for 2 h at 37 °C, 5% CO2. Propidium iodide (PI, Invitrogen) solution was added at a final concentration of 2 μg/ml, and PI positive cells were detected with FACS Celesta.
Preparation of human and mouse macrophages
Primary human monocyte-derived macrophages (MDMs) were prepared as previously described [46]. Briefly, PBMCs were incubated for 1 h in 10 cm tissue culture dishes (Corning Incorporated, Corning, NY) at 37 °C, 5% CO2 in 1% FCS RPMI1640, and the non-attached cells were removed by gentle washing. The adherent monocytes were further cultured in 10% FCS RPMI1640 for 7 days in the presence of 100 ng/ml recombinant human M-CSF (Morinaga Milk Co., Tokyo, Japan).
Mouse peritoneal macrophages were isolated from 10-week-old BALB/c Rag-2−/− Jak3−/− (BRJ) mice [47] that had been injected intraperitoneally with 2 ml of 4% (w/v) thioglycollate medium (BD Diagnostic Systems, Sparks, MD) three days prior to peritoneal lavage with 10 ml PBS [46]. The percentage of mouse CD11b positive cells was more than 95%.
Antibody-dependent cell phagocytosis (ADCP) assay
The phagocytic assay was performed as previously described [48]. Briefly, macrophages were plated in a glass-bottomed dish (Greiner Bio-One, Frickenhausen, Germany) overnight. PEL cells were labeled with CFSE according to the manufacturer’s protocol. CFSE-labeled BCBL-1 cells were incubated with either DARA or human IgG1 for 15 min at room temperature, added to the wells of macrophages (E: T ratio = 1:5), and incubated for 2 h at 37 °C, 5% CO2. Wells were gently washed five times with PBS to remove non-phagocytosed cells. The nuclei of the adherent cells were stained with Hoechst 33342 (Dojindo, Kumamoto, Japan). Images were taken with a BZ-8100 Biozero fluorescent microscope (Keyence, Osaka, Japan). Data are presented as the phagocytic index calculated as the number of phagocytosed CFSE+Hoechst 33342+ cells per 100 macrophages.
The phagocytic activity was measured by flow cytometry as previously described [46]. Macrophages were plated into 24-well tissue culture plates in 10% FCS containing RPMI1640 media and incubated for 24 h. CFSE-labeled BCBL-1 cells were incubated with either DARA or human IgG1 for 15 min at room temperature. The antibody coated CFSE-labeled BCBL-1 cells were added to the wells of macrophages (E:T ratio = 1:2) and further incubated for 2 h at 37 °C, 5% CO2. The cells were harvested from each well using trypsin/EDTA. Cells were then stained with anti-CD11b-APC and analyzed with FACS Celesta. Phagocytosed BCBL-1 cells were defined as CD11b+CFSE+ cells.
CD38 cross-linking assay in vitro
The cross-linking assay was modified from a previous study [49]. Briefly, PEL cells were seeded at 5 × 104 cells per well in 48-well plates and pre-incubated for 15 min with varying concentrations of DARA followed by overnight incubation in the presence of either 25 μg/ml of rabbit anti-human IgG F(ab’)2 fragments or goat anti-mouse IgG (H + L). The morphologic changes were visualized using a light microscope. MTT assay was done to detect viability of cells.
Tetrazolium dye methylthiotetrazole (MTT) assay
The viability of PEL cells was measured by the MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) assay (Sigma-Aldrich, St. Louis, MO), as previously described [50]. Briefly, PEL cells were seeded at 3 × 104 cells per well in 96-well plates and pre-incubated for 15 min with varying concentrations of DARA followed by 24 h of incubation in the presence of either 25 μg/ml of rabbit anti-human IgG F(ab’)2 fragments or goat anti-mouse IgG (H + L) in a final volume of 100 μl. Subsequently, MTT solution was added to each well at a final concentration of 0.5 mg/ml. After 4 h of additional incubation, 100 μl of 0.04 N HCl/isopropanol was added to dissolve the formazan crystals. Finally, dual absorbance was detected at 595/630 nm using a plate reader (iMark; Bio-Rad Laboratories Inc., Hercules, CA). Percentage cell viabilities compared with controls were calculated using the following formula:
PEL xenograft mouse model
BRJ mice [47] were housed and monitored at the Center for Animal Resources and Development (CARD), Kumamoto University, according to institutional guidelines. Water and food were provided ad libitum. All experiments were approved by Kumamoto University animal ethics committee (A2019-041). BCBL-1 cells (1 × 107 cells per mouse) were subcutaneously injected into both flanks of 10-to-12-week-old female BRJ mice (6 mice per group). Three days after cell transplantation, DARA (100 μg/mouse) was intraperitoneally injected into the mice twice a week. Body weights were monitored, and the mice were sacrificed to remove and measure the tumor weights.
Statistical analysis
Data are presented as mean values ± standard errors (SE) or standard deviation (SD). For statistical analysis, the data were analyzed by one-way analysis of variance (ANOVA) with the Turkey’s multiple-comparison test or Student's paired t test using GraphPad Prism Ver. 6 (GraphPad Software, San Diego, CA) as indicated in the figure legends. P values less than 0.05 were taken to indicate significance.
Results
Expression of surface CD38 on PEL cells
We analyzed the surface expression of CD38 on PEL cells to evaluate the potential of CD38 as a therapeutic molecular target. As shown in Fig. 1, all PEL cell lines (BCBL-1, BC-1, BC-2, BC-3, BCP-1, GTO, RM-P1, and TY-1 cells) expressed CD38 on their surfaces. Collectively, the results suggest that DARA could be an effective therapeutic mAb against PEL.
Fig. 1.

CD38 expression on PEL cell lines. Expression of CD38 on PEL cell lines was examined by flow cytometry. The MFI values are indicated at the upper left corner
DARA-mediated ADCC activity toward PEL cell lines
We evaluated the ADCC activity of DARA by using an indicator: ADCC recombinant Jurkat cells expressing firefly luciferase gene under the control of NFAT responsive elements with constitutive expression of human FcγIIIa, high affinity (V158) variant and Fcγ chain. To assess ADCC activity, we varied the concentration of DARA from 0.01–10,000 ng/ml. The results demonstrated dose-dependent ADCC activity in BCBL-1, GTO, and TY-1 cell lines (Fig. 2A).
Fig. 2.

DARA-mediated ADCC based on CD16 expressing NK cells. A BCBL-1, GTO, and TY-1 cells were incubated with various concentrations of DARA, and co-cultured with ADCC-indicating Jurkat cells (E:T ratio = 1:5) for 18 h. Luciferase activity was measured. B, C, D, E) CFSE-labeled BCBL-1 cells were treated with IgG1 or DARA, co-cultured with CD16 + NK (N6) or CD16- NK (KHYG-1) cell lines at different E:T ratio for 4 h, and analyzed for dead BCBL-1 (CFSE+Ghost Dye+) cells by flow cytometry. F Up-regulation of CD107a after DARA treatment was analyzed by flow cytometry. G Percentages of CD56+CD107a+ cells after stimulation with DARA (n = 3). Data are presented as mean values ± standard error (SE). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 significant levels
A CD16-negative human NK cell line, KHYG-1 [51], and V158 variant CD16 expressing KHYG-1 cell line, N6 [52], were used to detect ADCC activity of DARA. The KHYG-1 cell lines did not express CD16, whereas N6 NK cell lines showed positive CD16 expression (Supplement Fig. 2A). The killing activities of KHYG-1 and N6 NK cell lines were tested by co-culturing with K562 cells at different E:T ratios. The results showed that both CD16− NK and CD16+ NK cell lines demonstrated killing activities toward K562 cells. KHYG-1 cells showed a greater level of killing activity (Supplementary Fig. 2B and 2C).
To test the ADCC activity of NK cell lines toward PEL cells, we selected BCBL-1 cells as a representative PEL. We then varied the E:T ratio of NK cell lines and BCBL-1 cells in the presence or absence of 10 μg/ml DARA.
Interestingly, both CD16− and CD16+ NK cell lines did not show killing activity toward BCBL-1 cells in the absence of DARA (Fig. 2B-2E). DARA-induced killing activity of N6 NK cells in an E:T ratio-dependent manner (Fig. 2B and D), whereas KHYG-1 did not induce killing activity against BCBL-1 (Fig. 2C and E).
Lysosomal-associated membrane protein-1 (LAMP-1 or CD107a) has been described as a marker of degranulation and killing activity of active-state NK cells [53]. Surface expression of CD107a in N6 NK cells was increased by co-culture with BCBL-1 cells, and administration of DARA increased the surface expression of CD107a (Fig. 2F and G).
Isolation and expansion of CD16-expressing NK cells
We isolated NK cells from PBMCs using the NK Cell Isolation Kit (Miltenyi Biotec). Isolated NK cells were co-cultured with MHC class I negative K562 cells in the presence of IL-2 and IL-15 (Fig. 3A) for expansion [40]. The majority of isolated and expanded NK cells were CD56+CD16 NK+ cells, which play major roles in both direct NK activity and ADCC through CD16 (FcγRIII) (Fig. 3B). The fold expansion of NK cells varied by donor (5–22 × 106 cells from 1 × 105 NK cells) (data not shown).
Fig. 3.

NK cell expansion and the phenotypes of NK cells. DARA-elevated NK cells mediated ADCC toward primary effusion lymphoma A Schematic diagram of methodological NK cell expansion. B Phenotypes of NK cells. Flow cytometric analysis of NK cell subsets. Two major subsets of fresh isolated NK cells (left) and expanded NK cells (right). Cells were gated as CD3− PI− populations. C Calcein-AM-labeling BCBL-1, GTO, and TY-1 cells were incubated with various concentrations of DARA, and co-cultured with expanded NK cells from a representative healthy donor (E:T ratio = 5:1) for 4 h. Fluorescence was measured by plate reader. D Calcein-AM-labeling BCBL-1, GTO, and TY-1 cells were coated with either control IgG1 (10 μg/ml) or DARA (10 μg/ml) for 15 min at room temperature. The mAb coated cells were co-cultured with expanded NK cells from a representative healthy donor at different E:T ratios for 4 h. Fluorescence was measured by plate reader. Percentages of PEL lysis are shown and were analyzed by using student t test. Data are presented as mean values ± standard error (SE). The data were analyzed by one-way analysis of variance (ANOVA) with the Turkey’s multiple-comparison test. E and F CFSE- labeling BCBL-1 cell line was coated with either control IgG1 (10 μg/ml) or DARA (10 μg/ml) for 15 min at room temperature. The mAb coated cells were co-cultured with expanded NK cells from a healthy donor at different E:T ratios for 4 h. The dead cells were determined using Ghost dye 780 staining. (G and H) BCBL-1, GTO, and TY-1 were pre-incubated with DARA at room temperature for 15 min and co-cultured with expanded NK cells from a representative healthy donor (E:T ratio = 5:1). Anti-CD107a was simultaneously added into the mixture. The reaction was next incubated for 4 h. CD56+CD107a+ cells were detected and analyzed by flow cytometry. G Up-regulation of CD107a after DARA treatment was analyzed by flow cytometry. H Percentages of CD56+CD107a+ cells after stimulation with DARA. Data are presented as mean values ± standard error (SE). * P < 0.05, ** P < 0.01, ***P < 0.001, ****P < 0.0001 significant levels
NK cells possess direct killing activity against PEL, and DARA enhances NK cytotoxicity through ADCC
Antibody-dependent cytotoxicity via FcR-bearing effector cells is an important mechanism of action for therapeutic mAbs [54, 55]. We showed DARA dose response in 3 PEL cells using a classic calcein-AM releasing method and expanded NK cells from a healthy donor, and we observed dose-dependent fashion of ADCC activity (Fig. 3C) and E:T dependent manner, in which DARA-mediated ADCC (Fig. 3D). We investigated the direct killing activity of expanded NK cells and the DARA-mediated ADCC activity using FACS analysis. CSFE labeled BCBL-1 cells were co-cultured with expanded NK cells in the presence or absence of DARA (10 μg/ml). As shown in Fig. 3E and F, we confirmed that the expanded NK cells could directly kill PEL cells in an E:T ratio-dependent manner, and DARA but not control human IgG1 enhanced the cytotoxicity of NK cells in an E:T ratio-dependent manner. The ADCC results were consistent by both a classic calcein-AM releasing method and FACS analysis in 3 PEL; BCBL-1, GTO, and TY-1. The surface expression of CD107a in expanded NK cells was increased by co-culture with PEL cells, and administration of DARA further increased the surface expression of CD107a (Fig. 3G and H, Supplement Fig. 1A and B).
DARA-mediated CDC activity toward PEL cells
Tumor cell killing by CDC is also considered an important mechanism of action for therapeutic Abs. To assess the CDC activity of DARA, we used rabbit serum as a source of complement. As shown in Fig. 4A, addition of rabbit serum also killed BCBL-1, GTO, TY-1 cells, demonstrating an alternative pathway of the complement (Fig. 4B), and dead (PI+) cells increased in a DARA dose-dependent manner. Heat-inactivated rabbit serum (56 °C, 30 min) used as a negative control did not induce cell death (Fig. 4B). The complement by itself induced death of PEL cells in a dose-dependent manner. These results indicate that the complement has anti-PEL activity and that DARA enhanced the CDC activity.
Fig. 4.

DARA exerts CDC toward primary effusion lymphoma. A BCBL-1, GTO, and TY-1 cells were incubated with different concentrations of DARA for 15 min at room temperature. 10% of serum (HI; heat-inactivated serum by 56 °C for 30 min, NA; native serum as the source of complement) was added to the reaction and further incubated for 2 h. The percentage of PI+ cells was analyzed by flow cytometry. Data are presented as mean values ± standard error (SE). B BCBL-1, GTO, and TY-1 cells were incubated with 10 μg/ml DARA for 15 min at room temperature. The serum (HI; heat-inactivated serum by 56 °C for 30 min, NA; native serum as the source of complement) at various concentrations was added to the reaction and further incubated for 2 h. The percentage of PI+ cells was analyzed by flow cytometry. Data are presented as mean values ± standard error (SE). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 significant levels
DARA-mediated ADCP activity toward PEL cells
Antibody-dependent cell phagocytosis (ADCP) is considered to be an effective mechanism to boost macrophage activities and enhance phagocytosis of malignant cells. We tested the effect of DARA on phagocytosis by co-culturing DARA-coating CFSE-labeling BCBL-1 cells and human monocyte-derived macrophages (MDMs). As shown in Fig. 5A and B, DARA enhanced phagocytosis, indicated by the significantly higher phagocytic index, compared to the IgG1 control. We also assessed the percentage of phagocytotic cells based on FACS analysis. DARA boosted phagocytosis from 19% to 26.6% (Fig. 5C). DARA elicited ADCP against BCBL-1, GTO, and TY-1 (Supplement Fig. 4A and B). The results indicated the same tendency for both donors (Fig. 5D). We also used mouse thioglycolate-induced peritoneal macrophages as effector cells. As shown in Fig. 5E and F, DARA significantly increased phagocytosis as compared to the IgG1 control. These results demonstrated that DARA augments phagocytosis activity in both MDMs and peritoneal macrophages.
Fig. 5.

DARA demonstrates ADCP toward primary effusion lymphoma. A MDMs from one representative healthy donor were plated into a glass-bottomed dish. CFSE-labeled BCBL-1 cells were incubated with either DARA or IgG1 antibody for 15 min at room temperature. Next, antibody coating CFSE-labeled BCBL-1 cells were added to the wells of MDMs (E: T ratio = 1:5) and further incubated for 2 h. The nuclei of the MDMs cells were stained with Hoechst 33342, shown in blue, whereas BCBL-1 cells were seen as green fluorescence. Phagocytic cells are indicated by red arrows. B Data are presented as a phagocytic index, calculated as the number of phagocytic CFSE+Hoechst 33342+ per 100 MDMs. C MDMs from one representative healthy donor were plated into a well. CFSE-labeled BCBL-1 cells were incubated with either DARA or IgG1 antibody for 15 min at room temperature. The antibody coating CFSE-labeled BCBL-1 cells were added to the well of MDMs (E: T ratio = 1:2) and further incubated for 2 h. Phagocytic cells were defined as CD11b+CFSE+ cells. D Data are presented as the % phagocytosis (CD11b+CFSE+ cells) of two healthy donors. The data are presented as mean values ± the standard error (SE). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. E Peritoneal macrophages from BRJ mice were plated into a well. CFSE-labeled BCBL-1 cells were incubated with either DARA or IgG1 antibody for 15 min at room temperature. The antibody coating CFSE-labeled BCBL-1 cells were added to the well of macrophages at the indicated ratios and further incubated for 2 h. Phagocytic cells were defined as CD11b+CFSE+ cells. F Data are presented as the % phagocytosis (CD11b+CFSE+ cells). The data are presented as mean values ± the standard error (SE). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 significant levels
FcR-mediated cross-linking of DARA induces cell death in PEL cell lines.
Ab-mediated cross-linking of antigens, not related to the death receptor family, may also induce programmed cell death (PCD), but not via the classical apoptotic pathway [56–59]. This pathway is specifically characterized by homotypic aggregation of cells involving cytoskeleton reorganization, lysosomal activation, and production of reactive oxygen species [60]. DARA-induced cell death can be enhanced by Fc cross-linking secondary Ab or by FcγR-expressing cells [49]. BCBL-1, GTO, and TY-1 cells were cultured with various doses of DARA in the presence of anti-human Fc receptor (anti-huFc) or anti-mouse Fc receptor (anti-moFc) Ab, and DARA-induced cell death in the presence of anti-huFc Ab (Fig. 6A), indicating that FcR-mediated cross-linking of DARA induces death in PEL cells. In addition, clumping and morphological changes were observed by microscopic analysis (Fig. 6B).
Fig. 6.

Induction of cell death and clustering by fcɤR-mediated cross-linking of DARA. BCBL-1, GTO, TY-1 cells were pre-incubated for 15 min with varying concentrations of DARA followed by an overnight incubation in the presence of either 25 μg/ml of rabbit anti-human IgG F(ab’)2 fragments or goat anti-mouse IgG (H + L). A The percentage of cell viability was assessed by MTT assay. B DARA cross-linking-mediated clustering of BCBL-1 cells. Bright-field images of BCBL-1 after 18 h of incubation with a range of DARA concentration in the presence of 25 μg/ml Fc cross-linking secondary Ab
DARA suppresses tumor growth in PEL xenograft mice.
To evaluate the in vivo efficacy of DARA, BCBL-1 cells were subcutaneously xenografted into BRJ mice. Tumor growth was monitored as tumor volume for two weeks after PEL xenografting. As shown in Fig. 7A and B, the tumor size and volume in DARA treated mice (143.2 ± 97.6 mm3, n = 12) were significantly lower than those in non-treated mice (781.3 ± 438.0 mm3, n = 12) (P < 0.05). The tumor weight of DARA treated mice (0.28 ± 0.12 g, n = 12) was also significantly lower compared with non-treated mice (0.85 ± 0.51 g, n = 12) (P < 0.05).
Fig. 7.
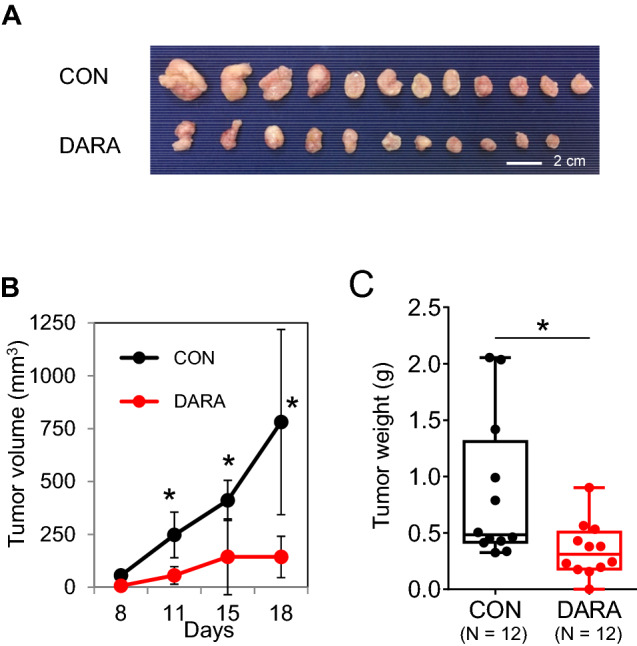
DARA treatment reduces tumor growth in a mouse model. DARA suppresses tumor growth in PEL xenograft mice. 107 BCBL-1 cells were subcutaneously injected into both flanks of mice (6 mice per group, in total 12 nodules). Three days after cell transplantation, 100 μg of DARA/mouse was intraperitoneally injected into the mice twice per week. The tumor size and morphology are shown in A. Tumor volumes were monitored B and weighed C. The data are presented as mean values ± standard deviation (SD). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Discussion
In this study, we demonstrated that PEL cells highly express CD38, suggesting that CD38 could be a promising target for PEL antibody immunotherapy treatment and used DARA to develop a CD38-targeting toward PEL. We confirmed that DARA confers multiple mechanisms including ADCC, CDC, ADCP and induction of PCD and cell death via a cross-linking antibody against PEL, both in vitro and in vivo. We also showed that DARA suppressed tumor growth in a PEL xenograft model.
By using the Jurkat ADCC indicator, CD16+ KHYG-1, NK cells from healthy donors, our results showed that ADCC-mediated by DARA against PEL. The potent activity of mAb for increasing ADCC activity depends on the antigen expression on target cell surfaces [61, 62], and higher affinity type FcγRIIIα 158 V/V compared with low affinity type FcγRIIIα 158F/F carriers [55]. Reduction of the core fucose content has been shown to induce higher binding affinity to FcγRIIIα [63]. Most PEL have lymphocytes activation markers such as CD30 and CD38 without normal B cell markers [6], indicating that DARA can be used for the treatment of PEL [64]. The level of antigen expression on target cells is critical for treatments using therapeutic monoclonal antibodies. As shown in Fig. 1, TY-1 showed lower CD38 expression compared to other PEL cell lines. Notably, even though TY-1 showed mediocre expression of CD38, ADCC activity was positive in the platform of classic calcein-AM-releasing assay and an indicator FcR expressing Jurkat cells (Figs. 2A, 3C and D).
DARA was reported to induce CDC of tumor cell samples from MM patients [15]. CDC has been highlighted as a representative and important mechanism for the therapeutic efficacy of monoclonal antibodies. As presented in many antibody immunotherapy studies, the in vivo effect of rituximab (anti-CD20 Ab) is largely reduced by depletion of complement [65]. Furthermore, ofatumumab (anti-CD20 Ab) efficiently kills CLL cells and shows a positive correlation with potent CDC [66]. Moreover, alemtuzumab (anti-CD52 Ab) kills B cell malignancies, mainly via CDC [67]. C1q is the main initiator of the classical complement cascade, binding to the complement-binding site of the antibody to form an antigen–antibody complex [68]. An alternative, antibody-independent pathway of complement activation also exists, as shown in Fig. 4A. However, BCBL-1 was the most sensitive to complement cascades compared to GTO and TY-1 cells, which are more resistant. The results suggested that the possibilities of complement inhibitory molecules- expression such as CD46, CD55, and CD59 on each PEL might be different. Taken together, our results showed potent DARA-mediated CDC toward PEL.
Regarding the ADCP property of DARA, it has been reported that DARA shows anti-tumor activity and hampers leukemia-microenvironment interaction in the poor-prognosis CD38+ CLL subtype, with potent and efficient lysis of patient-derived CLL cells and cell lines by ADCC in vitro and ADCP, both in vitro and in vivo, contrasting with negligible CDC [38].
One study showed that DARA induces PCD of CD38-expressing cells via FcγR-mediated cross-linking, resulting in clustering of cells, phosphatidylserine translocation, loss of mitochondrial membrane potential, and loss of membrane integrity followed by cell death [49]. Our study revealed that DARA induces clustering of cells and induction of cell death in PEL.
We observed that DARA monotherapy drastically suppressed tumor growth in a PEL xenograft mice model. BRJ mice lack mature B, T, and NK cells (47), and have a low level of complement [69]. As shown in Fig. 5E and F, peritoneal macrophages obtained from BRJ mice demonstrated potent ADCP activity. It has been shown that DARA reacts to effector cells of SCID mice and mediates ADCP in vivo [70]. A study of elotuzumab (humanized anti-SLAMF7 antibody), which has a human Fc portion, in SCID and NSG mice revealed that elotuzumab enables mouse macrophages to prevent MM tumor growth and progression through the activation of mouse FcγR [71]. ADCP is considered to be the main mechanism of DARA in our BRJ mouse model. Thus, mouse macrophages induce ADCP via human antibody. Moreover, our BRJ xenograft mice model is a useful tool for clarifying the ADCP activity of human antibodies. The further studying on NK cell expansion and adoptive transfer together with DARA treatment [72] can be eventually considered in our PEL xenograft mice model.
In conclusion, we demonstrated that CD38 was highly expressed on PEL cells, and DARA-induced cell death of PEL cells by ADCC, CDC, and ADCP activities, and by cross-linking in vitro. DARA confers PEL tumor inhibitory effect in vivo. Our data provide preclinical evidence for antibody targeting of CD38 as a novel effective therapeutic strategy for treating PEL.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We are grateful to Ms. Sawako Fujikawa for technical assistance and Ms. Michiko Teramoto for secretarial assistance.
Abbreviations
- ADCC
Antibody-dependent cell cytotoxicity
- ADCP
Antibody-dependent cell phagocytosis
- CDC
Complement-dependent cytolysis
- DARA
Daratumumab
- MM
Multiple myeloma
- MDMs
Human monocyte-derived macrophages
- NHL
Non-Hodgkin’s lymphoma
- PEL
Primary effusion lymphoma
Author contributions
JP contributed to conceptualization, methodology, formal analysis, investigation, and writing—original draft; RK contributed to investigation and methodology; and SO contributed to conceptualization, resources, writing—review and editing, supervision, project administration, and funding acquisition.
Funding
This work was supported in part by the Research Program on HIV/AIDS (Grant No. 18fk0410008h003) of the Japan Agency for Medical Research and Development and Grants-in-Aid for Science Research (16K08742) from the Ministry of Education, Science, Sports, and Culture of Japan.
Declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
Mice were bred and cared for in the animal research facility according to institutional guidelines. All experimental procedures in this study were approved by the Institutional Animal Care and Use Committee at Kumamoto University, Japan.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Simonelli C, Spina M, Cinelli R, Talamini R, Tedeschi R, Gloghini A, Vaccher E, Carbone A, Tirelli U. Clinical features and outcome of primary effusion lymphoma in HIV-infected patients: a single-institution study. J Clin Oncol. 2003;21:3948–3954. doi: 10.1200/JCO.2003.06.013. [DOI] [PubMed] [Google Scholar]
- 2.Chen YB, Rahemtullah A, Hochberg E. Primary effusion lymphoma. Oncologist. 2007;12:569–576. doi: 10.1634/theoncologist.12-5-569. [DOI] [PubMed] [Google Scholar]
- 3.Ota Y, Hishima T, Mochizuki M, et al. Classification of AIDS-related lymphoma cases between 1987 and 2012 in Japan based on the WHO classification of lymphomas. Cancer Med. 2014;3:143–153. doi: 10.1002/cam4.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okada S, Goto H, Yotsumoto M. Current status of treatment for primary effusion lymphoma. Intractable Rare Dis Res. 2014;3:65–74. doi: 10.5582/irdr.2014.01010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimada K, Hayakawa F, Kiyoi H. Biology and management of primary effusion lymphoma. Blood. 2018;132:1879–1888. doi: 10.1182/blood-2018-03-791426. [DOI] [PubMed] [Google Scholar]
- 6.Hu Z, Pan Z, Chen W, et al. Primary Effusion Lymphoma: A Clinicopathological Study of 70 Cases. Cancers (Basel) 2021 doi: 10.3390/cancers13040878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castillo JJ, Shum H, Lahijani M, Winer ES, Butera JN. Prognosis in primary effusion lymphoma is associated with the number of body cavities involved. Leuk Lymphoma. 2012;53:2378–2382. doi: 10.3109/10428194.2012.694075. [DOI] [PubMed] [Google Scholar]
- 8.Boulanger E, Gerard L, Gabarre J, Molina JM, Rapp C, Abino JF, Cadranel J, Chevret S, Oksenhendler E. Prognostic factors and outcome of human herpesvirus 8-associated primary effusion lymphoma in patients with AIDS. J Clin Oncol. 2005;23:4372–4380. doi: 10.1200/JCO.2005.07.084. [DOI] [PubMed] [Google Scholar]
- 9.Coiffier B. Monoclonal antibodies in the management of newly diagnosed, aggressive B-cell lymphoma. Curr Hematol Rep. 2003;2:23–29. [PubMed] [Google Scholar]
- 10.Czuczman MS, Gregory SA. The future of CD20 monoclonal antibody therapy in B-cell malignancies. Leuk Lymphoma. 2010;51:983–994. doi: 10.3109/10428191003717746. [DOI] [PubMed] [Google Scholar]
- 11.Subramanian J, Cavenagh J, Desai B, Jacobs I. Rituximab in the treatment of follicular lymphoma: the future of biosimilars in the evolving therapeutic landscape. Cancer Manag Res. 2017;9:131–140. doi: 10.2147/CMAR.S120589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freeman CL, Sehn LH. A tale of two antibodies: obinutuzumab versus rituximab. Br J Haematol. 2018;182:29–45. doi: 10.1111/bjh.15232. [DOI] [PubMed] [Google Scholar]
- 13.Lim ST, Rubin N, Said J, Levine AM. Primary effusion lymphoma: successful treatment with highly active antiretroviral therapy and rituximab. Ann Hematol. 2005;84:551–552. doi: 10.1007/s00277-005-1040-6. [DOI] [PubMed] [Google Scholar]
- 14.Bhatt S, Ashlock BM, Natkunam Y, Sujoy V, Chapman JR, Ramos JC, Mesri EA, Lossos IS. CD30 targeting with brentuximab vedotin: a novel therapeutic approach to primary effusion lymphoma. Blood. 2013;122:1233–1242. doi: 10.1182/blood-2013-01-481713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Weers M, Tai YT, van der Veer MS, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. 2011;186:1840–1848. doi: 10.4049/jimmunol.1003032. [DOI] [PubMed] [Google Scholar]
- 16.Fishwild DM, O'Donnell SL, Bengoechea T, et al. High-avidity human IgG kappa monoclonal antibodies from a novel strain of minilocus transgenic mice. Nat Biotechnol. 1996;14:845–851. doi: 10.1038/nbt0796-845. [DOI] [PubMed] [Google Scholar]
- 17.Lonberg N, Taylor LD, Harding FA, et al. Antigen-specific human antibodies from mice comprising four distinct genetic modifications. Nature. 1994;368:856–859. doi: 10.1038/368856a0. [DOI] [PubMed] [Google Scholar]
- 18.Bhatnagar V, Gormley NJ, Luo L, et al. FDA Approval summary: daratumumab for treatment of multiple myeloma after one prior therapy. Oncologist. 2017;22:1347–1353. doi: 10.1634/theoncologist.2017-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mikhael J, Richardson P, Usmani SZ, et al. A phase 1b study of isatuximab plus pomalidomide/dexamethasone in relapsed/refractory multiple myeloma. Blood. 2019;134:123–133. doi: 10.1182/blood-2019-02-895193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raab MS, Engelhardt M, Blank A, et al. MOR202, a novel anti-CD38 monoclonal antibody, in patients with relapsed or refractory multiple myeloma: a first-in-human, multicentre, phase 1–2a trial. Lancet Haematol. 2020;7:e381–e394. doi: 10.1016/S2352-3026(19)30249-2. [DOI] [PubMed] [Google Scholar]
- 21.Dhillon S. Isatuximab: first approval. Drugs. 2020;80:905–912. doi: 10.1007/s40265-020-01311-1. [DOI] [PubMed] [Google Scholar]
- 22.Liu Q, Kriksunov IA, Graeff R, Munshi C, Lee HC, Hao Q. Crystal structure of human CD38 extracellular domain. Structure. 2005;13:1331–1339. doi: 10.1016/j.str.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 23.Lee HC. Structure and enzymatic functions of human CD38. Mol Med. 2006;12:317–323. doi: 10.2119/2006-00086.Lee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malavasi F, Funaro A, Roggero S, Horenstein A, Calosso L, Mehta K. Human CD38: a glycoprotein in search of a function. Immunol Today. 1994;15:95–97. doi: 10.1016/0167-5699(94)90148-1. [DOI] [PubMed] [Google Scholar]
- 25.Deaglio S, Mallone R, Baj G, et al. Human CD38 and its ligand CD31 define a unique lamina propria T lymphocyte signaling pathway. FASEB J. 2001;15:580–582. doi: 10.1096/fj.00-0522fje. [DOI] [PubMed] [Google Scholar]
- 26.Funaro A, Spagnoli GC, Ausiello CM, Alessio M, Roggero S, Delia D, Zaccolo M, Malavasi F. Involvement of the multilineage CD38 molecule in a unique pathway of cell activation and proliferation. J Immunol. 1990;145:2390–2396. [PubMed] [Google Scholar]
- 27.Deaglio S, Mallone R, Baj G, Arnulfo A, Surico N, Dianzani U, Mehta K, Malavasi F. CD38/CD31, a receptor/ligand system ruling adhesion and signaling in human leukocytes. Chem Immunol. 2000;75:99–120. doi: 10.1159/000058765. [DOI] [PubMed] [Google Scholar]
- 28.Deaglio S, Aydin S, Grand MM, Vaisitti T, Bergui L, D'Arena G, Chiorino G, Malavasi F. CD38/CD31 interactions activate genetic pathways leading to proliferation and migration in chronic lymphocytic leukemia cells. Mol Med. 2010;16:87–91. doi: 10.2119/molmed.2009.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aarhus R, Graeff RM, Dickey DM, Walseth TF, Lee HC. ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J Biol Chem. 1995;270:30327–30333. doi: 10.1074/jbc.270.51.30327. [DOI] [PubMed] [Google Scholar]
- 30.Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, Vaisitti T, Aydin S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. 2008;88:841–886. doi: 10.1152/physrev.00035.2007. [DOI] [PubMed] [Google Scholar]
- 31.Billadeau D, Ahmann G, Greipp P, Van Ness B. The bone marrow of multiple myeloma patients contains B cell populations at different stages of differentiation that are clonally related to the malignant plasma cell. J Exp Med. 1993;178:1023–1031. doi: 10.1084/jem.178.3.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin P, Owens R, Tricot G, Wilson CS. Flow cytometric immunophenotypic analysis of 306 cases of multiple myeloma. Am J Clin Pathol. 2004;121:482–488. doi: 10.1309/74R4-TB90-BUWH-27JX. [DOI] [PubMed] [Google Scholar]
- 33.Santonocito AM, Consoli U, Bagnato S, Milone G, Palumbo GA, Di Raimondo F, Stagno F, Guglielmo P, Giustolisi R. Flow cytometric detection of aneuploid CD38(++) plasmacells and CD19(+) B-lymphocytes in bone marrow, peripheral blood and PBSC harvest in multiple myeloma patients. Leuk Res. 2004;28:469–477. doi: 10.1016/j.leukres.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 34.Koullouros M, Chouari TAM, Stewart A, Kerr K, Dawson D. Isolated cardiac desminopathy. Eur Heart J Cardiovasc Imaging. 2017 doi: 10.1093/ehjci/jex049. [DOI] [PubMed] [Google Scholar]
- 35.Mistry JJ, Moore JA, Kumar P, et al. Daratumumab inhibits acute myeloid leukaemia metabolic capacity by blocking mitochondrial transfer from mesenchymal stromal cells. Haematologica. 2021;106:589–592. doi: 10.3324/haematol.2019.242974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naik J, Themeli M, de Jong-Korlaar R, et al. CD38 as a therapeutic target for adult acute myeloid leukemia and T-cell acute lymphoblastic leukemia. Haematologica. 2019;104:e100–e103. doi: 10.3324/haematol.2018.192757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bride KL, Vincent TL, Im SY, et al. Preclinical efficacy of daratumumab in T-cell acute lymphoblastic leukemia. Blood. 2018;131:995–999. doi: 10.1182/blood-2017-07-794214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matas-Cespedes A, Vidal-Crespo A, Rodriguez V, et al. The human CD38 monoclonal antibody daratumumab shows antitumor activity and hampers leukemia-microenvironment interactions in chronic lymphocytic leukemia. Clin Cancer Res. 2017;23:1493–1505. doi: 10.1158/1078-0432.CCR-15-2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goto H, Kojima Y, Nagai H, Okada S. Establishment of a CD4-positive cell line from an AIDS-related primary effusion lymphoma. Int J Hematol. 2013;97:624–633. doi: 10.1007/s12185-013-1339-3. [DOI] [PubMed] [Google Scholar]
- 40.Harada H, Saijo K, Watanabe S, Tsuboi K, Nose T, Ishiwata I, Ohno T. Selective expansion of human natural killer cells from peripheral blood mononuclear cells by the cell line. HFWT Jpn J Cancer Res. 2002;93:313–319. doi: 10.1111/j.1349-7006.2002.tb02174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hattori S, Matsuda K, Kariya R, Harada H, Okada S. Proliferation of functional human natural killer cells with anti-HIV-1 activity in NOD/SCID/Jak3(null) mice. Microbiol Immunol. 2016;60:106–113. doi: 10.1111/1348-0421.12355. [DOI] [PubMed] [Google Scholar]
- 42.Parekh BS, Berger E, Sibley S, Cahya S, Xiao L, LaCerte MA, Vaillancourt P, Wooden S, Gately D. Development and validation of an antibody-dependent cell-mediated cytotoxicity-reporter gene assay. MAbs. 2012;4:310–318. doi: 10.4161/mabs.19873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghose J, Viola D, Terrazas C, et al. Daratumumab induces CD38 internalization and impairs myeloma cell adhesion. Oncoimmunology. 2018;7:e1486948. doi: 10.1080/2162402X.2018.1486948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamashita M, Kitano S, Aikawa H, Kuchiba A, Hayashi M, Yamamoto N, Tamura K, Hamada A. A novel method for evaluating antibody-dependent cell-mediated cytotoxicity by flowcytometry using cryopreserved human peripheral blood mononuclear cells. Sci Rep. 2016;6:19772. doi: 10.1038/srep19772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baig NA, Taylor RP, Lindorfer MA, Church AK, Laplant BR, Pavey ES, Nowakowski GS, Zent CS. Complement dependent cytotoxicity in chronic lymphocytic leukemia: ofatumumab enhances alemtuzumab complement dependent cytotoxicity and reveals cells resistant to activated complement. Leuk Lymphoma. 2012;53:2218–2227. doi: 10.3109/10428194.2012.681657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goto H, Kojima Y, Matsuda K, Kariya R, Taura M, Kuwahara K, Nagai H, Katano H, Okada S. Efficacy of anti-CD47 antibody-mediated phagocytosis with macrophages against primary effusion lymphoma. Eur J Cancer. 2014;50:1836–1846. doi: 10.1016/j.ejca.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 47.Ono A, Hattori S, Kariya R, Iwanaga S, Taura M, Harada H, Suzu S, Okada S. Comparative study of human hematopoietic cell engraftment into BALB/c and C57BL/6 strain of rag-2/jak3 double-deficient mice. J Biomed Biotechnol. 2011;2011:539748. doi: 10.1155/2011/539748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vaeteewoottacharn K, Kariya R, Pothipan P, et al. Attenuation of CD47-SIRPalpha signal in cholangiocarcinoma potentiates tumor-associated macrophage-mediated phagocytosis and suppresses intrahepatic metastasis. Transl Oncol. 2019;12:217–225. doi: 10.1016/j.tranon.2018.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Overdijk MB, Jansen JH, Nederend M, Lammerts van Bueren JJ, Groen RW, Parren PW, Leusen JH, Boross P. The therapeutic CD38 monoclonal antibody daratumumab induces programmed cell death via fcgamma receptor-mediated cross-linking. J Immunol. 2016;197:807–813. doi: 10.4049/jimmunol.1501351. [DOI] [PubMed] [Google Scholar]
- 50.Panaampon J, Kudo E, Kariya R, Okada S. Ephedrine enhances HIV-1 reactivation from latency through elevating tumor necrosis factor receptor II (TNFRII) expression. Heliyon. 2019;5:e02490. doi: 10.1016/j.heliyon.2019.e02490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yagita M, Huang CL, Umehara H, Matsuo Y, Tabata R, Miyake M, Konaka Y, Takatsuki K. A novel natural killer cell line (KHYG-1) from a patient with aggressive natural killer cell leukemia carrying a p53 point mutation. Leukemia. 2000;14:922–930. doi: 10.1038/sj.leu.2401769. [DOI] [PubMed] [Google Scholar]
- 52.Alpert MD, Heyer LN, Williams DE, Harvey JD, Greenough T, Allhorn M, Evans DT. A novel assay for antibody-dependent cell-mediated cytotoxicity against HIV-1- or SIV-infected cells reveals incomplete overlap with antibodies measured by neutralization and binding assays. J Virol. 2012;86:12039–12052. doi: 10.1128/JVI.01650-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods. 2004;294:15–22. doi: 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 54.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 55.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99:754–758. doi: 10.1182/blood.v99.3.754. [DOI] [PubMed] [Google Scholar]
- 56.Alduaij W, Ivanov A, Honeychurch J, et al. Novel type II anti-CD20 monoclonal antibody (GA101) evokes homotypic adhesion and actin-dependent, lysosome-mediated cell death in B-cell malignancies. Blood. 2011;117:4519–4529. doi: 10.1182/blood-2010-07-296913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cerisano V, Aalto Y, Perdichizzi S, et al. Molecular mechanisms of CD99-induced caspase-independent cell death and cell-cell adhesion in Ewing's sarcoma cells: actin and zyxin as key intracellular mediators. Oncogene. 2004;23:5664–5674. doi: 10.1038/sj.onc.1207741. [DOI] [PubMed] [Google Scholar]
- 58.Ivanov A, Beers SA, Walshe CA, et al. Monoclonal antibodies directed to CD20 and HLA-DR can elicit homotypic adhesion followed by lysosome-mediated cell death in human lymphoma and leukemia cells. J Clin Invest. 2009;119:2143–2159. doi: 10.1172/JCI37884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mateo V, Brown EJ, Biron G, Rubio M, Fischer A, Deist FL, Sarfati M. Mechanisms of CD47-induced caspase-independent cell death in normal and leukemic cells: link between phosphatidylserine exposure and cytoskeleton organization. Blood. 2002;100:2882–2890. doi: 10.1182/blood-2001-12-0217. [DOI] [PubMed] [Google Scholar]
- 60.Honeychurch J, Alduaij W, Azizyan M, Cheadle EJ, Pelicano H, Ivanov A, Huang P, Cragg MS, Illidge TM. Antibody-induced nonapoptotic cell death in human lymphoma and leukemia cells is mediated through a novel reactive oxygen species-dependent pathway. Blood. 2012;119:3523–3533. doi: 10.1182/blood-2011-12-395541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Velders MP, van Rhijn CM, Oskam E, Fleuren GJ, Warnaar SO, Litvinov SV. The impact of antigen density and antibody affinity on antibody-dependent cellular cytotoxicity: relevance for immunotherapy of carcinomas. Br J Cancer. 1998;78:478–483. doi: 10.1038/bjc.1998.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lustig HJ, Bianco C. Antibody-mediated cell cytotoxicity in a defined system: regulation by antigen, antibody, and complement. J Immunol. 1976;116:253–260. [PubMed] [Google Scholar]
- 63.Ferrara C, Stuart F, Sondermann P, Brunker P, Umana P. The carbohydrate at FcgammaRIIIa Asn-162. An element required for high affinity binding to non-fucosylated IgG glycoforms. J Biol Chem. 2006;281:5032–5036. doi: 10.1074/jbc.M510171200. [DOI] [PubMed] [Google Scholar]
- 64.Shah NN, Singavi AK, Harrington A. Daratumumab in primary effusion lymphoma. N Engl J Med. 2018;379:689–690. doi: 10.1056/NEJMc1806295. [DOI] [PubMed] [Google Scholar]
- 65.Cragg MS, Glennie MJ. Antibody specificity controls in vivo effector mechanisms of anti-CD20 reagents. Blood. 2004;103:2738–2743. doi: 10.1182/blood-2003-06-2031. [DOI] [PubMed] [Google Scholar]
- 66.Teeling JL, French RR, Cragg MS, et al. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood. 2004;104:1793–1800. doi: 10.1182/blood-2004-01-0039. [DOI] [PubMed] [Google Scholar]
- 67.Zent CS, Secreto CR, LaPlant BR, Bone ND, Call TG, Shanafelt TD, Jelinek DF, Tschumper RC, Kay NE. Direct and complement dependent cytotoxicity in CLL cells from patients with high-risk early-intermediate stage chronic lymphocytic leukemia (CLL) treated with alemtuzumab and rituximab. Leuk Res. 2008;32:1849–1856. doi: 10.1016/j.leukres.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20:34–50. doi: 10.1038/cr.2009.139. [DOI] [PubMed] [Google Scholar]
- 69.Kotimaa J, Klar-Mohammad N, Gueler F, Schilders G, Jansen A, Rutjes H, Daha MR, van Kooten C. Sex matters: Systemic complement activity of female C57BL/6J and BALB/cJ mice is limited by serum terminal pathway components. Mol Immunol. 2016;76:13–21. doi: 10.1016/j.molimm.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 70.Vidal-Crespo A, Matas-Cespedes A, Rodriguez V, et al. Daratumumab displays in vitro and in vivo anti-tumor activity in models of B-cell non-Hodgkin lymphoma and improves responses to standard chemo-immunotherapy regimens. Haematologica. 2020;105:1032–1041. doi: 10.3324/haematol.2018.211904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kurdi AT, Glavey SV, Bezman NA, et al. Antibody-dependent cellular phagocytosis by macrophages is a novel mechanism of action of elotuzumab. Mol Cancer Ther. 2018;17:1454–1463. doi: 10.1158/1535-7163.MCT-17-0998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thangaraj JL, Ahn SY, Jung SH, et al. Expanded natural killer cells augment the antimyeloma effect of daratumumab, bortezomib, and dexamethasone in a mouse model. Cell Mol Immunol. 2021;18:1652–1661. doi: 10.1038/s41423-021-00686-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.