

Abstract
Innovations in viral vaccine manufacturing are crucial for pandemic preparedness and to meet ever‐rising global demands. For influenza, however, production still mainly relies on technologies established decades ago. Although modern production shifts from egg‐based towards cell culture technologies, the full potential has not yet been fully exploited. Here, we evaluate whether implementation of state‐of‐the‐art technologies for cell culture‐based recombinant protein production are capable to challenge outdated approaches in viral vaccine process development. For this, a fully automated single‐cell cloning strategy was established to generate monoclonal suspension Madin‐Darby canine kidney (MDCK) cells. Among selected cell clones, we could observe distinct metabolic and growth characteristics, with C59 reaching a maximum viable cell concentration of 17.3 × 106 cells/mL and low doubling times in batch mode. Screening for virus production using a panel of human vaccine‐relevant influenza A and B viruses in an ambr15 system revealed high titers with yields competing or even outperforming available MDCK cell lines. With C113, we achieved cell‐specific virus yields of up to 25,000 virions/cell, making this cell clone highly attractive for vaccine production. Finally, we confirmed process performance at a 50‐fold higher working volume. In summary, we present a scalable and powerful approach for accelerated development of high‐yield influenza virus production in chemically defined medium starting from a single cell.
Keywords: ambr15, cell culture‐based viral vaccine manufacturing, cell line development, CellCelector, chemically defined medium, influenza virus
Abbreviations
- µ
cell‐specific growth rate
- CLD
cell line development
- CSVY
cell‐specific virus yield
- DO
dissolved oxygen
- HA
hemagglutination activity
- HCD
high cell density
- hpi
hours post infection
- IAV
influenza A virus
- IBV
influenza B virus
- IIV
inactivated influenza vaccine
- MDCK
Madin‐Darby canine kidney
- MOI
multiplicity of infection
- qs
cell‐specific substrate consumption/production rate
- STR
stirred tank bioreactor
- TCID50
50 % tissue culture infective dose
- tD
doubling time
- VCC
viable cell concentration
- wv
working volume
1. INTRODUCTION
Expanding viral vaccine production capacity is a necessity to overcome global supply shortages, become prepared for pandemics and build a more sustainable future. Yet, the vast majority (approximately 84.5%) of inactivated influenza vaccine (IIV) production capacity still relies on a process using embryonated chicken eggs that dates back to the forties of the last century [1]. Although this process is well established, several drawbacks that result in restricted protection and limited supply of vaccines have been reviewed extensively in the past [2, 3]. Accordingly, the use of animal cell technology has become a progressively popular alternative to traditional egg‐based production methods for human influenza vaccines. Research from both, academia and industry, could convincingly demonstrate that cell culture‐based production processes can circumvent current limitations and have the potential to serve as the most effective mode for IIV manufacturing [4, 5, 6].
In contrast, sophisticated cell‐based production platforms are available for the manufacturing of recombinant proteins, that allow to rapidly bring new products into the clinic [7, 8]. Such production processes start usually from a clonally derived CHO cell bank carrying the product‐specific transgene, followed by a cell clone screening and production run under pre‐defined conditions with only minor adjustments. Here, various cutting‐edge innovations have been made over the past two to three decades that can technologically facilitate CHO cell line development (CLD).
For virus production, so far, “one‐size‐fits‐all” solutions do not exist and usually, comprehensive cell line screenings have to be performed to identify a suitable host cell substrate, followed by extensive process development [9]. For production of influenza viruses, a long list of traditional and designer cell lines, such as MDCK, Vero, HEK293, AGE1.CR, PER.C6, EB66, CAP, DuckCelt‐T17, or PBG.PK2.1 cells have been evaluated [10, 11, 12, 13, 14, 15, 16, 17, 18, 19].
Although monoclonal cell banks are not a regulatory requirement in vaccine manufacturing, there are several reports in literature describing a high heterogeneity inherent in the virus production capacities of influenza infected MDCK cells [20, 21, 22]. Thus, we hypothesized that we can take advantage of this variability by identifying high‐yield cell clones and implemented novel approaches and technologies in CLD and process development for viral vaccine production.
In the present study, we established a sophisticated workflow for CLD via single‐cell cloning, identification of high‐yield cell clones, and propose a new paradigm in process development in cell culture‐based influenza virus production. We generated monoclonal suspension MDCK cell cultures in a chemically defined medium using a CellCelector and analyzed their growth characteristics, aiming for doubling times between 20 and 30 h and a low accumulation of metabolic waste products [23]. Moreover, we screened selected cell clones for influenza virus production in an ambr15 system, and demonstrate that standard virus titration assays allow capturing significant differences among cell clones. For biological relevance, high yields of several influenza subtypes were obtained with the two top cell clones in batch mode. We show that virus production in selected cell clones can compete with or even outcompete other adherent or suspension cell lines for processes implemented at different scales (15 mL to 1 L) that were previously reported in literature. Although similar approaches are conventionally used in cell culture‐based recombinant protein manufacturing, such a strategy has to our knowledge not yet been evaluated for viral vaccine production.
PRACTICAL APPLICATION
Influenza vaccine manufacturing, still mainly relies on outdated processes using embryonated chicken eggs, and even cell culture‐based processes leave room for improvement. In contrast, the field of protein production offers advanced technologies that considerably support cell line development and with that also approval by governmental authorities. Despite their benefits, scientific reports on the implementation of such state‐of‐the‐art technologies for viral vaccines process development are rarely found. We present a novel workflow for automated single‐cell cloning using a CellCelector, followed by clone screening in an ambr15 system. Our results demonstrate not only a successful technology transfer, but also present an animal‐component‐free process that could potentially meet GMP‐standards with a high‐yield cell clone, suitable to challenge yields obtained from existing cell lines. We anticipate that this approach possesses significant dissemination potential to enhance the broader field of viral vaccine process development and may ultimately enable faster and more efficacious manufacturing of next‐generation vaccines.
2. MATERIALS AND METHODS
2.1. Generation of monoclonal suspension cultures
Adherent MDCK cells (#CCL‐34, ATCC) were thawed in chemically defined MDXK medium (Xell Sartorius, Germany) and cultured for 8 days in 6‐well plates to initially adapt to the serum‐free conditions. Next, cells were adapted to suspension growth in non‐baffled shake flasks (#431143, Corning). After 30 days, a single‐cell cloning strategy was performed using the CellCelector (ALS Sartorius, Germany). To this end, cells were strained and seeded into the nanowell plate. Monoclonality was reviewed using the integrated imaging system and cells were monitored daily over 4 days. Subsequently, selected colonies were picked and automatically transferred into a 384‐well plate. When a confluency of about 70% was reached, cells were detached using trypsin and transferred into a 24‐well plate, a 6‐well plate, then spin tubes (#11361724, ThermoFisher), and finally, shake flasks. Cell banks were prepared after 33 and 70 passages.
2.2. Cell lines and cultivation conditions
The obtained monoclonal suspension MDCK cell cultures were cultivated in MDXK medium (Xell Sartorius, Germany) supplemented with 8 mM L‐glutamine (Sigma–Aldrich, USA). As a reference, the suspension MDCK cell line MDCK.Xe.A (Xe.A) was used and cultivated in 4Cell MDCK CD medium (Sartorius, Germany; previously Xeno‐CDM, Shanghai BioEngine Sci‐Tech) [10, 24]. All cells were maintained in non‐baffled shake flasks (#431143, Corning) with 30 mL working volume (wv). A Multitron Pro incubator (Infors AG, Switzerland) was controlled at 37°C and 5% CO2 atmosphere with a shaking frequency of 120 rpm (50 mm throw). Cells were passaged 2–3 times a week, and inoculated using viable cell concentrations (VCCs) between 2 and 8 × 105 cells/mL. VCC, diameter, and viability was measured with a Vi‐CELL XR automated cell counter (#731050, Beckman Coulter). Prior to seeding, cells were centrifuged at 300 × g for 5 min at room temperature. For batch and infection experiments involving a growth phase, cells were seeded at 5 × 105 cells/mL in an ambr15 vessel (Sartorius, Germany) or stirred DASGIP bioreactor (#76DS0700ODSS, Eppendorf) with a starting wv of 15 and 350 mL, respectively. To keep the tip speed for both systems constant (at 0.21 m/s), agitation was set to 350 rpm for the ambr15 system and 80 rpm for the DASGIP system. An air‐oxygen mixture was sparged for aeration and dissolved oxygen (DO) was controlled at 40% saturation. Culture temperature was maintained at 37°C and pH value controlled at 7.15 during the cell growth phase; during the virus production phase all cultivations were shifted to 34°C and pH 7.2. For pH control, CO2 sparging and addition of 1 M sodium bicarbonate (NaHCO₃) was used. If needed, antifoam C (3% stock solution) was added to dissolve accumulating bubbles. For screening experiments without a growth phase, cells from a preculture in shake flasks were centrifuged (300 × g, 3 min), seeded at 2 × 106 cells/mL in fresh medium, and directly infected at inoculation time under the same conditions. Cell‐specific growth rate (µ), biomass yield based on substrate consumption (YX/S), cell‐specific substrate consumption and by‐product production rate (qs), and doubling time (tD) was determined using the following equations:
| (1) |
| (2) |
| (3) |
| (4) |
with X: viable cell concentration, t: cultivation time, n: sampling time point, and Cs : substrate concentration.
2.3. Influenza virus production
Several influenza A and B viruses (IAV, IBV) were used that are listed in Table 1. All seed viruses were propagated in adherent MDCK cells (#84121903, ECACC). Infection experiments were carried out in shake flasks, ambr15 vessels, or in DASGIP bioreactors. For infection, either a full or a partial medium exchange was performed. For the latter case, the wv was decreased by half (ambr15) and filled up with infection medium; in the DASGIP system the wv was directly increased to achieve a 1:2 dilution. In all cases, the infection medium contained recombinant trypsin (TrypLE Select Enzyme (10×), #A1217701, ThermoFisher) at a final concentration of 20 USP U/mL. Seed viruses were diluted with phosphate‐buffered saline and added to the cell suspension using a multiplicity of infection (MOI) of 10−3 infectious virions/cell. Virus samples were taken from the supernatant and centrifuged at 3000 × g for 5 min at room temperature to remove cell debris, then aliquoted and stored at −80°C until further analysis.
TABLE 1.
List of influenza viruses used for infection studies.
| Strains | Subtype/lineage | Origin | Antibody | Stock titer [TCID50/mL] |
|---|---|---|---|---|
| A/Puerto Rico/8/34 (A/PR8) | H1N1 | RKI | NIBSC, #03/242 | 9.9 × 107 |
| A/Cambodia/e0826360/2020 | H3N2 | NIBSC | NIBSC, #21/118 | 4.8 × 106 |
| B/Brisbane/60/2008 | Victoria | NIBSC | NIBSC, #13/254 | 6.6 × 107 |
| B/Phuket/3073/2013 | Yamagata | NIBSC | NIBSC, #14/248 | 3.0 × 107 |
Note: All strains were adapted and produced in adherent MDCK cells (#84121903, ECACC).
Abbreviations: NIBSC, National Institute for Biological Standards and Control, UK; RKI, Robert Koch Institute, Germany.
2.4. Sample analysis and calculations
Concentrations of lactate, ammonium, glutamine, glutamate, and glucose were determined in single measurements with the Cedex Bio Analyzer (Roche, Switzerland). The measurement range was previously validated for each metabolite and samples were diluted accordingly when out of range. To determine the virus titer of the samples, two established assays were used. Firstly, the total influenza virus content was assessed based on a hemagglutination activity (HA) assay as described by Kalbfuss et al. [25]. Additionally, for the quantification of infectious particles, a 50% tissue culture infectious dose (TCID50) assay using the influenza strain specific antibodies (Table 1) was performed as described by Genzel et al. The maximum standard deviation of the HA assay was ± 0.03 log10(HAU/100 µL); the dilution error of the TCID50 assay was ± 0.3 log10 [26]. The concentration of total virus particles Cvir in the supernatant and the cell‐specific virus yield (CSVYTCID, CSVYHA) was determined using the following equation derived from [25, 27]:
| (5) |
| (6) |
| (7) |
with VCCmax: maximum viable cell concentration obtained post infection, Cvir,max: maximum concentration of virus particles according to the HA assay, CEry : concentration of chicken erythrocytes used in the HA assay (2 × 107 cells/mL).
3. RESULTS AND DISCUSSION
With this proof of concept study, we aim to transfer technologies and knowledge from recombinant protein production (platform) processes into the field of viral vaccines. Thus, we generated monoclonal suspension MDCK cells using a CellCelector and following, challenged whether cell growth and viral yields are competitive to an existing well‐performing suspension MDCK cell line. Lastly, we evaluated the applicability of an ambr15 system for cell culture‐based influenza virus production.
3.1. The use of a CellCelector to generate monoclonal suspension MDCK cells
Heterogeneous adherent MDCK cells were thawed and adapted to suspension growth in chemically defined MDXK medium. Following, a hundred cells were seeded into a nanowell‐plate mounted in a CellCelector. After settling of cells, the integrated imaging system scanned the plate to identify wells containing a single cell (Figure 1, d0) to provide an image‐based monoclonality proof. In the following days, selected wells were monitored to follow the outgrowth of cell populations (Figure 1, d1‐d4). Wells containing more than one cell on d0 and wells for which no growth was observed, were excluded from further evaluation. After 4 days, the robot arm automatically transferred selected cell colonies (113 in total) from the nanowells into a 384‐well plate using a glass capillary (Figure 1, picked). Further manual expansion in differently sized culture formats finally resulted in five monoclonal suspension MDCK cell cultures. Cultures labeled C15 and C86 were split into two subcultures (C15‐1, C15‐2, and C86‐1, C86‐2, respectively) to investigate whether different growth patterns can occur despite their monoclonal origin.
FIGURE 1.
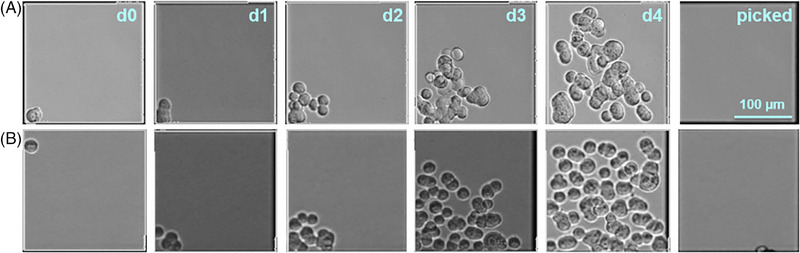
Generation of monoclonal MDCK suspension cell cultures using a CellCelector. After seeding and settling of cells in MDXK medium, the integrated imaging system was used to scan nanowells on day 0 (d0) to identify wells containing single cells. Although separated in nanowells, the cells were overlaid with the same medium and growth was monitored d1‐d4. After 4 days, 113 colonies were picked by a robot arm using a glass capillary and transferred into a 384‐well plate. Cloning history of (A) C59 and (B) C113 is exemplarily depicted. Scale bar is valid for all pictures.
As MDCK cells are well‐known to vary in their virus production capacities, cloning studies have been performed before [20, 21]. Yet, laborious serial dilution protocols were used and resulted in adherent cell lines cultivated in serum‐containing medium. In an attempt, to adapt MDCK cells to serum‐free conditions, most cells died within in a few passages [20]. Owing to the implementation of a CellCelector and choosing an eligible medium, our approach is not only faster but adaptation issues that might arise later can be circumvented, as cells are kept in a co‐culture. The number of developed cell lines was limited to five to serve as a proof of concept, and a higher cloning efficiency should easily be achievable.
3.2. Passaging of cell clones demonstrate robust growth properties
After the successful transfer into shake flasks, most cell clones immediately showed cell viabilities between 75% and 90% (Figure 2, C59, C86‐1, C86‐2). However, doubling times of some cell clones were high and exceeded 50 h. Other cultures started with poor growth performance in shake flasks, but could be adapted stepwise to the new conditions and cell viabilities increased steadily (Figure 2, C15‐2, C26, C113). Interestingly, cell clone C59 showed good growth characteristics in the beginning, then viability decreased significantly around 80 days, but could be recovered when lower inoculation densities were used. Following, C59 showed an outstanding growth performance with cell viability consistently above 95%, and doubling times of around 30 h. About 100–120 days after the single‐cell cloning process, cell viabilities and VCC of most clonal cultures stabilized at a certain level. For all other clonal cultures, high variations and an oscillation in cell viabilities (± 10%–20%), especially after a 3‐day split, were observed. This indicated that the passaging regime (every 2–3 days, inoculation at VCC 1 × 106 cells/mL) seemed not to be optimal. Thus, lower seeding densities were implemented later, which helped to further stabilize the growth performance of cell cultures. Overall, the growth performance of C15‐1 (not shown) and C15‐2, and both cultures of C86 were very similar. Cell banks of all clonal cultures were prepared after 33 passages and for selected cell clones again after 70 passages (C59‐p70, C113‐p70). To assess biological stability, multiple further cell banks were prepared from C59‐p70 and C113‐p70 and tested against each other for batch growth and virus production, here, no significant differences in growth or titer could be observed (data not shown).
FIGURE 2.
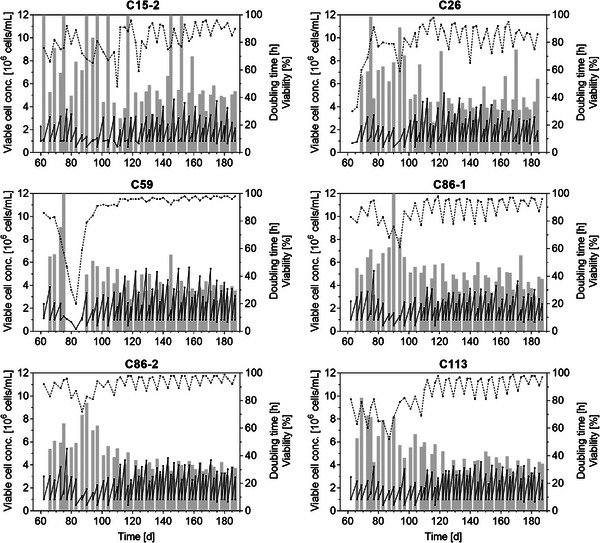
Adaptation of monoclonal suspension MDCK cells to shake flask cultivation. About 60 days after single‐cell cloning (t = 0), cells were transferred into non‐baffled shake flasks in MDXK medium. Viable cell concentration (black, solid line), cell viability (black, dashed line) and doubling time (gray bars) over the first 6 months (about 50 passages in shake flasks) are depicted. Doubling times exceeding 100 h are not shown.
Consistent cell culture performance is an important hallmark of any biopharmaceutical manufacturing process to achieve desired productivity and product quality attributes. Here, the extended passaging of cell cultures before cell banking may raise concerns about the evolution of the clonal cultures, as individual MDCK cells can show chromosomal alterations upon long‐term culture [28]. However, the requirement for a certain stabilization period to achieve consistent cell growth properties was also described as a typical phenomenon in (CHO) CLD. Clearly, during extended passaging, every clonal culture will accumulate genetic and phenotypic heterogeneity but this plasticity can even elevate cell line performance [29, 30]. In previous studies on suspension MDCK cells, more than 50 passages (180 days) were required to achieve a full adaptation from Smif8 medium to Xeno medium [11]. Moreover, the cGMP cell bank of the clonal MDCK 9B9‐1E4 cell line developed by MedImmune (now AstraZeneca) for the pandemic influenza H5N1 vaccine was established after about 100 passages [20]. In contrast to our study, they first developed an adherent clonal cell line and only top cell clones were then adapted to serum‐free conditions over more than 25 passages. Furthermore, the clonal adherent MDCK 9B9‐1E4 cell line as well as the suspension MDCK‐33016PF (developed by Novartis, and now used for the manufacture of seasonal influenza vaccines by CSL Seqirus) displayed no/very low tumorigenic and oncogenic potential in extensive investigations and was found to be safe for human vaccine manufacturing [20, 31, 32]. Together, it seems reasonable to assume that the MDCK cell clones developed with our approach could pass licensing tests. However, cell line tumorigenicity and product safety have to be tested thoroughly for compliance with FDA and EMA guidelines.
3.3. Batch cultivation of monoclonal cultures to characterize cell growth
To further characterize the cell growth behavior of the monoclonal cultures, cells were seeded into shake flasks with fresh medium using a VCC of 8 × 105 cells/mL. After a short lag‐phase, monoclonal cell cultures grew exponentially reaching a maximum viable cell concentration (VCCmax) of 4–9 × 106 cells/mL (Figure 3A). Doubling times ranged between 26 and 47 h (Table 2); cell diameters were distinct between cultures and varied by about 3–4 µm (Figure 3B). After 2 days of cultivation, cell viability decreased in most cultures. In particular, the viability of cells of both C15 cultures dropped significantly (under 60%). Cell clone C59 reached the highest VCCmax of 8.8 × 106 cells/mL with a tD of 26 h, while maintaining a high cell viability (above 90 %) over the whole cultivation time.
FIGURE 3.
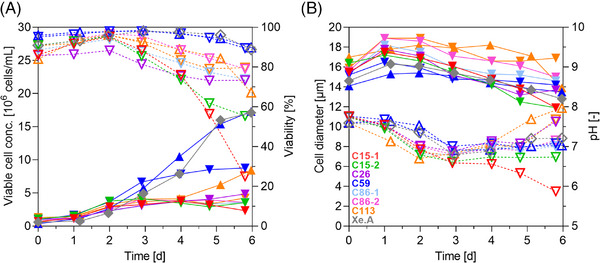
Batch cultivation of clonal MDCK cultures in non‐baffled shake flasks (30 mL wv MDXK medium). (A) Viable cell concentration (full symbols, solid lines), cell viability (dashed lines, empty symbols), (B) cell diameter (full symbols, solid lines) and pH value (dashed lines, empty symbols). Downward‐pointing triangles: cell clone from early cell bank (passage 33); Upward‐pointing triangles: C59 and C113 from later cell bank (passage 70), respectively; diamond‐shape, gray: Xe.A grown in 4Cell MDCK CD medium.
TABLE 2.
Growth characteristics of monoclonal suspension MDCK cells.
| Cell line/clone | VCCmax [106 cells/mL] | D [µm] | tD [h] | qGlc [fmol/(cell × h)] | qGln [fmol/(cell × h)] | qLac [fmol/(cell × h)] | qNH4 [fmol/(cell × h)] | Yglc/lac [−] | YNH4/gln [−] |
|---|---|---|---|---|---|---|---|---|---|
| C15‐1 | 3.8 | 15.5–17.5 | 31 | −135 | −48 | 240 | 49 | 0.6 | 1.0 |
| C15‐2 | 4.2 | 15.5–17.2 | 37 | −114 | −38 | 189 | 40 | 0.6 | 1.0 |
| C26 | 3.8 | 15.2–17.8 | 46 | −178 | −50 | 197 | 43 | 0.9 | 0.9 |
| C59 | 8.8 | 14.8–16.5 | 26 | −78 | −26 | 102 | 15 | 0.8 | 0.6 |
| C59‐p70 | 17.3 | 13.8–15.4 | 20 | −66 | −24 | 75 | 17 | 0.9 | 0.8 |
| C86‐1 | 3.7 | 15.3–18.2 | 42 | −143 | −35 | 216 | 35 | 0.7 | 1.0 |
| C86‐2 | 4.1 | 17.1–18.9 | 33 | −130 | −39 | 180 | 34 | 0.7 | 0.9 |
| C113 | 4.2 | 15.9–18.9 | 47 | −140 | −44 | 174 | 41 | 0.8 | 0.9 |
| C113‐p70 | 5.5 | 17.0–19.6 | 22 | −217 | −72 | 262 | 79 | 0.8 | 1.1 |
| Xe.A | 17.1 | 13.7–16.3 | 20 | −54 | −18 | 72 | 12 | 0.7 | 0.6 |
Note: Maximum viable cell concentrations (VCCmax), cell diameter (d), doubling time (tD) for a growth period of 72 h for all cell clones and 96 h for reference Xe.A cells and C59‐p70 cells, respectively; consumption rates (qs) of the main metabolites glucose, glutamine, lactose and ammonium; negative values indicate uptake and positive values release (for profiles see Figure S1); Y represents the yield coefficient for the conversion of the respective metabolites determined from the absolute difference over time.
Although showing best growth properties, the glucose consumption rate (qGlc) for C59 was about twofold lower in comparison to the other clonal cultures. In addition, glutamine consumption rate (qGln) was also low (Table 2). In contrast, glutamine was fully depleted for C113 within 3 days of cultivation and glucose after 5 days. This was followed by a significant drop in lactate levels, indicating a metabolic shift from lactate release towards consumption, what has been observed before for suspension MDCK cells (see also Figure S1) [11]. Batch cultivation of cell clones C59 and C113 was later repeated from a cell bank prepared with a history of more than 70 passages with improved cell growth characteristics (C59‐p70, C113‐p70). Then, cell clone C59‐p70 reached a VCCmax of 17.3 × 106 cells/mL with a tD of 20 h, and the cell clone C113‐p70 achieved 5.5 × 106 cells/mL with a tD of 22 h, respectively. With that, C59‐p70 showed almost identical growth properties as the reference Xe.A that grew to 17.1 × 106 cells/mL with a tD of 20 h and displayed very similar metabolite consumption rates and yield coefficients (although cultivated in a different medium). Results obtained for Xe.A cells within this study, also matched previous results obtained for this cell line [33]. Overall, cell growth to high cell densities within doubling times of 20–30 h are ideal for vaccine manufacturing using continuous cell lines [23]. Moreover, the release of secondary by‐products such as ammonium and lactate should preferably be low, as their accumulation was shown to adversely impact cell growth and virus production [34]. Yet, specific tolerance levels towards inhibiting metabolic waste products are known to vary widely between cell lines [35]. Over a growth period of 6 days, ammonium levels in C59 cell cultures did not exceed 5 mM, while ammonium chloride concentrations above 7 mM were shown to drastically decrease cell growth rates and, at 20 mM, even prevented influenza virus entry into the cell [35, 36]. Finally, lactate levels for C59 cells were lower than those described for MDCK cells in a previous study [37]. Different growth profiles for cell clones as observed in this study are also commonly known in CHO CLD. Even subcloning of clonally‐derived CHO cell lines further yielded widely diverse cell clones differing in growth properties, productivity, and even product quality [38]. However, the shown growth profiles and maximum cell concentrations match typical batch cultivations of several cell lines considered for influenza vaccine manufacturing, including MDCK cells, but also avian cell lines as, for instance, AGE1.CR or DuckCelt‐T17 cells [10, 11, 18, 39]. In summary, we identified C59 as top growing cell clone with a growth behavior that is competitive to Xe. A cells and other suspension cell lines.
3.4. Screening for IAV production in ambr15 to identify high‐yield cell clone
Next, we evaluated virus yields in batch cultures. In a preliminary shake flask cultivation, we identified a suitable concentration of a recombinant trypsin alternative for establishment of an animal component‐free production process. In addition, we could confirm the production of an A(H1N1) virus, precisely A/PR8 (see Table 1). Here, all cell clones reached titers ranging from 2.0 to 2.6 log10(HAU/100 µL) and 2 × 107 to 3 × 108 TCID50/mL within less than 3 days (data not shown). After those pre‐tests, cell clones C15‐1 and C26 were excluded from further experiments due to aggregate formation and poor growth properties (data not shown).
However, in such an uncontrolled shake flask environment, pH values can drop rapidly, which may adversely affect virus titer and stability. Moreover, cell growth under shaken conditions can vary from stirred conditions in a STR. To overcome this, we transferred the screening experiment into an ambr15 system that allows for tight process control due to integrated probes, sparging and automated base addition and the comparison of 24 vessels in parallel. Process parameters were matched across scales according to previously determined conditions for the Xe.A cell line [27]. Furthermore, all set points were fixed, except for the impeller speed. Here, agitation was increased from 80 rpm in the 1 L STR to 350 rpm in the ambr15 to achieve a constant tip speed of 0.21 m/s across scales. For maximal comparability, cells were seeded in trypsin‐containing medium into the ambr15 vessels (at 2.0 × 106 cells/mL) and were directly infected.
As expected, cell growth was rather limited due to infection and the reduced temperature, but viability remained high during the first hours post infection (hpi) (Figure 4A). Between 36 to 48 hpi, maximum titers were reached and cell viabilities declined as infected cells started to die (Figure 4A,B). Interestingly, strong differences in cell metabolism could be observed for infected cell clones. Within 2 days, for C113, glucose and glutamine were almost completely depleted, which resulted in a strong accumulation of lactate and ammonium. In contrast, cell metabolism of C59 seemed to be less demanding, and substrate levels remained high (Figure S2).
FIGURE 4.
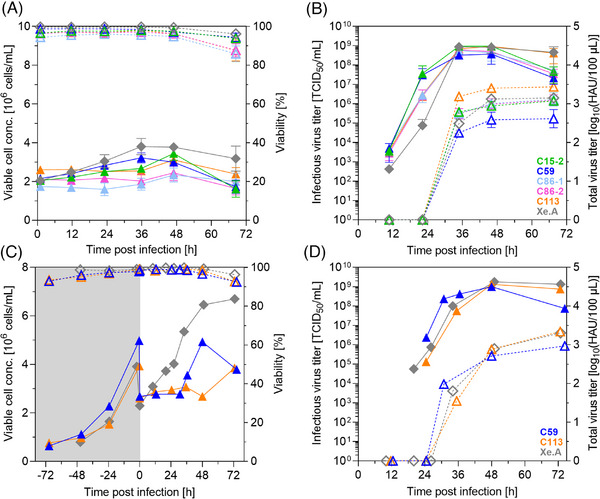
Screening of MDCK cell clones for A(H1N1) production in an ambr15 system and in 1 L STRs. Cultures with 2 × 106 cells/mL in trypsin‐containing medium were infected with A/PR8 at MOI 10−3 under standard conditions (triangles: clones in MDXK medium, diamond‐shape, gray: Xe.A in 4Cell medium). Cells were (A) directly seeded with target VCC in the ambr15 vessels, or (C) first grown in a 1 L STR system and subsequently diluted at time of infection. VCC (full symbols, solid lines) and cell viability (dashed lines, empty symbols); infectious (full symbols, solid lines) and total virus titer (dashed lines, empty symbols) determined by TCID50 and HA assay, respectively. (A, B) Mean and standard deviation of three culture vessels; 1 L STR (C, D): single runs. Gray area in (C) represents cell growth phase not shown in the other graphs.
All clonal cultures reached total and infectious virus titers close to or even above 3 log10(HAU/100 µL) and 3 × 108 TCID50/mL that we consider an internal benchmark for the H1N1 virus. In particular, requirements defined by Genzel and Reichl for a suitable influenza virus producing continuous cell line were achieved [23]. Titers were also similar as those obtained for the Xe.A cell line used as a reference [27]. Such high titers for all cell clones were rather surprising, as a high variability in CSVY of at least 20‐fold among different cell clones of MDCK cells was described previously in literature [20]. Similar observations were also made by Heldt et al. in single‐cell infection studies, where progeny virus titers in the range 1–970 plaque‐forming units and intracellular viral RNA levels spanning three orders of magnitude were obtained [22]. Considering virus yields, A/PR8 production in C113 achieved highest CSVYHA of 17,771 virions/cell, which is more than twofold in comparison to the 7694 virions/cells obtained from reference Xe.A (Table 3). In contrast, C59, which was previously identified as top growing cell clone, displayed the lowest titers; with 2277 virions/cells an 8‐fold lower CSVYHA compared to C113 and a 3.5‐fold lower CSVYHA compared to the Xe.A cell line. The highest TCIDmax and CSVYTCID was achieved for C15‐2 with 1.1 × 109 TCID50/mL and 321 infectious virions/cell, respectively. However, HA is the most important immunogenic antigen in influenza vaccines, and its concentration is used to standardize vaccine doses (usually 15 µg of HA per strain) [40]. Therefore, C59 and C113 were rated as the most promising cell clones for further scale‐up studies.
TABLE 3.
Summary of A(H1N1) production in different monoclonal suspension MDCK cell lines in ambr15 vessels and 1 L STR (MDXK medium).
| System | Cell line/clone | VCCmax [106 cells/mL] | HAmax [log10(HAU/100 µL)] | TCIDmax [109 TCID50/mL] | CSVYHA [virions/cell] | CSVYTCID [virions/cell] |
|---|---|---|---|---|---|---|
| ambr15 | C15‐2 | 3.6 ± 0.3 | 3.08 ± 0.08 | 1.1 ± 0.3 | 6977 | 321 |
| C59 | 3.2 ± 0.3 | 2.55 ± 0.16 | 0.4 ± 0.3 | 2277 | 135 | |
| C86‐1 | 2.5 ± 0.4 | 3.13 ± 0.17 | 0.6 ± 0.3 | 11,342 | 236 | |
| C86‐2 | 2.6 ± 0.4 | 3.08 ± 0.15 | 0.7 ± 0.3 | 9599 | 281 | |
| C113 | 3.1 ± 0.3 | 3.43 ± 0.03 | 0.9 ± 0.1 | 17,771 | 294 | |
| Xe.A | 4.0 ± 0.3 | 3.17 ± 0.06 | 1.0 ± 0.2 | 7694 | 258 | |
| 1 L STR | C59 | 4.93 | 2.97 | 1.0 | 3900 | 203 |
| C113 | 3.07 | 3.34 | 1.3 | 14,680 | 346 | |
| Xe.A | 6.46 | 3.31 | 1.8 | 6436 | 265 |
Note: Maximum viable cell concentration (VCCmax), maximum virus titers (HAmax and TCIDmax), and cell‐specific virus yields (HA‐based: CSVYHA, TCID50‐based: CSVYTCID). For the ambr15 system, mean and standard deviation of three vessels inoculated from same pre‐culture. STR results are from a single run. Xe.A: reference cultivated in 4Cell medium.
3.5. Process scale‐up from ambr15 vessels to 1 L benchtop bioreactor system
Scaling‐up production from miniaturized (10−2 L) to industrial scale manufacturing (about 105 L) can be challenging and requires thoughtful considerations. In this feasibility study, however, we only transferred the process for top cell clones C59 and C113 to a 1 L benchtop STR system using previously determined parameters for the reference Xe.A (also see Section 4.4) [27]. C59 was selected as the top growing cell clone with highly efficient metabolism, whereas cell clone C113 provided high CSVYs but lower cell growth performance and a less efficient metabolism.
Cells were seeded at a VCC of about 0.8 × 106 cells/mL and grown to concentrations of at least 4 × 106 cells/mL. Subsequently, cells were diluted two‐fold with fresh medium and infected with A/PR8 at a MOI of 10−3 at 34°C (Figure 4C,D). In line with the previous experiment in an ambr15 vessel, C113 reached the highest CSVYHA of 14,680 virions/cell, whereas the reference cells Xe.A and the cell clone C59 yielded a considerably lower value of 6436 virions/cell and 3900 virions/cell, respectively. Infectious and total virus yields were slightly lower in the 1 L STR in comparison to the ambr15 vessels, yet in a comparable range (Table 3). The small size (87 mm in height) and geometry of an ambr15 vessel (cuboid) may rise concerns about the scalability, as this results in a distinct fluid dynamic profile and requires a relatively high specific power input [41]. However, for protein production processes, several studies have shown that this does not relate to biological divergence and that production yields remain similar across scales (up to 15,000 L) [41, 42, 43, 44, 45]. Clearly, pH and oxygen control is beneficial for the ambr15 system compared to shake flasks and stirring as opposed to shaking can potentially yield advantageous outcomes during later scale‐up processes. Together, this can enable the reproducibility of cell performance under conventional bioreactor production conditions and mitigate the typical disparities encountered when transitioning from shake flask to STR [42, 43, 44, 45]. Currently, the ambr15 system is predominantly used in CHO CLD and to a lesser extent in the field of cell and gene therapy. For vaccine manufacturing, we are only aware of a SARS‐CoV‐2 production process simulation involving adherent Vero cells on macrocarriers, a virus‐like particle vaccine against Chikungunya virus and a cancer vaccine [46, 47, 48, 49]. With our study, we can add another example for virus production and showcase the usefulness of the system for screenings. Furthermore, we can confirm scalability up to a 50‐fold higher working volume, as we reached yields that were comparable or even increased compared to experiments performed previously using A/PR8‐infected MDCK cells. For suspension MDCK cells, a maximum CSVYHA of 8200 virions/cell was shown in batch mode, whereas in a study using adherent MDCK cells cultured in a single‐use hollow fiber bioreactor a CSVYHA between 2262 and 8110 virions/cell was reached, and in microcarrier cultures about 23,000 virions/cells [11, 50, 51]. As process parameters were adapted from the reference cell line Xe.A, it may raise the question whether process optimization and intensification studies for the newly identified cell clones can further improve yields. This may comprise the implementation of advanced feeding strategies, such as fed‐batch or perfusion that allows for higher cell densities by providing deprived nutrients and reducing toxic side‐products [11, 19, 24, 52, 53]. In previous studies, (semi) perfusion cultivations have been conducted using suspension MDCK cells that resulted in maximum CSVYs of 13,600 virions/cell and 11,690 virions/cell, respectively [11, 24]. Yet, yields obtained in this study with cell clone C113 were already higher than those intensified suspension MDCK processes.
3.6. Evaluation of further influenza vaccine‐relevant strains in top cell clones
Following WHO recommendations, seasonal influenza vaccines are formulated from bulks of three or four different influenza A and B virus strains. This usually comprises an influenza A(H1N1) and A(H3N2), as well as selected strains of the influenza B(Victoria) and B(Yamagata) lineages. Therefore, we next evaluated our top cell clone candidates for the yields of corresponding strains. Following a 3 days growth phase, cells were two‐fold diluted with trypsin‐containing medium and infected with one of the four different viruses at a MOI of 10−3 (Table 1). Infection with either B(Victoria) or B(Yamagata) resulted in good titers comparable to infections with A(H1N1) (Figure 5, Table 4). C113 cells infected with B(Yamagata) even resulted in a CSVYHA of 25,353 virions/cell, whereas C59 cells yielded 5140 virions/cell and the reference Xe.A cell line 8542 virions/cell. Infection with A(H3N2), however, showed a rather low infectious virus titer for both cell clones, and also for the Xe.A cell line; values were in the range of 0.8–1.3 × 106 TCID50/mL. In line with the low infectious titer, HA titers were also rather low. For the cell clone C59, the obtained value was even below the detection limit of the HA assay (Figure 5). However, these observations go in line with reports from literature, where influenza B virus (IBV) strains are described to be the least demanding to the cell properties, whereas some, that is, A(H3N2) strains, seem to be in general more difficult to propagate [20, 21]. Nevertheless, A(H3N2) strains play a crucial role in vaccine formulation, as they traditionally result in the most severe seasonal epidemics with the highest rates of infection, hospitalization, and mortality compared to outbreaks related to strains of other subtypes [54]. Thus, further evaluation and process optimization of such strains should be performed but this was considered out of the scope of this study.
FIGURE 5.
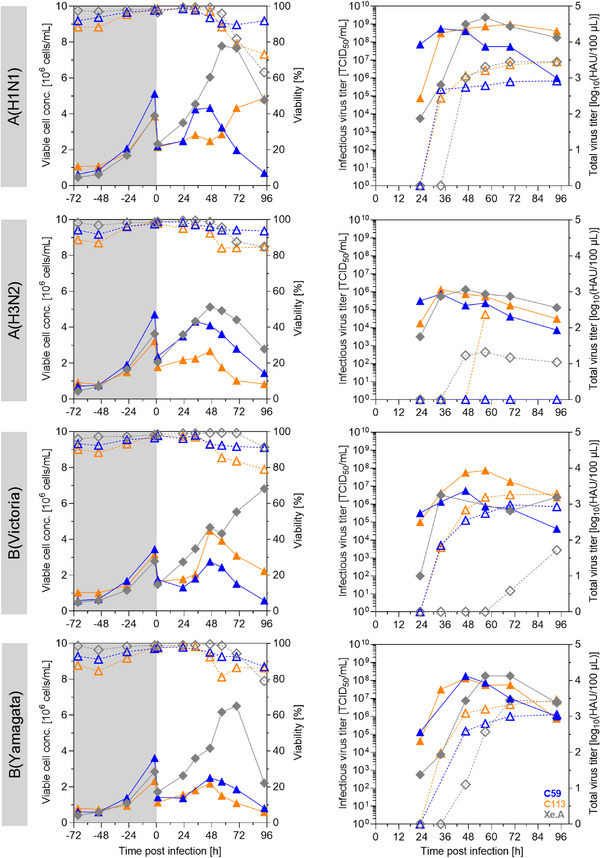
Production of influenza vaccine‐relevant virus strains using top cell clones in an ambr15 system. Cells were seeded into an ambr15 vessel and grown for 3 days (triangles: C59 and C113 in MDXK medium, diamond‐shape, gray: Xe.A in 4Cell medium). Subsequently, cells were two‐fold diluted in trypsin‐containing medium to about 2 × 106 cells/mL and infected with A(H1N1), A/(H3N2), B(Victoria), or B(Yamagata) at a MOI of 10−3. Left panel: Viable cell concentration (solid lines, full symbols) and cell viabilities (dashed lines, empty symbols), gray background: cell growth phase. Right panel: Infectious (solid lines, full symbols) and total (dashed lines, empty symbols) virus titers determined by HA and TCID50 assay, respectively.
TABLE 4.
Production of different influenza vaccine‐relevant strains in C59, C113, and Xe.A cells in ambr15 vessels.
| Virus | Cell line/clone | VCCmax [106 cells/mL] | HAmax [log10(HAU/100 µL)] | TCIDmax [TCID50/mL] | CSVYHA [virions/cell] | CSVYTCID [virions/cell] |
|---|---|---|---|---|---|---|
| A(H1N1) | C59 | 4.3 | 2.92 | 5.6 × 108 | 3784 | 130 |
| C113 | 2.9 | 3.45 | 1.0 × 109 | 19,413 | 350 | |
| Xe.A | 7.8 | 3.45 | 2.4 × 109 | 7137 | 305 | |
| A(H3N2) | C59 | 4.3 | n.d. | 7.5 × 105 | n.d. | <1 |
| C113 | 2.7 | 2.37 | 1.3 × 106 | 1730 | <1 | |
| Xe.A | 5.1 | 1.32 | 1.3 × 106 | 80 | <1 | |
| B(Victoria) | C59 | 2.8 | 2.98 | 5.6 × 106 | 6841 | 2 |
| C113 | 4.5 | 3.28 | 7.5 × 107 | 8398 | 17 | |
| Xe.A | 6.8 | 1.72 | 3.2 × 106 | 152 | <1 | |
| B(Yamagata) | C59 | 4.3 | 3.05 | 1.8 × 108 | 5140 | 41 |
| C113 | 2.2 | 3.45 | 1.3 × 108 | 25,353 | 61 | |
| Xe.A | 6.5 | 3.45 | 1.8 × 108 | 8542 | 27 |
Note: Maximum viable cell concentration (VCCmax), maximum titers (HAmax and TCIDmax), and cell‐specific virus yields (HA assay‐based: CSVYHA, TCID50 assay‐based: CSVYTCID). n.d.: not detectable, HA value below limit of detection (0.45 log10(HAU/100 µL)). All results depicted are from a single cultivation in ambr15 vessels.
In general, maximum titers for all three cell lines tested were very similar. However, viruses were released earlier for both clonal cultures (12 h) in comparison to Xe.A. This suggests not only a certain variability in virus production capacity among the different cell clones, but also differences in virus replication kinetics. Nevertheless, advanced process strategies could be applied to further maximize production (as described in Section 4.5). Additionally, virus adaptation by serial passaging could be carried out [12, 18]. Several studies support, that this should preferably be done in MDCK cells instead of embryonated chicken eggs, to avoid an antigenic drift towards avian cell receptors, which could lead to poor protection against virus strains circulating in the human population [2]. Together, our proof of concept study demonstrated that the selected cell clones enable the propagation of influenza strains relevant for human vaccine production with titers at a promising level.
4. CONCLUDING REMARKS
With the ever‐present need to efficiently produce influenza vaccines, the pursuit of an optimal host cell line has become a paramount objective, driving both academic and industrial efforts towards a responsive solution, particularly essential for pandemic strain vaccine production. Robust suspension growth, high viability, scalable processes, and rapid virus production may characterize such an optimal host cell line. In the field of recombinant protein production, CLD starting from single‐cell clones is a common practice, which has not only resulted in high‐yield cell lines but has also led to numerous technological innovations and platform processes. Yet, implementation of such state‐of‐the‐art technologies in viral vaccine process development is rarely seen. With our study, we are the first to demonstrate accelerated cloning of MDCK suspension cells in a chemically defined medium using a CellCelector. As a proof of concept, we generated five monoclonal cell lines and the follow‐up screenings could be easily handled when applying a microbioreactor systems such as the ambr15 system. Already at 15 mL scale, remarkable differences in virus replication dynamics, titers, and yields of the cell clones were detected. Based on the high‐quality data obtained, process performance for top cell clones could successfully be confirmed at 1 L scale. All selected cell clones allowed to propagate A/PR8 to high titers and even outperformed (C113) those of a heterogeneous suspension cell line (Xe.A) used as reference. Moreover, replication of three other influenza strains could be demonstrated at promising levels. Overall, this approach could make a paradigm shift for future cell culture‐based process development and optimization strategies in viral vaccine manufacturing.
AUTHOR CONTRIBUTIONS
Conceptualization: T.Z., D.A., K.T., U.R., and Y.G. Methodology: T.Z., N.B., K.T., and Y.G. Investigation: T.Z. and N.B. Writing—original draft: T.Z. Writing—review and editing: T.Z., N.B., D.A., K.T., U.R., and Y.G. Supervision: Y.G. and U.R. Project administration: T.Z., D.A., and Y.G.
CONFLICT OF INTEREST STATEMENT
Y.G. and U.R. have declared no conflict of interest. T.Z. was supported by a grant from Sartorius Stedim Biotech GmbH. N.B., D.A., and K.T. were employed at Sartorius Stedim Biotech GmbH.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The authors would like to thank Angelika Hinkelmann, Corina Siewert, and Emelie Wicke for their excellent technical support and Sven Göbel for scientific advice. Initiation and support of this project by Carole Langlois is highly appreciated. Moreover, we would like to thank Sartorius Stedim Biotech GmbH for providing equipment, such as an ambr15 system and respective consumables as well as 4Cell and MDXK medium.
Zinnecker T, Badri N, Araujo D, Thiele K, Reichl U, Genzel Y. From single‐cell cloning to high‐yield influenza virus production – implementing advanced technologies in vaccine process development. Eng Life Sci. 2024;24:e2300245. 10.1002/elsc.202300245
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Sparrow E, Wood JG, Chadwick C, et al. Global production capacity of seasonal and pandemic influenza vaccines in 2019. Vaccine. 2021;39: 512‐520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rockman S, Laurie K, Ong C, et al. Cell‐based manufacturing technology increases antigenic match of influenza vaccine and results in improved effectiveness. Vaccines (Basel). 2022;11: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hegde NR. Cell culture‐based influenza vaccines: a necessary and indispensable investment for the future. Hum Vaccin Immunother. 2015;11: 1223‐1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Silva CAT, Kamen AA, Henry O. Recent advances and current challenges in process intensification of cell culture‐based influenza virus vaccine manufacturing. Can J Chem Eng. 2021;99: 2525‐2535. [Google Scholar]
- 5. Doroshenko A, Halperin SA. Trivalent MDCK cell culture‐derived influenza vaccine Optaflu®(Novartis Vaccines). Expert Rev Vaccines. 2014;8: 679‐688. [DOI] [PubMed] [Google Scholar]
- 6. Rajaram S, Wojcik R, Moore C, et al. The impact of candidate influenza virus and egg‐based manufacture on vaccine effectiveness: literature review and expert consensus. Vaccine. 2020;38: 6047‐6056. [DOI] [PubMed] [Google Scholar]
- 7. Tihanyi B, Nyitray L. Recent advances in CHO cell line development for recombinant protein production. Drug Discov Today Technol. 2020;38: 25‐34. [DOI] [PubMed] [Google Scholar]
- 8. Wright C, Alves C, Kshirsagar R, Pieracci J, Estes S. Leveraging a CHO cell line toolkit to accelerate biotherapeutics into the clinic. Biotechnol Prog. 2017;33: 1468‐1475. [DOI] [PubMed] [Google Scholar]
- 9. Göbel S, Kortum F, Chavez KJ, et al. Cell‐line screening and process development for a fusogenic oncolytic virus in small‐scale suspension cultures. Appl Microbiol Biotechnol. 2022;106: 4945‐4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang D, Peng WJ, Ye Q, et al. Serum‐free suspension culture of MDCK cells for production of influenza H1N1 vaccines. Plos One. 2015;10(11): e0141686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bissinger T, Fritsch J, Mihut A, et al. Semi‐perfusion cultures of suspension MDCK cells enable high cell concentrations and efficient influenza A virus production. Vaccine. 2019;37: 7003‐7010. [DOI] [PubMed] [Google Scholar]
- 12. Genzel Y, Dietzsch C, Rapp E, Schwarzer J, Reichl U. MDCK and Vero cells for influenza virus vaccine production: a one‐to‐one comparison up to lab‐scale bioreactor cultivation. Appl Microbiol Biotechnol. 2010;88: 461‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Petiot E, Jacob D, Lanthier S, Lohr V, Ansorge S, Kamen AA. Metabolic and kinetic analyses of influenza production in perfusion HEK293 cell culture. BMC Biotechnol. 2011;11:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lohr V, Genzel Y, Jordan I, et al. Live attenuated influenza viruses produced in a suspension process with avian AGE1.CR.pIX cells. BMC Biotechnol. 2012;12:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pau MG, Ophorst C, Koldijk MH, Schouten G, Mehtali M, Uytdehaag F. The human cell line PER.C6 provides a new manufacturing system for the production of influenza vaccines. Vaccine. 2001;19:2716‐2721. [DOI] [PubMed] [Google Scholar]
- 16. Schuind A, Segall N, Drame M, Innis BL. Immunogenicity and safety of an EB66 cell‐culture‐derived influenza A/Indonesia/5/2005(H5N1) AS03‐adjuvanted vaccine: a Phase 1 randomized trial. J Infect Dis. 2015;212:531‐541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Genzel Y, Behrendt I, Rodig J, et al. CAP, a new human suspension cell line for influenza virus production. Appl Microbiol Biotechnol. 2013;97:111‐122. [DOI] [PubMed] [Google Scholar]
- 18. Petiot E, Proust A, Traversier A, et al. Influenza viruses production: evaluation of a novel avian cell line DuckCelt(R)‐T17. Vaccine. 2018;36:3101‐3111. [DOI] [PubMed] [Google Scholar]
- 19. Granicher G, Coronel J, Pralow A, et al. Efficient influenza A virus production in high cell density using the novel porcine suspension cell line PBG.PK2.1. Vaccine. 2019;37:7019‐7028. [DOI] [PubMed] [Google Scholar]
- 20. Liu J, Mani S, Schwartz R, Richman L, Tabor DE. Cloning and assessment of tumorigenicity and oncogenicity of a Madin‐Darby canine kidney (MDCK) cell line for influenza vaccine production. Vaccine. 2010;28:1285‐1293. [DOI] [PubMed] [Google Scholar]
- 21. Lugovtsev VY, Melnyk D, Weir JP. Heterogeneity of the MDCK cell line and its applicability for influenza virus research. PLoS ONE. 2013;8(9):e75014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heldt FS, Kupke SY, Dorl S, Reichl U, Frensing T. Single‐cell analysis and stochastic modelling unveil large cell‐to‐cell variability in influenza A virus infection. Nat Commun. 2015;6:8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Genzel Y, Reichl U. Continuous cell lines as a production system for influenza vaccines. Expert Rev Vaccines. 2009;8:1681‐1692. [DOI] [PubMed] [Google Scholar]
- 24. Wu Y, Bissinger T, Genzel Y, Liu X, Reichl U, Tan WS. High cell density perfusion process for high yield of influenza A virus production using MDCK suspension cells. Appl Microbiol Biotechnol. 2021;105:1421‐1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kalbfuss B, Knochlein A, Krober T, Reichl U. Monitoring influenza virus content in vaccine production: precise assays for the quantitation of hemagglutination and neuraminidase activity. Biologicals. 2008;36:145‐161. [DOI] [PubMed] [Google Scholar]
- 26. Genzel Y, Rodig J, Rapp E, Reichl U. Vaccine production: upstream processing with adherent or suspension cell lines. Methods Mol Biol. 2014;1104:371‐393. [DOI] [PubMed] [Google Scholar]
- 27. Bissinger T, Wu Y, Marichal‐Gallardo P, et al. Towards integrated production of an influenza A vaccine candidate with MDCK suspension cells. Biotechnol Bioeng. 2021;118:3996‐4013. [DOI] [PubMed] [Google Scholar]
- 28. Cassio D. Long term culture of MDCK strains alters chromosome content. BMC Res Notes. 2013;6:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frye C, Deshpande R, Estes S, et al. Industry view on the relative importance of “clonality” of biopharmaceutical‐producing cell lines. Biologicals. 2016;44:117‐122. [DOI] [PubMed] [Google Scholar]
- 30. Wurm MJ, Wurm FM. Naming CHO cells for bio‐manufacturing: genome plasticity and variant phenotypes of cell populations in bioreactors question the relevance of old names. Biotechnol J. 2021;16:e2100165. [DOI] [PubMed] [Google Scholar]
- 31. Vepachedu RS, Menon A, Hussain AI, Liu J. Evaluation of tumorigenic potential of high yielding cloned MDCK cells for live‐attenuated influenza vaccine using in vitro growth characteristics, metastatic gene expression and in vivo nude mice model. Biologicals. 2012;40:482‐494. [DOI] [PubMed] [Google Scholar]
- 32. Onions D, Egan W, Jarrett R, Novicki D, Gregersen JP. Validation of the safety of MDCK cells as a substrate for the production of a cell‐derived influenza vaccine. Biologicals. 2010;38:544‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bissinger T. Evaluation of MDCK suspension cell lines for influenza A virus production: media, metabolism, and process conditions. Doctoral dissertation, Otto‐von‐Guericke‐Universität Magdeburg. 2020.
- 34. Schneider M, Marison IW, von Stockar U. The importance of ammonia in mammalian cell culture. J Biotechnol. 1996;46:161‐185. [DOI] [PubMed] [Google Scholar]
- 35. Hassell T, Gleave S, Butler M. Growth inhibition in animal cell culture. The effect of lactate and ammonia. Appl Biochem Biotechnol. 1991;30:29‐41. [DOI] [PubMed] [Google Scholar]
- 36. Morris SJ, Price GE, Barnett JM, Hiscox SA, Smith H, Sweet C. Role of neuraminidase in influenza virus‐induced apoptosis. J Gen Virol. 1999;80(1):137‐146. [DOI] [PubMed] [Google Scholar]
- 37. Genzel Y, Behrendt I, Konig S, Sann H, Reichl U. Metabolism of MDCK cells during cell growth and influenza virus production in large‐scale microcarrier culture. Vaccine. 2004;22:2202‐2208. [DOI] [PubMed] [Google Scholar]
- 38. Ko P, Misaghi S, Hu Z, et al. Probing the importance of clonality: single cell subcloning of clonally derived CHO cell lines yields widely diverse clones differing in growth, productivity, and product quality. Biotechnol Prog. 2018;34:624‐634. [DOI] [PubMed] [Google Scholar]
- 39. Lohr V, Rath A, Genzel Y, Jordan I, Sandig V, Reichl U. New avian suspension cell lines provide production of influenza virus and MVA in serum‐free media: studies on growth, metabolism and virus propagation. Vaccine. 2009;27:4975‐4982. [DOI] [PubMed] [Google Scholar]
- 40. Yu ED, Grifoni A, Sutherland A, et al. Balanced cellular and humoral immune responses targeting multiple antigens in adults receiving a quadrivalent inactivated influenza vaccine. Vaccines. 2021;9(5):426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nienow AW, Rielly CD, Brosnan K, et al. The physical characterisation of a microscale parallel bioreactor platform with an industrial CHO cell line expressing an IgG4. Biochem Eng J. 2013;76:25‐36. [Google Scholar]
- 42. Hsu WT, Aulakh RP, Traul DL, Yuk IH. Advanced microscale bioreactor system: a representative scale‐down model for bench‐top bioreactors. Cytotechnology. 2012;64:667‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Arena TA, Chou B, Harms PD, Wong AW. An anti‐apoptotic HEK293 cell line provides a robust and high titer platform for transient protein expression in bioreactors. MAbs. 2019;11:977‐986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rameez S, Mostafa SS, Miller C, Shukla AA. High‐throughput miniaturized bioreactors for cell culture process development: reproducibility, scalability, and control. Biotechnol Prog. 2014;30:718‐727. [DOI] [PubMed] [Google Scholar]
- 45. Janakiraman V, Kwiatkowski C, Kshirsagar R, Ryll T, Huang YM. Application of high‐throughput mini‐bioreactor system for systematic scale‐down modeling, process characterization, and control strategy development. Biotechnol Prog. 2015; 31:1623‐1632. [DOI] [PubMed] [Google Scholar]
- 46. Jayson A, Goldvaser M, Dor E, et al. Application of Ambr15 system for simulation of entire SARS‐CoV‐2 vaccine production process involving macrocarriers. Biotechnol Prog. 2022;38:e3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rosen O, Jayson A, Goldvaser M, et al. Optimization of VSV‐DeltaG‐spike production process with the Ambr15 system for a SARS‐COV‐2 vaccine. Biotechnol Bioeng. 2022;119:1839‐1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen P, Demirji J, Ivleva VB, Horwitz J, Schwartz R, Arnold F. The transient expression of CHIKV VLP in large stirred tank bioreactors. Cytotechnology. 2019;71:1079‐1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gobel S, Jaen KE, Fernandes RP, et al. Characterization of a quail suspension cell line for production of a fusogenic oncolytic virus. Biotechnol Bioeng. 2023;120(11):3335‐3346. [DOI] [PubMed] [Google Scholar]
- 50. Tapia F, Vogel T, Genzel Y, et al. Production of high‐titer human influenza A virus with adherent and suspension MDCK cells cultured in a single‐use hollow fiber bioreactor. Vaccine. 2014;32:1003‐1011. [DOI] [PubMed] [Google Scholar]
- 51. Peschel B, Frentzel S, Laske T, Genzel Y, Reichl U. Comparison of influenza virus yields and apoptosis‐induction in an adherent and a suspension MDCK cell line. Vaccine. 2013;31:5693‐5699. [DOI] [PubMed] [Google Scholar]
- 52. Genzel Y, Vogel T, Buck J, et al. High cell density cultivations by alternating tangential flow (ATF) perfusion for influenza A virus production using suspension cells. Vaccine. 2014;32:2770‐2781. [DOI] [PubMed] [Google Scholar]
- 53. Coronel J, Granicher G, Sandig V, Noll T, Genzel Y, Reichl U. Application of an inclined settler for cell culture‐based influenza a virus production in perfusion mode. Front Bioeng Biotechnol. 2020;8:672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Barr IG, Donis RO, Katz JM, et al. Cell culture‐derived influenza vaccines in the severe 2017–2018 epidemic season: a step towards improved influenza vaccine effectiveness. NPJ Vaccines. 2018;3:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.