

Abstract
Tumors that develop in the genetic LSL-K-rasG12D murine lung cancer model are resistant to anti-PD-1 antibody treatment. Analysis of tumor-bearing lungs from anti-PD-1-treated mice revealed an up to 2.5-fold increase in IL-17-producing T-cells, with minimal change in CD8+ T-cell activity. Neutralization of IL-17 concurrent with anti-PD-1 treatment on the other hand, resulted in robust CD8+ T-cell activation and a threefold reduction in tumor burden. Loss-of-function studies demonstrated that anti-PD-1 driven activation of CD4+ and γδTCR+ T-cells contributed to IL-17-mediated de-sensitization of CD8+ cytotoxic T-cells (CTL) to therapy; and that CTL activation was critical to tumor eradication. Importantly, post-therapy lung Th17 cell prevalence and activity prognosticated treatment efficacy. Consistent with the murine data, analysis of tumor biopsy samples from non-small cell lung cancer (NSCLC) patients revealed that pre-therapy intratumoral CD8+/RORc+ cell ratio correlated with response to immune checkpoint blockade (ICB). These findings provide the initial evidence for a new mechanism of ICB resistance in lung cancer.
Electronic supplementary material
The online version of this article (10.1007/s00262-020-02795-2) contains supplementary material, which is available to authorized users.
Keywords: Anti-PD-1, NSCLC, IL-17, Checkpoint blockade resistance, CD4 T-cell, Th17 cell
Introduction
Immune checkpoint inhibitors, in particular anti-PD-1 antibodies, represent a new paradigm in the management of NSCLC [1]. At the same time, while effective in a subset of patients as first or second line therapy, anti-PD-1 still fails in 50–75% of NSCLC patients [1]. High tumor PD-L1 expression and neoantigen burden correlate with anti-PD-1 responsiveness, and in part may explain the suboptimal response rates [1, 2]. However, 30–40% of PD-L1-positive and high neoantigen burden tumors still do not respond while up to 14% of low neoantigen tumors do respond [3] suggesting that other, yet unidentified, factors contribute to resistance. To this end, the broader tumor immune signature, the functional ontogeny of tumor-infiltrating CD8+ T-cells as well as the commensal microbiota have been identified as other contributors to responsiveness, but are yet to provide specific measurable prognostic markers in the clinical setting [2].
To gain further insight into this conundrum we investigated the therapeutic potential of anti-PD-1 antibody in the checkpoint blockade-resistant, low-neoantigen burden LSL-K-rasG12D murine spontaneous lung cancer model [4–6]. Anti-PD-1 antibody had no detectable therapeutic benefit in LSL-K-rasG12D mice, confirming previous findings [6]. Phenotypic and functional analyses of lung T-cell populations revealed that anti-PD-1-mediated activation of lung-intrinsic Type 17 T-cells (T17 cells) directly interfered with the ability of the antibody to activate antitumor CD8+ T-cells, and that post-therapy lung Th17 cell prevalence was predictive of therapeutic outcome in individual mice. Consistent with these findings, preliminary analysis of NSCLC patient tumor biopsies revealed a correlation between intratumoral CD8+/RORc+ cell ratio and tumor responsiveness to ICB. This is the first report of a potential role for anti-PD1-mediated T17 cell activation in resistance to ICB.
Materials and methods
Mice and tumor model
LSL-K-rasG12D (B6.129S4-Krastm4Tyj/J) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Tumors were induced as described previously [7]. All experiments were approved by the University IACUC.
Patient samples
De-identified NSCLC patient tumor biopsy specimens were obtained from the Brown Cancer Center Biorepository, University of Louisville. Tissue sections prepared from formalin-fixed, paraffin-embedded tumor samples were processed for RNAscope. The study was approved by the University Institutional Review Board.
Microsphere preparation and treatment
Anti-PD-1 antibody-encapsulated biodegradable polylactic acid microspheres were prepared as previously described [7]. Two formulations were produced: (1) control (no antibody) and (2) anti-mouse CD279 (PD-1) antibody (clone J43, BioXCell, West Lebanon, NH) with a loading of 8 μg antibody/mg of particles. Control or anti-PD-1 microspheres (0.125 mg particles in 35 μl sterile water) were administered via intubation-mediated intratracheal instillation (IMIT) 2x/week for 4 weeks starting 6 weeks after adenoviral infection [7]. Soluble antibody (200 µg in 0.2 ml saline) was administered i.p. 3x/week.
In vivo antibody-mediated leukocyte subset depletion and cytokine neutralization
Depletion was performed by i.p. injection of 250 μg of anti-mouse CD4 (clone GK1.5), CD8 (clone 53-6.72, BioXCell) or via i.v. injection of γδTCR antibody (clone UC7-13D5, BioXCell or Leinco Technologies) 3x/week for 4 weeks. For in vivo neutralization of IL-17, 100 μg anti-mouse IL-17A (clone 17F3; BioXCell) was administered i.p. 3x/week for 4 weeks.
Tumor quantification
Lung tumor burden was quantified by digital imaging analysis of H&E-stained serial lung sections as described previously [7]. QuPath open source software was used to quantify lesion vs total lung area per section.
Isolation of lung mononuclear cells
Lung mononuclear cells were isolated as previously described [7].
Antibodies and flow cytometry
Fluorescence-conjugated anti-CD4 (RM4-5), anti-CD8a (53-6.7), anti-γδTCR (GL3), anti-CD11b (M1/70), anti-Gr-1 (RB6-8C5), anti-Ly-6G (1A8), anti-IL-17A (TC11-18H10.1), anti-IFN-γ (XMG1.2) and anti-RORγt (Q31-378), were purchased from BioLegend (San Diego, CA), eBioscience (Waltham, MA) or BD Biosciences (San Jose, CA). Foxp3 (PJK-16s; eBioscience) was quantified by intracellular staining performed according to the manufacturer’s protocol. For intracellular cytokines, cells were stimulated for 4 h with PMA and ionomycin (Sigma-Aldrich, St. Louis, MO) in the presence of brefeldin A (Sigma-Aldrich) and stained with antibodies. For CD107a degranulation assay, cells were cultured in RPMI1640 with anti-CD3 (10 µg/ml) and anti-CD28 (1 µg/ml) in 96-well plates for 24 h; were washed and re-stimulated with PMA and ionomycin in the presence of anti-CD107a (1D4B, BioLegend) for an additional 4 h.
Single-molecule RNA in situ hybridization
RNAscope [8] was performed at Advanced Cell Diagnostics (Newark, CA). Briefly, manual chromogenic staining was performed with paired double-Z oligonucleotide probes for CD8a green (cat. no. 560391) and RORc red (cat. no. 556991) using RNAscope® 2.5 HD Duplex Reagent Kit (cat. no. 322430) per manufacturer’s instructions. Each sample was quality controlled for RNA integrity with a probe specific to peptidylpropyl isomerase B. Negative control background staining was evaluated using a probe specific to the bacterial dapB gene. Stained slides were scanned with Aperio ScanScope slide scanner at 40 × objective resolution (Aperio Technologies, Vista, CA). High-resolution images taken with ObjectiveView (Digital Pathology Image Viewer) and TIFF images were then subjected to computerized analysis with Fuji software to quantify positive cells per field.
Statistical analysis
Student’s t test was used to determine the significance of the differences between control and experimental groups in pairwise comparisons. In experiments with multiple groups, homogeneity of intergroup variance was analyzed by one-way ANOVA with multiple pairwise comparisons using Tukey or Holm–Sidak analyses. A p value of < 0.05 was considered significant.
Results
Failure of anti-PD-1 therapy is associated with exacerbation of T17 cell activity
We previously demonstrated that intratracheal (i.t.) delivery of a sustained-release IL-10 formulation, but not systemic bolus cytokine, suppressed lung tumorigenesis in the LSL-K-rasG12D model [7]. To this end, we wanted to determine whether a similar formulation of anti-PD-1 antibody could overcome ICB-resistance in these mice. Animals with established lung adenomas were administered either the slow-release formulation or soluble anti-PD-1 i.t. or intraperitoneally (i.p.), respectively. Analysis of lung tumor burden in post-therapy mice demonstrated that treatment failed with either approach (Fig. 1a). To gain further insight into the observed lack of effect, global analysis of lung immune cell infiltrates was undertaken. Immune phenotyping of lung lymphocytes in treated vs control mice revealed a variable effect on CD8+ T-cell cytotoxicity, which did not reach statistical significance (Fig. 1b). In contrast, we observed significant twofold and 1.3-fold increases in Th17 and γδT17 cell activity, respectively, in mice treated with i.t., but not i.p., antibody (Fig. 1b). A minor but significant reduction in Th1 cells was also observed in mice receiving encapsulated antibody while no changes were detected in the prevalence of Treg or the myeloid cell subsets (Supplementary Fig. 1).
Fig. 1.
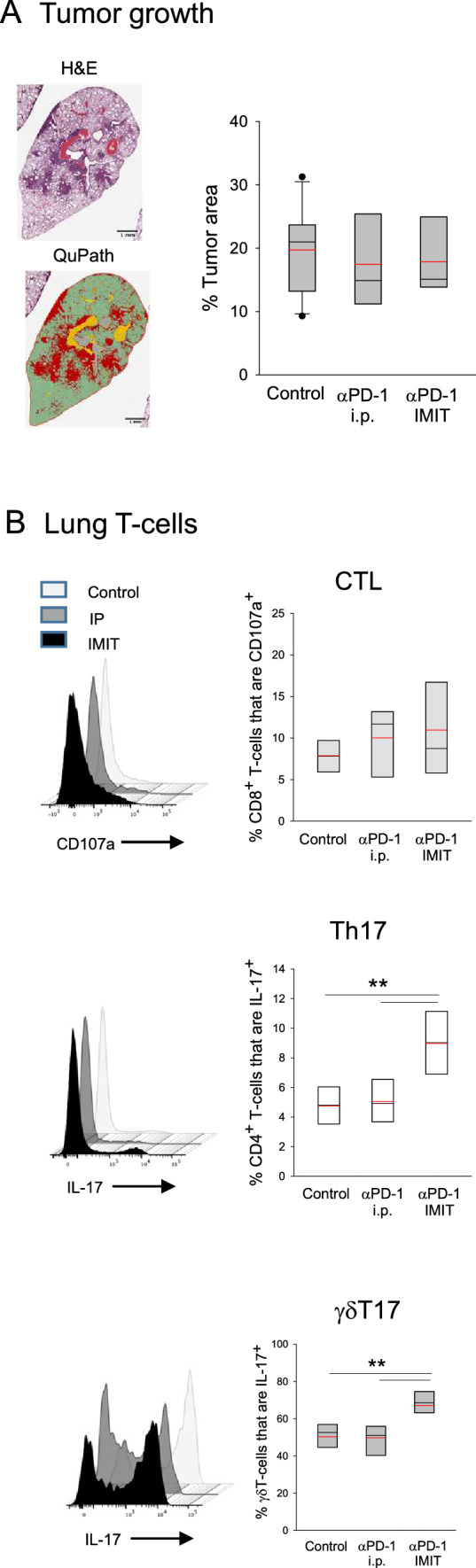
Effect of anti-PD-1 treatment on tumor growth and lung T-lymphocyte activity. a Tumor burden. Tumor-bearing mice were treated with soluble (i.p) or encapsulated (IMIT intubation-mediated intratracheal instillation) anti-PD-1 and lungs were analyzed for tumor burden. Representative lung histology (left upper) with accompanying QuPath analysis identifying tumor areas (left lower, in red) and quantitative data (right) are shown. There were no significant differences between the groups (n = 8–10 per group). b T-cell activity. Single cell suspensions prepared from the lungs were analyzed for cytotoxic CD8+ T-lymphocytes (CD8+CD107a+), Th17 cells (CD4+RORγt+IL-17+) and γδT17 cells (γδTCR+RORγt+IL-17+). Representative flow cytometry panels and quantitative data are shown (n = 5 per group). Boxes have lines at the median (black) and mean (red) showing lower (25%) and upper (75%) quartiles. Whiskers extend to show the 10th and 90th percentiles with symbols representing the extreme values (in groups with n ≥ 9). Significance: asterisks (**) denotes p < 0.01 (one-way ANOVA with pairwise multiple comparisons, Holm–Sidak)
Neutralization of IL-17 sensitizes LSL-K-rasG12D lung tumors to anti-PD-1 therapy
Next, we wanted to determine whether exacerbation of T17 cell immunity interfered with the ability of anti-PD-1 to stimulate CD8+ T-cell activity. Anti-PD-1 treatment in the presence of IL-17 neutralization resulted in effective tumor suppression in mice that received i.t., but not i.p. anti-PD-1 antibody (Fig. 2a). To gain further insight into the synergy, we examined the post-therapy lung T-cell landscape. Phenotypic analysis of single cell preparations revealed that, in contrast to anti-PD-1 monotherapy, which had no effect on CD8+ T-cells, anti-IL-17 + i.t. anti-PD-1 (but not i.p. anti-PD-1) treatment enhanced CD8+ T-cell membrane CD107a expression by twofold (Fig. 2a); suggestive of an antagonistic relationship between type 17 immunity and CTL reinvigoration.
Fig. 2.

Effect of IL-17 neutralization on anti-PD-1 therapy. a Effect of combined anti-PD-1 + anti-IL-17 antibody treatment on tumor growth and CD8+ T-cell cytotoxicity. Tumor-bearing mice were treated with anti-PD-1 alone i.p (soluble antibody) or via IMIT (slow-release formulation) in the presence or absence of IL-17 neutralization (αIL-17). Tumor burden (n = 8–14 per group) and quantitative cellular data for Th17 and CD8+CD107a+ cells including representative flow cytometry panels are shown (n = 4–5 per group). b Effect of IL-17 neutralization alone vs anti-PD-1 + anti-IL-17 antibody treatment on tumor growth and CD8+ T-cell cytotoxicity. Mice were treated with control blank microspheres, anti-IL-17 alone (soluble, i.p.), anti-PD-1 alone (encapsulated, via IMIT) or with anti-IL-17 + anti-PD-1. Tumor burden and cellular analysis data are shown (n = 5 per group). Significance: asterisks (*, **, ***) denote p ≤ 0.05, 0.01 and 0.001, respectively (one-way ANOVA with pairwise multiple comparisons, Dunn’s or Holm–Sidak)
Since neutralization of IL-17 alone can result in reduced tumor growth in the LSLKrasG12D lung cancer model [9], we next determined whether combinatorial treatment was superior to anti-IL-17 alone (using i.t. anti-PD-1 from this point on). Figure 2b data show that while IL-17 blockade alone resulted in a trend towards reduced tumor burden, statistical significance was reached only in the combination group with a > threefold reduction in tumor burden in comparison to controls. Consistent with this finding, analysis of lung T-cell subsets revealed that combination therapy resulted in more effective CD8+ T-cell activation than anti-IL-17 alone.
Th17 and γδT17 subsets contribute to anti-PD-1 resistance via distinct pathways
Next, we investigated the relative roles of Th17 and γδT17 cells in anti-PD-1 resistance. Tumor-bearing mice were treated with anti-PD-1 in the presence or absence of CD4+ or γδTCR+ T-cells and tumor burden was analyzed. Administration of anti-PD-1 antibody to CD4+ T-cell-depleted mice resulted in significant tumor suppression in comparison to CD4-sufficient mice (Fig. 3a), while depletion of CD4+ T-cells alone had no effect. We thus concluded that anti-PD-1-mediated activation of CD4+ T-cells played an important role in conferring resistance to therapy. In the case of γδTCR+ cell subset however, depletion alone was just as effective as depletion + anti-PD-1 treatment in comparison to anti-PD-1 alone, suggesting that constitutive IL-17 production by γδT-cells contributed to anti-PD-1 resistance (Fig. 3a).
Fig. 3.

Roles of T17 cell subsets and CD8+ T-cells in tumor responsiveness and prognosis. a Contribution of CD4+ and γδTCR+ cells to anti-PD-1 resistance. The roles of CD4+ T-cells (CD4+ T-cell depletion), γδT-cells (γδTCR+ T-cell depletion), or the two in combination (dual depletion) in resistance to anti-PD-1 were examined. Antibody treatments were as indicated on the abscissa for each plot. Control mice received blank microspheres (via IMIT) without any antibody treatment. Top and bottom panels display the effect of each specific treatment on tumor burden and CD8+ T-cell activity, respectively (n = 4–6 per group). b Role of CD8+ T-cells in tumor suppression. Mice were treated with anti-PD-1 + anti-IL-17 (denoted as Rx), in the presence or absence of CD8+ T-cell depletion. Control mice received blank microspheres (n = 8–13 mice per group). c Correlation of Th17 cell prevalence and tumor burden in post-therapy mice. Individual mice were analyzed for tumor burden and Th17 cell prevalence after anti-PD-1 + anti-IL-17 treatment (n = 12). Box plots: asterisks (*, **, ***) denote p ≤ 0.05, 0.01 and 0.001, respectively (one-way ANOVA with pairwise multiple comparisons, Holm–Sidak)
In parallel, analysis of CD8+ T-cells for membrane CD107a in mice that received anti-PD-1 in the absence or presence of CD4+ T-cells revealed that only the former group displayed significantly increased cytotoxicity (Fig. 3a, lower panels) while CD4+ T-cell depletion alone had no benefit. These data suggested that anti-PD-1-mediated CD4+ T-cell activation interfered with CTL reinvigoration and tumor kill, consistent with the tumor burden data. In the complementary study, analysis of post-therapy CD8+ T-cells in γδT-cell deficient vs sufficient mice indicated that anti-PD-1 induced significant CTL activation in the absence of γδTCR+ cells (Fig. 3a, lower panels). However, unlike what was observed with CD4+ T-cells, depletion of γδT-cells in the absence of anti-PD-1 also resulted in partial CTL activation, again consistent with a role for constitutive IL-17 production by these cells in anti-PD-1 resistance.
Based on these findings, treatment was undertaken in mice with dual depletion of CD4+ and γδTCR+ cells to determine whether this approach would replicate the IL-17-neutralization data. Treatment resulted in a ~ 2.5-fold reduction in tumor burden with a concurrent twofold increase in CD8+ T-cell cytotoxicity in CD4+ and γδTCR+ T-cell depleted mice (Fig. 3a, lower panels) consistent with the IL-17-blockade data. Combined depletion in the absence of treatment was also partially effective suggesting that constitutive IL-17 production, likely by γδT17 cells, maintained CD8+ T-cell suppression at steady-state.
CD8+ T-cells are critical to the ability of anti-PD-1 to induce tumor eradication in the presence of IL-17 neutralization
As the data supported a strong link between CD8+ T-cell activation and tumor suppression, we next performed a loss of function study to directly test this notion. Mice with established disease were treated with anti-IL-17 + anti-PD-1 antibodies in the presence or absence of CD8+ T-cells. Elimination of CD8+ T-cells led to a complete loss of therapeutic efficacy confirming that tumor eradication was strictly dependent on CD8+ T-cells (Fig. 3b).
Post-therapy Th17 cell prevalence is prognostic of treatment outcome in LSLKrasG12D mice
Above findings suggested that post-treatment T17 cell prevalence/activity could prognosticate treatment outcome. To this end, mice were analyzed individually for Th17, γδT17 and cytotoxic CD8+ T-cell prevalence and activity as well as tumor burden following combinatorial therapy. Regression analysis revealed a significant association between post-therapy Th17 cell prevalence and therapeutic outcome (Fig. 3c). In contrast, γδT17 and CTL prevalence, or the functional ratio of CTL to T17 subsets did not correlate with tumor burden (Supplementary Fig. 2a).
Tumor CD8+/RORc+ cell ratio correlates with checkpoint blockade responsiveness in NSCLC patients
To determine whether we could extend the murine findings to human, pre-treatment lung tumor biopsy samples of NSCLC patients who subsequently received ICB therapy (Table 1), were analyzed for CD8+ and RORc+ cell infiltrates by RNAscope (Fig. 4). Quantitative analysis revealed a statistically significant correlation between the ratio of tumor-infiltrating CD8+ to RORc+ cells and response to therapy. On the other hand, in contrast to the murine model, RORc+ cell prevalence alone was not predictive of ICB responsiveness (Supplementary Fig. 2b).
Table 1.
Individual data of study patients
| Case | Age at diagnosis | Sex | Smoking status | Pathology | Treatment line | ICB | PD-L1 expression (%) | CD8+/RORc+ cells per field‡ | Responder status* | PFS† | Status |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 67 | M | Ex-smoker | SCC | First | Pembrolizumab | Unknown | 1.75 | R | 385 | Expired |
| 2 | 63 | M | Ex-smoker | AC | First | Pembrolizumab | 50 | 3.18 | R | 987 | Alive |
| 3 | 74 | F | Current smoker | AC | First |
Pembrolizumab Atezolizumab |
Unknown | 1.87 | R | 420 | Expired |
| 4 | 66 | F | Ex-smoker | SCC | First | Pembrolizumab | 65 | 0.98 | NR | 168 | Expired |
| 5 | 65 | M | Ex-smoker | AC | First | Pembrolizumab | 0 | 0.48 | NR | 42 | Expired |
| 6 | 59 | F | Ex-smoker | AC | First | Pembrolizumab | 0 | 0.09 | NR | 126 | Expired |
| 7 | 61 | F | Current smoker | SCC | First | Pembrolizumab | 60 | 0.14 | NR | 21 | Expired |
| 8 | 62 | M | Ex-smoker | SCC | First |
Pembrolizumab Nivolumab |
0 | 0.3 | NR | 126 | Expired |
| 9 | 75 | F | Ex-smoker | AC | First | Pembrolizumab | 50 | 0.28 | NR | 126 | Expired |
‡Three distinct areas from each section were analyzed and the average numbers of CD8+ and RORc+ cells per representative field (400 ×) were determined
*Patients that maintained stable disease (SD) for at least 180 days post-treatment initiation were considered responders
†Progression-free survival indicates the time period (days) between first day of treatment and radiological assessment of progression
Fig. 4.

Analysis of patient tumor CD8+ and RORc+ cell infiltrates. Tissue sections prepared from tumor biopsies were analyzed for CD8a (blue-green) and RORc (red) mRNA by RNAscope. A representative image is shown on the left (magnification = × 400). Cells expressing CD8a or RORc are highlighted with circles of relevant color. Inset displays magnification of the area demarcated by the rectangle. The average ratio of CD8+ to RORc+ cell numbers were calculated for each patient sample and plotted according to response criteria (right panel). The difference between responders (progression free survival > 180 days, n = 3) and non-responders (n = 6) was significant (p < 0.01, Student’s t test)
Discussion
Our data establish that the failure of anti-PD-1 therapy in the LSL-K-rasG12D model is associated with an unexpected exacerbation of lung T17 cell activity that in turn antagonizes CD8+ T-cell reinvigoration. We further demonstrate that Th17 cells are the major responders to anti-PD-1 and that post-treatment Th17 cell prevalence is predictive of therapeutic efficacy in individual mice. Importantly, preliminary analysis of pre-therapy NSCLC patient lung samples revealed a statistically significant correlation between tumor CD8+/RORc+ cell ratio and subsequent ICB responsiveness, consistent with the murine data. We propose that the intensity of the anti-PD-1-Th17 cell axis may be an important determinant of ICB resistance.
The finding that both CD4+ and γδTCR+ T-cells contribute to IL-17-mediated CD8+ T-cell unresponsiveness, but that Th17 cells are the primary prognosticators of outcome for anti-PD-1 treatment is an intriguing observation. Our data suggest that constitutive production of IL-17 by γδT-cells vs anti-PD-1-dependent activation of quiescent Th17 cells may underlie this observation. One caveat is that some of our conclusions regarding the Th17 subset were derived from studies involving total CD4+ T-cell depletion, which cannot exclude potential contribution from T-regulatory cells. However, based on: (a) the identification of IL-17 as the central mediator of anti-PD-1 resistance, and (b) the nearly identical effects of IL-17 blockade and combined CD4+ + γδTCR+ cell depletion on anti-PD-1 efficacy, we expect T-regulatory cells to play a minor role in ICB resistance in this model.
ICB resistance of LSL-K-rasG12D lung tumors [6, 10, 11] has been attributed to low neoantigen burden [5] and suboptimal CTL activity [12]. At the same time, others have reported the presence of significant CD8+ T-cell infiltrates in the tumor-bearing lungs of LSL-K-rasG12D mice [13]. Our findings suggest that these tumors are intrinsically immunogenic and that the CD8+ T-cell infiltrates represent a bona fide antitumor response. Whether the observed CTL response in this model is driven by the G12D mutation [14–16] or involves other epitopes is yet to be determined.
IL-17 is a pleiotropic cytokine that can promote tumor growth directly or indirectly [17]. However, the mechanistic basis of the IL-17-CTL antagonism remains to be elucidated. Potential mechanisms include the IL-17-MDSC axis [18, 19], direct proliferative effects of IL-17 on dysplastic epithelial cells [20], and/or increased PD-L1 expression on epithelium [21].
An intriguing observation that arose from this study was that anti-PD-1 antibody was effective only when delivered locally as a sustained release formulation. A similar observation was previously made with IL-10 [7] suggesting that in this ICB resistant model, i.p. injection may not achieve the therapeutic antibody threshold in the lung, even in the presence of IL-17 blockade. Whether inhalable slow-release formulations represent a more effective alternative to i.v. antibody in individuals that are intrinsically resistant to ICB treatment is to be determined.
The prognostic data obtained in the murine model and the patient samples were conceptually consistent in that type 17 immunity was predictive of response in both. At the same time distinct markers, i.e., post-therapy Th17 cell prevalence in the case of mice and pre-therapy tumor CD8+/RORc+ cell ratio in patients, associated with responsiveness. Several factors including post- vs. pre-therapy analysis or total lung vs intratumoral assessment of cell populations (in mice vs humans, respectively); patient neoantigen and PD-L1 heterogeneity; and/or the limited patient cohort could all account for the differences observed. Regardless, our findings support a novel role for anti-PD-1-T17 axis in mediating resistance to ICB in lung cancer.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Author contributions
NKE: original concept, design and supervision of studies, data interpretation, manuscript preparation, funding. QL: study design, execution of experiments, data interpretation, manuscript preparation. PTN: analysis and interpretation of human data.
Funding
This work was supported by the Department of Defense Lung Cancer Research Program Concept award W81XWH1610185 (NKE) and Idea award W81XWH1910265 (NKE).
Data availability
All data will be made available upon request.
Compliance with ethical standards
Conflict of interest
N.K.E has ownership interest in Therapyx, Inc. The remaining authors declare no competing financial interests.
Ethical approval
Murine and human studies were approved by the University of Louisville IACUC and IRB, respectively.
Consent to participate
All authors consented to participate in these studies.
Consent for publication
All authors consent to publication of the findings.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Qingsheng Li, Email: qingsheng.li@louisville.edu.
Nejat K. Egilmez, Email: nejat.egilmez@louisville.edu
References
- 1.Proto C, Ferrara R, Signorelli D, et al. Choosing wisely first line immunotherapy in non-small cell lung cancer (NSCLC): what to add and what to leave out. Cancer Treat Rev. 2019;75:39–51. doi: 10.1016/j.ctrv.2019.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Tunger A, Sommer U, Wehner R, et al. The evolving landscape of biomarkers for anti-PD-1 or anti-PD-L1 therapy. J Clin Med. 2019 doi: 10.3390/jcm8101534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McFadden DG, Politi K, Bhutkar A, et al. Mutational landscape of EGFR-, MYC-, and Kras-driven genetically engineered mouse models of lung adenocarcinoma. Proc Natl Acad Sci USA. 2016;113:E6409–E6417. doi: 10.1073/pnas.1613601113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pfirschke C, Engblom C, Rickelt S, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity. 2016;44:343–354. doi: 10.1016/j.immuni.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Q, Anderson CD, Egilmez NK. Inhaled IL-10 suppresses lung tumorigenesis via abrogation of inflammatory macrophage-Th17 cell axis. J Immunol. 2018;201:2842–2850. doi: 10.4049/jimmunol.1800141. [DOI] [PubMed] [Google Scholar]
- 8.Wang F, Flanagan J, Su N, et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn. 2012;14:22–29. doi: 10.1016/j.jmoldx.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang SH, Mirabolfathinejad SG, Katta H, Cumpian AM, Gong L, Caetano MS, Moghaddam SJ, Dong C. T helper 17 cells play a critical pathogenic role in lung cancer. Proc Natl Acad Sci USA. 2014;111:5664–5669. doi: 10.1073/pnas.1319051111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ritzmann F, Jungnickel C, Vella G, et al. IL-17C-mediated innate inflammation decreases the response to PD-1 blockade in a model of Kras-driven lung cancer. Sci Rep. 2019;9:10353. doi: 10.1038/s41598-019-46759-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koyama S, Akbay EA, Li YY, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. doi: 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DuPage M, Cheung AF, Mazumdar C, Winslow MM, Bronson R, Schmidt LM, Crowley D, Chen J, Jacks T. Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell. 2011;19:72–85. doi: 10.1016/j.ccr.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Busch SE, Hanke ML, Kargl J, Metz HE, MacPherson D, Houghton AM. Lung cancer subtypes generate unique immune responses. J Immunol. 2016;197:4493–4503. doi: 10.4049/jimmunol.1600576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tran E, Ahmadzadeh M, Lu YC, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350:1387–1390. doi: 10.1126/science.aad1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cafri G, Yossef R, Pasetto A, et al. Memory T cells targeting oncogenic mutations detected in peripheral blood of epithelial cancer patients. Nat Commun. 2019;10:449. doi: 10.1038/s41467-019-08304-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tran E, Robbins PF, Lu YC, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. 2016;375:2255–2262. doi: 10.1056/NEJMoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuen DS, Kim BS, Chung Y. IL-17-producing cells in tumor immunity: friends or foes? Immune Netw. 2020;20:e6. doi: 10.4110/in.2020.20.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He D, Li H, Yusuf N, Elmets CA, Li J, Mountz JD, Xu H. IL-17 promotes tumor development through the induction of tumor promoting microenvironments at tumor sites and myeloid-derived suppressor cells. J Immunol. 2010;184:2281–2288. doi: 10.4049/jimmunol.0902574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akbay EA, Koyama S, Liu Y, et al. Interleukin-17A promotes lung tumor progression through neutrophil attraction to tumor sites and mediating resistance to PD-1 blockade. J Thorac Oncol. 2017;12:1268–1279. doi: 10.1016/j.jtho.2017.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang K, Kim MK, Di Caro G, et al. Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity. 2014;41:1052–1063. doi: 10.1016/j.immuni.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma YF, Chen C, Li D, Liu M, Lv ZW, Ji Y, Xu J. Targeting of interleukin (IL)-17A inhibits PDL1 expression in tumor cells and induces anticancer immunity in an estrogen receptor-negative murine model of breast cancer. Oncotarget. 2017;8:7614–7624. doi: 10.18632/oncotarget.1381919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data will be made available upon request.