

Abstract
Among diverse neurological immune-related adverse events (irAEs), autoimmune encephalitis, aseptic meningitis, Guillain-Barré syndrome (GBS), myasthenia gravis (MG), and myositis are particularly important. The clinical presentation may be different from that of patients with conditions unrelated to immune checkpoint inhibitors (ICIs). Many of the autoantibodies detected in patients’ sera are committed to the pathogenesis, while the clinical significance of such autoantibodies in cases of neurological irAEs is different from the significance in cases of typical neuronal disorders. A broad range of clinical symptoms complicates the diagnosis of autoimmune encephalitis. The clinical features of aseptic meningitis induced by classical drugs are different from those of aseptic meningitis induced by ICIs. Although autoantibodies against synaptic receptors or neuronal cell surface proteins are not detected, anti-Ma2 antibodies, which are onconeural antibodies against intracellular proteins, are detected in patients with autoimmune encephalitis associated with ICIs. GBS induced by ICIs sometimes shows gradual progression and a relapse of symptoms, suggesting chronic inflammatory demyelinating polyneuropathy. Bulbar symptoms and myasthenic crisis are frequently observed in ICI-induced MG. Anti-acetylcholine receptor antibodies are found in only half of patients with MG occurring as an irAE. ICI-induced myositis is accompanied by ocular muscle symptoms, such as ptosis and diplopia, which can suggest MG. Patients receiving ICI treatment present clinical features and laboratory findings that represent a mixture of MG and myositis. Anti-striational antibodies may act as biomarkers in cases in which MG and myositis overlap. A correct understanding of neurological adverse events is required to achieve the best management of patients.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00262-021-03053-9.
Keywords: Immune checkpoint inhibitors, Immune-related adverse events, Neurological disorders, Autoantibody
Introduction
Immunotherapy with monoclonal antibodies targeting cytotoxic T lymphocyte-associated antigen 4 and programmed cell death-1 and their ligand has become the standard cancer treatment for an increasing number of indications. As cancer treatment with these immune checkpoint inhibitors (ICIs) has become more common, the safety management of immune-related adverse events (irAEs) due to cancer immunotherapy has become more important. Today, cancer treatment is conducted in cooperation with organ specialists.
The American Society of Clinical Oncology (ASCO) has published a guideline for the management of neurological irAEs [1]. Since this guideline was published, further clinical findings with irAEs have been accumulated. Importantly, the clinical features of neurological irAEs are different from the usual clinical presentation. Both clinical oncologists and consulting neurologists are now expected to understand the disease as a new entity if it occurs as an irAE. The representative irAEs are encephalitis, aseptic meningitis, Guillain-Barré syndrome (GBS), myasthenia gravis (MG), and myositis. Many autoantibodies detected in the routine clinical settings are involved in the pathogenesis of these immune-mediated disorders.
The use of ICIs causes the activation of autoreactive T cell as well as CD8+ cytotoxic T cells against cancers. The activation of autoreactive CD4+ T cells modulate humoral immunity, enhancing preexisting autoantibodies or leading to the production of autoantibodies [2, 3]. Although the association between pathogenesis of these antibodies and irAEs is not fully elucidated, these autoantibodies are expected to be valuable diagnosis biomarkers (Table 1). The profiles of the autoantibodies in cases with irAEs are also different from those in routine settings. The significance of autoantibodies is gradually being elucidated.
Table 1.
Autoantibodies in neurological immune-related adverse events
| Autoantibodies | Associated diseases | Immune-related adverse events |
|---|---|---|
| Anti-synaptic receptors or neuronal cell surface proteins | Autoimmune encephalitis | Undetected; anti-N-methyl-D-aspartate receptor and anti-contactin-associated protein-like 2, rarely detected |
| Onconeural antibodies | Paraneoplastic syndrome | Anti-Ma2 more than anti-Hu, 40% of autoimmune encephalitis |
| Anti-ganglioside antibodies | Guillain-Barré syndrome | Undetected; anti-GM1, anti-GM2 and anti-GalNAc-GD1a, rarely detected |
| Anti-neuromuscular junction proteins | Myasthenia gravis | Anti-acetylcholine receptor, 50%; anti-muscle-specific kinase, undetected |
| Myositis-specific antibodies | Immune-mediated necrotizing myopathy, antisynthetase syndrome, and dermatomyositis | Undetected; anti-signal recognition particle, anti-aminoacyl transfer RNA synthetase and transcriptional intermediary factor 1γ, rarely detected |
| Anti-striational antibodies | Thymoma-associated myasthenia gravis | Anti-titin and/or anti-Kv1.4, 75% of cases with myasthenia and myositis overlap |
In this review, we focus on the associations between autoantibodies and representative neurological irAEs.
Autoimmune encephalitis
Autoimmune encephalitis is an increasingly recognized neuropsychiatric condition seen in patients of all ages. Diagnosing an occurrence of autoimmune encephalitis as an irAE can be a challenge, as patients present with a broad range of clinical symptoms including headache, fever, confusion, disorientation, memory impairment, somnolence, and gait ataxia. Tremors, seizure, and hallucination are also seen. Limbic encephalitis is a common phenotype of encephalitis related to ICIs [4]. Cerebellitis presenting gait ataxia, dysarthria, and opsoclonus-myoclonus syndrome is also described. However, unexpected clinical features can also be observed, such as those referred to as atypical syndrome [4]. Disease onset is typically acute to subacute over days to a few weeks. Since the fatality rate of encephalitis associated with ICIs is relatively high, it is important to make a prompt and correct diagnosis [5].
Brain MRI and cerebrospinal fluid (CSF) examinations are necessary to diagnose autoimmune encephalitis. T2-weighted MRI usually shows high intensity in the temporal lobes; however, there is no remarkable abnormality in patients with irAE encephalitis. Additionally, MRI examinations often demonstrate nonspecific findings or abnormal signals in uncommon regions such as the basal ganglia [6]. The CSF examination can reveal lymphocyte inflammation and elevated protein, but there may be no abnormalities. Some patients are difficult to diagnose with encephalitis due to the CSF and MRI findings being normal, and they are alternatively diagnosed simply with “encephalopathy.”
A wide range of disorders, such as encephalitis caused by a virus, tuberculosis, a fungus, or bacterial pathogens, metabolic or endocrine encephalopathy, and metastatic diseases must be excluded before the diagnosis of autoimmune encephalitis is made. Since elevated adenosine deaminase levels in the CSF are found in patients with ICI-induced encephalitis, special attention must be paid to exclude tuberculous meningitis [7].
Autoimmune encephalitis is associated with autoantibodies against synaptic receptors or neuronal cell surface proteins. Among these autoantibodies, those against N‐methyl‐D‐aspartic acid receptor, detected frequently in girls or young female patients with ovarian teratoma, are well known. However, these autoantibodies have been undetectable in ICI-induced encephalitis except in a few case reports [4, 8].
Autoimmune encephalitis appears as paraneoplastic syndrome in association with onconeural antibodies against intracellular proteins including anti‐Hu and anti‐Ma2 antibodies. Theoretically, the use of ICIs might increase the risk of paraneoplastic syndrome. Anti-Ma2 antibodies were detected in seven of 19 patients with autoimmune encephalitis during ICI treatment [4]. In addition, there was a clear association between ICI use and increased diagnosis of anti-Ma2 antibody-associated paraneoplastic syndrome [9]. Thus, differential diagnosis between ICI-induced encephalitis and paraneoplastic syndrome is potentially difficult [10].
An autopsy of a fatal case of encephalitis arising during ICI therapy showed vigorous inflammation characterized by approximately equal ratios of CD4 + and CD8 + T cells in the bilateral temporal lobes and striatum with surrounding gliosis and numerous macrophages [5]. Epstein–Barr virus-specific T cell receptors and Epstein–Barr virus-positive lymphocytes in the affected region were identified. This may suggest the presence of a viral infection that caused ICI-induced autoimmune encephalitis.
Aseptic meningitis
Aseptic meningitis is a clinical syndrome characterized by inflammation of the meninges accompanied by leukocytosis; however, microbiological testing of the CSF shows negative results in cases of aseptic meningitis. Therapeutic drugs that have been associated with aseptic meningitis include nonsteroidal anti-inflammation drugs, antimicrobial drugs, anticonvulsants, and intravenous immunoglobulin [11]. The clinical features of aseptic meningitis induced by the classical drugs and those of aseptic meningitis induced by ICIs are different.
Patients with aseptic meningitis present with fever, headache, photophobia, meningeal signs, nausea, and vomiting. In general, the interval between drug exposure and aseptic meningitis is usually 1–2 days. Symptoms are usually mild and improve within 1–5 days after discontinuation of the causative drugs. In contrast, aseptic meningitis tends to occur 10–30 days after the first treatment with ICIs. First, a fever appears, and then the disease progresses over several days.
Additional manifestations such as confusion, altered behavior, seizures, short-term memory loss, and aphasia suggest the spread of inflammation to the brain. Since brain MRI usually shows no specific findings, discriminating between meningitis and encephalitis is difficult. In irAEs, the term “meningoencephalitis” is more suitable than “aseptic meningitis” [4, 7]. Thus, diagnoses of aseptic meningitis and autoimmune encephalitis are usually indistinguishable when they occur as irAEs. In fact, the profiles of T cell and macrophage infiltration are commonly observed in the meninges and parenchymal regions, including gray matter and perivascular spaces [5].
Guillain-barré syndrome
GBS is a potentially life-threatening post-infectious disease characterized by rapidly progressive, symmetrical weakness of the extremities. GBS typically occurs after an infectious disease in which the immune response generates antibodies that cross-react with gangliosides at nerve membranes.
The occurrence of a preceding infection is less frequent in irAE GBS than in typical GBS [12]. Diarrhea in such cases is not due to Campylobacter jejuni gastroenteritis, but rather to gastrointestinal irAE. Limb weakness, which is observed symmetrically and predominantly in the legs, and a resulting gait disturbance are common symptoms [12–14]. Bulbar involvement and dyspnea are also reported. Nasogastric tubes are required due to severe dysphagia. Facial nerves are also involved in ICI-induced GBS and are likely to be vulnerable in patients receiving ICI treatment [15]. Motor weakness progresses rapidly within 14 days, and especially within 5 days, thus mimicking typical GBS.
Diagnosis can usually be made on clinical grounds, but lumbar puncture and electrophysiological studies can help to substantiate the diagnosis and to differentiate the demyelinating subtype from the axonal subtype of GBS. In cases with irAEs, conduction studies suggest that both demyelination and axonal involvement are present. The combination of elevated protein levels and normal cell counts in the CSF is considered a hallmark of GBS. However, CSF frequently shows pleocytosis with lymphocytes in patients with irAE GBS [12]. Spinal cord MRI may show hypertrophy and contrast enhancement of the nerve roots.
Anti-ganglioside antibody specificities are associated with particular subsets of GBS and related neurological deficits, reflecting the distribution of different gangliosides in human peripheral nerves. Autoantibodies against GM1, GM2, and GalNAc-GD1a gangliosides were reportedly found in GBS patients during ICI treatment [12, 16]. However, the clinical relevance of anti-ganglioside antibodies is likely to be limited.
The authors of previous reports preferred to apply the diagnosis of either GBS or chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) to patients with neuropathy related to ICIs based on the chronological changes in the disease progression [17, 18]. In some cases, it is more appropriate to diagnose CIDP based on the disease’s gradual progression and on the relapse of symptoms. However, the characteristics of the peripheral neuropathy observed in irAEs differ from those of both GBS and CIDP. The categorization of patients with neuropathy associated with ICI therapy in a preexisting disease subset of GBS or CIDP may interfere with the correct understanding of this disease subset; alternatively, it seems more appropriate to lump them together as “polyradiculoneuropathy” [12, 14].
Myasthenia gravis
MG is an organ-specific autoimmune disorder characterized by dysfunctional neuromuscular junctions of skeletal muscles targeted by pathogenic autoantibodies to acetylcholine receptor (AChR) or muscle-specific kinase (MuSK). MG is the most representative immune-mediated neurological disorder that occurs as an irAE, and its clinical features have been described by several authors [19, 20]. MG occurs in the early phase after ICI treatment with rapid deterioration. Bulbar symptoms and myasthenic crisis are observed more frequently than in idiopathic MG [20].
The screening of anti-AChR antibodies is required when the development of MG is suspected during ICI treatment. However, the positivity rate is estimated to be about 50% in patients with ICI-induced MG [21]. In addition, titers of anti-AChR antibodies usually present equivocal values, such as values under 1.0 nM, when measured by the standard radioimmunoassay [20]. In contrast, anti-MuSK antibodies are generally not detectable, but one case report showed borderline titers of both anti-AChR and anti-MuSK antibodies [22].
The diagnosis of MG cases occurring as irAEs is difficult when anti-AChR antibodies are undetectable. First, MG is characterized by the fluctuation of muscle weakness and easy fatigability. Since disease progression is relatively rapid, fluctuation is observed less in patients with irAE MG than in those with idiopathic MG. In addition, since cancer patients frequently experience fatigue, it is difficult to evaluate whether easy fatigability is present. Second, the assessment of edrophonium tests remains controversial. The response to cholinesterase inhibitors is likely to not be remarkable, as is observed in patients with idiopathic MG. Determining the results of an ice pack test that show the improvement of ptosis is also difficult. Third, an abnormal decrement in repetitive nerve stimulation is demonstrated in limited cases of irAE MG.
Myositis
Inflammatory myopathies (myositis) are a heterogeneous group of subacute, chronic, and acute systemic immune-mediated diseases of the skeletal muscles, and they can occur during ICI treatment [3, 23–25]. Patients exhibit objective limb and trunk muscle weakness relatively early after the initiation of ICI treatment. Myalgia tends to precede the muscle weakness. In addition to the common symptoms of typical myositis, ICI-induced myositis is accompanied by ocular muscle symptoms that are suggestive of MG, such as ptosis and diplopia. Bulbar symptoms are also frequent. Since extramuscular symptoms including interstitial pneumonitis and skin rash are infrequently seen in accompaniment with myositis, these symptoms are not specific for myositis but may be due to irAEs that are simultaneously occurring in other organs.
Serum creatine kinase (CK) levels are markedly increased to over 1000 IU/L [3]. Fat-suppression T2-weighted or gadolinium enhancement T1-weighted MR images show abnormal signals in the corresponding muscles. The muscle pathology is characterized by multifocal necrotic myofibers with scattered foci of endomysial inflammation and the expression of major histocompatibility complex class I. The findings are different from those typical of other subsets of myositis such as immune-mediated necrotizing myopathy [3, 23]. Further histological analysis has shown a lymphoid follicle-like structure with the expression of peripheral node addressin and chemokine ligand 21, suggesting the involvement of the tertiary lymphoid organs [26].
Myositis-specific autoantibodies are ideal biomarkers, not only for identifying homogeneous subsets of myositis, but also for more precisely exploring the potential environmental and genetic factors contributing to the disease. They are associated with three major subsets of inflammatory myopathies including immune-mediated necrotizing myopathy, antisynthetase syndrome, and dermatomyositis. The association between dermatomyositis and malignancy is particularly strong in those patients with antibodies to transcription intermediary factor 1γ or nuclear matrix protein 2. Myositis-specific autoantibodies are not detected in patients with myositis that occurs as an irAE, with some exceptional cases [25]. In addition, myositis-associated autoantibodies that are found in other rheumatic disorders are occasionally detected [24, 27].
Myasthenia and myositis overlap
Multiple neurological diseases can overlap in a single patient during ICI treatment [28]. Myasthenia and myositis are the conditions that most commonly overlap [3, 19, 20, 23–25]. In fact, it is difficult to diagnose MG or myositis independently when these conditions occur as irAEs. Patients can present both clinical manifestations and laboratory findings of a mixture of MG and myositis (Fig. 1).
Fig. 1.
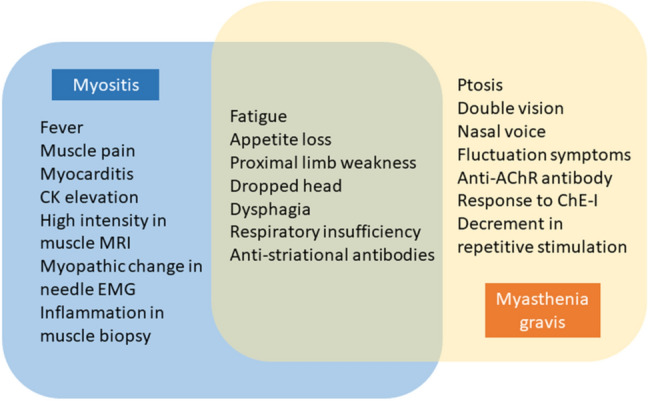
An outline of myasthenia and myositis overlap. Clinical manifestations and laboratory findings include a mixture of myasthenia gravis and myositis indications. AchR Acetylcholine receptor, ChE-I Cholinesterase inhibitors, EMG Electromyogram
Myocarditis is also observed as an overlapping condition and can cause both severe heart failure and lethal arrhythmias, resulting in a fatal outcome [29, 30]. In fact, many researchers have reported that patients with myocarditis have shown signs of cardiac involvement in the form of elevated myocardial enzymes, electrocardiogram changes, abnormal echocardiography, cardiac arrest, or cardiac symptoms prior to death. Careful observation is thus necessary to promptly diagnose and manage myocarditis. It is noted that respiratory insufficiency occurs not only due to myasthenic crisis, but also due to myocarditis. As a better clinical practice, it is recommended that serum CK measurement and electrocardiography should be monitored before and after ICI treatment in order to diagnose myocarditis.
Anti-striational antibodies are preferentially found in patients with thymoma-associated MG, although their pathogenetic roles are not fully elucidated. The standard method for detecting anti-striational antibodies is indirect immunofluorescence of animal skeletal muscle tissue. Since titin and Kv1.4 are representative autoantigens, cytometric cell-based assays for antibodies against these molecules have been established [31]. Many researchers have pointed out that anti-striational antibodies were detected in cases in which myasthenia and myositis overlap [3, 13, 20, 32–34]. Since the positivity rate of anti-striational antibodies seems to be approximately 75% in such cases, they are expected to be biomarkers that can be used to diagnose overlapping myasthenia and myositis.
With regard to the pathogenesis of these overlapping conditions, the T cell-mediated autoimmune mechanism against molecules in the skeletal and heart muscles may be crucial [3]. T cells that are autoreactive to muscle autoantigens including titin, Kv1.4, and others may be latent in the peripheral blood. ICI treatment causes the activation of cytotoxicity of autoreactive CD8 + T cells resulting in the development of myositis and myocarditis. In addition, the activation of autoreactive CD4 + T cells leads to the production of anti-AChR and anti-striational antibodies.
Practical management
Neurological irAEs are characterized by the following traits based on accumulating findings (Supplementary Table) [35–37]. Neurological irAEs affect the central nervous system, peripheral nerves, neuromuscular junction, and muscle, each with unique presentation. The estimated frequency of neurological irAEs is 3–5%, which is higher than previously recognized. Combination therapy with ICIs is expected to produce a remarkable antitumor response; however, the risk of severe neurological irAEs is upregulated. The prevalence of severe neurological irAEs that have a Common Terminology Criteria for Adverse Events (CTCAE) grade of 3 or greater was 1.5%, with such severe cases more likely to occur in patients receiving combination therapy [28]. Among the representative subsets, overlapping myasthenia and myositis were the most frequent, followed by autoimmune encephalitis and GBS in order.
Males are affected more than females. Though the association of human leucocyte antigens with neurological irAEs has been observed in a small number of patients, further investigation is necessary [3, 38]. No tendency for neurological irAEs to appear with certain types of underlying cancer has been observed. The brand of ICI is not associated with the risk of neurological irAEs. However, autoimmune encephalitis may develop in Asian cancer patients receiving atezolizumab with an unexpectedly high frequency [7, 38].
Disease onset occurs in the early phase after the start of ICI treatment. The duration from initial ICI administration to the onset of neurological irAEs is 6 weeks on average. Importantly, disease progression is rapid with a critical clinical course. Respiratory symptoms often require ventilatory support. Overlapping neurological manifestations produce a unique and distinct clinical spectrum. Appropriate differential diagnosis is indispensable for the proper management of neurological irAEs. There are marked differences in the clinical significance of autoantibodies between neuronal diseases in the typical clinical setting and those that occur as irAEs.
For all but cases with the minimum (CTCAE grade 1) neurological symptoms, ICI therapy should be withheld until the nature of the irAE is defined [1]. Corticosteroids and/or intravenous methylprednisolone are the principal treatment. Additional immune-modulating medication is also required in cases of severe neurological irAEs. Intravenous immunoglobulin therapy is also administered. In addition, plasma exchange, rituximab, or infliximab are potentially useful.
The neurological symptoms in patients with mild irAEs respond promptly and are usually relieved within several weeks. In contrast, the treatment response in patients with severe irAEs tends to show gradual improvement over several months. Some patients continue to suffer from severe neurological deficits. Thus, neurological irAEs impair patients’ activities and require long-term hospitalization. It is generally accepted that an oral corticosteroid should be tapered over several months [28]. A maintenance dose such as 5–10 mg/day may be required to prevent disease relapse.
The development of irAEs is associated with a survival benefit in cancer patients. Since irAEs are predictive of a better outcome of treatment with ICIs [39], restarting ICIs after the development of neurological irAEs would be expected to be effective. The readministration of ICIs is potentially associated with the occurrence of a second irAE [40]. However, the restarting of ICIs after the development of neurological irAEs may be considered possible when the proper diagnosis and management are performed.
Patients with preexisting autoimmune disorders should be given the benefit of cancer therapy with ICIs. The exacerbation of autoimmune conditions occurred in 38% of patients, but the most flares of autoimmune disorders were mild [41]. However, there are no data for patients with preexisting MG or myositis. The history of immune-mediated neurological diseases does not contraindicate the use of ICIs, but the practical risk–benefit ratio must be considered.
In conclusion, a correct understanding of neurological irAEs is necessary to provide the best oncological treatment.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by JSPS KAKENHI Grant No. JP20H03592.
Authors’ contributions
Conception and design were contributed by all authors. Collection and assembly of data were contributed by all authors. Data analysis and interpretation were contributed by all authors. Manuscript writing was contributed by Shigeaki Suzuki and Morinobu Seki. Final approval of manuscript was contributed by all authors. All authors were accountable for all aspects of the work.
Funding
This work was funded by JSPS KAKENHI Grant Number JP20H03592.
Declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Brahmer JR, Lacchetti C, Schneider BJ, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: american society of clinical oncology clinical practice guideline. J Clin Oncol. 2018;36:1714–1768. doi: 10.1200/jco.2017.77.6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. 2018;378:158–168. doi: 10.1056/NEJMra1703481. [DOI] [PubMed] [Google Scholar]
- 3.Seki M, Uruha A, Ohnuki Y, et al. Inflammatory myopathy associated with PD-1 inhibitors. J Autoimmun. 2019;100:105–113. doi: 10.1016/j.jaut.2019.03.005. [DOI] [PubMed] [Google Scholar]
- 4.Vogrig A, Muniz-Castrillo S, Joubert B, et al. Central nervous system complications associated with immune checkpoint inhibitors. J Neurol Neurosurg Psychiatry. 2020;91:772–778. doi: 10.1136/jnnp-2020-323055. [DOI] [PubMed] [Google Scholar]
- 5.Johnson DB, McDonnell WJ, Gonzalez-Ericsson PI, et al. A case report of clonal EBV-like memory CD4(+) T cell activation in fatal checkpoint inhibitor-induced encephalitis. Nat Med. 2019;25:1243–1250. doi: 10.1038/s41591-019-0523-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shah S, Dunn-Pirio A, Luedke M, Morgenlander J, Skeen M, Eckstein C. Nivolumab-induced autoimmune encephalitis in two patients with lung adenocarcinoma. Case Rep Neurol Med. 2018;2018:2548528. doi: 10.1155/2018/2548528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki S. Encephalitis as an immune-related adverse event. J Neurol Neurosurg Psychiatry. 2020;91:680. doi: 10.1136/jnnp-2020-323212. [DOI] [PubMed] [Google Scholar]
- 8.Williams TJ, Benavides DR, Patrice KA, Dalmau JO, de Avila AL, Le DT, Lipson EJ, Probasco JC, Mowry EM. Association of autoimmune encephalitis with combined immune checkpoint inhibitor treatment for metastatic cancer. JAMA Neurol. 2016;73:928–933. doi: 10.1001/jamaneurol.2016.1399. [DOI] [PubMed] [Google Scholar]
- 9.Vogrig A, Fouret M, Joubert B, et al. Increased frequency of anti-Ma2 encephalitis associated with immune checkpoint inhibitors. Neurol Neuroimmunol Neuroinflamm. 2019 doi: 10.1212/NXI.0000000000000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graus F, Dalmau J. Paraneoplastic neurological syndromes in the era of immune-checkpoint inhibitors. Nat Rev Clin Oncol. 2019;16:535–548. doi: 10.1038/s41571-019-0194-4. [DOI] [PubMed] [Google Scholar]
- 11.Yelehe-Okouma M, Czmil-Garon J, Pape E, Petitpain N, Gillet P. Drug-induced aseptic meningitis: a mini-review. Fundam Clin Pharmacol. 2018;32:252–260. doi: 10.1111/fcp.12349. [DOI] [PubMed] [Google Scholar]
- 12.Okada K, Seki M, Yaguchi H, Sakuta K, Mukai T, Yamada S, Oki K, Nakahara J, Suzuki S. Polyradiculoneuropathy induced by immune checkpoint inhibitors: a case series and review of the literature. J Neurol. 2020 doi: 10.1007/s00415-020-10213-x. [DOI] [PubMed] [Google Scholar]
- 13.Kao JC, Liao B, Markovic SN, et al. Neurological complications associated with anti-programmed death 1 (PD-1) Antibodies. JAMA Neurol. 2017;74:1216–1222. doi: 10.1001/jamaneurol.2017.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dubey D, David WS, Amato AA, et al. Varied phenotypes and management of immune checkpoint inhibitor-associated neuropathies. Neurology. 2019;93:e1093–e1103. doi: 10.1212/WNL.0000000000008091. [DOI] [PubMed] [Google Scholar]
- 15.Vogrig A, Muniz-Castrillo S, Joubert B, et al. Cranial nerve disorders associated with immune checkpoint inhibitors. Neurology. 2020 doi: 10.1212/WNL.0000000000011340. [DOI] [PubMed] [Google Scholar]
- 16.Fukumoto Y, Kuwahara M, Kawai S, Nakahama K, Kusunoki S. Acute demyelinating polyneuropathy induced by nivolumab. J Neurol Neurosurg Psychiatry. 2017 doi: 10.1136/jnnp-2017-316510. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka R, Maruyama H, Tomidokoro Y, et al. Nivolumab-induced chronic inflammatory demyelinating polyradiculoneuropathy mimicking rapid-onset Guillain-Barre syndrome: a case report. Jpn J Clin Oncol. 2016;46:875–878. doi: 10.1093/jjco/hyw090. [DOI] [PubMed] [Google Scholar]
- 18.de Maleissye MF, Nicolas G, Saiag P. Pembrolizumab-induced demyelinating polyradiculoneuropathy. N Engl J Med. 2016;375:296–297. doi: 10.1056/NEJMc1515584. [DOI] [PubMed] [Google Scholar]
- 19.Safa H, Johnson DH, Trinh VA, et al. Immune checkpoint inhibitor related myasthenia gravis: single center experience and systematic review of the literature. J Immunother Cancer. 2019;7:319. doi: 10.1186/s40425-019-0774-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suzuki S, Ishikawa N, Konoeda F, et al. Nivolumab-related myasthenia gravis with myositis and myocarditis in Japan. Neurology. 2017;89:1127–1134. doi: 10.1212/wnl.0000000000004359. [DOI] [PubMed] [Google Scholar]
- 21.Johansen A, Christensen SJ, Scheie D, Hojgaard JLS, Kondziella D. Neuromuscular adverse events associated with anti-PD-1 monoclonal antibodies: systematic review. Neurology. 2019;92:663–674. doi: 10.1212/WNL.0000000000007235. [DOI] [PubMed] [Google Scholar]
- 22.Mitsune A, Yanagisawa S, Fukuhara T, Miyauchi E, Morita M, Ono M, Tojo Y, Ichinose M. Relapsed myasthenia gravis after nivolumab treatment. Intern Med. 2018;57:1893–1897. doi: 10.2169/internalmedicine.9153-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Touat M, Maisonobe T, Knauss S, et al. Immune checkpoint inhibitor-related myositis and myocarditis in patients with cancer. Neurology. 2018;91:e985–e994. doi: 10.1212/wnl.0000000000006124. [DOI] [PubMed] [Google Scholar]
- 24.Aldrich J, Pundole X, Tummala S, Palaskas N, Andersen CR, Shoukier M, Abdel-Wahab N, Deswal A, Suarez-Almazor ME. Inflammatory myositis in cancer patients receiving immune checkpoint inhibitors. Arthritis Rheumatol. 2020 doi: 10.1002/art.41604. [DOI] [PubMed] [Google Scholar]
- 25.Kadota H, Gono T, Shirai Y, Okazaki Y, Takeno M, Kuwana M. Immune checkpoint inhibitor-induced myositis: a case report and literature review. Curr Rheumatol Rep. 2019;21:10. doi: 10.1007/s11926-019-0811-3. [DOI] [PubMed] [Google Scholar]
- 26.Matsubara S, Seki M, Suzuki S, Komori T, Takamori M. Tertiary lymphoid organs in the inflammatory myopathy associated with PD-1 inhibitors. J Immunother Cancer. 2019;7:256. doi: 10.1186/s40425-019-0736-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liewluck T, Kao JC, Mauermann ML. PD-1 inhibitor-associated myopathies: emerging immune-mediated myopathies. J Immunother. 2018;41:208–211. doi: 10.1097/CJI.0000000000000196. [DOI] [PubMed] [Google Scholar]
- 28.Dubey D, David WS, Reynolds KL, et al. Severe neurological toxicity of immune checkpoint inhibitors: growing spectrum. Ann Neurol. 2020 doi: 10.1002/ana.25708. [DOI] [PubMed] [Google Scholar]
- 29.Johnson DB, Balko JM, Compton ML, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. 2016;375:1749–1755. doi: 10.1056/NEJMoa1609214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anquetil C, Salem JE, Lebrun-Vignes B, et al. Immune checkpoint inhibitor-associated myositis. Circulation. 2018;138:743–745. doi: 10.1161/circulationaha.118.035898. [DOI] [PubMed] [Google Scholar]
- 31.Kufukihara K, Watanabe Y, Inagaki T, Takamatsu K, Nakane S, Nakahara J, Ando Y, Suzuki S. Cytometric cell-based assays for anti-striational antibodies in myasthenia gravis with myositis and/or myocarditis. Sci Rep. 2019;9:5284. doi: 10.1038/s41598-019-41730-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haddox CL, Shenoy N, Shah KK, Kao JC, Jain S, Halfdanarson TR, Wijdicks EF, Goetz MP. Pembrolizumab induced bulbar myopathy and respiratory failure with necrotizing myositis of the diaphragm. Ann Oncol. 2017;28:673–675. doi: 10.1093/annonc/mdw655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bilen MA, Subudhi SK, Gao J, Tannir NM, Tu SM, Sharma P. Acute rhabdomyolysis with severe polymyositis following ipilimumab-nivolumab treatment in a cancer patient with elevated anti-striated muscle antibody. J Immunother Cancer. 2016;4:36. doi: 10.1186/s40425-016-0139-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takamatsu K, Nakane S, Suzuki S, et al. Immune checkpoint inhibitors in the onset of myasthenia gravis with hyperCKemia. Ann Clin Transl Neurol. 2018;5:1421–1427. doi: 10.1002/acn3.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson DB, Manouchehri A, Haugh AM, Quach HT, Balko JM, Lebrun-Vignes B, Mammen A, Moslehi JJ, Salem JE. Neurologic toxicity associated with immune checkpoint inhibitors: a pharmacovigilance study. J Immunother Cancer. 2019;7:134. doi: 10.1186/s40425-019-0617-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spain L, Walls G, Julve M, et al. Neurotoxicity from immune-checkpoint inhibition in the treatment of melanoma: a single centre experience and review of the literature. Ann Oncol. 2017;28:377–385. doi: 10.1093/annonc/mdw558. [DOI] [PubMed] [Google Scholar]
- 37.Cuzzubbo S, Javeri F, Tissier M, et al. Neurological adverse events associated with immune checkpoint inhibitors: review of the literature. Eur J Cancer. 2017;73:1–8. doi: 10.1016/j.ejca.2016.12.001. [DOI] [PubMed] [Google Scholar]
- 38.Chang H, Shin YW, Keam B, Kim M, Im SA, Lee ST. HLA-B27 association of autoimmune encephalitis induced by PD-L1 inhibitor. Ann Clin Transl Neurol. 2020 doi: 10.1002/acn3.51213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haratani K, Hayashi H, Chiba Y, et al. Association of immune-related adverse events with nivolumab efficacy in non-small-cell lung cancer. JAMA Oncol. 2018;4:374–378. doi: 10.1001/jamaoncol.2017.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simonaggio A, Michot JM, Voisin AL, et al. Evaluation of readministration of immune checkpoint inhibitors after immune-related adverse events in patients with cancer. JAMA Oncol. 2019 doi: 10.1001/jamaoncol.2019.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Menzies AM, Johnson DB, Ramanujam S, et al. Anti-PD-1 therapy in patients with advanced melanoma and preexisting autoimmune disorders or major toxicity with ipilimumab. Ann Oncol. 2017;28:368–376. doi: 10.1093/annonc/mdw443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.