

Abstract
Background
Antibody-based therapies blocking the programmed cell death-1/ligand-1 (PD-1/PD-L1) axis have provided unprecedent clinical success in cancer treatment. Acquired resistance, however, frequently occurs, commonly associated with the upregulation of additional inhibitory molecules. Diacylglycerol kinase (DGK) α limits the extent of Ras activation in response to antigen recognition, and its upregulation facilitates hypofunctional, exhausted T cell states. Pharmacological DGKα targeting restores cytotoxic function of chimeric antigen receptor and CD8+ T cells isolated from solid tumors, suggesting a mechanism to reverse T cell exhausted phenotypes. Nevertheless, the contribution of DGKα downstream of the PD-1/PD-L1 inhibitory axis in human T cells and the consequences of combining DGKα and anti-PD-1/PD-L1 inhibitors are still unresolved relevant issues.
Materials and methods
We used a human triple parameter reporter cell line to investigate DGKα contribution to the PD-1/PD-L1 inhibitory pathway. We also addressed the impact of deleting DGKα expression in the growth dynamics and systemic tumor-derived effects of a PD-1-related tumor model, the MC38 colon adenocarcinoma.
Results
We identify DGKα as a contributor to the PD-1/PD-L1 axis that strongly limits the Ras/ERK/AP-1 pathway. DGKα function reinforces exhausted T cell phenotypes ultimately promoting tumor growth and generalized immunosuppression. Pharmacological DGKα inhibition selectively enhances AP-1 transcription and, importantly, cooperates with antibodies blocking the PD-1/PD-L1 interrelation.
Conclusions
Our results indicate that DGKα inhibition could provide an important mechanism to revert exhausted T lymphocyte phenotypes and thus favor proper anti-tumor T cell responses. The cooperative effect observed after PD-1/PD-L1 and DGKα blockade offers a promising strategy to improve the efficacy of immunotherapy in the treatment of cancer.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00262-021-02924-5.
Keywords: T Lymphocytes, Immunotherapy, Drug Therapy, Combination, Programmed Cell Death-1, Diacylglycerol Kinase
Précis
DGKα contributes to PD-1 inhibitory function, and thus its targeting opens new opportunities to reactivate exhausted T cell phenotypes in cancer. DGKα inhibitors could contribute to overcome resistances and cooperate with PD-1/PD-L1 blocking therapies
Introduction
Tumor cells hijack the same mechanisms that maintain peripheral tolerance to avoid generalized T cell-mediated destruction. The expression of programmed cell death-1 (PD-1) by activated T cells is one of the main mechanisms that guarantees protection from T cell-induced damage [1]. PD-1 recognition of its ligands (PD-L1 and 2) facilitates the association of SH2-containing phosphatases that attenuate T cell receptor (TCR) signals preventing the destruction of healthy organs during immune responses [2]. PD-L1 is commonly expressed by T and B lymphocytes, NK cells, macrophages, dendritic cells and epithelial and vascular endothelial cells [3]. The expression of PD-L1 by cancer cells limits the attack of PD-1-expressing tumor-targeted T cells, leading to their functional impairment and exhaustion [4, 5].
A major role described for PD-1 is that of limiting the formation c-Jun/c-Fos heterodimers that facilitate the activator protein-1 (AP-1) transcriptional program that is associated with productive T cell activation [6, 7]. According to the two-signal model, ligation of costimulatory molecules like CD28 facilitates activation of the AP-1 and nuclear factor κ B cells (NFκB) transcription factors that cooperate with the nuclear factor of activated T cells (NFAT), induced after TCR triggering [8]. PD-1 ligation inhibits CD28-elicited costimulatory signals [9] limiting the activation of AP-1 c-Jun/c-Fos heterodimers. In addition, PD-1 triggering favors the expression of additional AP-1 components like BATF, that cannot heterodimerize with c-Fos [6].
TCR triggering leads to a robust NFAT transcription, whereas that dependent on AP-1 is very transient [10]. This is partially the result of the activation of diacylglycerol (DAG) kinases (DGK) that transform DAG into phosphatidic acid, limiting the activation of the Ras/extracellular signal-regulated kinase (ERK) pathway, essential for AP-1 activation [11, 12]. Among the DGK family, the α isoform is predominantly expressed in the T cell lineage [13, 14]. DGKα is abundantly expressed in naïve T cells where is activated by Ca2+ in response to TCR triggering. DGKα regulation in response to antigen recognition confers T lymphocytes with a unique mechanism to limit the intensity of Ras-regulated signals through the control of DAG-dependent activation of Ras guanyl-releasing protein 1 (RasGRP1)- and protein kinase C (PKC)-regulated pathways [15]. Early studies identified DGKα as an anergy-induced gene, whose upregulation in mouse T lymphocytes facilitates the unbalance between the Ca2+/NFAT and the Ras/ERK/AP-1 axes [16]. In mouse T cells, DGKα expression decays very rapidly in response to signal 2 activation [17]. In human T lymphocytes, costimulatory signals, as those delivered by CD28 or the signaling lymphocytic activation molecule (SLAM), facilitate DGKα enzymatic inhibition, ensuring adequate DAG-dependent supply to sustain RasGRP1 and PKCθ activation [18]. Defective DGKα inhibition, as described in T cells deficient for the SLAM-associated protein in X-linked lymphoproliferative disease, accelerates DAG metabolism limiting activation-induced T cell death [19].
Studies in humans and mice suggest that upregulation of DGKα contributes to limit anti-tumor T cell functions; DGKα was found overexpressed in human renal cancer tumor-infiltrating lymphocytes (TIL) and in chimeric antigen receptor (CAR) T cells engineered against human mesothelioma [20, 21]. Together with DGKζ, another DGK isoform also expressed in T cells, DGKα was found as one of the negative regulators preventing T cell-dependent tumor elimination in mouse melanoma models [22]. Ex vivo treatment with DGK inhibitors restored the killing abilities of TIL and CAR T cells isolated from tumors [20, 21], suggesting that targeting DGKα activity may reverse the hypofunctional state of TIL. Despite these observations, no studies up to date have reported differences in tumor growth as the result of impaired DGKα expression. In contrast, mice deficient for DGKζ show marked reduction in the growth of implanted tumors [23–25].
In spite of its potential, both the contribution of DGKα to PD-1-driven signaling and the ability of DGKα inhibitors to cooperate with immune checkpoint-targeted therapies in human T cells have not been examined. Here we used a human triple parameter reporter (TPR) T cell line to demonstrate that DGKα contributes as a negative regulator of AP-1 transcription downstream of PD-1 triggering. The limitation of the PD-1 coinhibitory signals imposed by DGKα loss is conserved in mice, as shown by the limited growth of MC38 tumors implanted in DGKα-deficient mice. Finally, we used two different pharmacological inhibitors to demonstrate that dual PD-1/PD-L1 and DGKα targeting results in a synergized effect promoting AP-1 induction. Our results point the therapeutic potential of combinatorial treatments targeting this DGK isoform and the PD-1, or other immune checkpoints, in the restatement of exhausted anti-tumor T cell responses.
Materials and methods
Antibodies and reagents
For monoclonal antibody (mAb)-based stimulation, we used mouse anti-human anti-CD3 and -CD28 Ab (555,336, 555,725; BD Pharmingen). For flow cytometry analyses, we used LIVE/DEAD fixable red cell death staining kit (L23102, Invitrogen) and anti-mouse CD45-APC (L23102, eBioscience) or -Percp-Cy5.5, CD8-BV610, and CD25-PCy7 (103,132, 100,744, and 102,016, respectively; all from Biolegend), TCRβ-FITC (732,247, Beckman Coulter), and PD-1-APC780 (47,998,582, eBioscience), anti-human CD28-PE (IM2071U, Beckman Coulter), and paraformaldehyde (TedPella). For western blot analyses, we used anti-DGKα (11,547–1-AP, ProteinTech), pERK (Thr202/Tyr204) and prpS6 (Ser235/236) (9101, 2211; Cell Signaling), GFP (C163, Invitrogen), α tubulin (T5168, Sigma), GAPDH (G-9, Santa Cruz), horseradish peroxidase-conjugated anti-mouse and rabbit IgG (P0447, P0260; Dako), anti-rabbit IgG Dylight 800 (SA5-35,571, Invitrogen) and mouse IgG AlexaFluor 680 (175,775, AbcamLife). DGK inhibitor II (R59949) and ritanserin were from Sigma. Anti-PD-1 (nivolumab, nivo) humanized Ab was from BioVision. For TIL isolation, we used collagenase type I (Worthington), dispase II and DNase I (both from Roche). Trypsin was from Biowest.
Cell lines, culture and stimulation
Human Jurkat JE6.1-derived TPR cells, transduced to express NFAT-green (GFP), NFκB-cyan (CFP), and AP-1-red (Cherry) fluorescent proteins, were generated and selected as reported [26]. TPR cells retrovirally transduced with vectors encoding PD-1 were described [26]. T cell stimulator (TCS) cells, a murine thymoma cell line (Bw5147) engineered to stably express an anti-human CD3 single-chain fragment anchored to the cell membrane via a human CD14 stem, have been described [27]. TCS cells expressing no human costimulatory molecule were used as control TCS (TCS-control) and compared with cells retrovirally transduced to additionally express human CD86 (TCS-CD86), PD-L1 (TCS-PD-L1), or both (TCS-CD86/PD-L1). Murine MC38 adenocarcinoma cell line was a kind gift from Prof. Santos Mañes (CNB-CSIC). All cells were maintained in complete RPMI medium (cRPMI) consisting of RPMI (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Invitrogen), 1% HEPES (Sigma), 2 mM L-glutamine, and penicillin and streptomycin (100 U/ml; Biowest) (37ºC, 5% CO2).
For Ab-based stimulation, TPR cells (5 × 104) in 50 μl were stimulated in cRPMI medium with anti-CD3/CD28 mAb (1 μg/ml each) in a 96-well round-bottom plate for 24 h. For stimulation with TCS cells, TPR cells (5 × 104) were cocultured with TCS cells (2 × 104) in 100 μl medium in a 96-well round-bottom plate for 24/72 h (37ºC, 5% CO2). For western blot experiments with TCS cells, TPR cells (1 × 106) were cocultured with TCS cells (4 × 105) in a 1.5 ml tube for the indicated times (37ºC, 5% CO2). Where indicated, 15 μM R59949/ritanserin or 0.5 μg/ml nivolumab was added.
Flow cytometry analysis
TPR/TCS cocultures were stained with anti-mouse CD45-APC or -Percp-Cy5.5 to exclude TCS cells from analysis. Live cells were gated using forward and side scatter parameters within the CD45− subset. Expression of NFAT-GFP, NFκB-CFP and AP-1-Cherry was determined by flow cytometry analysis of the live cell population using a CytoFLEX S flow cytometer (Beckman Coulter). For GFP-DGKα-Kinase Dead (KD) experiments, expression of AP-1-Cherry was determined in the CD45− live GFP+ subset. Percentage of positive cells was determined from triplicate wells. Transcription factor activity was calculated by normalizing data to the maximal activation for each experiment.
Cells isolated from tumors were stained with LIVE/DEAD fixable red cell death staining kit, and stained with a mixture of Ab, using CD45 staining to exclude MC38 cells from analysis. CD8+ T cells were identified among total T cells (CD45+ TCR+) and the expression of CD25 and PD-1 evaluated with the corresponding Ab. Data were analyzed using FlowJo software (version 10.2, FlowJo LLC, Ashland, OR).
Western blot analysis
Cells were lysed and processed as in [28] and western blots probed with specific Ab. Protein bands were visualized by enhanced chemiluminescence (ECL detection, Amersham Bioscience) or with an Odyssey scanner (LI-COR). Densitometric analysis of proteins was performed using ImageJ.
Plasmids and transfections
TPR cells in logarithmic growth (4–5 × 105 cells/ml) were transfected with 15–20 μg plasmid DNA by electroporation using Gene Pulser II (BioRad) (270 mV, 975 μF). For DGK silencing experiments, a validated sequence for the α or ζ isoform was selected to generate the short hairpin interfering RNA (shRNAi) pSUPER construct [28]. Equivalent murine sequences were cloned and used as negative controls [29]. Expression was evaluated from days 1 to 4 post-transfection by western blot. Plasmid encoding GFP-DGKα-KD constructions were reported [30].
Tumor experiments and TIL isolation
C57BL/6 J-DGKα-deficient mice were reported previously [31]. All protocols were approved by the CNB-CSIC Ethics Committee on Animal Experimentation. MC38 cells were trypsinized from subconfluent monolayers, washed, and injected subcutaneously in 100 μl (5 × 105) into one flank of female 8–12-week-old C57BL/6 J wild type (wt) or DGKα-deficient mice. Tumor growth was monitored in a blind manner with calipers, and area and volume were estimated according to the formulas: area = a x b/volume = (a2 x b)/2, where a is tumor width and b is tumor length in mm. Mice were killed when wt tumors reached 1 cm3, at ~ 19 days post-injection, and tumors were excised, measured and weighed.
For TIL isolation, tumors were fragmented into 1 mm3 pieces using a scalpel. Fragments were suspended in DMEM culture medium (Invitrogen) supplemented with 20 mM HEPES, with 2 mg/ml collagenase type I, 2.5 mg/ml dispase II and 0.1 mg/ml DNase I, and incubated with gentle shaking (15 min, 37 °C). The resulting suspension was filtered with a 70-μm filter, washed with PBS + 5% FBS, and centrifuged (5 min, 300 X g, 4 °C). Resulting pellets were processed for flow cytometry analysis.
For blood parameters analyses, blood samples were collected by the time the tumor-bearing mice had to be killed by heart puncture in EDTA tubes and processed on a Sysmex XN-1000 blood analyzer 24 h after extraction.
Statistical analysis
Flow cytometry data were analyzed with GraphPad Prism 8 software. Data are shown as mean ± SEM. Samples were assumed to fit normality. When more than two conditions were analyzed, we applied analysis of variance (ANOVA) and Bonferroni post-test analysis. If not applicable, parametric unpaired t tests were performed. In kinetic experiments, we calculated area under curve (AUC) values. In all cases, differences were considered statistically not significant (ns) for p-values > 0.05, and significant for p-values *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Results
PD-1 ligation limits AP-1 induction by costimulatory signals
The TPR cell line was engineered to simultaneously evaluate the activation of NFAT, NFκB and AP-1 transcription factors through the analysis of GFP, CFP, and Cherry, respectively [26]. This analysis can be performed either in physiological conditions (TPR-control) or under constitutive PD-1 expression (TPR-PD-1) [26]. As antigen-presenting cells, we used TCS cells providing an anti-CD3 Ab fragment, either alone (TCS-control) or in combination with CD86 (TCS-CD86), PD-L1 (TCS-PD-L1), or both (TCS-CD86/PD-L1) [27]. Coincubation of TPR-PD-1 cells with the distinct TCS cells provides an excellent framework to investigate the effect of PD-1 ligation to PD-L1 in the context of TCR or costimulatory signals.
As previously reported [32], coculture of TPR-PD-1 cells with TCS-control cells revealed a noticeable increase compared to basal conditions in the activation of NFAT and NFκB reporters with a minor effect on AP-1 induction (Fig. 1a; Supp. Figure 1a). Costimulatory signals through CD28/CD86 ligation significantly enhanced the activation of the three transcription factors, with a marked increase for AP-1 (Fig. 1a, right; Supp. Figure 1a). Although its effects were also observed in TCR triggering conditions, PD-1/PD-L1 ligation noticeably decreased the input of costimulation on reporter induction (Fig. 1a; Supp. Figure 1a); the presence of PD-L1 in the surface of presenting cells markedly decreased NFAT activation, both in the absence or in the presence of costimulation, whereas PD-1 ligation only slightly affected NFκB activation in all cases (Fig. 1a, left and middle; Supp. Figure 1a). The effects of PD-1 triggering were specially observed in the TCS-CD86-induced AP-1 activation (Fig. 1a, right; Supp. Figure 1a), thus confirming the negative effect of the PD-1/PD-L1 axis on CD28 costimulatory functions.
Fig. 1.

PD-1 triggering strongly limits early- and long-term signaling toward AP-1 activation. a. NFAT-GFP (left), NFκB-CFP (middle) or AP-1-Cherry (right) induction was analyzed. TPR-PD-1 cells were stimulated using TCS-control, -PD-L1, -CD86, or -CD86/PD-L1 cells for 24 h. b–c. ERK (b) or rpS6 (c) phosphorylation was analyzed; α tubulin was used as loading control. TPR-PD-1 cells were stimulated using TCS-CD86 or -CD86/PD-L1 for the indicated times. Graphs shown in the right part of the figures correspond to the normalization of pERK or prpS6 to α tubulin. In a–c, transcription factor activity or normalized ERK/rpS6 phosphorylation was calculated by normalizing data to the maximal activation. d. CD28 expression was analyzed. TPR-PD-1 cells were stimulated using TCS-CD86 or -CD86/PD-L1 for 72 h. In a, data were analyzed using parametric unpaired t test. In b–c, data were analyzed using two-way ANOVA and Bonferroni post-test; ns p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Results are representative of at least three independent experiments with similar results. ANOVA, analysis of variance; AP-1, activator protein-1; ERK, extracellular signal-regulated kinase; NFκB, nuclear factor κ B cells; NFAT, nuclear factor of activated cells; ns, not significant; PD-1/PD-L1, programmed cell death-1/ligand-1; rpS6, ribosomal protein S6; TCS, T cell stimulator; TPR, triple parameter reporter
Analysis of early signaling confirmed that PD-1 triggering in the context of the CD28 signals results in a profound attenuation of ERK phosphorylation (Fig. 1b), while mildly limiting that of ribosomal protein S6 (rpS6) (Fig. 1c), in agreement with the differential effects observed in AP-1 and NFκB induction, respectively.
PD-1 triggering was described to not only limit CD28-delivered signaling, but also to reduce CD28 expression in T cells [9]. In accordance, and further supporting the reliability of the TPR/TCS model, long-term analysis of CD28 surface presence in TPR-PD-1 cells showed a diminished expression for this receptor in those conditions of TCS-CD86/PD-L1 cells-driven stimulation (Fig. 1d, left).
DGKα silencing promotes AP-1 and NFκB activation in response to costimulatory and coinhibitory signals
DGKα limits DAG-dependent regulation of RasGRP1, dampening the intensity of Ras/ERK activation in human T cells [20]. While DGKα expression is transcriptionally repressed in activated mouse T cells [17], the expression of this DGK isozyme in human T lymphocytes is still observed upon strong activation, although its enzymatic activity results inhibited during costimulation [18]. In line with the PD-1-derived alteration of CD28 expression and signaling, we investigated the consequences of altering DGKα expression by transfection of a previously validated shRNAi plasmid against this DGK isoform [28]. DGKα protein expression decreased by 40% compared to controls (Fig. 2a). Although partial, this reduction in DGKα abundance enhanced NFAT activation in TCS-PD-L1-stimulated cells, and strongly increased NFκB and AP-1 activation under all stimulatory conditions, including those involving PD-1 triggering (Fig. 2b; Supp. Figure 1b). These results agree with the well-characterized function of DGKα as a negative regulator of DAG-based signals. Furthermore, they demonstrate that, in addition of acting as a negative regulator of TCR- and CD28-delivered signaling [11], DGKα also contributes to limit DAG-regulated signals in response to coinhibitory receptors.
Fig. 2.
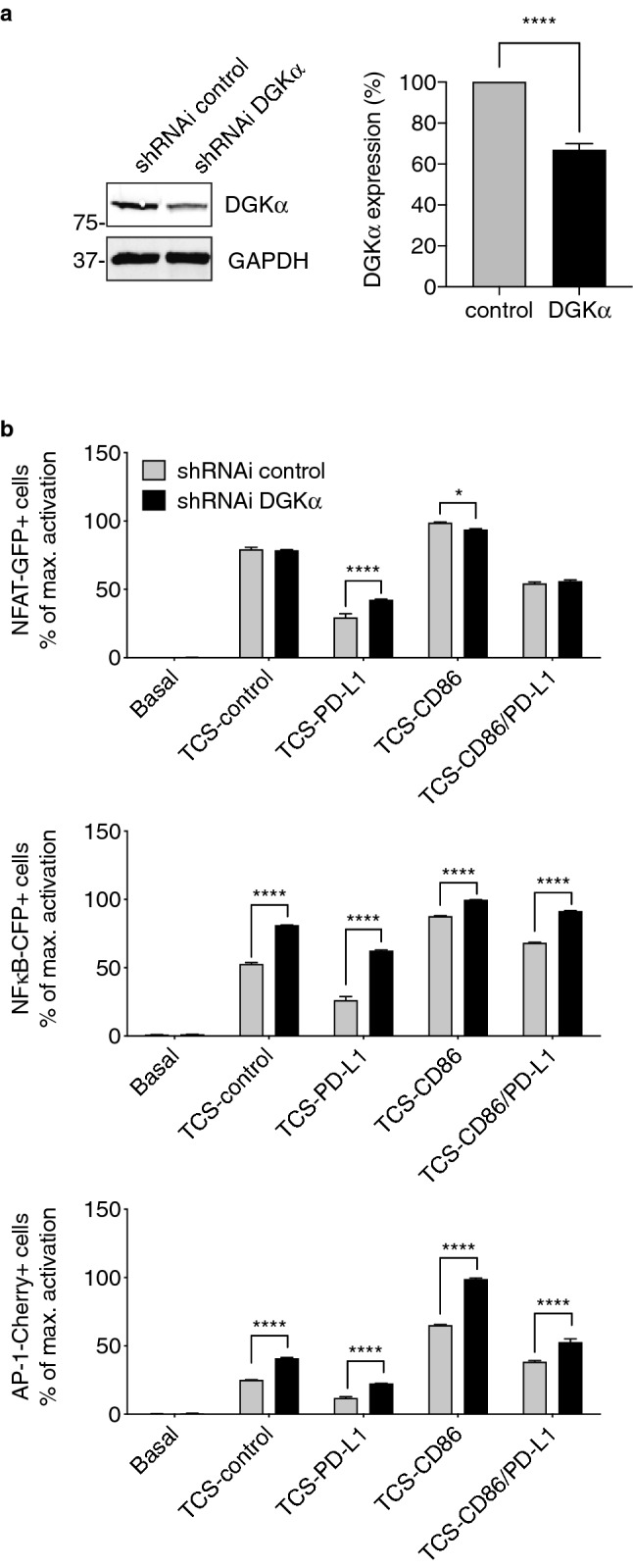
DGKα silencing strongly promotes AP-1 activation upon costimulatory and coinhibitory signals. a. DGKα was silenced in TPR-PD-1 cells and silencing confirmed by western blot (left); GAPDH was used as loading control. Quantification of DGKα expression in control and DGKα-silenced TPR-PD-1 cells was determined in more than three independent experiments (right). b. NFAT-GFP (top), NFκB-CFP (middle) or AP-1-Cherry (bottom) induction was analyzed. Control or DGKα-silenced TPR-PD-1 cells were stimulated using TCS-control, -PD-L1, -CD86, or -CD86/PD-L1 cells for 24 h. Transcription factor activity was calculated by normalizing data to the maximal activation. In a, right, data were analyzed using parametric unpaired t test. In b, data were analyzed using two-way ANOVA and Bonferroni post-test; *p < 0.05, ****p < 0.0001. Results are representative of at least three independent experiments with similar results. ANOVA, analysis of variance; AP-1, activator protein-1; DGK, diacylglycerol kinase; NFκB, nuclear factor κ B cells; NFAT, nuclear factor of activated cells; PD-1/PD-L1, programmed cell death-1/ligand-1; shRNAi, short hairpin interfering RNA; TCS, T cell stimulator; TPR, triple parameter reporter
Deletion of DGKα correlates with improved rejection of PD-1-related MC38 tumors
Our previous results indicate that diminishing DGKα expression enhances the concerted activation of NFAT, NFκB and AP-1 transcription factors, under conditions of PD-1/PD-L1 ligation. The coordinated activation of the three transcription factors determines the expansion potential and functional capacity of T lymphocytes. To examine if DGKα deletion might have relevant implications regarding anti-tumor potency of T cells, we used the MC38 colorectal adenocarcinoma tumor model, highly sensitive to the modulation of PD-1/PD-L1 axis [33].
To elucidate the contribution of DGKα limiting anti-tumor T cell functions, wt or DGKα-deficient mice were subcutaneously inoculated with a small number of MC38 cells to slow tumor growth and allow time for an adaptive immune response to develop. Analysis of MC38 growth kinetics showed a noticeable lag in DGKα-deficient mice, that developed significant smaller tumors than those observed in wt animals by the time at which these ones had to be killed (Fig. 3a–c). We recently reported that DGKζ deficiency limits the growth of MC38 tumors [32]. DGKζ absence correlates with increased tumor infiltration of effector CD8+ T cells, that display enhanced expansion features and reduced predisposition to exhaustion [32]. As described in DGKζ-deficient mice [32], flow cytometry analysis of TIL isolated from wt or DGKα-deficient mice revealed an increased trend of CD8+ T cell infiltration in mice lacking DGKα expression (Fig. 3c, left). In order to explore the activation status of these CD8+ T cells, we analyzed the expression of the α-chain interleukin-2 (IL-2) receptor CD25 and that of PD-1. These studies showed a marked increase in CD25-expressing cell percentage (Fig. 3c, middle). In agreement with previous studies that demonstrated IL-2-mediated modulation of PD-1 expression [32, 34], the analysis of the CD8+CD25+ T cell subset showed reduced PD-1 expression (Fig. 3c, right).
Fig. 3.

DGKα deficiency limits MC38 tumor growth favoring expansion of CD8+CD25+PD-1low T cell populations and limiting tumor-induced anemia. wt or DGKα-deficient mice received subcutaneous injections of 5 × 105 MC38 colon adenocarcinoma cells. a. Tumor progression was evaluated daily until wt animals had to be killed when standards imposed by CNB-CSIC Ethics Committee for Animal Experimentation were reached by day 19. Individual (left and middle) or summarized by genotype (right) tumor area (top) or volume (bottom) was analyzed. b. AUC of tumor area (top) or volume (bottom) was evaluated. c. Tumors from wt or DGKα-deficient mice were excised and cells analyzed by flow cytometry. The percentage of CD8+ (left) or CD8+CD25+ (middle) T cells was compared. PD-1 gMFI (right) was analyzed within the CD8+CD25+ or CD8+CD25− T cell subset. c. Blood samples were collected from wt or DGKα-deficient MC38 tumor-bearing mice by day 19 and processed for red blood cell count (left), hemoglobin (middle) and hematocrit (right) assessment. Dotted bars correspond to physiological values from The Jackson Laboratory. In a, c, right, data were analyzed using two-way ANOVA and Bonferroni post-test. In b, AUC were calculated and compared using parametric unpaired t test. In c, left and middle, d, data were analyzed using parametric unpaired t test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Results are representative of at least three independent experiments with similar results. ANOVA, analysis of variance; AUC, area under curve; DGK, diacylglycerol kinase; gMFI, geometric mean fluorescence intensity; PD-1, programmed cell death-1; wt, wild type
Subcutaneous injection of tumor cells translates into a systemic effect that ultimately results in anemia-like conditions and in the development of cell populations with strong immunosuppressive capacities [35, 36]. Assessment of red blood cell counts, hemoglobin and hematocrit indicated that these phenomena occurred in our tumor-bearing wt animals, that became anemic compared to the standard values reported in healthy mice (Mouse Phenome Database, The Jackson Laboratory) (Fig. 3d, dotted bars). On the contrary, and in correlation with the reduced MC38 tumor development, DGKα-deficient mice displayed similar values compared to the standards (Fig. 3d). These observations suggest that targeting DGKα might have a relevant impact not only in a local context as the tumor microenvironment, but also in a broad, systemic manner.
DGKα inhibition enhances AP-1 transcriptional activation
The studies in genetically modified mice confirm that DGKα absence favors enhanced immune infiltration in tumors that correlates with diminished tumor growth and tumor-related hematological disfunction. Silencing/deletion experiments nonetheless do not provide any information regarding the requirement for DGKα catalytic activity downstream of negative immune checkpoints. In particular, the contribution of DGKα catalytic function in the control of AP-1 activation has not been properly addressed.
Early experiments demonstrated that, following antigen recognition by the TCR, the combined effect of Ca2+ binding and tyrosine phosphorylation results in the transient translocation of DGKα to the membrane, in a mechanism controlled by its own activity [37, 38]. Mutations in the conserved -GGDG- motif in the DGKα catalytic site result in a kinase dead version of the enzyme (DGKα-KD) that, when overexpressed in T cells, does not metabolized DAG and remains membrane-bound, blocking endogenous DGKα translocation and functions [30]. We thus hypothesized that DGKα-KD overexpression in TPR cells would impair the function of the endogenous DGKα, thus providing information about the contribution of its catalytic activity. The use of a DGKα-KD mutant fused to GFP (GFP-DGKα-KD) allowed us to compare AP-1 activation exclusively in GFP-expressing populations (Fig. 4a). To better assess any effect on AP-1 induction upon different ranges of activation, we compared different stimulating conditions by using mAb against CD3 or CD28, or cell-based stimulation with TCS-control and TCS-CD86 cells, known to provide stronger NFAT activation signals [32].
Fig. 4.

DGKα catalytic activity limits AP-1 transcription. A GFP-fused kinase inactive version of DGKα (DGKα-KD) was overexpressed in TPR cells. DGKα-KD expression was confirmed by western blot (GFP, arrowhead) and compared to endogenous DGKα (a); α tubulin was used as loading control. b-c. AP-1-Cherry induction was analyzed. GFP+ control or GFP-DGKα-KD-expressing TPR cells were stimulated using anti-CD3 or anti-CD3/28 mAb, TCS-control, or -CD86 cells for 24 h. When indicated, R59949 or ritanserin was added. In b, transcription factor activity was calculated by normalizing data to the maximal activation. In c, values are normalized to each stimulation condition in the absence of inhibitors = 1.0. Data were analyzed using two-way ANOVA and Bonferroni post-test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Results are representative of at least three independent experiments with similar results. ANOVA, analysis of variance; AP-1, activator protein-1; DGK, diacylglycerol kinase; KD, kinase dead; TCS, T cell stimulator; TPR, triple parameter reporter
The percentage of AP1+ cells in the GFP+ control population was very low downstream of anti-CD3 mAb stimulation and increased upon costimulation (Fig. 4b, grey bars). GFP-DGKα-KD overexpression further increased the percentage of AP-1-expressing cells upon these stimuli, and even in non-stimulated cells (Fig. 4b, black bars), confirming DGKα-KD transdominant negative function. The enhanced AP-1 activation in TPR cells expressing GFP-DGKα-KD was significantly increased only in mAb-stimulated cells, with no significant effect after stimulation with TCS-control or TCS-CD86 cells (Fig. 4b).
Previous experiments already demonstrated that TCS cell-based stimulation delivers stronger Ca2+-dependent signals [32], suggesting enhanced activation of the endogenous DGKα. To examine additional contribution of endogenous DGKα, GFP- or GFP-DGKα-KD-expressing TPR cells were treated with either R59949 or ritanserin, two well-characterized DGKα inhibitors [11] and AP-1 activation calculated as fold induction respect to no inhibitor conditions. R59949 treatment significantly increased the percentage of AP-1+ cells in response to anti-CD3 mAb or TCS-control stimulation exclusively in the GFP+ control but not in the GFP-DGKα-KD+ population (Fig. 4c), confirming the transdominant function of the KD mutant under conditions of weak stimulation. At difference from that observed with anti-CD3 mAb stimulation, inhibitor treatment cooperated with the expression of DGKα-KD in cells stimulated with anti-CD3 and -CD28 mAb, suggesting additional modulation of the endogenous enzyme. The effect of the inhibitors was nonetheless minor in DGKα-KD-expressing cells, suggesting that a major part of endogenous DGKα was already inhibited by the presence of the inactive mutant (Fig. 4c). The effect of the inhibitors was lost in TCS-CD86-stimulated cells (Fig. 4c), confirming the endogenous DGKα blockade by strong costimulatory signals [18]. Interestingly, the comparison of the two inhibitors revealed differential actions, with ritanserin being effective only in response to anti-CD3/28 mAb stimulation (Fig. 4c).
DGKα inhibitors cooperate with anti-PD-1 blocking Ab nivolumab promoting AP-1 transcription
We next investigated if the used pharmacological DGKα inhibitors would restore the effect of PD-1 triggering, as well as their possible cooperation with blocking antibodies targeting the PD-1/PD-L1 inhibitory pathway. Compared to that observed in TCS-control-stimulated cells, R59949 single treatment of TPR-PD-1 cells stimulated with TCS-PD-L1 cells only restored NFκB induction, with partial but noticeable promoting effects regarding NFAT and AP-1 activation (Fig. 5a). As previously observed in Fig. 4c, ritanserin did not have any effect in conditions of TCR triggering in the absence of costimulation (Fig. 5a).
Fig. 5.

DGKα and PD-1 blockade cooperates in promoting AP-1 activation. a. NFAT-GFP (left), NFκB-CFP (middle) or AP-1-Cherry (right) induction was analyzed. TPR-PD-1 cells were stimulated using TCS-control or -PD-L1 cells for 24 h. b. DGKζ was silenced in TPR-PD-1 cells and AP-1-Cherry induction analyzed. Control or DGKζ-silenced TPR-PD-1 cells were stimulated using TCS-CD86 or -CD86/PD-L1 cells for 24 h. When indicated, R59949 (R59), ritanserin (rit) or nivolumab (nivo) was added. Transcription factor activity was calculated by normalizing data to the maximal activation. In a, data were analyzed using parametric unpaired t test. In b, data were analyzed using two-way ANOVA and Bonferroni post-test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Results are representative of at least three independent experiments with similar results. ANOVA, analysis of variance; AP-1, activator protein-1; DGK, diacylglycerol kinase; NFκB, nuclear factor κ B cells; NFAT, nuclear factor of activated cells; PD-1/PD-L1, programmed cell death-1/ligand-1; shRNAi, short hairpin interfering RNA; TCS, T cell stimulator; TPR, triple parameter reporter
Nivolumab is a human immunoglobulin G4 mAb that binds to PD-1 receptor and blocks its interaction with PD-L1 and 2 [39]. It is indicated for the treatment of melanoma, non-small cell lung cancer, renal cell carcinoma, Hodgkin lymphoma, squamous cell cancer of the head and neck, and urothelial carcinoma (European Medicines Agency). Nivolumab single treatment of TPR-PD-1 cells stimulated with TCS-PD-L1 cells fully restored NFAT and NFκB activation to that observed in TCS-control condition, and even further promoted a slight increase in the percentage of AP1+ cells (Fig. 5a). Combined treatment with R59949 and nivolumab yielded a marked increase solely in the percentage of AP-1+cells compared to the treatment with nivolumab alone (Fig. 5a, right); in this situation, and as previously observed for anti-CD3/28 costimulation, ritanserin mimicked R59949 effect (Fig. 5a, right). Our results suggest a cooperation between DGKα inhibition and PD-1 blockade on AP-1 activation in response to TCR triggering.
We already described that PD-1/PD-L1 ligation profoundly affects AP-1 activation in response to costimulatory signals [32], so we next investigated if DGKα inhibition would also cooperate with nivolumab restoring costimulatory functions. To assess possible redundant functions with other important DGK isozymes in T cells, we performed similar analysis in DGKζ-silenced cells. The analysis of each individual transcription factor did not reveal any major effect of DGKα inhibitors on NFAT or NFκB induction, while showing a modest cooperation between nivolumab, DGKα inhibitors and DGKζ silencing promoting AP-1 activation (data not shown). The analysis of triple positive cells nonetheless showed that DGKα blockade by R59949 or ritanserin similarly promoted the restoring effects of nivolumab treatment, further enhancing the activation threshold observed in the absence of PD-1 triggering (Fig. 5b). This gain-of-function effect observed upon DGKα inhibition was further noticed in DGKζ-silenced cells (Fig. 5b), suggesting non-redundant functions between the two DGK isoforms and the potential of their dual targeting in addition to nivolumab treatment.
Discussion
DGKα and DGKζ are two well-characterized negative regulators that contribute to maintain hyporesponsive T cell phenotypes in the context of the tumor microenvironment [20, 21]. Studies with mice deficient for each individual isoform suggested a predominant contribution for DGKζ limiting anti-tumor T cell functions. Nonetheless, the exact contribution of DGKα in the context of T cell exhaustion and the consequence of simultaneous targeting DGKα and other immune checkpoint inhibitors remained unanswered.
Here, using the human TPR cell platform [26] and the PD-1-related MC38 tumor model [33], we identify DGKα-dependent negative regulation of AP-1 transcription as a mechanism that contributes to PD-1 inhibitory functions. We demonstrate that targeting DGKα expression or catalytic activity strongly limits the inhibitory effects imposed by the PD-1/PD-L1 axis. DGKα inhibition translates into an enhanced T cell activation and, interestingly, cooperates with nivolumab [39], a PD-1/PD-L1 ligation blocker that is extensively used in the clinic.
The stimulation of the TPR-PD-1 cell line with different TCS cells showed the varied effects of PD-1 triggering over the activation of NFAT, NFκB and AP-1 transcription factors. These experiments confirmed the strict requirement of CD28-delivered costimulation for AP-1 transcription [40] that ensures effective clonal expansion and anergy/exhaustion avoidance [41, 42]. Our early- and long-term analyses of TPR-PD-1 cells stimulated in the presence of CD86 and PD-L1 also reproduce the strong limitation imposed by PD-1 on the CD28 activation of the Ras/ERK/AP-1 axis, as described for the dampened CD28 signaling [9] and for the alteration of the equilibrium between AP-1 components as a result of PD-1 triggering [6].
The impaired functionality of CD28 signaling after PD-1 triggering might have an impact on the CD28 negative modulation of DGKα activity [18]. This led us to a hypothesis defining this DGK isozyme as a contributor of the PD-1 inhibitory pathway. In agreement, DGKα silencing in TPR-PD-1 cells resulted in an increased activation both downstream of costimulatory and coinhibitory signals. This was especially noticeable regarding AP-1 induction, that was the most augmented after DGKα silencing, pointing to the strong limitation by DGKα on the activation of such an important transcription factor.
These observations correlate well with the diminished capacity of MC38 tumor cells to grow in DGKα-deficient mice, as a result of a major infiltration of activated CD8+ T cells, and with the systemic resistance to tumor-induced anemia and related immunosuppression [35, 36]. We recently described similar results for TIL in tumor-bearing DGKζ-deficient mice [32], which could suggest redundant functions between the two DGK isoforms. Nonetheless, we demonstrate that DGKα pharmacological inhibition cooperates with DGKζ silencing in the TPR cell line, in agreement with the differential effects coming from DGKα or ζ single deletion compared to the dual abrogation of DGKα and ζ expression, as reported in mouse or in human CAR T cells [43, 44]. Besides the T cell lineage, DGKα activity was also shown to limit NK cell attack on human solid tumors [45]. Enhancing NK cell function could contribute to the diminished tumor growth observed in DGKα-deficient mice. Although further analyses will clarify this issue, previous studies have shown that, at difference from those lacking DGKζ expression, NK cells from DGKα-deficient mice do not show enhanced functions [46].
DGKα inhibition in mouse and human T cells provides a gain-of-function effect that is not reproduced by its genetic silencing or deletion [11]. The use of the currently available DGK inhibitors in mice is nonetheless still limited as a result of their poor pharmacokinetic properties and their potential off-target effects [11, 47, 48]. This manifests the need for reliable, reproducible human T cell platforms which optimize these and other compounds targeting modulators of T lymphocyte responses.
The use of a kinase inactive DGKα construct with a trans-dominant effect over endogenous DGKα confirms that DGKα enzymatic activity limits AP-1 transcriptional activation. Overexpression in TPR cells of the kinase defective mutant only promoted AP-1 transcription under anti-CD3/28 mAb stimulating conditions while these effects were lost in TCS-CD86-stimulated cells. The extent of NFAT activation, that is much stronger under cell–cell contact-derived stimulation [32], suggests enhanced mobilization of endogenous DGKα fully correlating with the described DGKα blockade upon strong costimulatory signals [18].
In agreement with this massive inhibition of endogenous DGKα, pharmacological inhibitors only provided gain-of-function effects on AP-1 transcription under weak stimulatory conditions (anti-CD3/28 mAb or TCS-control) or in the presence of coinhibitory signals (TCS-PD-L1). Combined inhibition of DGKα and PD-1/PD-L1 further enhanced AP-1 transcription; this suggests maximal restoration of the activation program in human T cells, and correlates with the recently reported role for DGKα in promoting T cell dysfunction during anti-PD-1 therapy in mouse T cells [49]. This effect was observed even in DGKζ-silenced cells, as shown by the increase in percentage of cells coexpressing NFAT, NFκB and AP-1 reporters that fully correlate with IL-2-producing cells [32].
Our experiments suggest the existence of mechanistical differences between two previously characterized DGKα inhibitors: R59949 and ritanserin. Only R59949, but not ritanserin, enhanced AP-1 activation after TCR stimulation, either after stimulation with mAb or TCS-control cells. In contrast, the two compounds displayed similar effects following anti-CD3/28 mAb stimulation. Ritanserin interacts very strongly with the DGKα catalytic domain and C1 regions [50], while R59949 preferentially recognizes the enzyme in its active, ATP-binding conformation [51]. We thus hypothesize that weak costimulatory signals may modify DGKα conformation, which would have an impact on the ability of the inhibitors to bind the enzyme. A different non-excluding alternative would be that of ritanserin releasing some specific protein interactions that would facilitate costimulatory-like signals. A better knowledge of DGKα structure and its interacting partners is thus necessary to fully exploit its potential as a modulator of immune anti-tumor functions.
Although some of our findings might be restricted to the TPR experimental system, our results offer important conclusions of therapeutic interest. Both DGKα single inhibition and, importantly, its combination with PD-1/PD-L1 blocking Ab, constitute a promising therapeutic approach for reinvigorating anti-tumor T cell responses in the context of immunotherapy.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We thank all members of the Merida Lab for discussion and suggestions and C. Mark for expert editorial assistance
Abbreviations
- Ab
Antibody
- ANOVA
Analysis of variance
- AP-1
Activator protein-1
- AUC
Area under curve
- CAR
Chimeric antigen receptor
- DAG
Diacylglycerol
- DGK
DAG kinase
- ERK
Extracellular signal-regulated kinase
- GFP/CFP/Cherry
Green/cyan/red fluorescent protein
- gMFI
Geometric mean fluorescence intensity
- IL-2
Interleukin-2
- KD
Kinase Dead
- NFκB
Nuclear factor κ B cells
- NFAT
Nuclear factor of activated T cells
- Nivo
Nivolumab
- PD-1/PD-L1
Programmed cell death-1/ligand-1
- PKC
Protein kinase C
- RasGRP1
Ras guanyl-releasing protein 1
- rpS6
Ribosomal protein S6
- shRNAi
Short hairpin interfering RNA
- TCR
T cell receptor
- TCS
T cell stimulator cell
- TIL
Tumor-infiltrating lymphocyte
- TPR
Triple parameter reporter cell
- wt
Wild type
Authors’ contributions
JA-N and IM were involved in conceptualization. JL and PS were involved in generation of TPR and TCS cells. JA-N and IA-B were involved in TPR cell experiments. MM-S and RL were involved in MC38 tumor cell engraftment. RL and AA-F were involved in TIL isolation experiments. MCM-O and AA-F were involved in development and optimization of flow cytometry protocols for TIL studies. JA-N and IM were involved in writing manuscript. All authors read and gave input on the manuscript.
Funding
This work was supported in part by grants from the MINECO (PID2019-108357RB-I00) to AA-F and IM and MINECO (BFU2016-77207-R), the Spanish Association Against Cancer (AECC-1518), the Madrid regional government (IMMUNOTHERCAM Consortium B2017/BMD-3733) and the Aplastic Anemia and MDS International Foundation (AAMDSIF OPE01644), to IM. IA-B hold a graduate fellowship of the Fundación Ramón Areces-UAM.
Data availability
All data relevant to the study are included in the article or uploaded as online supplemental information.
Declarations
Conflicts of interest
Not applicable.
Ethics approval
Protocols approved by the CNB-CSIC Ethics Committee on Animal Experimentation.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Javier Arranz-Nicolás, Email: jarranz@cnb.csic.es.
Isabel Merida, Email: imerida@cnb.csic.es.
References
- 1.Hamanishi J, Mandai M, Matsumura N, et al. PD-1/PD-L1 blockade in cancer treatment: perspectives and issues. Int J Clin Oncol. 2016;21(3):462–473. doi: 10.1007/s10147-016-0959-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patsoukis N, Duke-Cohan JS, Chaudhri A, et al. Interaction of SHP-2 SH2 domains with PD-1 ITSM induces PD-1 dimerization and SHP-2 activation. Commun Biol. 2020 doi: 10.1038/s42003-020-0845-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48(3):434–452. doi: 10.1016/j.immuni.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14(4):847–856. doi: 10.1158/1535-7163.MCT-14-0983. [DOI] [PubMed] [Google Scholar]
- 5.Ahmadzadeh M, Johnson LA, Heemskerk B, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009 doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quigley M, Pereyra F, Nilsson B, et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat Med. 2010 doi: 10.1038/nm.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lynn RC, Weber EW, Sotillo E, et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature. 2019 doi: 10.1038/s41586-019-1805-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edmead CE, Patel YI, Wilson A, et al. Induction of activator protein (AP)-1 and nuclear factor-κB by CD28 stimulation involves both phosphatidylinositol 3-kinase and acidic sphingomyelinase signals. J Immunol. 1996;157(8):3290–3297. [PubMed] [Google Scholar]
- 9.Hui E, Cheung J, Zhu J, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017 doi: 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baine I, Abe BT, Macian F. Regulation of T-cell tolerance by calcium/NFAT signaling. Rev: Immunol; 2009. [DOI] [PubMed] [Google Scholar]
- 11.Arranz-Nicolás J, Ogando J, Soutar D, et al. Diacylglycerol kinase α inactivation is an integral component of the costimulatory pathway that amplifies TCR signals. Cancer Immunol Immunother. 2018 doi: 10.1007/s00262-018-2154-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhong XP, Hainey EA, Olenchock BA, et al. Regulation of T cell receptor-induced activation of the Ras-ERK pathway by diacylglycerol kinase ζ. J Biol Chem. 2002 doi: 10.1074/jbc.M203818200. [DOI] [PubMed] [Google Scholar]
- 13.Goto K, Kondo H. Diacylglycerol kinase in the central nervous system-molecular heterogeneity and gene expression. Chem Phys Lipids. 1999;98(1–2):109–117. doi: 10.1016/s0009-3084(99)00023-7. [DOI] [PubMed] [Google Scholar]
- 14.Shulga YV, Topham MK, Epand RM. Regulation and functions of diacylglycerol kinases. Rev: Chem; 2011. [DOI] [PubMed] [Google Scholar]
- 15.Mérida I, Andrada E, Gharbi SI, Ávila-Flores A. Redundant and specialized roles for diacylglycerol kinases α and ζ in the control of T cell functions. Sci Signal. 2015 doi: 10.1126/scisignal.aaa0974. [DOI] [PubMed] [Google Scholar]
- 16.Macián F, García-Cózar F, Im SH, et al. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002 doi: 10.1016/S0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- 17.Martinez-Moreno M, Garcia-Lievana J, Soutar D, et al. FoxO-dependent regulation of diacylglycerol kinase gene expression. Mol Cell Biol. 2012 doi: 10.1128/mcb.00654-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baldanzi G, Pighini A, Bettio V, et al. SAP-mediated inhibition of diacylglycerol kinase α regulates TCR-induced diacylglycerol signaling. J Immunol. 2011 doi: 10.4049/jimmunol.1002476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruffo E, Malacarne V, Larsen SE, et al. Inhibition of diacylglycerol kinase α restores restimulation-induced cell death and reduces immunopathology in XLP-1. Sci Transl Med. 2016 doi: 10.1126/scitranslmed.aad1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prinz PU, Mendler AN, Masouris I, et al. High DGK-α and disabled MAPK pathways cause dysfunction of human tumor-infiltrating CD8 + T cells that is reversible by pharmacologic intervention. J Immunol. 2012 doi: 10.4049/jimmunol.1103028. [DOI] [PubMed] [Google Scholar]
- 21.Moon EK, Wang LC, Dolfi DV, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. 2014 doi: 10.1158/1078-0432.CCR-13-2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou P, Shaffer DR, Alvarez Arias DA, et al. In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature. 2014 doi: 10.1038/nature12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riese MJ, Wang LCS, Moon EK, et al. Enhanced effector responses in activated CD8+ T cells deficient in diacylglycerol kinases. Cancer Res. 2013 doi: 10.1158/0008-5472.CAN-12-3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andrada E, Liébana R, Merida I. Diacylglycerol kinase ζ limits cytokine-dependent expansion of CD8+ T cells with broad antitumor capacity. EBioMedicine. 2017 doi: 10.1016/j.ebiom.2017.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wesley EM, Xin G, McAllister D, et al. Diacylglycerol kinase ζ (DGKζ) and casitas b-lineage proto-oncogene b–deficient mice have similar functional outcomes in T cells but DGKζ-deficient mice have increased T cell activation and tumor clearance. Immuno Horizons. 2018 doi: 10.4049/immunohorizons.1700055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jutz S, Leitner J, Schmetterer K, et al. Assessment of costimulation and coinhibition in a triple parameter T cell reporter line: simultaneous measurement of NF-κB, NFAT and AP-1. J Immunol Methods. 2016 doi: 10.1016/j.jim.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 27.Leitner J, Kuschei W, Grabmeier-Pfistershammer K, et al. T cell stimulator cells, an efficient and versatile cellular system to assess the role of costimulatory ligands in the activation of human T cells. J Immunol Methods. 2010 doi: 10.1016/j.jim.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ávila-Flores A, Arranz-Nicolás J, Andrada E, et al. Predominant contribution of DGKζ over DGKα in the control of PKC/PDK-1-regulated functions in T cells. Immunol Cell Biol. 2017 doi: 10.1038/icb.2017.7. [DOI] [PubMed] [Google Scholar]
- 29.Gharbi SI, Rincón E, Avila-Flores A, et al. Diacylglycerol kinase ζ controls diacylglycerol metabolism at the immunological synapse. Mol Biol Cell. 2011 doi: 10.1091/mbc.E11-03-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanjuán MA, Jones DR, Izquierdo M, Mérida I. Role of diacylglycerol kinase α in the attenuation of receptor signaling. J Cell Biol. 2001 doi: 10.1083/jcb.153.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olenchock BA, Guo R, Carpenter JH, et al. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat Immunol. 2006 doi: 10.1038/ni1400. [DOI] [PubMed] [Google Scholar]
- 32.Arranz-Nicolás J, Martin-Salgado M, Rodríguez-Rodríguez C, et al. Diacylglycerol kinase ζ limits IL-2-dependent control of PD-1 expression in tumor-infiltrating T lymphocytes. J Immunother Cancer. 2020 doi: 10.1136/jitc-2020-001521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Juneja VR, McGuire KA, Manguso RT, et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med. 2017 doi: 10.1084/jem.20160801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meng X, Liu X, Guo X, et al. FBXO38 mediates PD-1 ubiquitination and regulates anti-tumour immunity of T cells. Nature. 2018;564(7734):130–135. doi: 10.1038/s41586-018-0756-0. [DOI] [PubMed] [Google Scholar]
- 35.Liu M, Jin X, He X, et al. Macrophages support splenic erythropoiesis in 4T1 tumor-bearing mice. PLoS ONE. 2015 doi: 10.1371/journal.pone.0121921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao L, He R, Long H, et al. Late-stage tumors induce anemia and immunosuppressive extramedullary erythroid progenitor cells. Nat Med. 2018 doi: 10.1038/s41591-018-0205-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanjuán MA, Pradet-Balade B, Jones DR, et al. T cell activation in vivo targets diacylglycerol kinase α to the membrane: a novel mechanism for Ras Attenuation. J Immunol. 2003 doi: 10.4049/jimmunol.170.6.2877. [DOI] [PubMed] [Google Scholar]
- 38.Merino E, Ávila-Flores A, Shirai Y, et al. Lck-dependent tyrosine phosphorylation of diacylglycerol kinase α regulates its membrane association in T Cells. J Immunol. 2008 doi: 10.4049/jimmunol.180.9.5805. [DOI] [PubMed] [Google Scholar]
- 39.Mahoney KM, Freeman GJ, McDermott DF. The next immune-checkpoint inhibitors: Pd-1/pd-l1 blockade in melanoma. Ther: Clin; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rincón M, Flavell RA. AP-1 transcriptional activity requires both T-cell receptor-mediated and co-stimulatory signals in primary T lymphocytes. EMBO J. 1994 doi: 10.1002/j.1460-2075.1994.tb06757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kang SM, Beverly B, Tran AC, et al. Transactivation by AP-1 is a molecular target of T cell clonal anergy. Science. 1992 doi: 10.1126/science.257.5073.1134. [DOI] [PubMed] [Google Scholar]
- 42.Stelekati E, Chen Z, Manne S, et al. Long-term persistence of exhausted CD8 T cells in chronic infection is regulated by MicroRNA-155. Cell Rep. 2018 doi: 10.1016/j.celrep.2018.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo R, Wan CK, Carpenter JH, et al. Synergistic control of T cell development and tumor suppression by diacylglycerol kinase α and ζ. Proc Natl Acad Sci U S A. 2008 doi: 10.1073/pnas.0711856105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jung IY, Kim YY, Yu HS, et al. CRISPR/Cas9-mediated knockout of DGK improves antitumor activities of human T cells. Cancer Res. 2018 doi: 10.1158/0008-5472.CAN-18-0030. [DOI] [PubMed] [Google Scholar]
- 45.Prinz PU, Mendler AN, Brech D, et al. NK-cell dysfunction in human renal carcinoma reveals diacylglycerol kinase as key regulator and target for therapeutic intervention. Int J Cancer. 2014 doi: 10.1002/ijc.28837. [DOI] [PubMed] [Google Scholar]
- 46.Yang E, Singh BK, Paustian AMS, Kambayashi T. Diacylglycerol kinase ζ Is a target to enhance NK cell function. J Immunol. 2016 doi: 10.4049/jimmunol.1600581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boroda S, Niccum M, Raje V, et al. Dual activities of ritanserin and R59022 as DGKα inhibitors and serotonin receptor antagonists. Biochem Pharmacol. 2017 doi: 10.1016/j.bcp.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Velnati S, Ruffo E, Massarotti A, et al. Identification of a novel DGKα inhibitor for XLP-1 therapy by virtual screening. Eur J Med Chem. 2019 doi: 10.1016/j.ejmech.2018.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fu L, Li S, Xiao W, et al. DGKA mediates resistance to PD-1 blockade. Cancer Immunol Res. 2021 doi: 10.1158/2326-6066.CIR-20-0216. [DOI] [PubMed] [Google Scholar]
- 50.Franks CE, Campbell ST, Purow BW, et al. The ligand binding landscape of Diacylglycerol kinases. Cell Chem Biol. 2017 doi: 10.1016/j.chembiol.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang Y, Sakane F, Kanoh H, Walsh JP. Selectivity of the diacylglycerol kinase inhibitor 3-{2-(4-[bis-(4-fluorophenyl)methylene]-1-piperidinyl)ethyl}-2,3-dihydro-2-thioxo-4(1H)quinazolinone (R59949) among diacylglycerol kinase subtypes. Biochem Pharmacol. 2000 doi: 10.1016/S0006-2952(99)00395-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data relevant to the study are included in the article or uploaded as online supplemental information.