

Abstract
The clinical success of immune checkpoint blockade against diverse human cancers highlights the critical importance of insightful understanding into mechanisms underlying PD-L1 regulation. IFN-γ released by intratumoral lymphocytes regulates PD-L1 expression in tumor cells through JAK-STAT-IRF1 pathway, while the molecular events prime IRF1 to translocate into nucleus are still obscure. Here we identified STXBP6, previously recognized involving in SNARE complex assembly, negatively regulates PD-L1 transcription via retention of IRF1 in cytoplasm. IFN-γ exposure stimulates accumulation of cytosolic IRF1, which eventually saturates STXBP6 and triggers nuclear translocation of IRF1. Nuclear IRF1 in turn inhibits STXBP6 expression and thereby liberates more IRF1 to migrate to nucleus. Therefore, we identified a novel positive feedback loop between STXBP6 and IRF1 in regulation of PD-L1 expression in cancer. Furthermore, we demonstrate STXBP6 overexpression significantly inhibits T cell activation both in vitro and in vivo. These findings offer new insight into the complexity of PD-L1 expression in cancer and suggest a valuable measure to predict the response to PD-1/PD-L1-based immunotherapy.
Electronic supplementary material
The online version of this article (10.1007/s00262-020-02678-6) contains supplementary material, which is available to authorized users.
Keywords: STXBP6, IRF1, PD-L1, Positive feedback loop, Cancer.
Introduction
The PD-1/PD-L1 antibody-based tumor immunotherapy is prosperous currently [1, 2], the clinical outcomes of which vary greatly depending on tumor types and individual conditions. Cell surface PD-L1 abundance is well-recognized as one of the critical determinants of therapeutic responses [3, 4]. Therefore, the comprehensive understanding into the regulatory mechanisms underlying PD-L1 expression is of essential importance to decipher tumor immune evasion and instruct immunotherapy [5, 6]. So far multiple signaling and mechanisms have been unraveled in control of PD-L1 expression. In nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma, chromosome 9p24.1 is selectively amplified which links to aberrant high expression of PD-L1 along with PD-L2 and JAK2 [7, 8]. Oncogenic transcription factors, including MYC [9], HIF1α [10, 11] and NF-κB [12, 13] involving in PD-L1 transcription are identified along with their respective responsive elements. More recently, the post-transcriptional regulation of PD-L1 by multiple microRNAs has also been documented [14, 15], and its importance has been underlined by the finding of structural variations (SVs) in 3′ region of PD-L1 [16], which associates with elevated PD-L1 transcripts and sophisticated immune evasion highly likely through disrupting the interaction between PD-L1 and unidentified microRNAs. At protein level, type III transmembrane protein CMTM6 increases cell surface PD-L1 through either promoting recycling endosomes [17] or inhibiting ubiquitination-mediated degradation [18]. Likely, cell cycle kinase cyclin D/CDK4-dependent phosphorylation stabilizes SPOP, which thereby promotes PD-L1 degradation via ubiquitin/proteasome system [19].
In addition to the intrinsic regulatory mechanisms, interplays between immune cells and cancer cells in tumor microenvironment constitute a fundamental dimension in control PD-L1 abundance [20, 21]. The interferons released from tumor infiltrating T lymphocytes trigger PD-L1 induction in cancer cells and thereby inhibit the anti-tumor immune response [22]. Among which, type II IFN-γ binds to IFNGR1/2 receptors and stimulates phosphorylation of JAK1/2, subsequently phosphorylated receptors recruit and phosphorylate STAT1/3 [23]. Activated STAT dimers migrate into nucleus and transcriptionally activate IFN-γ inducible genes, such as IRF1. The systematic mapping of JAK1/JAK2-STAT1/STAT1/STAT2/STAT3-IRF1 axis with shRNA screening has highlighted its importance for clinical responses to PD-1 blockade therapy [24]. IRF1-deficient tumor fails to up-regulate PD-L1 and is more susceptible to T cell-mediated killing, which is rescued by induced expression of PD-L1 [25]. However, as a critical interferon response mediator, the molecular events underlying nuclear transportation of IRF1 largely remains elusive. Here we unravel a novel mechanism that STXBP6, a protein previously recognized involving in SNARE complex formation [26, 27], controls IRF1 subcellular distribution and therefore negatively regulates PD-L1 expression in multiple cancers. We further identify a positive feedback loop between STXBP6 and IRF1 in response to IFN-γ stimulation. Our study offers new insight into the complexity of PD-L1 expression in tumor.
Materials and methods
Cell culture
HCT116, HepG2, 769p, A2780, HeLa, MCF7 and MDA-MB-231 cells were obtained from ATCC and cultured in modified RPMI medium (Gibco) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin and 10% fetal bovine serum. Cell identity was authenticated by STR profiling method by SYSU and possible mycoplasma contamination was regularly excluded by PCR analysis. Cell transfection was performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instruction. For IFN-γ exposure, cells were incubated with 500 IU/ml IFN-γ (Pepprotech) for 48 h unless indicated.
Plasmid constructs
The shSTXBP6-1 sequence AACTTATATCTGCCTGTCAGTGACAAACA and shSTXBP6-2 sequence AGATCCTCCACCATACCTGCCAGAGGTAC were cloned into HuSH shRNA tGFP Cloning Vector (pGFP-V-RS, TR30007, OriGene) to generate STXBP6-specific shRNAs, and HuSH 29-mer Non-Effective Scrambled pGFP-V-RS (TR30013, OriGene) was used as scrambled control. STXBP6 transcript NM_014178 was cloned into pcDNA vector for expressing purpose.
Real-time PCR
Total RNA was extracted from cells with TRIzol reagent (Invitrogen) following the manufacturer’s instruction. cDNA was prepared from each 1 μg of RNA with Transcriptor First Strand cDNA Synthesis Kit (Roche). Real-time PCR was performed with PowerUp SYBR Green Master Mix (Thermo Fisher) on CFX96 Touch PCR Detection System (Bio-Rad). β-actin was employed as internal reference gene and relative expression was calculated by 2− △△Ct method. Primer sequences were as follows:
PD-L1 Forward 5′-GCTGCACTAATTGTCTATTGGGA-3′, reverse 5′-AATTCGCTTGTAGTCGGCACC-3′; mPD-L1 Forward 5′-GCTCCAAAGGACTTGTACGTG-3′, reverse 5′-TGATCTGAAGGGCAGCATTTC-3′; STXBP6 Forward 5′-TTCTTGGCAACTGGAGGTCAA-3′, reverse 5′-TCGATACCATTAACCTGGCGAA-3′; mSTXBP6 Forward 5′-CTGGGAGCCATCCAAGTCAAG-3′, reverse 5′-TGACCTTCGTGATAGATGCCT-3′; IRF1 Forward 5′-ATGCCCATCACTCGGATGC-3′, reverse 5′-CCCTGCTTTGTATCGGCCTG-3′; β-actin Forward 5′-CATGTACGTTGCTATCCAGGC-3′, reverse 5′-CTCCTTAATGTCACGCACGAT-3′; mβ-actin Forward 5′-GTGACGTTGACATCCGTAAAGA-3′, reverse 5′-GCCGGACTCATCGTACTCC-3′.
Western blots
Cells were lysed in RIPA buffer containing proteinase inhibitor cocktail (Roche) on ice for 30 min and debris was removed by refrigerated centrifugation. The supernatant was quantitated with BCA Protein Assay Kit (Thermo Fisher) and denatured in SDS sample buffer at 100 ℃ for 5 min. The protein samples were resolved by SDS-PAGE and transferred to PVDF membrane (Millipore). Membranes were briefly blocked with 5% skim milk and incubated with indicated antibodies (mouse anti-HSP90, 610418, BD Biosciences; mouse anti-PD-L1, UMAB228, Origene; mouse anti-STXBP6, sc-271959, Santa Cruz; mouse anti-Lamin B1, 3C10G12; mouse anti-α-Tubulin, 1E4C11, Proteintech; rabbit anti-IRF1, D5E4; goat anti-rabbit, 7074; horse anti-mouse, 7076; Cell Signaling Technology), and blots were visualized using ECL (Millipore).
Immunoprecipitation
Cells were lysed in 1% Triton X-100 for 30 min at 4 ℃. After refrigerated centrifugation for 15 min, the supernatant was transferred into new tubes and incubated with indicated primary antibodies (mouse anti-STXBP6, sc-271959; mouse anti-IRF1, sc-514544; Santa Cruz) on rotator overnight at 4 ℃, followed by addition of Dynabeads Protein G and incubation for another 2 h at 4 ℃. Beads were washed with 0.2% Triton X-100 three times and co-immunoprecipitated protein species were eluted with SDS sample buffer containing 50 mM DTT for 10 min at 70 ℃, and subjected to western blots detection as previously described.
Orthotopic 4T1 xenograft animal model
Animal study was approved by the Sun Yat-sen University Institutional Animal Care and Use Committee (SYSU IACUC). Female BALB/c mice (6-week-old) were purchased from Vital River Laboratory. 4T1 cells (5 × 106 eV or STXBP6-overexpressing) were s.c. inoculated into the mammary gland and allowed to grow for 15 days. Tumor progression and body weight of tumor-bearing mice was regularly monitored, and xenograft tumor volume was estimated as length × width2 × 0.5.
Micro-metastasis evaluation
Lung and liver tissues from aforementioned 4T1 tumor mice were collected, washed with 1 × HBSS buffer, and minced into small pieces in 6-cm petri dish. After transfer into a new 15 ml tubes, lung tissues were digested with collagenase type IV/elastase (1 mg/ml collagenase type IV and 6 U/ml elastase, Worthington) on rotator at 4 ℃ for 75 min, and liver tissues were digested with collagenase type I/hyaluronidase (1 mg/ml collagenase type I from Worthington and 1 mg/ml hyaluronidase from Sigma-Aldrich) on rotator at 37 ℃ for 30 min. Enzyme-digested samples were filtered with 70 μm cell strainer and washed with 1 × HBSS buffer. After brief centrifugation, the sample pellets were resuspended in complete modified RPMI medium supplemented with 60 μM 6-thioguanine (Sigma-Aldrich), and followed by consecutive culture for 2 weeks. The clonogenic 4T1 cells were stained with crystal violet and counted.
Immunofluorescence
Mice were euthanized at day 15 post-implantation following SYSU IACUC guideline, xenograft tumors were collected in base molds pre-filled with Tissue-Tek CRYO-OCT (SAKURA) and subjected to cryosection with NX50 Cryostat (Thermo Fisher). Tissue sections were fixed with 4% paraformaldehyde (PFA) plus 0.3% Triton X-100, and sequentially blocked with 3% BSA/0.05% Tween-20 and normal rat serum (Sigma). Incubation with anti-CD8 primary antibody (MCA609G, Bio-Rad) was performed at 4 ℃ overnight, and followed by incubation with goat anti-rat AF488 secondary antibody (A-11006, Invitrogen) for another h at room temperature. Slides were then mounted with ProLong Gold/DAPI (P36931, Invitrogen) and analyzed under (LSM800, Carl Zeiss).
Flow cytometry
Cells (pre-treated with either vehicle or 500 IU/ml IFN-γ for 48 h) were trypsinized and harvested, followed by PBS wash to remove any residual components from culture medium. Cell pellets were resuspended in staining buffer (2% BSA in PBS) and 100 μl aliquot was incubated with PE-labeled anti-PD-L1 antibody (329705, BioLegend) at 4 ℃ in the dark for 15 min. After wash with staining buffer twice, cells were resuspended in staining buffer for flow cytometry analysis on Gallios (Beckman Coulter).
PD-1 binding assay
HCT116 (EV and STXBP6-overexpressing) and HepG2 (control and STXBP6-knockdown) cells were placed on coverslips and pre-treated with IFN-γ for 48 h. After fixation with 4% PFA for 15 min at room temperature, cells were sequentially incubated with recombinant human PD-1 Fc protein (1086-PD, R&D Systems) for 1 h and anti-human AF594 (A-11014, Thermo Fisher) for another 1 h at room temperature. Coverslips were mounted with ProLong Gold/DAPI (Invitrogen) and representative images were captured under confocal microscope (LSM800, Carl Zeiss).
Jurkat co-culture IL-2 production
HCT116 (EV and STXBP6-overexpressing) and HepG2 (shNT, shSTXBP6-1 and shSTXBP6-2) cells were pre-treated with IFN-γ for 24 h and then co-cultured with Jurkat cells at the ratio of 2:1 Jurkat: HCT116/HepG2. Jurkat cells were activated in advance with PMA (25 ng/ml, P1585, Sigma-Aldrich) and PHA (1 μg/ml, L2769, Sigma-Aldrich) for 24 h. Secreted IL-2 was determined at 48 and 72 h later with IL-2 Human ELISA Kit (Invitrogen) as recommended by provider.
Luciferase assays
Both PD-L1 (HPRM40139), STXBP6 (HPRM43060) promoter reporter plasmids and secreted alkaline phosphatase (SEAP)-driven Gaussia luciferase (reference) were obtained from GeneCopoeia. The truncate mutations were generated by PCR splicing method and scrambled mutations were generated by mutagenesis PCR method, respectively. PD-L1 promoter reporter (wild type or truncate mutations) plasmids were co-transfected with either empty control or STXBP6-overexpressing plasmids into HCT116 for 6 h, and followed by IFN-γ exposure for 24 h. STXBP6 promoter reporter (wild type or scrambled mutations) plasmids were transfected into HCT116 cells for 6 h and subjected to IFN-γ exposure for 24 h. The culture medium was collected for relative luciferase activity determination with Secrete-Pair Luminescence Assay Kit (GeneCopoeia) in accordance with the manufacturer’s manual.
Immunohistochemistry
Human liver tumor tissue array was obtained from AlenaBio. The immunohistochemistry procedure was performed with Biotin-Streptavidin HRP Detection Systems (ZSGB-BIO) and strictly complied with the provider’s instruction. Briefly, tissue section was first heated at 60 ℃ for 30 min, and followed by deparaffinization with xylene and rehydration with gradient ethanol solution. Antigen retrieval was performed in sodium citrated solution (0.01 M, pH 6.0) at 98 ℃ for 15 min in microwave oven. The endogenous peroxidase activity was blocked with 3% H2O2 in methanol at room temperature for 10 min, and tissue sections were blocked with 10% FBS in a humidified chamber at room temperature for 1 h. The primary antibody (mouse anti-PD-L1, 405.9A11, Cell Signaling Technology; rabbit anti-STXBP6, HPA003552, Sigma-Aldrich) was then incubated at 4 ℃ overnight, and followed by 15 min of incubation with biotin-labelled secondary antibody. After incubation with HRP-streptavidin at room temperature for 15 min, the section was detected with diaminobenzidine (DAB) and counterstained with hematoxylin. Images were captured under DMi8 Inverted Microscope (Leica).
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were prepared from indicated cells using Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime). Oligo probes (with or without biotin labelling) were synthesized by Synbio. EMSA was performed with Chemiluminescent EMSA Kit (GS009, Beyotime) following the manufacturer’s instruction. For binding reaction, 10 μg nuclear extracts, 20 fmole biotin-labeled probes, 4 pmol competitor probes and 1 μg IRF1 antibody (D5E4, Cell Signaling Technology) were used. The mixtures were then resolved by 4% non-denaturing TBE-PAGE gel and transferred to nylon membrane (Beyotime). After crosslinking in UV-light cross-linker (Fisher Scientific) for 45 s (254 nm, 120 mJ/cm2), the bands were detected with ECL Reagent (Millipore) and visualized using ChemiDoc Touch Imaging System (Bio-Rad).
Chromatin immunoprecipitation (ChIP)
ChIP assay was performed using EZ-Magna ChIP A/G Chromatin Immunoprecipitation Kit (17–10086, Millipore) in accordance with the manufacturer’s protocol. Crosslinked chromatin was extracted from 1 × 106 cells and sheared into 200 ~ 1000 bp fragments by ultrasonication. 2 μg IRF1 antibody (sc-514544x, Santa Cruz) and 20 μl protein A/G magnetic beads were then applied for incubation at 4 ℃ overnight with rotation. Immunoprecipitated complex was eluted and DNA fragments were recovered with Proteinase K digestion at 62 ℃ for 2 h. After purification using spin column, 2 μl DNA species were subjected to real-time PCR analysis as previously described.
Statistical analysis
We employed unpaired, two-tailed Student’s t test for comparison with GraphPad Prism 7.0, and p < 0.05 was defined as significant difference.
Results
STXBP6 negatively regulates PD-L1 expression
We first evaluated the potential impact of STXBP6 on PD-L1 expression in human colorectal carcinoma cell, HCT116 and hepatocellular carcinoma cell, HepG2. Stable cell lines of HCT116 with STXBP6-overexpression and HepG2 with STXBP6-knockdown were established in view of their endogenous expression levels and validated by both real-time PCR and western blots (Fig. 1a, Huh7 and HT29 cells in Figure S1). PD-L1 induction by IFN-γ was significantly compromised in STXBP6-procificient HCT116 cells, while tremendously augmented in two individual STXBP6-silenced HepG2 cells (Fig. 1b, similar observations were obtained in Huh7 and HT29 cell shown in Figure S2). The inhibitory effect of STXBP6 on IFN-γ-stimulated PD-L1 expression was further validated at protein level (Fig. 1c). Likewise, cell surface PD-L1 interrogated with both affinity antibody (Fig. 1d, S3) and recombinant human PD-1 Fc protein (Fig. 1e) exhibited consistent changes in both cell lines exposed to IFN-γ treatment. The functional influences of both STXBP6-proficiency and -deficiency on T cell were evaluated by co-culture system, and secretory IL-2 was measured which represented a key player in the cell-mediated immune response in allograft rejection and indicated local immune activation [28]. STXBP6-overexpression significantly stimulated the secretion of IL-2 in Jurkat: HCT116 co-culture medium, and opposite effects were observed in STXBP6-silenced HepG2 cells (Fig. 1f, S4). In addition, we also noticed the negative correlation between STXBP6 and PD-L1 transcripts in panel of cell lines originating from breast, renal, cervical, ovarian cancers (Fig. 1g). The in vivo relevance was further confirmed by immunohistochemical analysis, in which strong STXBP6 signaling was detected in the PD-L1-negative tissue section, and vice versa (Fig. 1h), and significantly negative correlation was observed based on IHC scores. Transcriptome analysis of clinical tumor samples from The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC) on UCSC Xena platform revealed weak but significant correlation between STXBP6 and PD-L1 mRNAs in range of cancer types (Fig. 1i), indicating a universal mode-of-action of STXBP6 on PD-L1 regulation.
Fig. 1.

STXBP6 negatively regulates PD-L1 expression. a Stable cell lines derived from HCT116 with over-expression of STXBP6 and HepG2 with knockdown of STXBP6, STXBP6 was measured with both real-time PCR (normalized to β-actin) and western blots (normalized to HSP90). b Relative expression of PD-L1 in HCT116 (EV and STXBP6) with or without exposure to IFN-γ (left); Relative expression of PD-L1 in HepG2 (shNT, shSTXBP6-1, -2) with or without exposure to IFN-γ (right). c Western blot analysis of STXBP6 and PD-L1 expression in HCT116 (EV and STXBP6, upper) and HepG2 (shNT, shSTXBP6-1, -2, lower). d Cell surface PD-L1 determined by flow cytometry in HCT116 (EV and STXBP6, left) and HepG2 cells (shNT, shSTXBP6-1, -2, right) exposed to IFN-γ treatment. e Binding of recombinant human PD-1 Fc on the cytomembrane of HCT116 (EV and STXBP6, left) and HepG2 (shNT and shSTXBP6, right) analyzed by immunofluorescence, scale bar, 50 μm. f IL-2 production in Jurkat: HCT116/HepG2 co-culture system. Jurkat cells were pre-activated with PMA and PHA for 24 h, HCT116 (EV and STXBP6) and HepG2 (shNT, shSTXBP6-1 and shSTXBP6-2) cells were pre-treated with IFN-γ for 24 h. Co-culture was performed at the ratio of 2:1 Jurkat: HCT116/HepG2, and IL-2 levels in medium were measured by ELISA after 48 and 72 h, respectively. g Correlation analysis of endogenous STXBP6 and PD-L1 mRNA in 769p, A2780, HCT116, HeLa, HepG2, MCF7 and MDA-MB-231. h Representative images of immunohistochemical analysis on PD-L1 and STXBP6 expression in clinical liver tumor samples (left). Correlation analysis was performed based on IHC scores (R2 = 0.5072, p < 0.0001, right). i Correlation analysis of STXBP6 and PD-L1 mRNA in human cancers with Xena. Pearson’s correlation coefficients are shown along with unadjusted p values. CESC, Cervical Cancer; COADREAD, Colon and Rectal Cancer; ESCA, Esophageal Cancer; GBM, Glioblastoma; HNSC, Head and Neck Cancer; LGG, Lower Grade Glioma; LIHC, Liver Cancer; PACA-AU; PCPG, Pheochromocytoma and Paraganglioma; SKCM, Melanoma; STAD, Stomach Cancer; THCA, Thyroid Cancer; UCEC, Endometrioid Cancer; TCGA Pan-Cancer; ICGC-donor, International Cancer Genome Consortium-donor. **p < 0.01, ***p < 0.001, ****p < 0.0001
The inhibitory effect of STXBP6 on PD-L1 is dependent on IRF1
We started the mechanistic investigation into inhibitory effect of STXBP6 on PD-L1 transcription with identification of the critical cis-element on PD-L1 promoter. To this purpose, we generated serially truncate mutants of PD-L1 promoter luciferase reporter plasmids, and co-transfection with STXBP6-expressing vector significantly inhibited wild-type PD-L1 promoter-driven luciferase activity upon IFN-γ exposure (Fig. 2a). All truncations except for the -292 to -92 nt deletion were shown to be subjected to the inhibition by STXBP6. Meanwhile, the induction of luciferase expression by IFN-γ was almost completely abrogated by destruction of -292 to -92 nt, which was in agreement with previously identified IRF1 responsive sites in this region. To pinpoint the sequences responding to STXBP6, we further conducted two-round EMSA assay with biotin-labelled overlapping probes spanning the region -292 to -92 nt. Incubation with nuclear extracts from HCT116 significantly retarded the mobility of fragments -215 to -148 nt, and prominent shift was observed with probe 3–2 (− 187 to – 148 nt, Fig. 2b). Notably, this shift was greatly abolished in the STXBP6-overexpressing cells in comparison with control (Fig. 2c), suggesting the inhibitory effect of STXBP6 was highly likely mediated by disruption of interaction between trans factors with their cis-elements. Close inspection of this region yielded three putative IRF1 binding sites (JASPAR [29], Fig. 2d), which prompted us to clarify whether IRF1 involved in STXBP6-regulated PD-L1 expression. Addition of IRF1-specific antibody in binding reaction led to remarkable super shift of retarded band, indicating that IRF1 was indeed complexed with probe 3–2 (Fig. 2e). The IRF1 expression remained constant at both transcript and protein levels in the context of either STXBP6-overexpression or -knockdown (Fig. 2f), which excluded the possibility that STXBP6 inhibited PD-L1 transcription through down-regulation of IRF1. However, we did detect the decrease of IRF1 occupancy on PD-L1 promoter in STXBP6-proficient HCT116 cells and increase in STXBP6-deficient HepG2 while compared to control cells (Fig. 2g), suggesting that association of IRF1 with responsive element was compromised by STXBP6.
Fig. 2.

The inhibitory effect of STXBP6 on PD-L1 is dependent on IRF1. A. PD-L1 promoter luciferase reporter assay with serial truncations conducted in HCT116 cells with co-transfection of either empty vector or STXBP6-expressing plasmids. b Two-round EMSA assay performed with indicated probes and HCT116 nuclear extracts. c EMSA behaviors of probe M3-1 and M3-2 are examined in HCT116 EV (left) and HCT116-STXBP6 cells (right). d Illustration of PD-L1 promoter underlining the putative IRF1 binding sites and highlighting the ChIP primer sequences. e EMSA performed with probe M3-2 in presence of IRF1 binding antibody. f Relative expression of IRF1 in response to IFN-γ stimulation in HCT116 (EV and STXBP6-overexpressing) and HepG2 (control, shSTXBP6-1 and shSTXBP6-2) is quantified by real-time PCR (left) and western blots (right). g ChIP analysis of IRF1 binding to PD-L1 promoter in HCT116 (EV and STXBP6-overexpressing) and HepG2 (control, shSTXBP6-1 and shSTXBP6-2). n.s no significance, *p < 0.05, **p < 0.01, ****p < 0.0001
Retention of IRF1 in cytoplasm by STXBP6
Next, we analyzed the subcellular localization of IRF1 and the absolute majority of intrinsic IRF1 was found in cytoplasm as indicated by immunofluorescence. With IFN-γ exposure, the small portion of induced IRF1 translocated into nuclear (circled) while the rest stayed outside nuclei (Fig. 3a). Interestingly, STXBP6 abundance remarkably influenced the re-distribution of IRF1 upon IFN-γ treatment, and intranuclear IRF1 levels were tremendously decreased in STXBP6-overexpressing HCT116 and induced in STXBP6-silenced HepG2 (Fig. 3b, c). We further conducted subcellular fractionization analysis to validate the translocation of IRF1, western blots consolidated the immunofluorescence results (Fig. 3d). Notably, STXBP6 and IRF1 manifested evident co-localization in naïve HCT116 and HepG2 (Fig. 3e), which prompted us to speculate that cytosolic STXBP6 might restrain translocation of IRF1 into nucleus directly. To verify this hypothesis, we performed reciprocal immunoprecipitation with both anti-IRF1 and anti-STXBP6 antibodies followed by western blot detection. The presence of STXBP6 in IRF1-immunoprecipitate was confirmed in lysates derived from both HCT116-STXBP6 and HepG2 cells, and IRF1 was detectable in STXBP6-immunoprecipitate as well (Fig. 3f). The interaction with STXBP6 might serve as fundamental mechanism to prevent unexpected entrance of IRF1 into nucleus and therefore abnormal activation of interferon signaling.
Fig. 3.

Retention of IRF1 in cytoplasm by STXBP6. a Subcellular localization of IRF1 is determined by immunofluorescence in HCT116 cells with or without exposure to IFN-γ. Subcellular localization of IRF1 in IFN-γ-treated HCT116 (EV and STXBP6-overexpressing, b) and HepG2 (shNT and shSTXBP6-1, c). d Subcellular distribution of IRF1 in aforementioned cells is analyzed by fractionized western blots. e Colocalization of STXBP6 and IRF1 is examined by confocal microscopy in HepG2 and HCT116 cells. f Reciprocal immunoprecipitation of IRF1 and STXBP6 in HepG2 (left) and HCT116-STXBP6 cells (right)
Nuclear IRF1 in turn inhibits STXBP6 expression
Surprisingly, we noticed that STXBP6 transcripts were significantly down-regulated along with PD-L1 up-regulation by IFN-γ in both HCT116 and HepG2 cells (Fig. 4a, S5 left). Consistently, STXBP6 proteins accordingly decreased upon IFN-γ treatment (Fig. 4b, S5 right). Next, we employed two independent JAK2-specific inhibitors, AZD1480 and Fedratinib, to simultaneously dose HCT116 and HepG2 cells while exposed to IFN-γ. As expected, both AZD1480 and Fedratinib potently inhibited IRF1 induction by IFN-γ at both transcript (Fig. 4c, S6) and protein (Fig. 4d) levels. Most importantly, the repressed STXBP6 in IFN-γ-exposed cells was almost completely restored by co-treatment with either AZD1480 or Fedratinib (Fig. 4e, S7), hinting the dependence of STXBP6 inhibition on JAK/STAT signaling. The assembling evidences were in support of the divergent roles of IRF1 in gene transcriptional regulation as either activator or repressor [30–34]. With aid of online bioinformatics algorithm (JASPAR), we identified two putative IRF1 binding sites (-1573 to -1562 nt and –1360 to -1349 nt) on STXBP6 promoter (Fig. 4f). STXBP6 promoter reporter luciferase was significantly inhibited by IFN-γ exposure in HCT116 cells, while scrambled mutation introduced in site 1 rather than site 2 completely abrogated this effect (Fig. 4g), which indicated the region -1573 to -1562 nt was the responsive element upon JAK/STAT signaling activation. We further analyzed the direct binding of IRF1 to STXBP6 promoter with ChIP assay. Results showed that STXBP6 promoter was greatly enriched in the IRF1 immunoprecipitated complex while exposed to IFN-γ for 24 h in both HCT116 and HepG2 cells (Fig. 4h). We noticed that despite of abundantly intrinsic IRF1 without IFN-γ exposure, there was not evident enrichment of STXP6 promoter in this context which was mainly due to cytosolic subcellular localization of non-induced IRF1. Upon stimuli like IFN-γ, IRF1 was dramatically induced and a small fraction translocated into nucleus to activate downstream signaling pathway. Meanwhile, intranuclear IRF1 specifically bound to STXBP6 promoter and suppressed its transcription. The down-regulated STXBP6 further facilitated the translocation of cytosolic IRF1 to augment the IFN-γ response.
Fig. 4.

IRF1 in turn inhibits STXBP6 expression. a Relative expressions of PD-L1 and STXBP6 in HCT116 (left) and HepG2 (right) with or without exposure to IFN-γ. b Western blot analysis of STXBP6 expression in HCT116 (upper) and HepG2 (lower) with or without IFN-γ treatment. c Relative expression of IRF1 mRNA in HCT116 (left) and HepG2 (right) with or without exposure to IFN-γ and simultaneously JAK2-specific inhibitors, AZD1480 and Fedratinib (Fed). d Western blots examination IRF1 protein in response to aforementioned treatments. e Relative expression of STXBP6 mRNA in HCT116 (left) and HepG2 (right) cells subjected to vehicle, IFN-γ alone or in combinations with AZD1480 or Fed. f Promoter sequences of human STXBP6 with underlined putative IRF1 binding sites/scrambled mutations and highlighted ChIP primers. g STXBP6 promoter luciferase reporter assay including mutations introduced into putative IRF1 binding sites. h ChIP analysis of IRF1 binding on STXBP6 promoter in response to IFN-γ exposure in HCT116 (left) and HepG2 (right) cells. n.s no significance, **p < 0.01, ***p < 0.001, ****p < 0.0001
STXBP6 promotes tumor specific T cell activity via suppression of PD-L1 expression
To examine the influences of STXBP6 on tumor progression in vivo, we established STXBP6-overexpressing stable cell line derived from mice mammary carcinoma 4T1. The tumor growth was significantly suppressed in STXBP6-proficient group in comparison with control mice in the orthotopic xenograft tumor model with BALB/c mice (Fig. 5a). Continuous monitoring up to 15 days post-implantation consistently displayed remarkable retardation of tumor progression in STXBP6-overexpressing mice than control (Fig. 5b) while with comparable body weights were recorded during experimental window (Fig. 5c). Mechanistically, we demonstrated that STXBP6 over-expression significantly enhanced CD8+ T infiltration and accumulation in tumor mass (Fig. 5d). In view of the highly metastatic nature, we further evaluated the distant dissemination of 4T1 cells in response to STXBP6 overexpression with quantitation of the clonogenic metastatic colonies in 6-thioguanine-containing medium. Likewise, both lung and liver metastasis were greatly compromised by ectopic STXBP6 (Fig. 5e). The persistently stable overexpression of STXBP6 and therefore inhibited PD-L1 levels in 4T1 xenograft tumor was validated at the endpoint of experiment (Fig. 5f).
Fig. 5.

STXBP6 promotes tumor specific T cell activity via suppression of PD-L1 expression. a Representative macroscopic images of 4T1 xenograft tumors in BALB/c mice expressing either empty vector (n = 7) or STXBP6 (n = 7). b Tumor growth of 4T1 cells expressing either empty vector or STXBP6 in BALB/c mice (unpaired t-test). c Body weight of xenograft tumor (4T1-EV and -STXBP6) bearing BALB/c mice. d Relative expression of both mPD-L1 and mSTXBP6 in 4T1 xenograft tumors. e Representative CD8+ T cell infiltration indicated by immunofluorescence staining in orthotopic xenograft tumor (4T1-EV and -STXBP6, scale bars: 200 μm.). f Colony formation assay for lung and liver metastatic 4T1 cells (empty or STXBP6-overexpressing). Representative crystal violet staining images are shown in left pane, and counting results for lung and liver micro-metastasis cells are provided in middle and right panes. *p < 0.05, **p < 0.01, ***p < 0.001
Discussion
The clinical successes of immunotherapy against number of human cancers are heavily relied on the increasingly insightful understanding of tumor immunology and especially mechanisms underlying immune escape of tumor cells. Here we unveiled that STXBP6, which was previously characterized involving in SNARE complex formation and exocytosis, potently retained IRF1 in cytoplasm and therefore inhibited PD-L1 expression. In response to IFN-γ stimuli, IRF1 increasingly accumulated in cytoplasm and eventually saturated STXBP6, which consequently led to nuclear translocation of IRF1. Nuclear IRF1 then exerted transcription repressor role on STXBP6 expression in addition to transcriptional activation of interferon stimulated genes. The decrease in STXBP6 further liberated more IRF1 to migrate to nucleus. Therefore, cytosolic STXBP6 constitutively blocked unexcepted entrance of IRF1 to nucleus, and upon IFN-γ stimuli, the positive feedback loop between induced IRF1 and STXBP6 predominantly instructed PD-L1 transcription (Fig. 6).
Fig. 6.
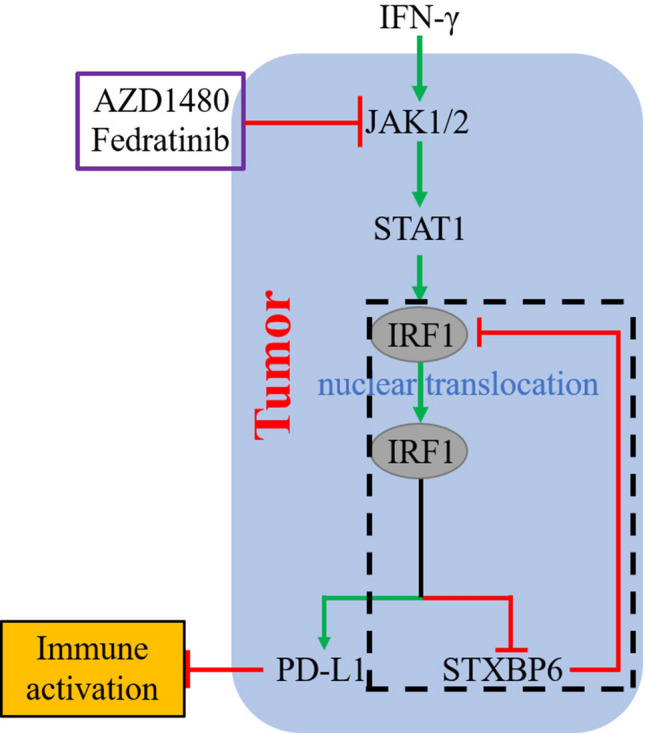
Graphic mode of STXBP6-IRF1-PD-L1 involved in tumor immune suppression. Cytosolic STXBP6 constitutively blocked unexcepted entrance of IRF1 to nucleus, and upon IFN-γ stimuli, the positive feedback loop (black dotted rectangle) between induced IRF1 and STXBP6 predominantly instructed PD-L1 transcription
So far, the molecular events prime IRF1 for translocation into nucleus were relatively obscure. Based on frequent pattern mining and linear motif scoring algorithm (seqNLS from Machine Learning & Evolution Laboratory, USC) [35], a possible nuclear localization signal (120–139 aa) was predicted positioning in the middle of IRF1 amino acid sequence. This prediction was consistent with a previous functional domain analysis performed by Schaper et al., who showed the NLS segment (116–139) was sufficient for nucleus localization of fused reporter GFP [36]. Accordingly, we hypothesized the intrinsic nuclear trafficking feature of IRF1, which was concealed by interaction with STXBP6 to retain IRF1 in cytoplasm and exert yet unrecognized roles. IFN-γ stimulated accumulation of cytosolic IRF1 and eventually led to active nuclear translocation of unbound IRF1 and activation of positive feedback loop between IRF1 and STXBP6. An alternate mechanism governed IRF1 translocation has been propose by Negishi et al. in elucidation of synergistic effect between IFN-γ and Toll-like receptors (TLRs) in host defense against pathogens [37]. It’s suggested that via complexation with MyD88 adaptor, IRF1 was licensed and more effectively translocated to nucleus as indicated by photoconversion assay, which then enhanced target genes expression through cooperation with NF-κB in granulocyte/macrophage colony-stimulating factor-cultured bone marrow-derived dendritic cells (GM-DCs). The authors also inferred that licensing of IRF1 might involve undefined modification such as phosphorylation while unlicensed IRF1 was distributed mainly in cytoplasm and slowly migrated into the nucleus. This was in line with our data in respect of the cytosolic localization of constitutive and vast majority of IFN-γ-induced IRF1 via interaction with STXBP6. We have not experimentally addressed further whether MyD88 was involved in this scenario and instead proposed a competitive relation between MyD88 and STXBP6 in determination of IRF1 subcellular distributions in the context of TLR activation. Low abundance of IRF1 was primarily retained in cytoplasm by STXBP6, while accumulated IRF1 was competitively liberated through interaction with MyD88. The positive feedback loop between IRF1 and STXBP6 proposed here was further endorsed by investigation from Luo et al., whose results showed that sequential treatments with two doses of all-trans retinoic acid (ATRA) significantly promoted the nuclear concentration of IRF1 albeit predominantly cytoplasmic localization after first round of treatment [38]. It’s reasonable to speculate that initially brief exposure to ATRA down-regulated STXBP6 and therefore released the inhibitory effects on nuclear migration of induced IRF1 by subsequent stimulation.
Involvement in SNARE complex assembly was the solely recognized biological role of STXBP6 so far. The subsequent investigation provided suggestive evidence of STXBP6 SNP rs749373 for systemic lupus erythematosus (SLE) risk along with susceptible IRF8 loci [39]. Emerging evidences supported the critical involvements of IRF1 as an important inflammation mediator in SLE as well, as IRF1 fundamentally interacted with multiple chromatin modifying enzymes in dysregulation of epigenome in this disease [40, 41]. Liu et al. demonstrated that enhanced inflammasome activity in monocyte derived from SLE patients was mediated by type I interferon-induced up-regulation of IRF1 [42], and higher activation of the interferon-gamma signaling pathway was characterized in SLE with a high type I IFN score [43]. Based on our results in cancer, here we offered a hypothesis that STXBP6 and IRF1 might cooperate in the similar way in SLE in controlling IRF1 subcellular localization and the downstream signaling, which warranted further experimental clarification. Besides, given the functional diversity of IRF1 in host response to viral and bacterial infections, cell proliferation, apoptosis, DNA damage and repair, which necessarily depended on nuclear localization, the potential involvements of STXBP6 in these processing were to be defined in the future.
Summarily, we identified a positive feedback loop between IRF1 and STXBP6 in regulation PD-L1 expression in diverse human cancers, and suggested the anti-tumoral properties of STXBP6 via negative control of PD-L1 expression and therefore activation of tumor immune response. This was partially supported by three independent DNA methylation studies. Fernandez et al. first adopted an in vitro model of human breast epithelial cell transformation and identified DNA methylation and down-regulation of STXBP6 in transformed cells [44]. The following study performed by Lenka et al. analyzed the differentially methylated regions and differentially expressed genes in lung tumor with both methylated DNA immunoprecipitation combined with microarray (MeDIP-chip) and gene expression microarray, and identified 4 candidate genes (STXBP6, BCL6B, FZD10, and HSPB6) that were significantly hypermethylated and downregulated [45]. The higher percentage of methylation in STXBP6 promoter was subsequently validated by Wang et al. in lung adenocarcinoma and adjacent normal lung tissue in non-smoking women [46]. In this regard, DNA demethylation would be a promising strategy to treat STXBP6-low cancers. Our findings highlighted that STXBP6 level might be a valuable measure to predict the response to PD-1/PD-L1-based immunotherapy.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abbreviations
- ATCC
American Tissue Culture Collection
- ATRA
All-trans retinoic acid
- BSA
Bovine serum albumin
- ChIP
Chromatin immunoprecipitation
- DAB
Diaminobenzidine
- DAPI
4′,6-Diamidino-2-phenylindole
- DMEM
Dulbecco’s modified Eagle medium
- DTT
Dithiothreitol
- ECL
Enhanced chemiluminescence
- ELISA
Enzyme-linked immunosorbent assay
- EMSA
Electrophoretic mobility shift assay
- FBS
Fetal bovine serum
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- HBSS
Hank's Balanced Salt Solution
- IHC
Immunohistochemistry
- IL-2
Interleukin-2
- PBS
Phosphate-buffered saline
- PCR
Polymerase chain reaction
- PFA
Paraformaldehyde
- PVDF
Polyvinylidene fluoride
- RIPA
Radioimmunoprecipitation assay
- TLRs
Toll-like receptors
- SD
Standard deviation
- SLE
Systemic lupus erythematosus
- STR
Short tandem repeat
- SV
Structural variation
Author contributions
Y.B.L. conceived this project and interpreted data. Y.B.L. and H.H.W. wrote the manuscript. Y.B.L., Z.C.H., Y.L.W., M.M.Z. and X.Z.L. performed experiments and analyzed data. Y.B.L., H.H.W. and S.L.Y. supervised the whole study.
Funding
This work was supported by National Natural Science Foundation of China, No. 81672796 to Y.B.L. and No. 81171947 to H.H.W.
Data availability
All data generated or analyzed during this study are included in this published article.
Compliance with ethical standards
Competing interests
All other authors have no conflicts of interest to disclose.
Ethical approval
This study was approved by the Sun Yat-sen University Institutional Review Board.
Consent for publication
All authors are aware and agree to the content of the paper and their authorships.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yanbin Liu, Email: yanbliu@hotmail.com.
Shulan Yang, Email: yangshl3@mail.sysu.edu.cn.
Haihe Wang, Email: wanghaih@mail.sysu.edu.cn.
References
- 1.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 2.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;17:e542–e551. doi: 10.1016/S1470-2045(16)30406-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pitt JM, Vetizou M, Daillere R, et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity. 2016;44:1255–1269. doi: 10.1016/j.immuni.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 5.Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19:133–150. doi: 10.1038/s41568-019-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–287. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green MR, Monti S, Rodig SJ, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116:3268–3277. doi: 10.1182/blood-2010-05-282780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goodman AM, Piccioni D, Kato S, et al. Prevalence of PDL1 amplification and preliminary response to immune checkpoint blockade in solid tumors. JAMA Oncol. 2018;4:1237–1244. doi: 10.1001/jamaoncol.2018.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casey SC, Tong L, Li Y, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352:227–231. doi: 10.1126/science.aac9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014;74:665–674. doi: 10.1158/0008-5472.CAN-13-0992. [DOI] [PubMed] [Google Scholar]
- 11.Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211:781–790. doi: 10.1084/jem.20131916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang G, Wen Q, Zhao Y, Gao Q, Bai Y. NF-kappaB plays a key role in inducing CD274 expression in human monocytes after lipopolysaccharide treatment. PLoS ONE. 2013;8:e61602. doi: 10.1371/journal.pone.0061602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gowrishankar K, Gunatilake D, Gallagher SJ, Tiffen J, Rizos H, Hersey P. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-kappaB. PLoS ONE. 2015;10:e0123410. doi: 10.1371/journal.pone.0123410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen L, Gibbons DL, Goswami S, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014;5:5241. doi: 10.1038/ncomms6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pyzer AR, Stroopinsky D, Rosenblatt J, et al. MUC1 inhibition leads to decrease in PD-L1 levels via upregulation of miRNAs. Leukemia. 2017;31:2780–2790. doi: 10.1038/leu.2017.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kataoka K, Shiraishi Y, Takeda Y, et al. Aberrant PD-L1 expression through 3'-UTR disruption in multiple cancers. Nature. 2016;534:402–406. doi: 10.1038/nature18294. [DOI] [PubMed] [Google Scholar]
- 17.Burr ML, Sparbier CE, Chan YC, et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature. 2017;549:101–105. doi: 10.1038/nature23643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mezzadra R, Sun C, Jae LT, et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature. 2017;549:106–110. doi: 10.1038/nature23669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang J, Bu X, Wang H, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018;553:91–95. doi: 10.1038/nature25015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 Checkpoint. Immunity. 2018;48:434–452. doi: 10.1016/j.immuni.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, Dang F, Ren J, Wei W. Biochemical aspects of PD-L1 regulation in cancer immunotherapy. Trends Biochem Sci. 2018;43:1014–1032. doi: 10.1016/j.tibs.2018.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee SJ, Jang BC, Lee SW, et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7–H1 (CD274) FEBS Lett. 2006;580:755–762. doi: 10.1016/j.febslet.2005.12.093. [DOI] [PubMed] [Google Scholar]
- 23.Mimura K, Teh JL, Okayama H, et al. PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. 2018;109:43–53. doi: 10.1111/cas.13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia-Diaz A, Shin DS, Moreno BH, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017;19:1189–1201. doi: 10.1016/j.celrep.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shao L, Hou W, Scharping NE, et al. IRF1 Inhibits antitumor immunity through the upregulation of PD-L1 in the tumor cell. Cancer Immunol Res. 2019;7:1258–1266. doi: 10.1158/2326-6066.CIR-18-0711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scales SJ, Hesser BA, Masuda ES, Scheller RH. Amisyn, a novel syntaxin-binding protein that may regulate SNARE complex assembly. J Biol Chem. 2002;277:28271–28279. doi: 10.1074/jbc.M204929200. [DOI] [PubMed] [Google Scholar]
- 27.Constable JR, Graham ME, Morgan A, Burgoyne RD. Amisyn regulates exocytosis and fusion pore stability by both syntaxin-dependent and syntaxin-independent mechanisms. J Biol Chem. 2005;280:31615–31623. doi: 10.1074/jbc.M505858200. [DOI] [PubMed] [Google Scholar]
- 28.Spolski R, Li P, Leonard WJ. Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat Rev Immunol. 2018;18:648–659. doi: 10.1038/s41577-018-0046-y. [DOI] [PubMed] [Google Scholar]
- 29.Fornes O, Castro-Mondragon JA, Khan A, et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2019 doi: 10.1093/nar/gkz1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pizzoferrato E, Liu Y, Gambotto A, et al. Ectopic expression of interferon regulatory factor-1 promotes human breast cancer cell death and results in reduced expression of survivin. Cancer Res. 2004;64:8381–8388. doi: 10.1158/0008-5472.CAN-04-2223. [DOI] [PubMed] [Google Scholar]
- 31.Xie RL, Gupta S, Miele A, Shiffman D, Stein JL, Stein GS, van Wijnen AJ. The tumor suppressor interferon regulatory factor 1 interferes with SP1 activation to repress the human CDK2 promoter. J Biol Chem. 2003;278:26589–26596. doi: 10.1074/jbc.M301491200. [DOI] [PubMed] [Google Scholar]
- 32.Fragale A, Gabriele L, Stellacci E, et al. IFN regulatory factor-1 negatively regulates CD4+ CD25+ regulatory T cell differentiation by repressing Foxp3 expression. J Immunol. 2008;181:1673–1682. doi: 10.4049/jimmunol.181.3.1673. [DOI] [PubMed] [Google Scholar]
- 33.Ramsauer K, Farlik M, Zupkovitz G, Seiser C, Kroger A, Hauser H, Decker T. Distinct modes of action applied by transcription factors STAT1 and IRF1 to initiate transcription of the IFN-gamma-inducible gbp2 gene. Proc Natl Acad Sci U S A. 2007;104:2849–2854. doi: 10.1073/pnas.0610944104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao J, Senthil M, Ren B, Yan J, Xing Q, Yu J, Zhang L, Yim JH. IRF-1 transcriptionally upregulates PUMA, which mediates the mitochondrial apoptotic pathway in IRF-1-induced apoptosis in cancer cells. Cell Death Differ. 2010;17:699–709. doi: 10.1038/cdd.2009.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin JR, Hu J. SeqNLS: nuclear localization signal prediction based on frequent pattern mining and linear motif scoring. PLoS ONE. 2013;8:e76864. doi: 10.1371/journal.pone.0076864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaper F, Kirchhoff S, Posern G, Koster M, Oumard A, Sharf R, Levi BZ, Hauser H. Functional domains of interferon regulatory factor I (IRF-1) Biochem J. 1998;335(Pt 1):147–157. doi: 10.1042/bj3350147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Negishi H, Fujita Y, Yanai H, et al. Evidence for licensing of IFN-gamma-induced IFN regulatory factor 1 transcription factor by MyD88 in Toll-like receptor-dependent gene induction program. Proc Natl Acad Sci U S A. 2006;103:15136–15141. doi: 10.1073/pnas.0607181103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo XM, Ross AC. Retinoic acid exerts dual regulatory actions on the expression and nuclear localization of interferon regulatory factor-1. Exp Biol Med (Maywood) 2006;231:619–631. doi: 10.1177/153537020623100517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lessard CJ, Adrianto I, Ice JA, et al. Identification of IRF8, TMEM39A, and IKZF3-ZPBP2 as susceptibility loci for systemic lupus erythematosus in a large-scale multiracial replication study. Am J Hum Genet. 2012;90:648–660. doi: 10.1016/j.ajhg.2012.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leung YT, Shi L, Maurer K, Song L, Zhang Z, Petri M, Sullivan KE. Interferon regulatory factor 1 and histone H4 acetylation in systemic lupus erythematosus. Epigenetics. 2015;10:191–199. doi: 10.1080/15592294.2015.1009764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Z, Shi L, Song L, Ephrem E, Petri M, Sullivan KE. Interferon regulatory factor 1 marks activated genes and can induce target gene expression in systemic lupus erythematosus. Arthritis Rheumatol. 2015;67:785–796. doi: 10.1002/art.38964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu J, Berthier CC, Kahlenberg JM. Enhanced inflammasome activity in systemic lupus erythematosus is mediated via type I interferon-induced up-regulation of interferon regulatory factor 1. Arthritis Rheumatol. 2017;69:1840–1849. doi: 10.1002/art.40166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu M, Liu J, Hao S, Wu P, Zhang X, Xiao Y, Jiang G, Huang X. Higher activation of the interferon-gamma signaling pathway in systemic lupus erythematosus patients with a high type I IFN score: relation to disease activity. Clin Rheumatol. 2018;37:2675–2684. doi: 10.1007/s10067-018-4138-7. [DOI] [PubMed] [Google Scholar]
- 44.Fernandez SV, Snider KE, Wu YZ, Russo IH, Plass C, Russo J. DNA methylation changes in a human cell model of breast cancer progression. Mutat Res. 2010;688:28–35. doi: 10.1016/j.mrfmmm.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lenka G, Tsai MH, Lin HC, et al. Identification of methylation-driven, differentially expressed STXBP6 as a novel biomarker in lung adenocarcinoma. Sci Rep. 2017;7:42573. doi: 10.1038/srep42573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J, Duan Y, Meng QH, Gong R, Guo C, Zhao Y, Zhang Y. Integrated analysis of DNA methylation profiling and gene expression profiling identifies novel markers in lung cancer in Xuanwei. China PLoS One. 2018;13:e0203155. doi: 10.1371/journal.pone.0203155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article.