

Abstract
Thyroid carcinoma (THCA) is the most common endocrine malignancy, and its incidence is increasing worldwide. Several studies have explored whether the tumor immune microenvironment and immune-related genes (IRGs) influence the prognosis of patients with THCA and can be used to predict the response to immune checkpoint inhibitors (ICIs). We developed an IRG prognostic/risk signature using a bioinformatics method, and its predictive capacity was validated in patients in the test set and the total set. Subsequently, we analyzed the correlation between this IRG prognostic signature and tumor-infiltrating immune cells, tumor mutation burden (TMB), and immune checkpoint protein expression in patients with THCA. With a multivariate analysis, the IRG prognostic signature, which comprised eight IRGs, was identified as an independent prognostic factor. High-risk patients had poor overall survival compared with low-risk patients. Plasma cells, monocytes, and dendritic cells infiltrated differently according to the IRG prognostic signature. The low-risk group had a higher TMB and immunophenoscore (IPS), which indicated a better response to ICIs. The qRT-PCR validated eight IRGs with differential expression in thyroid cancer and normal tissues. We conclude that the IRG prognostic signature may be a useful tool to predict survival and response to ICIs. However, further testing is required to assess the predictive capacity of this IRG prognostic signature.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00262-021-03020-4.
Keywords: THCA, Immune signature, Immune checkpoint, Prognosis, TMB
Introduction
Thyroid carcinoma (THCA), which derives from follicular thyroid cells, is the most common endocrine malignancy worldwide [1, 2]. The incidence of THCA morbidity has continued to rise in recent decades [3–5]. Papillary thyroid carcinoma (PTC), which is the most common subtype of THCA, accounts for approximately 85% of all THCA cases [6, 7]. After surgery and 131 treatment, numerous cases of PTC are cured and have good rates of survival; nevertheless, 10% of patients still suffer from advanced tumor recurrence and die from poorly differentiated [8]. Therefore, further studies are required to determine the underlying mechanisms of tumorigenesis and identify sensitive biomarkers to improve the prognosis of THCA.
In the clinic, immune checkpoint inhibitors (ICIs) targeting programmed cell death protein 1 (PD-1), programmed cell death ligand 1 (PD-L1), T cell immunoglobulin and ITIM domain (TIGIT), T cell immunoglobulin mucin-3 (TIM-3), and cytotoxic T lymphocyte antigen 4 (CTLA4) have successfully treated various types of tumor in recent years [9]. To date, only a few studies have reported immunotherapy targeting PD-1/PD-L1 in patients with THCA [10]. Evidence shows that cancer immunology and immunotherapy are hopeful in the field of cancer. Several immune-related prognostic signatures have been used to predict the prognosis of lung adenocarcinoma [11], hepatocellular carcinoma [12], endometrial carcinoma [13], epithelial ovarian cancer [14], and cervical cancer [15]. However, there are limited studies on using prognostic signatures based on IRGs to predict the prognosis and survival of THCA patients.
In our study, we aimed to construct an IRG prognostic signature according to The Cancer Genome Atlas (TCGA) database. We thoroughly explored the association between prognostic signature and tumor-infiltrating immune cells, mutation data, and the immunophenoscore (IPS) of patients with THCA. This may help to predict the response to immunotherapy for patients in different risk categories.
Materials and methods
Data sources and clinical samples
We downloaded 568 gene expression profiles (58 non-diseased samples and 510 tumor samples) of patients with THCA and clinical information on overall survival (OS) from TCGA (https://portal.gdc.cancer.gov/) [16]. The clinical characteristics of patients with THCA are shown in Table 1. Moreover, a list of IRGs were obtained from the Immunology Database and Analysis Portal (ImmPort) (https://immport.niaid.nih.gov) [17].
Table 1.
Clinical characteristics of THCA patients in TCGA
| Characteristics | Samples (n = 502) | Percentage (%) |
|---|---|---|
| Age | ||
| ≤ 60 | 389 | 77.49 |
| > 60 | 113 | 22.51 |
| Gender | ||
| Female | 367 | 73.11 |
| Male | 135 | 26.89 |
| Stage | ||
| Stage I-II | 333 | 66.33 |
| Stage III-IV | 167 | 33.27 |
| NA | 2 | 0.4 |
| T | ||
| T1-2 | 307 | 61.16 |
| T3-4 | 193 | 38.45 |
| NA | 2 | 0.4 |
| M | ||
| M0 | 282 | 56.18 |
| M1 | 9 | 1.79 |
| NA | 211 | 42.03 |
| N | ||
| N0 | 229 | 45.62 |
| N1 | 223 | 44.42 |
| NA | 50 | 9.96 |
Differentially expressed genes (DEGs)
We identified DEGs in tumor and non-diseased samples using the “limma” package of R (version 4.0.4). An adjusted p value of < 0.05 and a |log2 fold change |> 1) were set as the screening criterion [18]. DE-IRGs were obtained from the intersection of DEGs and IRGs.
Functional enrichment analysis
Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were conducted in the R software with the “clusterProfiler” package to explore the molecular mechanisms of DE-IRGs [19]. A p value of < 0.05 was considered statistically significant.
Construction of the IRG prognostic signature
We randomly divided 502 patients (the entire set) with THCA with complete OS information into a training set (n = 251) and a test set (n = 251) at a ratio of 1:1. Data from the training set were used to construct the prognostic model, whereas prognostic model performance was evaluated in the test set and in the entire set. We performed a univariate Cox proportional hazards regression analysis to identify prognostic IRGs with a p value of < 0.05. Subsequently, a least absolute shrinkage and selection operator (LASSO) Cox regression analysis with the “glmnet” package was used to minimize overfitting [20]. Finally, after a multivariate Cox regression analysis, we employed screening genes to construct the IRG prognostic risk signature. Risk score was calculated as follows: risk score = expression of gene a × coefficient a + expression of gene b × coefficient b + expression of gene c × coefficient c + …… + expression of gene n × coefficient n [15]. After that, all patients were classified into a high-risk and a low-risk groups according to median risk scores. We used Kaplan–Meier survival curves, log-rank tests, and time-dependent receiver operating characteristic (ROC) curves to evaluate the predictive capability of prognostic signatures for OS in the training set, the test set, and the entire set. Univariate and multivariate Cox regression analyses were also performed to identify independent prognostic variables for OS.
Estimation of tumor-infiltrating immune cells
CIBERSORT is a deconvolution algorithm based on RNA-Seq data to estimate the composition ratio of immune cells [21]. We examined the abundance of 21 types of infiltrating immune cell in all tumor samples according to THCA transcriptional profiles.
Single-sample GSEA (ssGSEA)
We performed ssGSEA to determine infiltration and activity of 29 IRG cell signatures in the high-risk and low-risk groups using the “GSVA” package of R [25]. We also compared the expression of the HLA gene between the high- and low-risk groups.
Mutation analysis
Mutation data from patients with THCA were also downloaded from TCGA data portal (https://portal.gdc.cancer.gov/). Data were further analyzed using the “maftools” package [22]. We calculated the tumor mutation burden (TMB) score of every patient as follows: (total mutation ÷ total covered bases) × 106 [23].
IPS analysis
The four determining components of immunogenicity were effector cells, immunosuppressive cells, major histocompatibility complex (MHC) molecules, and immunomodulators. Each patient’s IPS was obtained using machine learning without bias [24]. IPS ranged from 0–10 according to representative cell-type gene expression Z-scores. The IPS positively correlated with tumor immunogenicity. The IPS values of patients with THCA were downloaded from The Cancer Immunome Atlas (https://tcia.at/home). We used IPS values to compare the response to ICIs between the high-risk and low-risk groups.
Quantitative PCR
Ninety-eight matched tumorous and non-tumorous tissue specimens of thyroid cancer were collected from The First Affiliated Hospital of China Medical University. Total RNA was extracted from tissue samples using RNAiso (Takara, Dalian, China), and then, RNA was reverse transcribed into cDNA with the QuantiTect Reverse Transcription Kit (Takara, Shiga, Japan). Quantitative real-time PCR (qRT-PCR) analyses were performed by SYBR-Green (Takara, Shiga, Japan) to validate gene expression, and the level of GAPDH served as an internal control. The cycle threshold (Ct) values were calculated based on the comparative Ct (2−ΔΔCt) method. The premier sequences for qRT-PCR are shown in Supplementary Table 1.
Results
Identification DE-IRGs
According to the adjusted p value of < 0.05 and a |log2 (fold change) of |> 1, a total of 2,413 DEGs were identified, consisting of 1,559 upregulated and 854 downregulated genes (Fig. 1A). Subsequently, 262 DE-IRGs were extracted (Fig. 1B). GO enrichment analysis showed that the enriched biological process (BP) mainly included cell chemotaxis and the humoral immune response; the most enriched cellular component (CC) was the external side of the plasma membrane; and receptor–ligand activity and signaling receptor activation were significantly enriched in terms of molecular function (MF) (Figs. 1C). The top three highly enriched KEGG pathways were cytokine–cytokine receptor interactions, neuroactive ligand–receptor interactions, and viral protein interactions with cytokines and cytokine receptors (Figs. 1D).
Fig. 1.

Identification DE-IRGs and functional enrichment analyses of DE-IRGs. A Volcano plot of DEGs in THCA. B Venn diagram for the intersections of DEGs and IRGs. C Gene ontology analysis. D KEGG pathway enrichment analysis
Construction of IRG risk models
We obtained 502 OS samples by integrating messenger RNA expression profiles and clinical information of patients with THCA. A total of 502 OS samples (the entire set) were randomly divided into a training set (n = 251) and a test set (n = 251). In the training and test sets, there were no significant differences in clinical baseline characteristics (p > 0.05) (Table 2).
Table 2.
Clinical variables in training and test sets
| Variables | Group | Total set (n = 502) | Training set (n = 251) | Test set (n = 251) | p value |
|---|---|---|---|---|---|
| Age | ≤ 60 | 389(77.49%) | 194(77.29%) | 195(77.69%) | 1 |
| > 60 | 113(22.51%) | 57(22.71%) | 56(22.31%) | ||
| Gender | Female | 367(73.11%) | 184(73.31%) | 183(72.91%) | 1 |
| Male | 135(26.89%) | 67(26.69%) | 68(27.09%) | ||
| Stage | Stage I-II | 333(66.33%) | 172(68.53%) | 161(64.14%) | 0.2826 |
| Stage III-IV | 167(33.27%) | 77(30.68%) | 90(35.86%) | ||
| NA | 2(0.4%) | 2(0.8%) | 0(0%) | ||
| T | T1-2 | 307(61.16%) | 153(60.96%) | 154(61.35%) | 1 |
| T3-4 | 193(38.45%) | 96(38.25%) | 97(38.65%) | ||
| NA | 2(0.4%) | 2(0.8%) | 0(0%) | ||
| M | M0 | 282(56.18%) | 148(58.96%) | 134(53.39%) | 0.8916 |
| M1 | 9(1.79%) | 4(1.59%) | 5(1.99%) | ||
| NA | 211(42.03%) | 99(39.44%) | 112(44.62%) | ||
| N | N0 | 229(45.62%) | 119(47.41%) | 110(43.82%) | 0.8566 |
| N1 | 223(44.42%) | 113(45.02%) | 110(43.82%) | ||
| NA | 50(9.96%) | 19(7.57%) | 31(12.35%) |
To further explore the prognostic value of DE-IRGs, we performed a univariate Cox regression analysis in the training set. Forty-one genes were highly associated with the OS (Fig. 2C). Then, LASSO Cox regression analysis and multivariate Cox regression analysis were conducted to establish prognostic signatures (Fig. 2A and B). We identified eight genes (HSPA6, PPBP, S100A11, AZU1, SEMA6B, VGF, IL20RA, and FYN) to establish an immune predictive model in the training set (Fig. 2D). The risk scores of OS were calculated using multivariate Cox regression coefficients as follows: risk score = (0.76331 × HSPA6 expression) + (1.16703 × PPBP expression) + (− 2.59161 × S100A11 expression) + (1.85680 × AZU1 expression) + (0.76205 × SEMA6B expression) + (0.57494 × VGF expression) + (0.66903 × IL20RA expression) + (− 1.45947 × FYN expression). Subsequently, we calculated the risk score of every patient in the training set. All patients in the training set were divided into high-risk and low-risk group based on the median risk score. There was a significant difference in OS between the two groups (p = 0.008) (Fig. 3D). High-risk patients had a poorer OS compared with low-risk patients. The area under the curve (AUC) for this OS risk signature was 0.996 at 1 year, 0.973 at 3 years, and 0.932 at 5 years (Fig. 3E). We ranked the risk scores of patients with THCA and analyzed their distribution and survival status in the training set (Fig. 3A and B). The expressions of eight IRGs for OS in patients with THCA were shown as a heatmap in Fig. 3C.
Fig. 2.

Construction of immune-related prognostic signature. C Forest plot presenting the univariate Cox model results. A, B LASSO Cox regression analysis and multivariate Cox regression analysis. D Forest plot illustrating the multivariate Cox regression analysis of eight prognostic immune-related genes
Fig. 3.

Identification of eight immune-related genes prognostic signature in training, test, and entire sets. A The distribution of risk scores, B survival status, C the expression of eight prognostic IRGs in high- and low-risk groups, D Kaplan–Meier curve analysis of overall survival of THCA in high- and low-risk groups, E time-dependent ROC analysis in the training set. F The distribution of risk scores, G survival status, H 8-IRGs expression patterns in high- and low-risk groups, I Kaplan–Meier curve analysis of overall survival, J time-dependent ROC analysis in the test set. K The distribution of risk scores, L survival status, M 8-IRGs expression patterns in high- and low-risk groups, N Kaplan–Meier curve analysis of overall survival, O time-dependent ROC analysis in the entire set
Evaluating the predictive capacity of the 8-IRG prognostic signatures
We performed test set and entire set to evaluate the stability and prognostic value of prognostic signatures. Patients were also classified into high- and low-risk groups according to the expression of eight IRGs in the test set. We analyzed the distribution of risk scores, survival status, and the expression of eight IRGs in the test set (Fig. 3F, G and H). A Kaplan–Meier curve analysis showed significant difference between two predicted risk groups, and the low-risk group had a better OS compared with the high-risk group (p < 0.001) (Fig. 3I). The risk signature for OS showed ROC curve values of 1.000, 0.900, and 0.943 at 1, 3, and 5 years, respectively, in the test cohort (Fig. 3J).
Besides, patients were divided into high- and low-risk group in the total set. The prognostic predictions were similar to the previous two sets. Patients with high-risk scores had a lower OS (p < 0.001) (Fig. 3N). In the total set, the three years AUC was 0.930, and the AUC of 5-year OS was 0.938 (Fig. 3O). The distribution of the risk score, survival status, and the expression of 8-IRG in the total set are displayed in Fig. 3K, L and M.
Association between prognosis and risk signatures
Subsequently, we sequentially performed univariate Cox regression analysis and multivariate Cox regression analysis to determine whether risk score could independently predict OS of patients with THCA in the training, test, and entire sets. The univariate Cox regression analysis indicated that age, stage, and risk score were closely associated with OS. The multivariate Cox regression analysis showed that age and risk score correlated with OS (Table 3). These results indicate that risk score could be an independent predictor in THCA patients. Additionally, the AUC of the risk score for OS was 0.998 at 1 year, 0.930 at 3 years, and 0.937 at 5 years (Figs. 4A, B and C). These results indicate that risk score has considerable prognostic value compared with other clinical factors and demonstrate that risk score is an independent prognostic risk factor.
Table 3.
Univariate and multivariate Cox regression analysis in training, test, and entire sets
| Variables | Univariate analysis | Multivariate analysis | ||||||
|---|---|---|---|---|---|---|---|---|
| HR | HR.95L | HR.95H | P | HR | HR.95L | HR.95H | P | |
| Training set | ||||||||
| Age | 1.15 | 1.06 | 1.24 | 0.00 | 1.22 | 1.06 | 1.40 | 0.00 |
| Gender | 6.00 | 1.09 | 33.02 | 0.04 | 19.58 | 2.01 | 190.65 | 0.01 |
| Stage | 3.18 | 1.38 | 7.30 | 0.01 | 0.43 | 0.15 | 1.21 | 0.11 |
| Risk score | 1.00 | 1.00 | 1.00 | 0.00 | 1.00 | 1.00 | 1.00 | 0.01 |
| Test set | ||||||||
| Age | 1.16 | 1.09 | 1.25 | 0.00 | 1.18 | 1.05 | 1.33 | 0.01 |
| Gender | 0.83 | 0.18 | 3.93 | 0.81 | 0.04 | 0.00 | 1.34 | 0.07 |
| Stage | 2.02 | 1.18 | 3.48 | 0.01 | 0.68 | 0.29 | 1.58 | 0.37 |
| Risk Score | 1.00 | 1.00 | 1.01 | 0.00 | 1.01 | 1.00 | 1.01 | 0.00 |
| Entire set | ||||||||
| Age | 1.16 | 1.10 | 1.22 | 0.00 | 1.13 | 1.07 | 1.19 | 0.00 |
| Gender | 1.92 | 0.69 | 5.30 | 0.21 | 1.11 | 0.31 | 4.00 | 0.88 |
| Stage | 2.42 | 1.53 | 3.80 | 0.00 | 1.11 | 0.59 | 2.08 | 0.75 |
| Risk Score | 1.00 | 1.00 | 1.00 | 0.00 | 1.00 | 1.00 | 1.00 | 0.00 |
Fig. 4.

The relationships between immune-related prognostic signature and clinicopathological factors. Time-dependent ROC curves analysis according to risk score and clinical factors. The AUC of 1 (A), 3 (B) and 5 year (C) to predict overall survival for THCA patients. (D) Age, (E) gender, (F) clinical stage, (G) T stage, (H) N stage, and (I) M stage. (J) Heatmap showed the expression of the prognostic IRGs and clinicopathologic factors in high- and low-risk groups
Association between clinicopathological factors and IRG risk signature
The association between risk signatures and clinicopathological factors was performed in OS. The 8-IRG risk score was significantly different in N stage (p = 0.00024) and age (p = 5.3e-05) (Figs. 4D and H). The risk score was higher in patients with N0 stage or aged > 60. Nevertheless, there were no significant differences in gender, clinical stage, T stage, or M stage (Figs. 4E, F, G and I). Consequently, the prognostic signatures of OS had better prognostic value in patients with THCA. In addition, all samples were stratified based on age, gender, clinical stage, and tumor–node–metastasis stage to evaluate the prognostic capacity of the risk signature for OS. With the risk signature, most high-risk groups had poorer OS compared with low-risk groups (Supplementary Fig. 1). The correlation between the expression of prognostic genes and clinicopathological factors is shown in a heatmap (Fig. 4J). These results indicate that the risk signature for OS has independent prognostic capacity without considering other clinicopathological factors.
Association between tumor-infiltrating immune cells and risk signature
Immune cells are closely associated with the tumor immune microenvironment (TME). We therefore analyzed RNA sequencing data to explore the correlation between the risk signature and immune cells. The bar plot shows the relative proportions of 21 immune cells in each THCA sample. The correlation matrix shows the correlation of different tumor-infiltrating immune cells. An analysis of the differences in 21 immune cell types between normal and tumor samples is shown in violin plot (Supplementary Fig. 2). Subsequently, we analyzed the differences between risk scores and tumor-infiltrating immune cells with a violin plot for OS. Plasma cells (p = 0.001), monocytes (p < 0.001), and resting dendritic cells (p = 0.024) infiltrated differently according to the OS risk signature (Fig. 5A). Among them, monocytes and resting dendritic cells were positively correlated with risk score; however, plasma cells were negatively correlated with risk score. Furthermore, high plasma cells level was closely associated with better OS (p = 0.005) (Fig. 5B).
Fig. 5.

The relationships between tumor-infiltrating immune cells and immune-related risk signature. A The association between immune cells infiltration and the signature. B The association between immune cells infiltration (Plasma cells) and overall survival for THCA patients. The immune status in high- and low-risk groups. The immune status C, 16 immune cells D, 13 immune-related functions E and the distribution of HLA-related genes F in high- and low-risk groups
Additionally, we calculated the enrichment scores with ssGSEA to further study the association between risk signature and immune status. The heatmap showed the immune status of 29 immune signature gene sets in the high-risk and low-risk groups (Fig. 5C). The scores for aDCa, CD8+ T cells, DCs, iDCs, macrophages, neutrophils, natural killer cells, pDCs, type 1 T helper cells, type 2 T helper cells, TILs, and regulatory T cells were significantly higher in the low-risk group compared with the high-risk group (p < 0.01) (Fig. 5D). Meanwhile, APC co-inhibition, APC co-stimulation, CCR, checkpoint, cytolytic activity, HLA, inflammation promoting, MHC class I, parainflammation, T cell co-inhibition, co-stimulation, and type I interferon response, which were enriched in functional analyses, were significantly different between the high-risk and low-risk groups (p < 0.01) (Fig. 5E). Moreover, the expression of HLA was significantly different between the high-risk and low-risk groups (Fig. 5F).
IRG risk signature and mutation profile
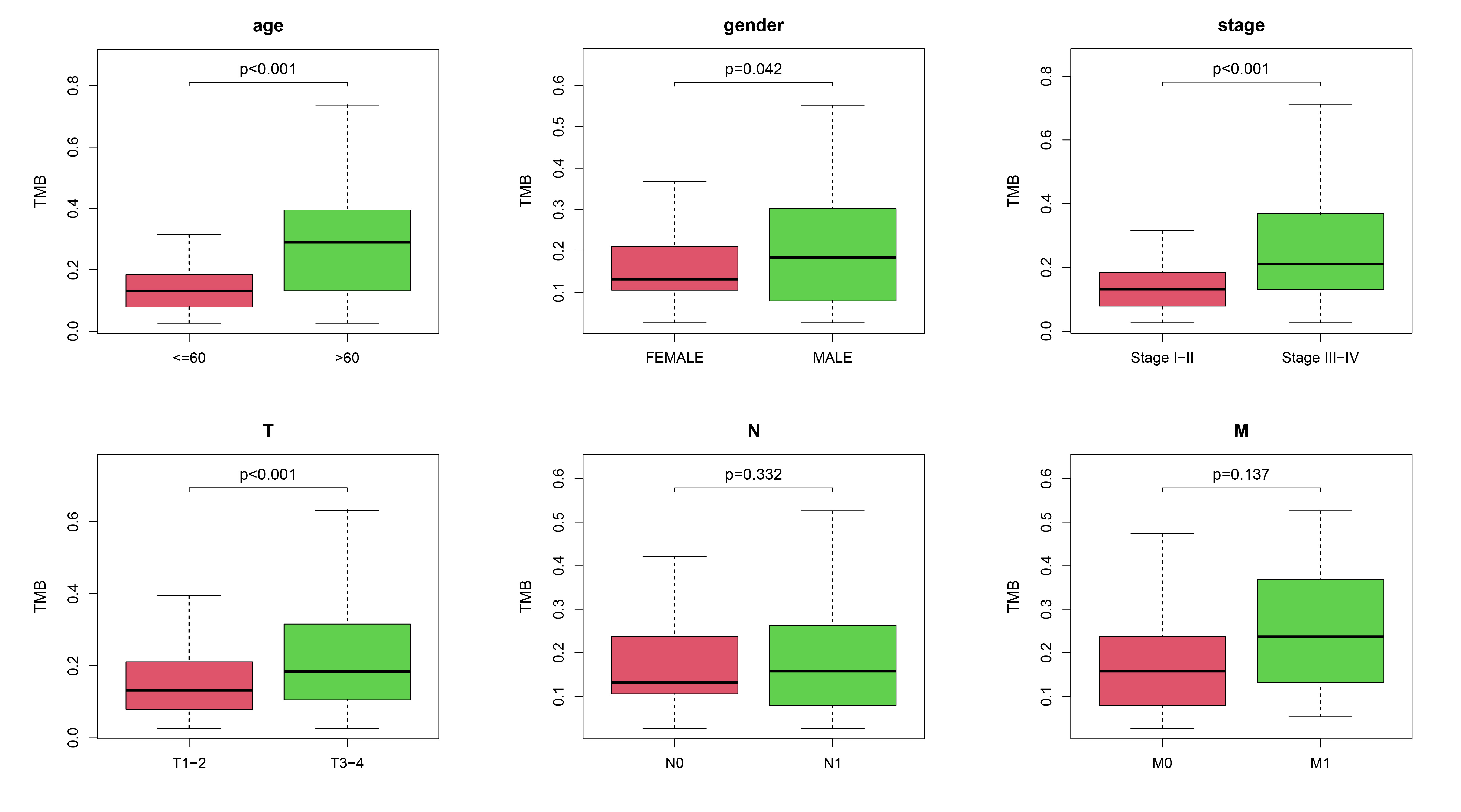
We evaluated the relationship between the risk signature and mutation profile in patients with THCA using somatic mutation data. In the high-risk group, the top ten mutated genes were BRAF, NRAS, HRAS, TG, EIF1AX, PPN, MUC16, USP9X, JMJD1C, and RYR1. In the low-risk group, the most frequently mutated genes were BRAF, NRAS, TTN, TG, and PDZD2 (Fig. 6A). TMB was higher in the high-risk group, and there was a significant difference in the OS model (p < 0.001) (Fig. 6B). In addition, the difference in OS was statistically significant between TMB and OS (p = 0.033) and progression-free interval (PFI) (p = 0.005) (Fig. 6C). We further explored the correlation between TMB and clinicopathological features in patients with THCA. A statistically significant difference was observed between TMB and age (p < 0.001), clinical stage (p < 0.001), and T stage (p < 0.001) for OS signature. However, no significant differences were found in the relationship between TMB and M stage or N stage (Supplementary Fig. 3).
Fig. 6.

The relationships between mutation profile and TMB in high- and low-risk groups. A Mutation profile of THCA patients in high- and low-risk groups. B The difference of TMB in high- and low-risk groups. C The association of TMB and OS and PFI of THCA patients. D The association between IPS and the immune-related prognostic signature. E The expression of PD-1, PD-L1, PD-L2, CTLA4, TIGIT, and TIM-3 in high- and low-risk groups
IRG risk signature and response to ICIs
IPS plays an important role in predicting the response to ICIs in patients with THCA. We therefore investigated the relationship between IPS and IRG risk signature. We used IPS, IPS-PD-1/PD-L1/PD-L2, IPS-CTLA4, and IPS-PD-1/PD-L1/PD-L2 + CTLA4 scores to evaluate the response to ICIs in patients with THCA. IPS, IPS-PD-1/PD-L1/PD-L2, IPS-CTLA4, and IPS-PD-1/PD-L1/PD-L2 + CTLA4 scores were significantly higher in the low-risk group for OS (p < 0.001) (Fig. 6D). The results indicated that patients with low-risk score appeared to be more sensitive to ICI treatment. Meanwhile, the expression of PD-L1, PD-L2, TIGIT, TIM-3, and CTLA4 was higher in the low-risk group (p < 0.001); and PD1 was higher in the low-risk group, although the difference was not statistically significant (p = 0.087) (Fig. 6E). Thus, low-risk patients demonstrated a better response to ICIs.
Validation of the expression levels of eight immune-related genes in thyroid cancer tissues
We examined the expression of the eight IRGs (HSPA6, PPBP, S100A11, AZU1, SEMA6B, VGF, IL20RA, and FYN) in 98 pairs of clinical samples from patients with THCA using qRT-PCR analysis according to the bioinformatics analysis results. Supplementary Table 2 displayed the clinical characteristics of these THCA patients from our hospital. The results of qRT-PCR showed that the mRNA expression of PPBP, S100A11, AZU1, SEMA6B, VGF, and FYN was higher in thyroid cancer tissues compared to normal tissues (p < 0.05). However, the expression of HSPA6 and IL20RA was decreased in thyroid cancer samples (p < 0.05) (Fig. 7), which was consistent with the results of bioinformatic analysis. Furthermore, we investigated the association between the distribution of the expression of 8-IRGs and clinicopathological features. Significantly differences of the expression of AZU1 were observed in clinical stage, N stage, and extrathyroidal extension (Fig. 8A, B and C). The expression level of S100A11 in stage I-II patients or patients aged ≤ 60 was higher than that in stage III-IV patients or patients aged > 60 (Fig. 8D and E). The expression level of PPBP was negatively correlated with T stage and N stage (Fig. 8F and G). The expression level of VGF was significantly different between ages ≤ 60 and > 60 (Fig. 8H). The expression level of SEMA6B showed the negative correlation with N classification of TNM stages (Fig. 8J).
Fig. 7.

The expression of eight IRGs in thyroid cancer tissues. The qRT-PCR results showed the mRNA expression of HSPA6 A, PPBP B, S100A11 C, AZU1 D, SEMA6B E, VGF F, IL20RA G, and FYN H in normal tissues and thyroid cancer tissues
Fig. 8.

The correlation between the RT-qPCR data and clinical features. A–C The correlation between AZU1 and clinical stage, N stage and extrathyroidal extension. D–E The association between S100A11 and clinical stage, age. F–G The correlation between the PPBP and T, N stage. H The relationship between VGF and age. I–J The association between SEMA6B and age, N stage
Discussion
Nowadays, immunotherapy is a promising method in the field of cancer treatment. ICIs are agents that inhibit the mechanisms of immune escape [26, 27]. The success of immunotherapy using ICIs has elucidated the important role of the TME in cancer progression and has attracted the attention of physicians [28]. Therefore, it is necessary to identify an IRG risk signature to predict survival and identify which patients are suitable for immunotherapy and can obtain good clinical results.
In our study, we identified 2413 DEGs (1559 upregulated DEGs and 854 downregulated DEGs) from TCGA in patients with THCA. Moreover, we extracted 262 DE-IRGs and performed GO and KEGG analyses of these genes. GO enrichment analysis showed that IRGs significantly enriched in cell chemotaxis, the external side of the plasma membrane, receptor ligand activity, and signaling receptor activator activity. The highly enriched KEGG pathway was cytokine–cytokine receptor interactions. These functional annotations were all immune-related, which indicated possible functional or pathways imbalanced in the development of THCA. Subsequently, we constructed an IRG risk signature using 262 DE-IRGs and univariate and multivariate Cox regression analyses, which comprised eight prognostic genes (HSPA6, PPBP, S100A11, AZU1, SEMA6B, VGF, IL20RA, and FYN) in the OS model. S100A11 expression was upregulated in PTC and gastric cancer. Overexpression of S100A11 enhanced the transforming capacity and affected cell proliferation. Thus, S100A11 is regarded as a possible target in THCA and gastric cancer therapy [29, 30]. SEMA6B and FYN also promoted cell proliferation and invasion via the NOTCH signal pathway, suggesting that SEMA6B and FYN may also be viable immunotherapeutic targets in THCA [31, 32]. FYN is a target for therapeutic intervention of breast, prostate, and colon cancers [33]. HSPA6 is associated with hepatocellular carcinoma recurrence, and overexpression of HSPA6 enhances the inhibitory effect of GE-mediated proliferation, migration, and invasion in bladder cancer [34, 35]. PPBP is significantly downregulated in patients with gastric cancer; thus, it may also be a useful prognostic marker [36]. PPBP was also identified as a reliable molecular biomarker in colorectal cancer. In addition, AZU1 plays a part in innate immunity through encoding multifunctional inflammatory mediators [37]. IL20RA is highly expressed in breast cancer and promotes the tumor-initiating ability of cancer in vivo, as well as lung metastasis. Moreover, IL20RA is involved in formation of the TME and is a novel therapeutic target for breast cancer [38]
There were obvious differences in the survival analysis with the IRG risk signature in the present study. High-risk patients had a poorer prognosis compared with low-risk patients. The results show that the IRG risk signature is significantly related to the prognosis of patients with THCA. When associated with other clinicopathologic features, univariate and multivariate Cox regression analyses indicated that the risk score was always an independent prognostic factor. In addition, the results of ROC curve analyses demonstrated that risk score can accurately predict the prognosis of patients when combined with other clinical factors. These results prove that the IRG risk signature is a dependable prognostic tool. The relationships between the signature and several clinicopathological features including age and N stage were validated.
Furthermore, to study the correlation between tumor-infiltrating immune cells and IRG risk signature, we evaluated the relative infiltration of 21 immune cells in high-risk and low-risk groups. Plasma cells, monocytes, and activated dendritic cells infiltrated differently in the high-risk and low-risk groups in the risk model. The ratios of monocytes and resting dendritic cells were significantly higher in the low-risk score group; however, plasma cells were negatively correlated with risk score. Meanwhile, plasma cell infiltration was associated with longer survival of patients with THCA. These results could explain the predictive capacity of the IRG risk signature.
We also calculated TMB to analyze the possible mechanism of the risk model. Previous studies have indicated that TMB and PD-L1 expression can predict the response to ICIs. In our study, the low-risk group had a higher TMB compared with the high-risk group. The low-risk group also had a better prognosis. Furthermore, TMB was significantly associated with OS and PFI, suggesting that patients with a higher TMB might respond better to immunotherapy.
We explored the association between IPS and 8-IRG risk signature in patients with THCA. PD-1/PD-L1 inhibitors play an important role in the therapy of some refractory tumors by altering the status of immune surveillance [39]. IPS, IPS-PD-1/PD-L1/PD-L2, IPS-CTLA4, and IPS-PD-1/PD-L1/PD-L2 + CTLA4 scores were remarkably increased in the low-risk group using the model of eight IRGs. We also analyzed the expression of immune checkpoint proteins, including PD-L1, PD-L2, CTLA4, TIGIT, and TIM-3, which were markedly higher in the low-risk group. Our study suggested that the risk signature low-risk score groups may represent the immunogenic tumor microenvironment. We inferred that THCA patients with low-risk score might respond better to checkpoint inhibitors of PD-L1, PD-L2, CTLA4, TIGIT, and TIM-3. The good response of ICIs might be one of the reasons for the promising survival outcome in the low-risk group.
Although our IRG risk signature could efficiently predict disease progression and survival of patients with THCA, the study has some limitations. Our signature was constructed based on TCGA, which is a public database; thus, external databases will need to be studied and basic experiments will need to be performed to validate the predictive capacity of the signature and identify the biological functions of IRGs.
Conclusions
In conclusion, the IRG risk signature presented in this study can be used to predict survival and the response to ICIs in patients with THCA. The model may provide a reliable prediction tool for patients with THCA in the future.
Supplementary Information
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We appreciate the linguistic assistance provided by TopEdit (www.topeditsci.com) during the preparation of this manuscript.
Author contributions
WP performed the data analysis and wrote the manuscript. SW and ZH reviewed and revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (81902726), China Postdoctoral Science Foundation (2018M641739).
Data availability
Gene expression profiles, clinical information, and mutation data of THCA in this study are available from the public database (TCGA, https://portal.gdc.cancer.gov/). The IPS values are downloaded from The Cancer Immunome Atlas (TCIA, https://tcia.at/home). The immune- related genes are acquired from the Immunology Database and Analysis Portal database (ImmPort, https://immport.niaid.nih.gov).
Declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
The research was approved by the Institutional Research Ethics Committees of the First Affiliated Hospital of China Medical University. Informed consent for publication was obtained from all patients for collection of tissue samples prior to the surgery.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Pu Wu and Wei Sun authors have contributed equally to do this work
References
- 1.Coelho M, et al. The potential of metabolomics in the diagnosis of thyroid cancer. Int J Mol Sci. 2020 doi: 10.3390/ijms21155272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bray F, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 3.Cabanillas ME, McFadden DG, Durante C. Thyroid cancer. The Lancet. 2016;388:2783–2795. doi: 10.1016/s0140-6736(16)30172-6. [DOI] [PubMed] [Google Scholar]
- 4.Schneider DF, Chen H. New developments in the diagnosis and treatment of thyroid cancer. CA Cancer J Clin. 2013;63:374–394. doi: 10.3322/caac.21195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta S, et al. International trends in the incidence of cancer among adolescents and young adults. J Natl Cancer Inst. 2020;112:1105–1117. doi: 10.1093/jnci/djaa007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morris LG, Tuttle RM, Davies L. Changing trends in the incidence of thyroid cancer in the United States. JAMA Otolaryngol Head Neck Surg. 2016;142:709–711. doi: 10.1001/jamaoto.2016.0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer. 2013;13:184–199. doi: 10.1038/nrc3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albero A, Lopez JE, Torres A, de la Cruz L, Martin T. Effectiveness of chemotherapy in advanced differentiated thyroid cancer: a systematic review. Endocr Relat Cancer. 2016;23:R71–84. doi: 10.1530/ERC-15-0194. [DOI] [PubMed] [Google Scholar]
- 9.Schachter J, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006) Lancet. 2017;390:1853–1862. doi: 10.1016/S0140-6736(17)31601-X. [DOI] [PubMed] [Google Scholar]
- 10.Mehnert JM, et al. Safety and antitumor activity of the anti-PD-1 antibody pembrolizumab in patients with advanced, PD-L1-positive papillary or follicular thyroid cancer. BMC Cancer. 2019;19:196. doi: 10.1186/s12885-019-5380-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yi M, et al. Immune signature-based risk stratification and prediction of immune checkpoint inhibitor's efficacy for lung adenocarcinoma. Cancer Immunol Immunother. 2021 doi: 10.1007/s00262-020-02817-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y, Wang Z, Li F. Survival prediction and response to immune checkpoint inhibitors: a prognostic immune signature for hepatocellular carcinoma. Transl Oncol. 2021;14:100957. doi: 10.1016/j.tranon.2020.100957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu J, et al. Exploration of a novel prognostic risk signatures and immune checkpoint molecules in endometrial carcinoma microenvironment. Genomics. 2020;112:3117–3134. doi: 10.1016/j.ygeno.2020.05.022. [DOI] [PubMed] [Google Scholar]
- 14.Liu J, et al. Identification of a prognostic signature of epithelial ovarian cancer based on tumor immune microenvironment exploration. Genomics. 2020;112:4827–4841. doi: 10.1016/j.ygeno.2020.08.027. [DOI] [PubMed] [Google Scholar]
- 15.Yang S, et al. Identification of a prognostic immune signature for cervical cancer to predict survival and response to immune checkpoint inhibitors. Oncoimmunology. 2019;8:e1659094. doi: 10.1080/2162402X.2019.1659094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Colaprico A, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44:e71. doi: 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhattacharya S, et al. ImmPort: disseminating data to the public for the future of immunology. Immunol Res. 2014;58:234–239. doi: 10.1007/s12026-014-8516-1. [DOI] [PubMed] [Google Scholar]
- 18.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simon N, Friedman J, Hastie T, Tibshirani R. Regularization paths for cox's proportional hazards model via coordinate descent. J Stat Softw. 2011;39:1–13. doi: 10.18637/jss.v039.i05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newman AM, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28:1747–1756. doi: 10.1101/gr.239244.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson DR, et al. Integrative clinical genomics of metastatic cancer. Nature. 2017;548:297–303. doi: 10.1038/nature23306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Charoentong P, et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2017;18:248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 25.Yu J, et al. Hierarchical clustering of cutaneous melanoma based on immunogenomic profiling. Front Oncol. 2020;10:580029. doi: 10.3389/fonc.2020.580029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez-Cerdeira C, et al. Advances in immunotherapy for melanoma: a comprehensive review. Mediators Inflamm. 2017;2017:3264217. doi: 10.1155/2017/3264217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steven A, Fisher SA, Robinson BW. Immunotherapy for lung cancer. Respirology. 2016;21:821–833. doi: 10.1111/resp.12789. [DOI] [PubMed] [Google Scholar]
- 28.Riley RS, June CH, Langer R, Mitchell MJ. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discov. 2019;18:175–196. doi: 10.1038/s41573-018-0006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anania MC, et al. S100A11 overexpression contributes to the malignant phenotype of papillary thyroid carcinoma. J Clin Endocrinol Metab. 2013;98:E1591–1600. doi: 10.1210/jc.2013-1652. [DOI] [PubMed] [Google Scholar]
- 30.Sung AK, Lee KH. HGF-mediated S100A11 overexpression enhances proliferation and invasion of gastric cancer. Am J Transl Res. 2018;10(11):3385. [PMC free article] [PubMed] [Google Scholar]
- 31.Lv XJ, et al. Aberrant expression of semaphorin 6B affects cell phenotypes in thyroid carcinoma by activating the Notch signalling pathway. Endokrynol Pol. 2021;72:29–36. doi: 10.5603/EP.a2020.0072. [DOI] [PubMed] [Google Scholar]
- 32.Zheng J, Li H, Xu D, Zhu H. Upregulation of tyrosine kinase fyn in human thyroid carcinoma: role in modulating tumor cell proliferation, invasion, and migration. Cancer Biother Radiopharm. 2017;32:320–326. doi: 10.1089/cbr.2017.2218. [DOI] [PubMed] [Google Scholar]
- 33.Yu B, et al. FYN is required for ARHGEF16 to promote proliferation and migration in colon cancer cells. Cell Death Dis. 2020;11:652. doi: 10.1038/s41419-020-02830-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang Z, et al. Upregulation of heat shock proteins (HSPA12A, HSP90B1, HSPA4, HSPA5 and HSPA6) in tumour tissues is associated with poor outcomes from HBV-related early-stage hepatocellular carcinoma. Int J Med Sci. 2015;12:256–263. doi: 10.7150/ijms.10735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shin SS, et al. HSPA6 augments garlic extract-induced inhibition of proliferation, migration, and invasion of bladder cancer EJ cells; Implication for cell cycle dysregulation, signaling pathway alteration, and transcription factor-associated MMP-9 regulation. PLoS ONE. 2017;12:e0171860. doi: 10.1371/journal.pone.0171860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su C, et al. Identification of plasma RGS18 and PPBP mRNAs as potential biomarkers for gastric cancer using transcriptome arrays. Oncol Lett. 2019;17:247–255. doi: 10.3892/ol.2018.9608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCabe D, Cukierman T, Gabay JE. Basic residues in azurocidin/HBP contribute to both heparin binding and antimicrobial activity. J Biol Chem. 2002;277:27477–27488. doi: 10.1074/jbc.M201586200. [DOI] [PubMed] [Google Scholar]
- 38.Gao W, et al. IL20RA signaling enhances stemness and promotes the formation of an immunosuppressive microenvironment in breast cancer. Theranostics. 2021;11:2564–2580. doi: 10.7150/thno.45280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yi M, et al. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol Cancer. 2018;17:129. doi: 10.1186/s12943-018-0864-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Gene expression profiles, clinical information, and mutation data of THCA in this study are available from the public database (TCGA, https://portal.gdc.cancer.gov/). The IPS values are downloaded from The Cancer Immunome Atlas (TCIA, https://tcia.at/home). The immune- related genes are acquired from the Immunology Database and Analysis Portal database (ImmPort, https://immport.niaid.nih.gov).