

Abstract
Despite the high sensitivity of renal cell carcinoma (RCC) to immunotherapy, RCC has been recognized as an unusual disease in which CD8+ T-cell infiltration into the tumor beds is related to a poor prognosis. To approach the inner landscape of immunobiology of RCC, we performed multiplexed seven-color immunohistochemistry (CD8, CD39, PD-1, Foxp3, PD-L1, and pan-cytokeratin AE1/AE3 with DAPI), which revealed the automated single-cell counts and calculations of individual cell-to-cell distances. In total, 186 subjects were included, in which CD39 was used as a marker for distinguishing tumor-specific (CD39+) and bystander (CD39−) T-cells. Our clear cell RCC cohort also revealed a poor prognosis if the tumor showed increased CD8+ T-cell infiltration. Intratumoral CD8+CD39+ T-cells as well as their exhausted CD8+CD39+PD-1+ T-cells in the central tumor areas enabled the subgrouping of patients according to malignancy. Analysis using specimens post-antiangiogenic treatment revealed a dramatic increase in proliferative Treg fraction Foxp3+PD-1+ cells, suggesting a potential mechanism of hyperprogressive disease after uses of anti-PD-1 antibody. Our cell-by-cell study platform provided spatial information on tumors, where bystander CD8+CD39− T-cells were dominant in the invasive margin areas. We uncovered a potential interaction between CD8+CD39+PD-1+ T-cells and Foxp3+PD-1+ Treg cells due to cell-to-cell proximity, forming a spatial niche more specialized in immunosuppression under PD-1 blockade. A paradigm shift to the immunosuppressive environment was more obvious in metastatic lesions; rather the infiltration of Foxp3+ and Foxp3+PD-1+ Treg cells was more pronounced. With this multiplexed single-cell pathology technique, we revealed further insight into the immunobiological standing of RCC.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00262-021-03006-2.
Keywords: Renal cell carcinoma, Kidney, CD8, CD39, PD-1, Single cell, Heterogeneity
Introduction
The advent of cancer immune-oncology focusing on specific immune-related factors has revolutionized cancer treatment. The schematized immune cycle clarified seven immune steps [1] that enabled effective immunotherapy and promoted the development of immune checkpoint inhibitors targeting PD-1, PD-L1, and CTLA-4 [2–4]. In particular, renal cell carcinoma (RCC) is at the center of susceptibility to immunotherapy, and the clinical success of nivolumab (targeting PD-1) is still a recent breakthrough. With the substantial survival benefits of anti-PD-1/PD-L1 treatment alone or in combination with CTLA-4 [5, 6], RCC is a hallmark of immune-oncology treatment in this decade [7–9].
However, the immune landscape in the tumor microenvironment (TME) of RCC differs from that of other cancer types. One typical representative example is the prognostic relevance of CD8+ T-cells infiltrating into tumor beds. Measuring CD8+ T-cell densities in primary tumors revealed that elevated CD8+ T-cell infiltration is associated with poor prognosis in RCC, with opposite results in most other cancers [10–12]. Further, this paradoxical phenomenon was maintained in the comparison of primary vs metastatic RCC lesions, and the infiltration of CD8+ T-cells is remarkable in tumor metastasis [13]. Although little is known about the precise mechanisms underlying this process, the immunobiology of RCC must be considered unique, unlike other immunotherapy-sensitive tumors.
In the heterogeneous population of CD8+ T-cells, which subgroup of CD8+ T-cells infiltrating RCC tumors is involved in tumorigenesis and progression? Are there any differences in the tumor area infiltrated by these specific CD8+ T-cells? The answers to these questions may be key to solving the mystery of CD8+ T-cells in RCC in this era of immuno-oncology. Recent single-cell sequencing studies [14, 15] showed that intratumoral CD8+ T-cells, which have been considered to be uniform, phenotypically include heterogeneous cell-by-cell characteristics [16–18]. In fact, not all infiltrated CD8+ T-cells react with cancer cells, and existing so-called bystander CD8+ T-cells are evident [19]. Further, in addition to PD-1 expression associated with exhaustion in tumor-reactive CD8+ T-cells, the levels of PD-L1 expression derived from cancer cells and suppressive Treg cells in tumors complicate the immunobiology of RCC. In this regard, technical advances in single-cell sequencing have been essential for exploring cellular phenotypes; however, the tumor samples used in sequencing are dissociated into a form of cell suspension, thereby destroying all of the spatial information, e.g., the localization of individual cells and the distance between cells [20, 21].
Herein, we applied a new imaging system to analyze the immune landscape in the TME of RCC, phenotypically and spatially characterizing the intratumoral heterogeneity of CD8+ T-cells at the single-cell level. Using a novel tyramide signal amplification technique [22, 23], we assessed multiplexed fluorescent immunohistochemistry, which allows intact tumors to be dyed in seven colors simultaneously in a high-throughput manner. Analyzing various primary tumors and metastatic organs of RCC, we revealed the automated single-cell counts and calculations of individual cell-to-cell distances, thereby providing more extensive and detailed information about the inner landscape of the immunobiology of RCC.
Materials and methods
Human samples
After approval by the Institutional Review Board, formalin-fixed paraffin-embedded (FFPE) RCC tumor samples from 1999 to 2017 were randomly collected at Keio University Hospital (Tokyo, Japan) with regard to histological type, pathological T stage, and systemic therapy used. The Union for International Cancer Control (UICC) tumor-node-metastasis (TNM) system was used for tumor staging, and nuclear grading was carried out according to the World Health Organization (WHO)/International Society of Urological Pathology grading system. In total, 186 tumor samples were included and were divided into four groups prior to the analysis: (1) primary clear cell RCC (ccRCC) tumors treated surgically (n = 97, COHORT A); (2) primary ccRCC tumors treated with antiangiogenic TKIs prior to surgery (n = 8, COHORT B); (3) ccRCC tumor metastases diagnosed histologically (n = 43; bone: 16, lung: 8, viscera: 9, and others: 10, COHORT C); and (4) primary non-ccRCC tumors (n = 38; papillary: 12, chromophobe: 11, sarcomatoid: 8, and Xp11.2 translocation: 7, COHORT D). No statistical methods were used to predetermine the sample size. The tissue microarray (TMA) blocks used in this study were created as follows. First, a well-experienced pathologist (S.M.), who is board certified and specializes in genitourinary malignancies, evaluated the suitability of the tissue sections for TMA construction. Tumor central and invasive margin areas were then selected. Second, 3-mm tumor cores were punched from the tumor center and invasive margin area. All samples were deciphered with numbers to avoid investigator bias during tissue preparation and data analysis. All procedures were performed in compliance with the 1964 Helsinki Declaration and present ethical standards, and informed consent for experimental use of the samples was obtained from the patients according to the hospital's ethical guidelines.
Multiplex staining and image analysis
Multiplex immunohistochemistry was performed with the Opal seven-color Manual IHC Kit (PerkinElmer, Waltham, Massachusetts, the USA). The TMA samples were cut into 5-μm-thick sections and placed on silane-coated glass slides. Each slide was baked in the oven at 65 °C for 1 h. Then, the slides were deparaffinized with xylene and rehydrated through a graded series of ethanol solutions. Thereafter, the slides were fixed in 0.3% hydrogen peroxide in methanol for 20 min. After antigen retrieval in a microwave, the slides were washed in TBST wash buffer (25 mM TRIS HCl (pH 7.5), 150 mM NaCl, 0.05% Tween®20). After blocking, the sections were incubated with primary antibodies for 4 h and then incubated with polymer HRP Ms + Rb as the secondary antibody for 10 min at room temperature. Opal fluorophores were pipetted onto each slide for 10 min at room temperature, and the slides were microwaved to strip the primary and secondary antibodies (Step 1, see Fig. 1a-b). Then, we repeated the same protocol using the next primary antibody targets (Steps 2–6, see Fig. 1b). Finally, DAPI was pipetted onto each slide for 5 min at room temperature (Step 7). The slides were covered with ProLong® Diamond Antifade Mountant (Thermo Fisher Scientific).
Fig. 1.

Quantitative seven-color multiplexed single-cell analysis of human ccRCC reveals CD8+ T-cell heterogeneity along with immunosuppressive cells. a Workflow of the present multiplexed immunofluorescence staining technique. b The antibodies used and protocol of sequential multiplex staining. AR, antigen retrieval. CST, cell signaling technology. c Representative image of ccRCC displaying multispectral seven-color fluorescence signals for CD8, CD39, PD-1, Foxp3, PD-L1, and AE1/AE3 together with DAPI. Scale bar, 100 μm. d–i Zoomed-in images of the indicated boxed regions in c showing each single fluorophore signal representing CD8 (d), CD39 (e), PD-1 (f), Foxp3 (g), PD-L1 (h), and AE1/AE3 (i). j Cellular phenotyping map of the indicated boxed regions in c. Scale bar, 20 μm. k Cell-by-cell images of each defined cell phenotype. l Summary of the percent distribution by cell phenotype in the tumor central area from 97 primary ccRCC specimens
Cellular image analysis and quantification in tissue sections were carried out using inForm software ver. 2.4 (PerkinElmer), allowing the separation and measurement of spectrally overlapping markers in current multiplex assays at the single-cell level. First, cell segmentation was performed by identifying individual locations of individual cell nuclei using a counterstain-based approach with DAPI. Multicolor phenotyping was then applied against six antigens using an intensity-based threshold that was determined manually in the inForm images. The distances between cells within individual tumors were analyzed as the closest proximity with an average nucleus-to-nucleus distance using the MATLAB function pdist2 (MATLAB version R2020, MathWorks).
Statistics
Values are presented as means with standard deviations or medians with interquartile ranges for continuous variables and as frequencies with percentages for categorical variables. Variables between groups were compared using the Chi-square test, the paired t test, the Mann–Whitney U test, Wilcoxon signed-rank test, and the Kruskal–Wallis H test, as appropriate. We performed receiver operating characteristic (ROC) curve analysis to define a potential cutoff for cell density in discriminating patient mortality, where an area under the curve (AUC) value of 1.0 represents perfect discrimination, and a value of 0.5 represents no discrimination. Univariate and multivariate Cox regression models with stepwise selection were used to evaluate variables for disease recurrence/progression and overall mortality. Survival curves were estimated using the Kaplan–Meier method and compared with the log-rank test. Radiographic responses to treatment were evaluated according to Response Evaluation Criteria in Solid Tumors (RECIST) v.1.1. Statistical significance was indicated when p < 0.05. All analyses were performed using R statistical language (R Foundation for Statistical Computing, Vienna, Austria), SPSS version 26.0 (IBM-SPSS Inc., Tokyo, Japan), and JMP version 15.0 (SAS Institute Inc., Cary, NC, the USA).
Results
Cellular characterization by multiplexed single-cell pathology
Applying a novel tyramide signal amplification technique, we first carried out a multiplex immunolabeling protocol for the simultaneous examination of six distinct markers with nuclei staining in a single thin section of FFPE tumors (Fig. 1a). In brief, a spectral signature obtained in each fluorophore by solo antigen staining (Fig. 1b) was captured simultaneously using dedicated multispectral fluorescence microscopy (PerkinElmer), creating one composite image from seven different fluorescent pictures (Fig. 1c).
To characterize CD8+ T-cell subpopulations in tumors, the marker CD8 was used to identify all cytotoxic T-cells. Subpopulations of CD8+ T-cells were then defined by the presence or absence of CD39, which discriminates tumor-specific (CD39+) and bystander (CD39−) CD8+ T-cells recruited to the tumor site [19]. PD-1 expression levels were used to determine tumor-specific T-cell exhaustion [24–26]. To uncover the relationship between cytotoxic CD8+ T-cells and representatives of immune suppression in tumors, Foxp3 expression levels were used to determine infiltrative Treg cells. Further, PD-1 expression levels on Foxp3+ cells were used to depict a fraction of proliferative Treg cells [27, 28]. PD-L1 expression levels on AE1/AE3+ cells were used as a phenotypic marker for immunosuppressive cancer cells. Automated cell segmentation and single-cell counts were performed using inForm software (PerkinElmer), in which nuclear staining by DAPI enabled the accurate discrimination between living cells and debris. The fluorescence intensity in every cell was recorded separately to further allow cell-by-cell phenotyping (Fig. 1j).
Then, we applied this multiplexed single-cell pathology to TMA sections from 97 primary ccRCC tumor samples in COHORT A (Table 1). In this automated analysis, all types of cells in the 3-mm core from central tumor areas were distinguished together with nuclei and counted per patient, as illustrated in Fig. 1k. In total, the number of all cells was 3115 ± 1039/mm2, and the cellular characterization by the percentage of cells is shown in Fig. 1l. Overall, these results demonstrate that our automated imaging platform can be applied to quantitatively examine and phenotypically characterize primary ccRCC tumor samples at the single-cell level.
Table 1.
Characteristics associated with the levels of CD8/CD39/PD-1 expression in 97 human primary ccRCC tumor samples from COHORT A
| Characteristics | All (n = 97) | Infiltrated CD8+ T-cell status | p value | |||
|---|---|---|---|---|---|---|
| Non-inflamedCD8low (n = 36) | Inflamed CD8high | |||||
| CD39low (n = 9) | CD39high PD-1low (n = 41) | CD39high PD-1high (n = 11) | ||||
| Gender, n (%) | 0.114 | |||||
| Male | 71(73.2%) | 24(66.7%) | 5(55.6%) | 34(82.9%) | 8(72.7%) | |
| Female | 26(26.8%) | 12(33.3%) | 4(44.4%) | 7(17.1%) | 3(27.3%) | |
| Age, n (%) | 0.029 | |||||
| < 60 years | 49(50.5%) | 23(63.9%) | 5(55.6%) | 16(39.0%) | 5(45.5%) | |
| ≥ 60 years | 48(49.5%) | 13(36.1%) | 4(44.4%) | 25(61.0%) | 6(54.5%) | |
| Pathological T stage, n (%) | 0.001 | |||||
| pT1 + pT2 | 58(59.8%) | 29(80.6%) | 6(66.7%) | 21(51.2%) | 2(18.2%) | |
| pT3 + pT4 | 39(40.2%) | 7(19.4%) | 3(33.3%) | 20(48.8%) | 9(81.8%) | |
| Histological grade, n (%) | < 0.001 | |||||
| G1 + G2 | 65(67.0%) | 32(88.9%) | 6(66.7%) | 25(61.0%) | 2(18.2%) | |
| G3 + G4 | 32(33.0%) | 4(11.1%) | 3(33.3%) | 16(39.0%) | 9(81.8%) | |
| Venous invasion, n (%) | 0.005 | |||||
| Yes | 40(41.2%) | 9(25%) | 5(55.6%) | 18(43.9%) | 8(72.7%) | |
| No | 57(58.8%) | 27(75%) | 4(44.4%) | 23(56.1%) | 3(27.3%) | |
| No of Foxp3+ cells/(mm2) | 19.3 ± 24.3 | 7.29 ± 14.0 | 15.5 ± 14.8 | 24.9 ± 23.2 | 40.7 ± 38.8 | < 0.001 |
| No of Foxp3+PD-1+ cells/(mm2) | 0.9 ± 3.4 | 0.2 ± 0.7 | 0.06 ± 0.1 | 0.7 ± 1.9 | 5.0 ± 8.7 | 0.001 |
| No of AE1/AE3+PD-L1+ cells/(mm2) | 1.49 ± 12.0 | 0.02 ± 0.11 | 0.04 ± 0.08 | 0.26 ± 0.86 | 12.1 ± 35.2 | 0.011 |
Association of phenotypic CD8+ T-cell heterogeneity with outcome
We next investigated whether phenotypic CD8+ T-cell subpopulations were associated with patient survival following surgery in primary ccRCC. We first sought to distinguish tumors in which CD8+ T-cells showed high infiltration, namely CD8high tumors, and then divided the 97 ccRCC patients into two groups, i.e., inflamed CD8high and non-inflamed CD8low tumor groups. Using the defined cutoff levels of infiltrated CD8+ T-cells (Supplementary Fig. 1a), Kaplan–Meier analyses revealed that patients in the CD8high tumor group exhibited disease recurrence (p = 0.001) and poor overall mortality (p = 0.013) compared with those in the CD8low tumor group (Fig. 2a). Then, we divided the patients in the CD8high tumor group into two groups, i.e., the CD8highCD39high and CD8highCD39low tumor groups. CD39 expression on CD8+ T-cells enables the discrimination of tumor-specific CD8+ T-cells and bystander CD8+ T-cells recruited to the tumor site. Using the defined cutoff levels of infiltrated CD8+CD39+ T-cells (Supplementary Fig. 1b), Kaplan–Meier analyses revealed that patients in the CD8highCD39high tumor group exhibited disease recurrence (p = 0.007) and poor overall mortality (p = 0.048) compared with those in the CD8highCD39low tumor group (Fig. 2b). Finally, using the T-cell exhaustion marker PD-1, we assessed CD8+CD39+PD-1+ T-cells in the CD8highCD39high tumor group (Supplementary Fig. 1c). Kaplan–Meier analyses revealed that patients in the CD8highCD39highPD-1high tumor group had disease recurrence (p = 0.011) and poor overall mortality (p = 0.006) compared with those in the CD8highCD39highPD-1low tumor group (Fig. 2c).
Fig. 2.

Relationships between CD8+ T-cell subpopulations and outcomes. a–c Kaplan–Meier survival curves for overall mortality and recurrence-free survival in 97 ccRCC patients following surgery according to the indicated CD8+ T-cell subpopulations: CD8low versus CD8high (a), CD8highCD39low versus CD8highCD39high (b), and CD8highCD39highPD-1low versus CD8highCD39highPD-1high (c). d–e Kaplan–Meier survival curves for the treatment response (d) and overall mortality (e) in 38 ccRCC patients receiving antiangiogenic TKIs based on the indicated CD8+ T-cell subpopulations. p values were obtained from the log-rank test. f Summary of the percent distribution by cell phenotype in the central tumor from eight primary ccRCC specimens post-TKI treatment. g–j Density of CD8+ T-cells (g), CD8+CD39+PD-1+ T-cells (h), Foxp3+ Treg cells (i), and Foxp3+PD-1+ Treg cells (j) in ccRCC tumors without (COHORT A)/with (COHORT B) TKI treatment prior to surgery. The data are presented using violin and box plots. The line within the box is the median, the upper and lower ends of the box are the upper and lower quartiles, and the bars are the minimum and maximum values. p values were obtained from the two-tailed Mann–Whitney U test
We then investigated the associations between CD8highCD39highPD-1high tumors and pathological features in primary ccRCC. Our results revealed significantly higher levels of CD8+CD39+PD-1+ T-cells in patients with advanced tumor stages, higher tumor grades, and venous invasion (Table 1). The CD8highCD39highPD-1high tumors correlated with representatives of tumor immune suppression such as Treg cells and PD-L1+ cancer cells infiltrated into the tumors (Table 1). Interestingly, multivariate analysis revealed that CD8highCD39highPD-1high tumors were a risk factor for predicting disease recurrence following surgery (hazard ratio referenced by CD8low tumor group, 3.262, p = 0.015) independent of representative clinical features (Table 2). On the other hands, CD8highCD39highPD-1high tumors were a significant risk factor for predicting overall mortality following surgery in the univariate analysis only (Table 2).
Table 2.
Parameters associated with post-operative disease recurrence and overall mortality in primary ccRCC patients after adjusting univariate and multivariate Cox regression analyses in COHORT A
| Characteristics | Disease recurrence | Overall mortality | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Univariate | Multivariate | Univariate | Multivariate | |||||||||
| Hazard ratio | 95% CI | p value | Hazard ratio | 95% CI | p value | Hazard ratio | 95% CI | p value | Hazard ratio | 95% CI | p value | |
| Gender (male vs. female) | 0.979 | 0.522–1.835 | 0.947 | 0.562 | 0.263–1.201 | 0.137 | ||||||
| Age ≥ 60 vs. < 60 | 1.366 | 0.791–2.358 | 0.263 | 2.948 | 1.344–6.468 | 0.007 | 2.947 | 1.230–7.058 | 0.015 | |||
| pT Stage (pT3,4 vs. pT1,2) | 3.273 | 1.850–5.792 | < 0.001 | 4.547 | 2.108–9.809 | < 0.001 | ||||||
| Grade (3,4 vs. 1,2) | 4.050 | 2.309–7.103 | < 0.001 | 2.299 | 1.159–4.559 | 0.017 | 5.553 | 2.619–11.773 | < 0.001 | |||
| Venous invasion (yes vs. no) | 2.936 | 1.679–5.135 | < 0.001 | 2.192 | 1.129–4.259 | 0.021 | 4.717 | 2.149–10.353 | < 0.001 | 3.086 | 1.092–8.715 | 0.033 |
| CD8+ T-cell heterogeneity | < 0.001 | 0.004 | 0.001 | |||||||||
| CD8low | Reference | Reference | Reference | |||||||||
| CD8highCD39low | 0.396 | 0.052–3.052 | 0.374 | 0.217 | 0.028–1.712 | 0.147 | 0.000 | 0.000 | 0.976 | |||
| CD8highCD39highPD-1low | 3.002 | 1.521–5.922 | 0.002 | 2.546 | 1.274–5.088 | 0.008 | 2.586 | 1.001–6.682 | 0.050 | |||
| CD8highCD39highPD-1high | 7.931 | 3.362–18.705 | < 0.001 | 3.262 | 1.260–8.446 | 0.015 | 8.063 | 2.836–22.925 | < 0.001 | |||
Among ccRCC patients in the CD8highCD39high tumor group, 38 (73%) received antiangiogenic tyrosine kinase inhibitors (TKIs), including sorafenib and sunitinib, following disease recurrence. We investigated the relationship between sensitivity to TKI treatment and CD8+ T-cell heterogeneity. Kaplan–Meier analyses revealed that the CD8highCD39highPD-1high tumor group was marginally associated with resistance to TKI treatment (p = 0.076, Fig. 2d) and significantly associated with overall mortality (p = 0.012, Fig. 2e) following TKI administration. Further, multivariate analysis revealed that CD8highCD39highPD-1high tumors were an independent risk factor for predicting overall mortality following TKI administration (p = 0.009) in this population receiving antiangiogenic therapy (Table 3).
Table 3.
Parameters associated with disease progression and overall mortality in primary ccRCC patients treated with TKI (n = 38) after adjusting univariate and multivariate Cox regression analysis in COHORT A
| Characteristics | Disease progression | Overall mortality | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Univariate | Multivariate | Univariate | Multivariate | |||||||||
| Hazard ratio | 95% CI | p value | Hazard ratio | 95% CI | p value | Hazard ratio | 95% CI | p value | Hazard ratio | 95% CI | p value | |
| Gender (male vs. female) | 0.529 | 0.217–1.291 | 0.162 | 0.529 | 0.224–1.251 | 0.147 | ||||||
| Age ≥ 60 vs. < 60 | 3.856 | 1.372–10.836 | 0.010 | 3.856 | 1.372–10.836 | 0.010 | 2.631 | 1.017–6.802 | 0.046 | |||
| pT Stage (pT3,4 vs. pT1,2) | 1.791 | 0.746–4.301 | 0.192 | 2.498 | 0.982–6.356 | 0.055 | ||||||
| Grade (3,4 vs. 1,2) | 1.737 | 0.763–3.956 | 0.188 | 2.851 | 1.174–6.923 | 0.021 | ||||||
| Venous invasion (yes vs. no) | 1.695 | 0.731–3.929 | 0.219 | 2.401 | 0.988–5.838 | 0.053 | ||||||
| CD8+ T-cell heterogeneity | 2.228 | 0.899–5.523 | 0.084 | 2.881 | 1.217–6.819 | 0.016 | 3.436 | 1.370–8.619 | 0.009 | |||
| (CD8highCD39highPD-1high vs. CD8highCD39highPD-1low) | ||||||||||||
In summary, these results demonstrated that tumor-specific CD8+CD39+PD-1+ T-cells are highly correlated with prognosis in ccRCC, suggesting that patients with tumors with a high infiltration of CD8+CD39+PD-1+ T-cells showed poor prognosis. Further, CD8highCD39highPD-1high tumors were linked to worse pathological features, intratumoral immune suppression by Treg cells, and PD-L1+ cancer cells.
CD8+ T-cell heterogeneity post-TKI treatment
We next explored how CD8+ T-cell heterogeneity changes after TKI treatment. The answer to this vital question should be the rationale for the second-line use of anti-PD-1 nivolumab after relapsing following TKI treatment or for the current first-line use of TKI and immuno-oncology combination in metastatic RCC [5, 29, 30]. We therefore applied our multiplexed single-cell pathology to eight ccRCC tumor samples treated with TKIs prior to surgery. With median period of TKI treatment of 9.4 (4.7–18) months, two patients were judged to have partial response by the RECIST criteria of tumor size and six had stable disease. The number of all cells was 3268 ± 870/mm2, and the cellular characterization by the percentage of cells is shown in Fig. 2f. No difference in CD8+ T-cell patterns was noted by any therapeutic responses following TKI treatments among patients in COHORT B (data not shown). However, the number of CD8+ T-cells was increased after TKI treatment (p = 0.014, Fig. 2g), and the increase in infiltrated CD8+CD39+PD-1+ T-cells was also observed (p = 0.065, Fig. 2h). No difference in PD-L1+ cancer cells was noted (p = 0.633); rather, drastic increase in Foxp3+ Treg cells was obvious (p = 0.010, Fig. 2i), accompanying by those proliferative fractions of Foxp3+PD-1+ Treg cells after TKI treatment (p = 0.001, Fig. 2j). This phenomenon suggests that blind uses of anti-PD-1 antibody after or under TKI treatment may induce rapid cancer progression, so-called hyperprogressive disease under PD-1 blockade [27, 28].
Intratumoral regional differences
We next explored whether CD8+ T-cell heterogeneity is affected by sample location, e.g., tumor central vs invasive margin area in individual ccRCC tumors. This question is essential in discussing the effects of intratumoral heterogeneity. Our multiplexed single-cell pathology was applied to match invasive margin areas per patient in COHORT A (Fig. 3a). The number of all cells was 3110 ± 802/mm2 in the invasive margin areas, and the cellular characterization by the percentage of cells in the invasive margin areas is shown in Fig. 3b. Compared with the central tumor areas, markedly increased numbers of CD8+ T-cells were found in the matched invasive margin areas of each ccRCC tumor (p = 0.016, Fig. 3c). However, the proportion of CD8+ T-cell subpopulations slightly differed, revealing that bystander CD8+CD39− T-cells were prominent in invasive margin areas (p < 0.001, Fig. 3d). On the other hands, no difference was noted in the levels of CD8+CD39+PD-1+ T-cells (p = 0.201, Fig. 3e).
Fig. 3.

Intratumoral regional differences and spatial niches underlying CD8+ T-cell heterogeneity along with immunosuppressive cells. a Schematic illustration of the regions of interest in multiplexed single-cell analysis. b Summary of the percent distribution by cell phenotype in ccRCC invasive margin samples. c–e Pairwise comparisons of the density of CD8+ T-cells (c), bystander CD8+CD39− T-cells (d), and CD8+CD39+PD-1+ T-cells (e) between the tumor central and invasive margins in primary ccRCC tumor samples. f Schematic illustration of the cell-to-cell distance analysis. g–h Pairwise comparisons of the minimum distance from the indicated T-cell subpopulations to the nearest Foxp3+PD-1+ Treg cells (n = 10); CD8+CD39− T-cells vs. CD8+CD39+ T-cells (g) and CD8+CD39+PD-1− T-cells vs. CD8+CD39+PD-1+ T-cells (h). i–j Pairwise comparisons of the minimum distance from the indicated T-cell subpopulations to the nearest PD-L1+ cancer cells (n = 9); CD8+CD39− T-cells vs. CD8+CD39+ T-cells (i) and CD8+CD39+PD-1− T-cells vs. CD8+CD39+PD-1+ T-cells (j). p values were obtained from the Wilcoxon signed-rank test
We then asked how the localization of CD8+ T-cell subpopulations shows heterogeneity within the same TME. We hypothesized a phenotypic interaction between a subset of CD8+ T-cells, Foxp3+PD-1+ Treg cells, and AE1/AE3+PD-L1+ cancer cells due to their proximity. First, the nucleus-to-nucleus distances in cells were analyzed (Fig. 3f, see Methods section). Second, we identified CD8+CD39+ T-cells in tumors located in the closer proximity to Foxp3+PD-1+ Treg cells compared with CD8+CD39− T-cells (Fig. 3g, p = 0.037). Further, we uncovered CD8+CD39+PD-1+ T-cells located in the closest proximity to Foxp3+PD-1+ Treg cells (Fig. 3h, p = 0.007). On the other hands, the distribution patterns of CD8+ T-cell subpopulations relative to AE1/AE3+PD-L1+ cancer cells in tumors slightly differ from those of Foxp3+ Treg cells, revealing there was no difference in their proximity to AE1/AE3+PD-L1+ cancer cells between CD8+CD39+PD-1+ and CD8+CD39+PD-1− T-cells (Fig. 3i-j). Collectively, these results indicate that CD8+CD39+PD-1+ T-cells, the vital target of PD-1 blockade, may live in close proximity to proliferative Foxp3+PD-1+ Treg cells in tumors, forming a spatial niche that possibly interferes with proper PD-1 blockade effects.
Metastasis-specific differences
We next explored the metastasis-specific differences in CD8+ T-cell heterogeneity by our multiplexed single-cell pathology. First, we analyzed 43 ccRCC tumor metastases, including lung, bone, viscera, and other lesions (COHORT C). The number of all cells was 3048 ± 1040/mm2, and the cellular characterization by the percentage of cells is shown in Fig. 4a. Compared with primary ccRCC tumors, markedly increased numbers of Foxp3+ Treg cells were observed (p = 0.004, Fig. 4b), accompanying by those proliferative fractions of Foxp3+PD-1+ Treg cells in ccRCC metastasis (p = 0.063, Fig. 4c). However, no drastic difference was found in the number of CD8+CD39+PD-1+ T-cells (p = 0.240) between 97 primary tumors and 43 metastases. Interestingly, analyzing by the types of metastatic organ (Fig. 4d-f) revealed no difference in cell phenotype distribution pattern between metastases to the bone (n = 16), lung (n = 8), and viscera (n = 9), respectively (Fig. 4g).
Fig. 4.

Metastasis-specific differences underlying CD8+ T-cell heterogeneity along with immunosuppressive cells by multiplexed single-cell analysis. a Summary of the percent distribution by cell phenotype in 43 metastatic ccRCC samples (COHORT C). b–c Density of Foxp3+ Treg cells (b) and Foxp3+PD-1+ Treg cells (c) between 97 primary tumors and 43 metastases in ccRCC. d–f Metastasis-specific differences in cell phenotype distribution obtained from the bone (n = 16, d), lung (n = 8, e), and viscera (n = 9, f) tumor samples. g Density of CD8+ T-cells, CD8+CD39+ T-cells, CD8+CD39+PD-1+ T-cells, Foxp3+ Treg cells, Foxp3+PD-1+ Treg cells, and AE1/AE3+PD-L1+ cancer cells between bone/lung/visceral metastasis. The data are presented using violin and box plots. The line within the box is the median, the upper and lower ends of the box are the upper and lower quartiles, and the bars are the minimum and maximum values. p values were obtained from the two-tailed Mann–Whitney U test or Kruskal–Wallis H test
Histological subtype-specific differences
We next explored histological subtype-specific differences in CD8+ T-cell heterogeneity by our multiplexed single-cell pathology. We analyzed 38 non-ccRCC samples, including papillary, chromophobe, sarcomatoid, and Xp11.2 translocation tumor samples (COHORT D). The number of all cells was 2736 ± 1108/mm2, and the cellular characterization by the percentage of cells is shown in Fig. 5a-e. In brief, compared with that in primary ccRCC tumors, significantly decreased numbers of CD8+CD39+PD-1+ T-cells were found in papillary RCC tumors (p = 0.026, Supplementary Fig. 2a). Non-inflamed CD8+ T-cell status was more frequent in chromophobe RCC tumors (Supplementary Fig. 2b). However, no drastic difference in either sarcomatoid (Supplementary Fig. 2c) or Xp11.2 translocation (Supplementary Fig. 2d) tumor samples was noted compared with primary ccRCC tumors. In summary, analysis by histological subtype revealed new insights into histological subtype-specific differences in the infiltration patterns and heterogeneity of CD8+ T-cells in non-ccRCC.
Fig. 5.
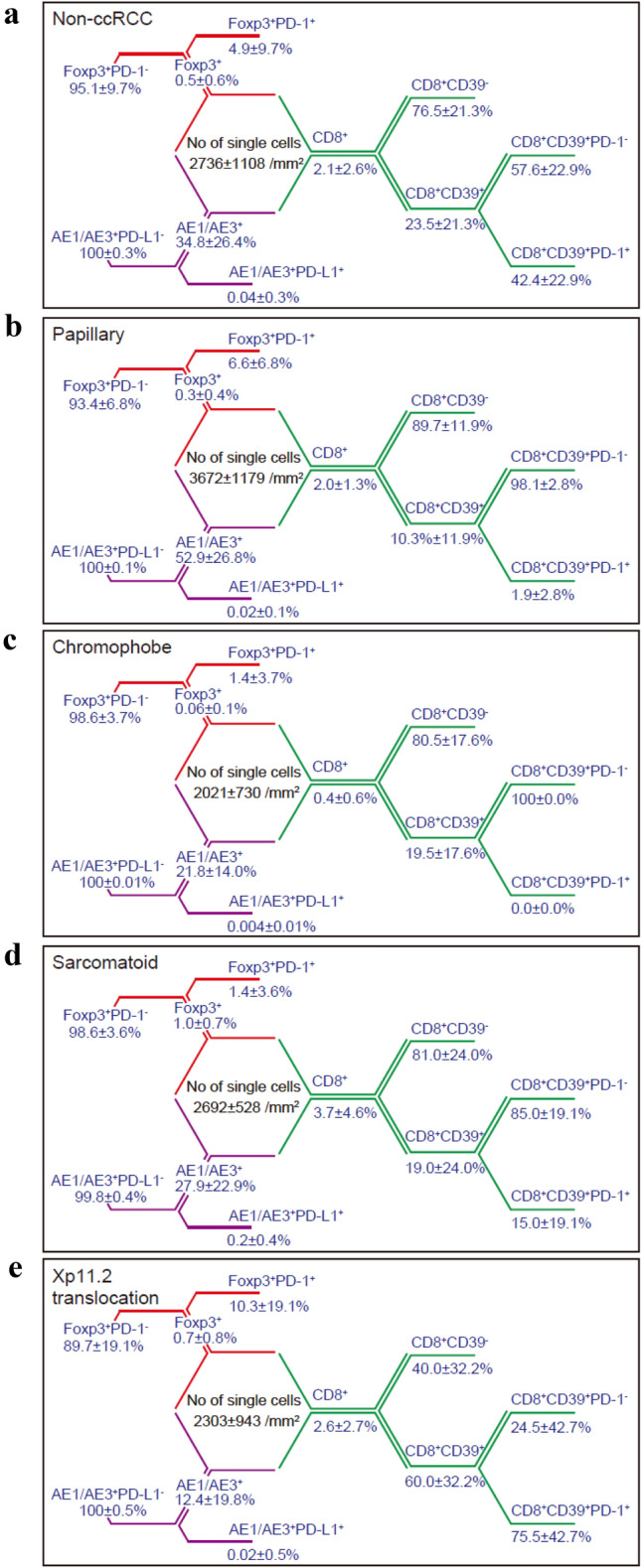
Histological subtype-specific differences in non-ccRCC underlying CD8+ T-cell heterogeneity along with immunosuppressive cells by multiplexed single-cell analysis. a Summary of the percent distribution by cell phenotype in 38 primary non-ccRCC specimens (COHORT D). b-e Histological subtype-specific differences in cell phenotype distribution obtained from the papillary (n = 12, b), chromophobe (n = 11, c), sarcomatoid (n = 8, d), and Xp11.2 translocation (n = 7, e) subtypes
Discussion
RCC has long been recognized as an unusual disease in which CD8+ T-cell infiltration into the tumor is related to a poor prognosis [12]. Despite the high sensitivity of RCC to immunotherapy, it differs from the characteristics of common solid tumors. The principle of immunotherapy in solid tumors is to induce cytotoxic T-cells into the tumor, which means maintaining the tumor immune environment as inflamed [1]. On the other hands, recent advances in single-cell RNA sequencing have revealed that host-derived immune cells are also diverse, and CD8+ T-cells, which have been considered uniform, have various phenotypes at the single-cell level [31, 32]. Thus, what kind of heterogeneity of CD8+ T-cells is shown in RCC, and which CD8+ T-cell subpopulation is involved in malignancy? How do these CD8+ T-cells interact with representative tumor immunosuppressive factors, i.e., Foxp3+ Treg cells and PD-L1+ cancer cells? Are there differences in CD8+ T-cell heterogeneity specific to organs or histological subtypes in RCC? We considered it important to clarify these questions to approach the paradoxical phenomenon of CD8+ T-cells in RCC. In this study, we applied a new multiplexed imaging platform and revealed three major findings.
First, our ccRCC cohort also tended to have a poor prognosis if the tumor revealed more CD8+ T-cell infiltration. When CD39 was used as a marker for distinguishing tumor-specific T-cells and bystander T-cells [19], excessive tumor-specific CD8+ T-cells within the central tumor enabled the subgrouping of the CD8high tumor group in ccRCC according to malignancy. These results were consistent with previous remarks in ccRCC, revealing tumor‑infiltrating CD8+CD39+ T-cells determine outcome of prognosis [33]. Furthermore, the stratification of tumor-specific CD8+ T-cells by multiplexed staining with the exhaustion marker PD-1 was useful for predicting the prognosis and effect of conventional antiangiogenic treatment in the CD8highCD39high tumor group. Interestingly, analysis using ccRCC specimens resected post-TKI treatment revealed a dramatic increase in proliferative fractions of Foxp3+PD-1+ Treg cells, accompanying by marginal CD8+CD39+PD-1+ T-cell infiltration. This means that the immunosuppressive environment expands after or under TKI treatment, and blind uses of anti-PD-1 antibody in clinical practice may have a potential cause of hyperprogressive disease if patients receive TKI treatment [27, 28].
Second, the strength of our cell-by-cell study platform provides spatial information on tumors, allowing us to quantitatively assess regional differences and cell-to-cell distances within the same tumor. The imaging of matched tumor central and invasive margin areas per patient revealed increased CD8+ T-cells in the invasive margin areas of each ccRCC tumor, where bystander CD8+CD39− T-cells were dominant. These results showed the regional heterogeneity of the CD8+ T-cell subpopulation in each tumor and added another layer of complexity that makes it difficult to simply divide the tumor immune environment into the “inflamed” and “excluded” states [34]. Further, our concept of single-cell pathology characterized the spatial niches of CD8+CD39+PD-1+ T-cells inside ccRCC tissues. For the first time, we uncovered a potential interaction between CD8+CD39+PD-1+ T-cells and Foxp3+PD-1+ Treg cells due to cell-to-cell proximity in tumors, forming a unique immunosuppressive environment that has the potential to interfere with proper PD-1 blockade effects in ccRCC.
Third, we also applied our multiplexed single-cell pathology to the metastatic ccRCC dataset, revealing that the infiltration of Foxp3+ and Foxp3+PD-1+ Treg cells was more pronounced in metastatic lesions than in primary renal tumor lesions. Although no difference was found in the infiltration of CD8+CD39+PD-1+ T-cells between primary tumors and metastases, however, overall, metastatic tumors may be more aggressive in malignancy, suggesting a paradigm shift to the immunosuppressive environment. In this regard, due to the limited number of cases, further discussion is needed regarding site-specific differences in the immune environment [35].
In sum, analyzing the immune landscape in the TME of RCC revealed phenotypic differences in CD8+ T-cells and clear CD8+ T-cell heterogeneity in tumors, where tumor-specific CD8+CD39+ T-cells and exhausted CD8+CD39+PD-1+ T-cells were central in malignancy. Further, our multiplexed single-cell pathology unraveled a unique interaction between CD8+CD39+PD-1+T-cells and Foxp3+PD-1+ Treg cells due to cell-to-cell proximity, forming a spatial niche more specialized in immunosuppression under PD-1 blockade. It is still discussing whether CD39 is the only marker that depict determines tumor-specific T-cells [36] and may require further consideration, however, our results could be used for developing new and efficient personalized treatment plans for patients with cancer and may provide further insight into the immunobiological standing of RCC differently apart from that of many other solid tumors.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
This study was supported by the Grant-in-Aid for Scientific Research (KAKENHI 19K18621 to T.M.; 18H04906, 18K19482, and 19H03792 to N.T.; 18K09150 to T.S.; 18H02939 to M.O.), the Takeda Science Foundation (N.T.), the Kobayashi Foundation for Cancer Research (N.T.), the SGH Foundation for Cancer Research (N.T.), and the Keio Gijuku Academic Development Funds (N.T.). The authors thank JKiC (JSR-Keio University Medical and Chemical Innovation Center) for special assistance to the multiplexed fluorescence imaging system.
Author contributions
NT, RM, and MO designed the study. TM, KT, KH, KF, MT, and SM performed the experiments. TS, KK, TT, KS, and TI provided conceptual advice. TM and NT wrote the manuscript.
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure) Ann Oncol. 2016;27(8):1492–1504. doi: 10.1093/annonc/mdw217. [DOI] [PubMed] [Google Scholar]
- 2.Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 3.Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389(10066):255–265. doi: 10.1016/S0140-6736(16)32517-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Motzer RJ, Tannir NM, McDermott DF, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. 2018;378(14):1277–1290. doi: 10.1056/NEJMoa1712126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Escudier B, Porta C, Schmidinger M, et al. Renal cell carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-updagger. Ann Oncol. 2019;30(5):706–720. doi: 10.1093/annonc/mdz056. [DOI] [PubMed] [Google Scholar]
- 8.Ljungberg B, Albiges L, Abu-Ghanem Y, et al. European association of urology guidelines on renal cell carcinoma: the 2019 update. Eur Urol. 2019;75(5):799–810. doi: 10.1016/j.eururo.2019.02.011. [DOI] [PubMed] [Google Scholar]
- 9.Motzer RJ, Jonasch E, Michaelson MD, et al. NCCN guidelines insights: kidney cancer version 2. J Natl Compr Canc Netw. 2019;17(11):1278–1285. doi: 10.6004/jnccn.2019.0054. [DOI] [PubMed] [Google Scholar]
- 10.Drake CG, Stein MN. The immunobiology of kidney cancer. J Clin Oncol. 2018;36:3547–3552. doi: 10.1200/JCO.2018.79.2648. [DOI] [PubMed] [Google Scholar]
- 11.Galon J, Bruni D. Tumor immunology and tumor evolution: intertwined histories. Immunity. 2020;52(1):55–81. doi: 10.1016/j.immuni.2019.12.018. [DOI] [PubMed] [Google Scholar]
- 12.Fridman WH, Zitvogel L, Sautes-Fridman C, et al. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14(12):717–734. doi: 10.1038/nrclinonc.2017.101. [DOI] [PubMed] [Google Scholar]
- 13.Remark R, Alifano M, Cremer I, et al. Characteristics and clinical impacts of the immune environments in colorectal and renal cell carcinoma lung metastases: influence of tumor origin. Clin Cancer Res. 2013;19(15):4079–4091. doi: 10.1158/1078-0432.CCR-12-3847. [DOI] [PubMed] [Google Scholar]
- 14.Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeisel A, Munoz-Manchado AB, Codeluppi S, et al. Brain structure. cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347(6226):1138–1142. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
- 16.Yu Y, Tsang JC, Wang C, et al. Single-cell RNA-seq identifies a PD-1(hi) ILC progenitor and defines its development pathway. Nature. 2016;539(7627):102–106. doi: 10.1038/nature20105. [DOI] [PubMed] [Google Scholar]
- 17.Dulken BW, Buckley MT, Navarro Negredo P, et al. Single-cell analysis reveals T cell infiltration in old neurogenic niches. Nature. 2019;571(7764):205–210. doi: 10.1038/s41586-019-1362-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gate D, Saligrama N, Leventhal O, et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer's disease. Nature. 2020;577(7790):399–404. doi: 10.1038/s41586-019-1895-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simoni Y, Becht E, Fehlings M, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557(7706):575–579. doi: 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka N, Kanatani S, Tomer R, et al. Whole-tissue biopsy phenotyping of three-dimensional tumours reveals patterns of cancer heterogeneity. Nat Biomed Eng. 2017;1(10):796–806. doi: 10.1038/s41551-017-0139-0. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka N, Kanatani S, Kaczynska D, et al. Three-dimensional single-cell imaging for the analysis of RNA and protein expression in intact tumour biopsies. Nat Biomed Eng. 2020;4:875–888. doi: 10.1038/s41551-020-0576-z. [DOI] [PubMed] [Google Scholar]
- 22.Stack EC, Wang C, Roman KA, et al. Multiplexed immunohistochemistry, imaging, and quantitation: a review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods. 2014;70(1):46–58. doi: 10.1016/j.ymeth.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 23.Huang YK, Wang M, Sun Y, et al. Macrophage spatial heterogeneity in gastric cancer defined by multiplex immunohistochemistry. Nat Commun. 2019;10(1):3928. doi: 10.1038/s41467-019-11788-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36(4):265–276. doi: 10.1016/j.it.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pauken KE, Wherry EJ. SnapShot: T cell exhaustion. Cell. 2015;163(4):1038–1038. doi: 10.1016/j.cell.2015.10.054. [DOI] [PubMed] [Google Scholar]
- 26.Day CL, Kaufmann DE, Kiepiela P, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443(7109):350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 27.Kamada T, Togashi Y, Tay C, et al. PD-1(+) regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci U S A. 2019;116(20):9999–10008. doi: 10.1073/pnas.1822001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumagai S, Togashi Y, Kamada T, et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat Immunol. 2020;21(11):1346–1358. doi: 10.1038/s41590-020-0769-3. [DOI] [PubMed] [Google Scholar]
- 29.Rini BI, Plimack ER, Stus V, et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380(12):1116–1127. doi: 10.1056/NEJMoa1816714. [DOI] [PubMed] [Google Scholar]
- 30.Motzer RJ, Penkov K, Haanen J, et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380(12):1103–1115. doi: 10.1056/NEJMoa1816047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Savas P, Virassamy B, Ye C, et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med. 2018;24(7):986–993. doi: 10.1038/s41591-018-0078-7. [DOI] [PubMed] [Google Scholar]
- 32.Guo X, Zhang Y, Zheng L, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med. 2018;24(7):978–985. doi: 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 33.Qi Y, Xia Y, Lin Z, et al. Tumor-infiltrating CD39(+)CD8(+) T cells determine poor prognosis and immune evasion in clear cell renal cell carcinoma patients. Cancer Immunol Immunother. 2020;69(8):1565–1576. doi: 10.1007/s00262-020-02563-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019;18(3):197–218. doi: 10.1038/s41573-018-0007-y. [DOI] [PubMed] [Google Scholar]
- 35.Salmon H, Remark R, Gnjatic S, et al. Host tissue determinants of tumour immunity. Nat Rev Cancer. 2019;19(4):215–227. doi: 10.1038/s41568-019-0125-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pauken KE, Shahid O, Lagattuta KA, et al. Single-cell analyses identify circulating anti-tumor CD8 T cells and markers for their enrichment. J Exp Med. 2021 doi: 10.1084/jem.20200920. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.