

Abstract
Photodynamic therapy (PDT) is an anti-tumor modality which employs three individually non-toxic substances, including photosensitizer, light and oxygen, to produce a toxic effect. Besides causing damage to blood vessels that supply oxygen and nutrients to the tumor and killing the tumor by a direct cytotoxic effect, PDT has also been known to trigger an anti-tumor immune response. For instance, our previous study showed that PDT with BAM-SiPc, a silicon(IV) phthalocyanine based-photosensitizer, can not only eradicate the mouse CT26 tumor cells in a Balb/c mouse model, but also protect the mice against further re-challenge of the tumor cells through an immunomodulatory mechanism. To understand more about the immune effect, the biochemical actions of BAM-SiPc-PDT on CT26 cells were studied in the in vitro system. It was confirmed that the PDT treatment could induce immunogenic necroptosis in the tumor cells. Upon treatment, different damage-associated molecular patterns were exposed onto the cell surface or released from the cells. Among them, calreticulin was found to translocate to the cell membrane through a pathway similar to that in chemotherapy. The activation of immune response was also demonstrated by an increase in the expression of different chemokines.
Keywords: Calreticulin, Chemokines, Damage-associated molecular patterns, Immunogenic cell death, Necroptosis, Photodynamic therapy
Introduction
Photodynamic therapy (PDT) employs three non-toxic components to produce a toxic effect. In the process, the photosensitizer, after activation by light of appropriate wavelength, converts oxygen to the toxic reactive oxygen species (ROS). PDT is considered as a promising treatment for cancer as it can eradicate tumor through direct killing, impair the tumor blood vessels supplying oxygen and nutrients and trigger immune response to prevent tumor relapse [1–3].
The cell death pathway takes a critical contribution in anti-tumor immunity. Immunogenic cell death (ICD) is a process which can initiate immune responses, for example recruitment and activation of dendritic cells (DCs), against the dying bodies [4]. About 2 decades ago, a regulated form of cell death, which is more similar to necrosis, was identified and named as necroptosis [5]. In this process, receptor interacting protein kinase (RIPK) 1, RIPK3 and mixed lineage kinase domain like (MLKL) proteins interact with each other to form a necroptotic complex, leading to the phosphorylation of MLKL, which promotes its oligomerization and migration to the plasma membrane to cause membrane permeabilization. During the process, caspase 8, which promotes apoptosis, has to be inhibited [6].
One characteristic of ICD is the exposure or secretion of damage-associated molecular patterns (DAMPs), for example, calreticulin (CRT), heat shock protein 90 (HSP90), high mobility group box 1 (HMGB1) and ATP, which are normally expressed and retained inside the cells. After translocation to the cell surface or released out of the cell during cell death, the DAMPs can stimulate immune response through promoting maturation of DCs and secretion of pro-inflammatory cytokines [6, 7]. Cell surface CRT is the most well-studied DAMP and is regarded as the “eat me” signal [8, 9]. The pathway of CRT translocation, which has been examined in chemotherapy and UVC-driven ICD, consists of three modules, viz. ER stress module, apoptotic module and translocation module. Briefly, upon ER stress, protein kinase RNA-like ER kinase (PERK) and eukaryotic translation initiation factor (eIF-2α) are phosphorylated and subsequently activate caspase 8 which, in turn, initiates the translocation of the CRT/ endoplasmic reticulum-resident protein 57 (ERp57) complex to the cell membrane through the anterograde ER-Golgi trafficking and soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE)-dependent exocytosis [10].
Apart from DAMPs, chemokines also contribute to immune cell activation. Inflammatory chemokines, C-X-C motif chemokine ligand (CXCL) 1 and 3, are found in necroptotic but not necrotic cells [11, 12]. CXCL2 has been shown to be involved in the translocation of CRT [13]. CXCL10 contributes to effector T cell generation [14] and recruits natural killer cells [15]. Expression of CXCL12 can inhibit tumor growth and metastasis [16] whereas CXCL13 can attract B cells [17]. Most prominently, it has been demonstrated that CXCL1 and 10 are released in chemotherapy- and hypericin-PDT-driven ICD and attract neutrophils to engulf cancer cells in ICD [18].
The development of photosensitizers plays a major role in the success of PDT. Different photosensitizers, like photofrin and 5-aminolevulinic acid (5-ALA), as well as different administration methods, like cellular (direct tumor cell damage) and vascular PDT (tumor vasculature damage), have been developed [19]. Other strategies utilizing nanoparticles for photosensitizer delivery have also been exploited to improve PDT efficacy [20]. Throughout the years, we have synthesized a number of novel photosensitizers, including BAM-SiPc, which is an unsymmetrical bisamino silicon(IV) phthalocyanine derivative [21]. The direct cytotoxicity of BAM-SiPc on the tumor cells has been studied in both in vitro cell culture system and in vivo animal model [22]. BAM-SiPc for vascular PDT could eradicate the tumor in the tumor-bearing BALB/c mice and more importantly, the cured mice could resist further re-challenge of the tumor cells [3]. To further understand the anti-tumor immunity, the biochemical actions of BAM-SiPc-PDT on the CT26 cells were investigated in the present study. The results showed that BAM-SiPc-PDT could induce immunogenic necroptosis. It caused the release of DAMPs from the tumor cells and triggered an increase in the expression of different chemokines.
Materials and methods
Preparation of BAM-SiPc, cell culture conditions and inhibitors
BAM-SiPc was synthesized according to the procedures of Lo et al. [21]. For in vitro assays, a stock solution of BAM-SiPc was solubilized in N,N-dimethylformamide followed by 1:10 dilution with 0.01 M Cremophor EL (Sigma-Aldrich). The stock solution was further diluted using complete RPMI-1640 medium (Thermo Fisher Scientific) to appropriate concentrations before being used.
The CT26 murine colon tumor cells (ATCC) were maintained in RPMI-1640 medium supplemented with 10% FBS (Thermo Fisher Scientific), 1% penicillin/ streptomycin mixture (Thermo Fisher Scientific), 1% glucose solution (Thermo Fisher Scientific) and 1 mM sodium pyruvate (Thermo Fisher Scientific).
Different inhibitors, including necrostatin-1 (20 μM, Abcam), zVAD-fmk (25 μM, Selleckchem), deferoxamine (10 μM, Selleckchem), brefeldin A (10 μM, Thermo Fisher Scientific), 16F16 (20 μM, Sigma-Aldrich), brefeldin A (10 μM, Thermo Fisher Scientific), 16F16 (20 μM, Sigma-Aldrich), GSK2606414 (10 μM, Calbiochem), wortmannin (10 μM, Selleckchem), LY294002 (50 μM, Selleckchem), salubrinal (20 μM, Calbiochem), Z-IETD-FMK (50 μM, Selleckchem) and glutathione (5 mM, Sigma-Aldrich), were used in various experiments to study the mode of cell death and the CRT translocation pathway.
In vitro photodynamic treatment
CT26 cells were incubated with 4 or 8 nM BAM-SiPc in the dark for 2 h. After washing, the cells were irradiated with a halogen lamp (300 W) at room temperature for 20 min. Light with λ < 610 nm was cut off using a red glass filter (Newport). The fluence rate used was 100 mW/cm2, giving a total fluence of 120 J/cm2 for the irradiation process [3]. “Control” refers to cells without any BAM-SiPc nor light treatment whereas “Dark” are cells treated with BAM-SiPc without any light illumination.
Cell proliferation assay
The PDT-treated CT26 cells were cultured for 20 h. After the addition of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Sigma-Aldrich), the cells were further incubated at 37 °C for 4 h. Dimethyl sulfoxide was added and the suspension was mixed well. The absorbance at 490 nm was measured using a plate reader (Tecan Spark 10 M Microplate Reader).
Detection of mode of cell death
After PDT, CT26 cells were incubated for 6 h with or without specific inhibitors before being stained with annexin V-GFP (a generous gift from Prof. SK Kong, School of Life Sciences, CUHK) together with propidium iodide (PI, Sigma-Aldrich) in staining buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2) at 4 °C for 15 min. The samples were subjected to flow cytometric analysis (BD Bioscience FACSVerse).
To detect the necroptotic complex RIPK1-RIPK3-MLKL, a co-immunoprecipitation method was used. After PDT, CT26 cells were incubated for 6 h and lysed using IP Lysis Buffer (Thermo Fisher Scientific) with 1% protease inhibitor cocktail (Sigma-Aldrich) and 1 mM phenylmethylsulfonyl fluoride at 4 °C for 10 min. Supernatants were collected after centrifugation and incubated with anti-MLKL antibody (Millipore) at 4 °C overnight. Protein G beads (Thermo Fisher Scientific) were added for an incubation of 2 h at room temperature. After washing with PBS, the samples were eluted with 0.1 M glycine–HCl (pH 2.8) and neutralized with 1/10 v/v Tris–HCl (pH 9.0). Western blot analyses were carried out and the samples were probed with anti-RIPK1 (BD Bioscience), anti-RIPK3 (Abcam), anti-MLKL (Millipore) and anti-β-actin (Sigma-Aldrich) as the primary antibody. The HRP conjugated secondary antibody (Thermo Fisher Scientific) was used and detection was carried out using Luminata Crescendo Western HRP substrate (Millipore).
Detection of DAMPs
CT26 cells were treated with BAM-SiPc, followed by incubation in the absence and presence of various inhibitors. Different methods, including simple biochemical assay, confocal microscopy, Western blot, and flow cytometry were used to follow the expression of DAMPs.
To detect the released ATP, the cell culture supernatant was used. Concentration of ATP was measured using the CellTiter-Glo Luminescent Cell Viability Assay (Promega). The luminescence was recorded using a plate reader.
To detect HMGB1 in the nucleus, confocal microscopy was used. Cells were collected 2 h after PDT and were fixed with 4% paraformaldehyde for 20 min followed by permeabilization in 0.1% Triton X-100 solution for 10 min. After washing and blocking with staining solution (10% heat-inactivated FBS in PBS), CT26 cells were stained with anti-HMGB1 (Abcam) and anti-CD44 (Abcam) antibodies at 4 °C overnight followed by incubation with the corresponding fluorescent-dye conjugated secondary antibodies (Thermo Fisher Scientific) together with Hoechst 33342 (Sigma-Aldrich) at room temperature for 1 h. Samples were examined under confocal microscopy (Leica TCS SP8 high speed imaging system).
The surface CRT was also detected using confocal microscopy. At 1 h after PDT, CT26 cells were first stained with anti-CRT (Abcam) and anti-CD44 (Abcam) antibodies at 4 °C for 1 h. Then they were washed and fixed with cold methanol at − 20 °C for 10 min. After washing, the cells were stained with the corresponding secondary antibodies together with Hoechst 33342 at 4 °C overnight before being visualized. To obtain a more quantitative result, flow cytometry was used. The PDT-treated CT26 cells were stained first with PI and anti-CRT antibody (Abcam) at 4 °C for 30 min and then with the fluorescent dye-conjugated secondary antibody before the analysis.
Western blot analyses were used to study the expression of CRT and its translocation pathway, as well as the expression of other DAMPs. Cell lysate (lysed in radioimmunoprecipitation assay buffer), crude culture supernatant (obtained after centrifugation) and cell membrane proteins (obtained by using the Cell Surface Protein Isolation Kit, Thermo Fisher Scientific) were used as the samples. To concentrate the samples for the detection of vasostatin, the cell supernatants were first precipitated with acetone at − 20 °C overnight and then re-dissolved in SDS loading buffer after washing. Different primary antibodies, including anti-caspase 8 (Novus), anti-Na/K-ATPase (Abcam), anti-CRT (Abcam), anti-CRT (vasostatin)(Abcam), anti-HMGB1 (Abcam), anti-ERp57 (Abcam), anti-HSP90 (Abcam), anti-CD47 (Abcam), anti-P-eIF2α (Cell Signaling Technology) and anti-PERK (Cell Signaling Technology), were used for the detections.
A gene knockdown technology was used to study the expression of CRT and its translocation pathway. Protein expression was suppressed with specific siRNA (siPERK-1: 5′-CAUACGGACUCAGUGCUUAtt-3′; siPERK-2: 5′- CGAAGAAUACAGUAAUGGUtt-3′; siCaspase 8–1: 5′- CAAGAACUAUAUUCCGGAUtt-3′; siCaspase 8–2: 5′- GGCGUGAACUAUGACGUGAtt-3′; Scramble siRNA-1; Scramble siRNA-2, Thermo Fisher Scientific) using RNAiMAX Transfection Reagent (Thermo Fisher Scientific). After 48 h, PDT was performed and cells were incubated for another 4 h. Having been stained with PI and anti-CRT antibody, samples were analyzed using flow cytometry.
Quantitation of chemokines
Total RNA of CT26 cells was isolated 8 h after PDT using the RNeasy Plus Mini Kit (Qiagen). cDNA synthesis was performed with the GoScript Reverse Transcriptase Kit (Promega). Chemokine expression was assessed with gene specific primers (CXCL1-F: 5′-CATGGCTGGGATTCACCTCAA-3′; CXCL1-R: 5′-CTTGGGGACACCTTTTAGCATCTT-3′; CXCL2-F: 5′-AGGCTACAGGGGCTGTTGT-3′; CXCL2-R: 5′-GCTTCAGGGTCAAGGCAAACT-3′; CXCL3-F: 5′-CAGGCTACAGGGGCTGTTGT-3′; CXCL3-R: 5′-GGTTGAGGCAAACTTCTTGACCAT-3′; CXCL10-F: 5′-CTCAAGGGATCCCTCTCGCA-3′; CXCL10-R: 5′-CGTGGCAATGATCTCAACACG-3′; CXCL12-F: 5′-ATCAGTGACGGTAAACCAGTCAGC-3′; CXCL12-R: 5′-GGTCAATGCACACTTGTCTGTTGT-3′; CXCL13-F: 5′-CTGGCCAGCTGCCTCTCTC-3′; CXCL13-R: 5′- CTTGGTCCAGATCACAACTTCAGT-3′) using SYBR Green Master Mix (Thermo Fisher Scientific). β-actin (β-actin-F: 5′-Gagaagctgtgctatgttgctctag-3′; and β-actin-R: 5′-Ccacaggattccatacccaagaagg-3′) was used as the reference gene. PCR was conducted using a real-time PCR system (BIO-RAD CFX96). Data were analyzed using the ΔΔCT method. Culture media were collected 24 h after PDT for determining the concentrations of CXCL1-3, 10, 12 and 13 using enzyme-linked immunosorbent assay (ELISA) kits (Thermo Fisher Scientific, Cusabio).
Statistical analyses
Graphs and statistical analyses were performed using Graphpad Prism 7.0. The data were analyzed using a student t test with p values < 0.05 considered as significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; n.s., not significant.
Results
BAM-SiPc-PDT induced cell death through necroptosis
The cytotoxic effects of BAM-SiPc-PDT on CT26 cells were examined in a cell proliferation assay. A dose-dependent cytotoxic effect was observed upon illumination with an IC50 value of ~ 4 nM while no cytotoxicity could be detected in the absence of light (Fig. 1a). To determine the types of cell death after PDT, different specific cell death inhibitors were used. In the flow cytometric analysis with dual annexin V and PI stainings, the percentage of viable cells (both annexin V and PI negative) was significantly increased when the cells were incubated with necrostatin-1 (an RIPK1 inhibitor) but not zVAD-fmk (a pan-caspase inhibitor) or deferoxamine (an iron chelator) (Fig. 1b). The expression of various proteins involved in the necroptosis pathway was studied using Western blot analysis. The expression of the cleaved form of caspase 8 was inhibited. The expression of the cleaved form of RIPK3 was also reduced in the first hour and became undetectable after 2 h (Fig. 1c). To follow the formation of the necroptotic complex RIPK1-RIPK3-MLKL, a co-immunoprecipitation method was employed. While all the three proteins were present and could be detected by their respective antibodies in the input, only after PDT, RIPK1 and RIPK3 could be pulled down by using antibodies against MLKL. No complex formation could be observed in the controls (Fig. 1d).
Fig. 1.
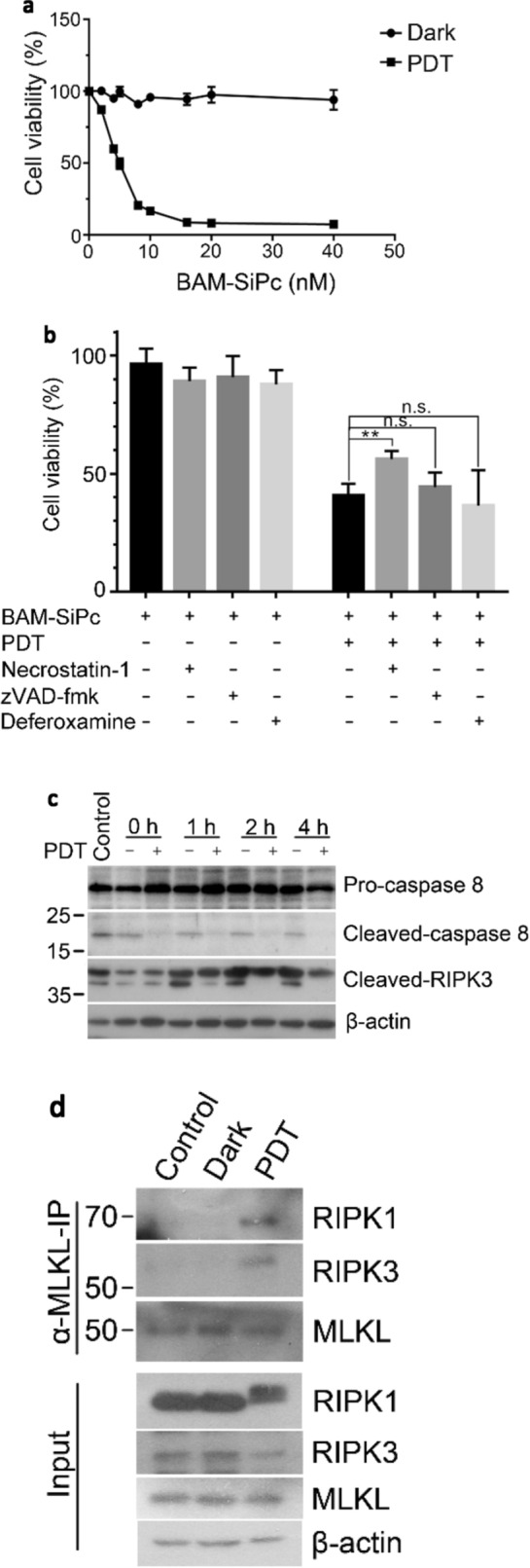
BAM-SiPc-PDT induced cell death through necroptosis. a Different concentrations of BAM-SiPc were incubated with CT26 cells. The cytotoxic effects with and without light illumination were determined. BAM-SiPc concentration of 8 nM was chosen for subsequent experiments. b The PDT-treated cells were incubated with necrostatin-1 (20 μΜ), zVAD-fmk (25 μM) and deferoxamine (10 μM). Cells were collected 6 h later for annexin V and PI staining and subjected to flow cytometric analysis. c The PDT-treated cells were collected at different time points for Western blot analysis of the indicated proteins. d The PDT-treated cells were incubated with anti-MLKL antibody for 6 h in a co-immunoprecipitation assay. The pull-downed proteins were subjected to Western blot analysis. Data shown are means ± SEM or representative results of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001; n.s. not significant
BAM-SiPc-PDT induced the exposure and release of DAMPs
After PDT treatment of the CT26 cells, the culture medium was used to detect the release of DAMPs. The concentration of ATP in the culture medium increased with time such that at 6 h post-PDT, the ATP concentration was ~ 15 times higher than that of the controls (Fig. 2a). Similarly, HMGB1 in the culture medium became detectable 4 h after PDT and its concentration continued to increase with time (Fig. 2b). At the same time, there was a decrease (~ 5.5 times) of nuclear HMGB1 after PDT, when examined under confocal microscopy (Fig. 2c). Confocal microscopy also showed the expression of CRT on the cell surface which could be easily identified by the positive CD44 staining (Fig. 2d). A more quantitative analysis of the expression level was obtained using flow cytometric analysis. The cell population with positive CRT staining but negative PI staining increased by more than five times after PDT when compared with the controls (Fig. 2e). Such increase in CRT and two other DAMPs, namely ERp57 and HSP90, was also obvious in the Western blot of the cell membrane protein preparation (Fig. 2f). There was also an increase in the level of secreted vasostatin (Fig. 2g). While the levels of various DAMPs, the so-called “eat me” signal, increased after PDT, in contrast, the level of CD47, the “don’t eat me” signal, decreased after PDT, as shown by both flow cytometric (Fig. 2e) and Western blot analyses (Fig. 2f).
Fig. 2.


BAM-SiPc-PDT induced the exposure and release of DAMPs. CT26 cells were first treated with 4 nM BAM-SiPc before different experiments were carried out. a, b The PDT-treated cells were incubated for various time periods before the culture supernatants were collected for the detection of a ATP (luminescence assay) and b HMGB1 (Western blot analysis). c The PDT-treated cells were incubated for 2 h before being observed under a confocal microscope. Cell membrane, nucleus and HMGB1 are shown in red, blue and green, respectively. The graph shows the mean fluorescence intensity (MFI) of HMGB1 in the nucleus. d The PDT- treated cells were incubated for 1 h. CRT the cell surface CD44 are shown in red and green respectively. The white arrow indicates the location of CRT. e The PDT-treated cells were incubated for 4 h and then stained with PI together with anti-CRT or anti-CD47 antibody for flow cytometric analysis. The population stained was firstly gated for cells of PI negative and displayed the corresponding antibody stained positive cells. f Cell membrane proteins were collected from the PDT-treated cells for Western blot analysis of the indicated proteins, with Na–K-ATPase serving as loading control. g Proteins in the culture supernatant of the PDT-treated cells were collected at various times, concentrated and probed for vasostatin in Western blot analysis. Data shown are means ± SEM or representative results of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001
BAM-SiPc-PDT induced the translocation of CRT through a classical pathway
To study the translocation pathway of CRT, the expression level of different protein markers and also the effects of different inhibitors on surface CRT expression in the BAM-SiPc-PDT treated CT26 cells were examined. After PDT, phosphorylation of PERK occurred as shown by the Western blot. Concurrently, phosphorylation of eIF-2α was boosted immediately after PDT but the level was reduced gradually such that at 4 h post-PDT, the level became similar to that of the controls (Fig. 3a). The cell population with surface CRT positive and PI negative determined using flow cytometry was ~ 7 times of that of the dark control. However, with the addition of any one of the inhibitors of the pathway, including glutathione (oxidative stress), 16F16 (ERp57), GSK2606414 (PERK), salubrinal (eIF-2α), Z-IETD-FMK (caspase 8), brefeldin A (ER to Golgi apparatus anterograde transport) and wortmannin and LY294002 (phosphoinositide-3-kinase dependent exocytosis), the surface expression of CRT was significantly reduced (Fig. 3b). Similar results were also observed when the cell membrane proteins were isolated for the detection of CRT level using Western blot (Fig. 3c). Knockdown of PERK and caspase 8 also showed a reduced expression of surface CRT (Fig. 3d, e).
Fig. 3.

BAM-SiPc-PDT induced the translocation of CRT through a classical pathway. CT26 cells were first treated with 4 nM BAM-SiPc before the following experiments. a The PDT-treated cells were incubated for the indicated times. Cell lysates were used to detect the phosphorylation of PERK and eIF-2α through Western blotting. b, c The PDT-treated cells were incubated with and without the various pathway inhibitors for 4 h. b The cells were stained with PI and anti-CRT antibody and subjected to flow cytometric analysis. The population stained represents cell population of PI negative and CRT positive. c The cell membrane proteins were isolated for Western blotting to probe for CRT. d CT26 cells were transfected with corresponding siRNA to restrain PERK and caspase 8 expressions. The suppressive effects were shown in Western blot analyses. e After transfection, PDT was performed 48 h later. Having been stained with PI and anti-CRT antibody, cells were subjected to flow cytometric analysis. Data shown are means ± SEM or representative results of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001
BAM-SiPc-PDT increased the expression of chemokines in CT26 cells
The total RNA of the PDT-treated cells was isolated for determining the mRNA levels of various chemokines, including CXCL1-3, 10, 12 and 13. Significant increase was observed for all the chemokines examined. The increase ranged from 3 to 70 times. Among them, CXCL10 (~ 70 times) and CXCL1 (~ 35 times) showed the highest increase followed by CXCL3 (~ 10 times) (Fig. 4a). Secretion of chemokines was confirmed with ELISA showing the increase of CXCL10 (~ 1.8 times), CXCL2 (~ 1.8 times) and CXCL1 (~ 1.6 times) (Fig. 4b).
Fig. 4.
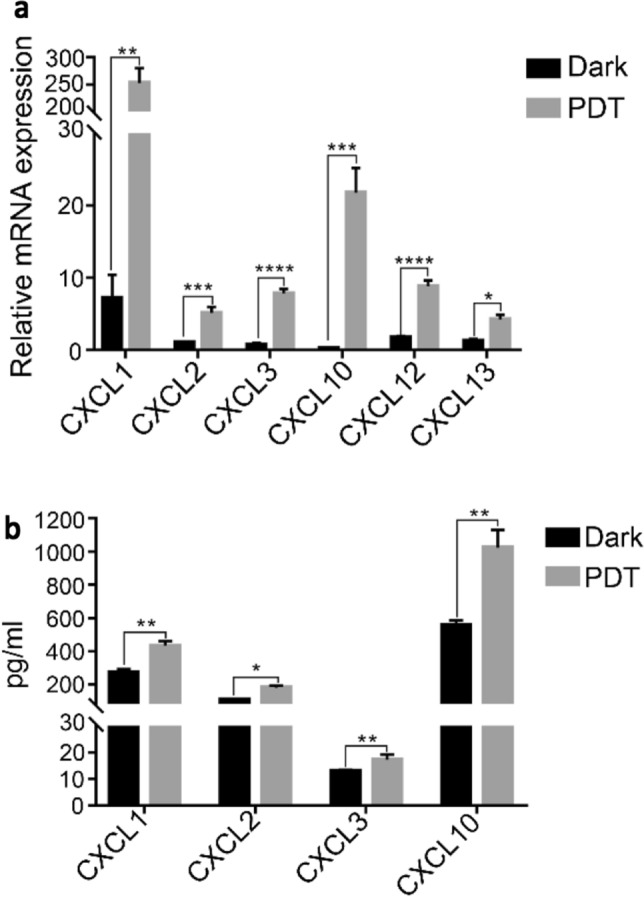
BAM-SiPc-PDT induced the expression of chemokines in CT26 cells. CT26 cells were treated with 8 nM BAM-SiPc and subjected to PDT. a After 8 h of incubation, total RNA was extracted from the cells and quantitative PCR was conducted to detect the level of various chemokines. b After 24 h of incubation, culture media were collected for cytokine concentration determination using ELISA kits. Data shown are means ± SEM of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Discussion
Photosensitizer is the most critical component in PDT. Throughout the years, its anti-tumor effect has been focused mainly on its direct cytotoxic effect. Nevertheless, a few photosensitizers have also been shown to trigger immune response. Among them is the ER residing photosensitizer, hypericin, which is exemplarily known to be able to generate immune response by causing immunogenic apoptosis [23]. This type of ICD is a regulated form of apoptosis which was first identified from studies of some anti-cancer drugs, such as anthracyclines and mitoxantrone [9, 24]. The finding revolutionarily overthrows the dichotomic classification of the mode of cell death in which apoptosis is regarded as a regulated and non-immunogenic collapse and necrosis as an immunogenic but pathological destruction [25]. Unlike hypericin-PDT, the cell death mechanism involved in BAM-SiPc-PDT is mainly necrosis rather than apoptosis. Such immunogenic necrosis has previously been named as necroptosis [5].
To confirm that BAM-SiPc-PDT induced necroptosis, three different approaches were used. First, the percentage of cell death was followed in the presence of the RIPK1 inhibitor, necrostatin-1. In cell biology, RIPK1 pertains to the induction of necrotic cell population [10]. The use of necrostatin-1 could reduce the population of dead cells. Second, the expressions of molecular markers of the pathway, including caspase 8 and RIPK3, were examined. The cleaved form of caspase 8 is known to cleave RIPK3 which subsequently can no longer interact with RIPK1 and MLKL to form the necroptotic complex [7, 26, 27]. Our result showed that the expressions of the cleaved forms of both caspase 8 and RIPK3 were suppressed after BAM-SiPc-PDT. Such decreases favor the occurrence of necroptosis. Third, the formation of the necroptotic complex RIPK1-RIPK3-MLKL was followed. The co-immunoprecipitation results revealed that RIPK1 and RIPK3 interact with MLKL to form the complex. In the present study, RIPK3 was detected in CT26 cells, similar to that observed in [28, 29], but in contrast with the findings in [11, 30]. Such discrepancy may be due to the variation of CT26 cells from different sources. With the positive results from these three crucial indicators, it was confirmed that BAM-SiPc-PDT, similar to the other two photosensitizers, 5-ALA [31] and talaporfin sodium [32], could trigger cell death through necroptosis.
The cell death mechanism in BAM-SiPc-PDT was immunogenic in nature, as shown by the release of DAMPs. Both ATP and HMGB1 were found to be secreted into the culture medium after PDT and more interestingly, vasostatin, a CRT fragment possessing anti-angiogenesis properties [33], could also be detected. Vasostatin has been proved for its potential to eradicate tumors [34], perhaps, by counteracting the secretion of proangiogenic factors in mesenchymal stromal cells in the vicinity of the PDT area [35]. However, whether the amount of vasostatin released in BAM-SiPc-PDT is sufficient to inhibit angiogenesis remains to be studied. Other DAMPs like CRT, ERp57 and HSP90, the “eat me” signals, were elevated, while CD47, the “don’t eat me” signal [8], was suppressed on the cell surface. Such changes could trigger different immune responses by interacting with different types of immune cells. For example, our recent study showed that the BAM-SiPc-PDT treated CT26 cells could be recognized and engulfed by DCs which, as a result, are activated, as shown by both phenotypic changes (increased expression of CD80, CD86 and MHCII) as well as functional stimulation (increased production of interleukin-12 and interferon γ) [36].
Both immunogenic apoptosis and immunogenic necroptosis trigger the release of DAMPs. It would be of interest to see whether the pathways of DAMPs translocation are identical or not. Translocation of CRT in immunogenic apoptosis has been examined in the hypericin-PDT study. It is independent of eIF-2α phosphorylation, caspase 8 activation, with a co-exposure of ERp57 [23, 37], in contrast with the classical ICD pathway triggered after chemotherapy [10]. In the present study, Western blot analyses were performed on the different markers in the pathways. Inhibitors and gene knockdown were also used to inhibit specific steps of the pathway. The results showed that blocking PERK, eIF-2α phosphorylation and caspase 8 activation resulted in diminution of CRT on the cell surface. These, together with exposure of ERp57 on cell surface upon treatment, suggested that translocation of CRT in necroptotic cells initiated by BAM-SiPc-PDT follows the chemotherapy-driven classical ICD pathway. Among the various steps, the role of caspase 8 is more obscure because translocation of CRT requires caspase 8 [10] but driving necroptosis requires silencing of caspase 8 to block apoptosis [27]. Our findings indicated that after PDT, caspase 8 was severely restrained but CRT could still be exposed (Figs. 1d and 2d–f). When caspase 8 was inhibited or its gene was suppressed, surface CRT was highly constricted but not entirely abolished (Fig. 3b, c, e). Based on the present results, it appears that caspase 8 might act in a dose-dependent manner. It could be suppressed down to a state that skews cell death toward necroptosis but still sufficient to expose CRT. In this regard, it will be interesting to study the translocation of CRT and the role of caspase 8 in the action of other necroptotic photosensitizer, for example, 5-ALA.
While ICD could trigger the interaction of the tumor cells with other immune cells, it could also up-regulate the production of chemokine directly in the tumor cells. In the present study, the mRNA and ELISA results suggested that BAM-SiPc-PDT could lead cells to release inflammatory chemokines (CXCL1-3, 10, 12 and 13) which are generally believed to play a role in promoting immune response. Among them, CXCL12 and 13 were detected only in transcript expression level but not the proteins. This may be attributed to the relatively lower sensitivity of ELISA or the low correlation in magnitude of gene activation and its gene product [38–40]. CXCL1-3 have been identified in previous necroptosis studies [11, 12, 41]. The dramatic increase in the expression of CXCL1 and 10 is consistent with the previous findings in chemotherapy- and hypericin-PDT driven ICD, implicating the potential of BAM-SiPc-PDT in recruiting neutrophils [18]. Nevertheless, there are also reports on the “pro-tumor” properties of chemokines [42, 43]. The scenario might be considered as a homeostasis of two circumstances and tumor cells usually turn the token into one side by overwhelming the balance favorable to angiogenesis and metastasis. BAM-SiPc-PDT, however, might break this one-sided game and turn the token into another situation. The generation of ROS causes tumor cells to die through a more gentle and regulated way of necroptosis. The elevated release of chemokines helps in recruiting neutrophils for innate immune response. With the aid of DAMPs to activate DCs, the situation becomes conducive to cancer killing by attracting more lymphocytes for DCs to elicit the adaptive immune response and finally initiate the tumor eradication process [3, 36]. In fact, necroptotic cancer cells have been exploited to act as a vaccine to protect mice from tumor cells challenge [11].
In conclusion, BAM-SiPc-PDT could cause CT26 tumor cell death through immunogenic necroptosis. DAMPs, for instance CRT, were released in a chemotherapy-driven ICD pathway. The production of inflammatory chemokines was up-regulated. The present findings on PDT-treated CT26 cells solidify our previous results in which in vitro BAM-SiPc-PDT can facilitate DC maturation [36] and in vivo BAM-SiPc-PDT could eradicate the tumor and protect the mice against further re-challenge [3].
Acknowledgements
This work was supported by a grant from the Research Grants Council of the Hong Kong Special Administrative Region (Ref. No. 14171717).
Abbreviations
- 5-ALA
5-Aminolevulinic acid
- CRT
Calreticulin
- CXCL
C-X-C motif chemokine ligand
- DAMPs
Damage-associated molecular patterns
- DCs
Dendritic cells
- eIF-2α
Eukaryotic translation initiation factor
- ELISA
Enzyme-linked immunosorbent assay
- ER
Endoplasmic reticulum
- ERp57
Endoplasmic reticulum-resident protein 57
- HSP90
Heat shock protein 90
- HMGB1
High mobility group box 1
- ICD
Immunogenic cell death
- MFI
Mean fluorescence intensity
- MLKL
Mixed lineage kinase domain like
- PDT
Photodynamic therapy
- PERK
Protein kinase RNA-like ER kinase
- PI
Propidium iodide
- RIPK
Receptor interacting protein kinase
- ROS
Reactive oxygen species
- SNARE
Soluble N-ethylmaleimide-sensitive factor attachment protein receptor
Author contributions
WPF designed the project. YZ and YKC conducted experiments. YZ, YKC, DKPN and WPF analyzed data. YZ, YKC and WPF wrote the manuscript. All authors assisted in editing the manuscript and approved its final version.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Cell line authentication
CT26 cells were purchased from the American Type Culture Collection (ATCC) and are free of mycoplasma infections. All CT26 cells used in the experiments were of passage numbers below 14.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Ying Zhang and Ying-Kit Cheung contributed equally to this work as first authors.
References
- 1.Preise D, Oren R, Glinert I, Kalchenko V, Jung S, Scherz A, Salomon Y. Systemic antitumor protection by vascular-targeted photodynamic therapy involves cellular and humoral immunity. Cancer Immunol Immunother. 2009;58(1):71–84. doi: 10.1007/s00262-008-0527-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fong WP, Yeung HY, Lo PC, Ng DKP. Photodynamic therapy. In: Ho AHP, K. D., Somekh MG, , editors. Handbook of photonics for biomedical engineering. Dordrecht: Springer; 2014. pp. 657–681. [Google Scholar]
- 3.Yeung HY, Lo PC, Ng DK, Fong WP. Anti-tumor immunity of BAM-SiPc-mediated vascular photodynamic therapy in a BALB/c mouse model. Cell Mol Immunol. 2017;14(2):223–234. doi: 10.1038/cmi.2015.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 5.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 6.Krysko O, Aaes T, Kagan VE, D'Herde K, Bachert C, Leybaert L, Vandenabeele P, Krysko DV. Necroptotic cell death in anti-cancer therapy. Immunol Rev. 2017;280(1):207–219. doi: 10.1111/imr.12583. [DOI] [PubMed] [Google Scholar]
- 7.Liu X, Shi F, Li Y, Yu X, Peng S, Li W, Luo X, Cao Y. Post-translational modifications as key regulators of TNF-induced necroptosis. Cell Death Dis. 2016;7(7):e2293. doi: 10.1038/cddis.2016.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123(2):321–334. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 9.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Métivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13(1):54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 10.Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, Durchschlag M, Joza N, Pierron G, van Endert P, Yuan J, Zitvogel L, Madeo F, Williams DB, Kroemer G. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009;28(5):578–590. doi: 10.1038/emboj.2009.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, Delrue I, Taminau J, Wiernicki B, De Groote P, Garg AD, Leybaert L, Grooten J, Bertrand MJ, Agostinis P, Berx G, Declercq W, Vandenabeele P, Krysko DV. Vaccination with necroptotic cancer cells induces efficient anti-tumor immunity. Cell Rep. 2016;15(2):274–287. doi: 10.1016/j.celrep.2016.03.037. [DOI] [PubMed] [Google Scholar]
- 12.Lin J, Kumari S, Kim C, Van TM, Wachsmuth L, Polykratis A, Pasparakis M. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature. 2016;540(7631):124–128. doi: 10.1038/nature20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sukkurwala AQ, Martins I, Wang Y, Schlemmer F, Ruckenstuhl C, Durchschlag M, Michaud M, Senovilla L, Sistigu A, Ma Y, Vacchelli E, Sulpice E, Gidrol X, Zitvogel L, Madeo F, Galluzzi L, Kepp O, Kroemer G. Immunogenic calreticulin exposure occurs through a phylogenetically conserved stress pathway involving the chemokine CXCL8. Cell Death Differ. 2014;21(1):59–68. doi: 10.1038/cdd.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol. 2002;168(7):3195–3204. doi: 10.4049/jimmunol.168.7.3195. [DOI] [PubMed] [Google Scholar]
- 15.Yuan J, Liu Z, Lim T, Zhang H, He J, Walker E, Shier C, Wang Y, Su Y, Sall A, McManus B, Yang D. CXCL10 inhibits viral replication through recruitment of natural killer cells in coxsackievirus B3-induced myocarditis. Circ Res. 2009;104(5):628–638. doi: 10.1161/CIRCRESAHA.108.192179. [DOI] [PubMed] [Google Scholar]
- 16.Williams SA, Harata-Lee Y, Comerford I, Anderson RL, Smyth MJ, McColl SR. Multiple functions of CXCL12 in a syngeneic model of breast cancer. Mol Cancer. 2010;9:250. doi: 10.1186/1476-4598-9-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gunn MD, Ngo VN, Ansel KM, Ekland EH, Cyster JG, Williams LT. A B-cell-homing chemokine made in lymphoid follicles activates Burkitt's lymphoma receptor-1. Nature. 1998;391(6669):799–803. doi: 10.1038/35876. [DOI] [PubMed] [Google Scholar]
- 18.Garg AD, Vandenherk L, Fang S, Fasche T, Van Eygen S, Maes J, Van Woensel M, Koks C, Vanthillo N, Graf N, de Witte P, Van Gool S, Salven P, Agostinis P. Pathogen response-like recruitment and activation of neutrophils by sterile immunogenic dying cells drives neutrophil-mediated residual cell killing. Cell Death Differ. 2017;24(5):832–843. doi: 10.1038/cdd.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donohoe C, Senge MO, Arnaut LG, Gomes-da-Silva LC. Cell death in photodynamic therapy: from oxidative stress to anti-tumor immunity. Biochim Biophys Acta Rev Cancer. 2019;1872(2):188308. doi: 10.1016/j.bbcan.2019.07.003. [DOI] [PubMed] [Google Scholar]
- 20.Sztandera K, Gorzkiewicz M, Klajnert-Maculewicz B. Nanocarriers in photodynamic therapy-in vitro and in vivo studies. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2020;12(3):e1509. doi: 10.1002/wnan.1599. [DOI] [PubMed] [Google Scholar]
- 21.Lo PC, Huang JD, Cheng DY, Chan EY, Fong WP, Ko WH, Ng DK. New amphiphilic silicon(IV) phthalocyanines as efficient photosensitizers for photodynamic therapy: synthesis, photophysical properties, and in vitro photodynamic activities. Chemistry. 2004;10(19):4831–4838. doi: 10.1002/chem.200400462. [DOI] [PubMed] [Google Scholar]
- 22.Leung SC, Lo PC, Ng DK, Liu WK, Fung KP, Fong WP. Photodynamic activity of BAM-SiPc, an unsymmetrical bisamino silicon(IV) phthalocyanine, in tumour-bearing nude mice. Br J Pharmacol. 2008;154(1):4–12. doi: 10.1038/bjp.2008.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, Rubio N, Firczuk M, Mathieu C, Roebroek AJ, Annaert W, Golab J, de Witte P, Vandenabeele P, Agostinis P. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012;31(5):1062–1079. doi: 10.1038/emboj.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao C, Han Y, Ren Y, Wang Y. Mitoxantrone-mediated apoptotic B16–F1 cells induce specific anti-tumor immune response. Cell Mol immunol. 2009;6(6):469–475. doi: 10.1038/cmi.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17(2):97–111. doi: 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 26.Feng S, Yang Y, Mei Y, Ma L, Zhu DE, Hoti N, Castanares M, Wu M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal. 2007;19(10):2056–2067. doi: 10.1016/j.cellsig.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 27.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471(7338):363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takemura R, Takaki H, Okada S, Shime H, Akazawa T, Oshiumi H, Matsumoto M, Teshima T, Seya T. PolyI:C-induced, TLR3/RIP3-dependent necroptosis backs up immune effector-mediated tumor elimination in vivo. Cancer Immunol Res. 2015;3(8):902–914. doi: 10.1158/2326-6066.CIR-14-0219. [DOI] [PubMed] [Google Scholar]
- 29.Yan G, Zhao H, Zhang Q, Zhou Y, Wu L, Lei J, Wang X, Zhang J, Zhang X, Zheng L, Du G, Xiao W, Tang B, Miao H, Li Y. A RIPK3-PGE2 circuit mediates myeloid-derived suppressor cell-potentiated colorectal carcinogenesis. Cancer Res. 2018;78(19):5586–5599. doi: 10.1158/0008-5472.CAN-17-3962. [DOI] [PubMed] [Google Scholar]
- 30.Yang H, Ma Y, Chen G, Zhou H, Yamazaki T, Klein C, Pietrocola F, Vacchelli E, Souquere S, Sauvat A, Zitvogel L, Kepp O, Kroemer G. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology. 2016;5(6):e1149673. doi: 10.1080/2162402X.2016.1149673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coupienne I, Fettweis G, Rubio N, Agostinis P, Piette J. 5-ALA-PDT induces RIP3-dependent necrosis in glioblastoma. Photochem Photobiol Sci. 2011;10(12):1868–1878. doi: 10.1039/c1pp05213f. [DOI] [PubMed] [Google Scholar]
- 32.Miki Y, Akimoto J, Moritake K, Hironaka C, Fujiwara Y. Photodynamic therapy using talaporfin sodium induces concentration-dependent programmed necroptosis in human glioblastoma T98G cells. Lasers Med Sci. 2015;30(6):1739–1745. doi: 10.1007/s10103-015-1783-9. [DOI] [PubMed] [Google Scholar]
- 33.Pike SE, Yao L, Jones KD, Cherney B, Appella E, Sakaguchi K, Nakhasi H, Teruya-Feldstein J, Wirth P, Gupta G, Tosato G. Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumor growth. J Exp Med. 1998;188(12):2349–2356. doi: 10.1084/jem.188.12.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Persano L, Crescenzi M, Indraccolo S. Anti-angiogenic gene therapy of cancer: current status and future prospects. Mol Aspects Med. 2007;28(1):87–114. doi: 10.1016/j.mam.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 35.Udartseva OO, Zhidkova OV, Ezdakova MI, Ogneva IV, Andreeva ER, Buravkova LB, Gollnick SO. Low-dose photodynamic therapy promotes angiogenic potential and increases immunogenicity of human mesenchymal stromal cells. J Photochem Photobiol B. 2019;199:111596. doi: 10.1016/j.jphotobiol.2019.111596. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Ng DKP, Fong WP. Antitumor immunity induced by the photodynamic action of BAM-SiPc, a silicon (IV) phthalocyanine photosensitizer. Cell Mol Immunol. 2019;16(7):676–678. doi: 10.1038/s41423-019-0239-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garg AD, Krysko DV, Vandenabeele P, Agostinis P. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol Immunother. 2012;61(2):215–221. doi: 10.1007/s00262-011-1184-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amsen D, de Visser KE, Town T. Approaches to determine expression of inflammatory cytokines. Methods Mol Biol. 2009;511:107–142. doi: 10.1007/978-1-59745-447-6_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13(4):227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y, Beyer A, Aebersold R. On the dependency of cellular protein levels on mRNA abundance. Cell. 2016;165(3):535–550. doi: 10.1016/j.cell.2016.03.014. [DOI] [PubMed] [Google Scholar]
- 41.Zhu K, Liang W, Ma Z, Xu D, Cao S, Lu X, Liu N, Shan B, Qian L, Yuan J. Necroptosis promotes cell-autonomous activation of proinflammatory cytokine gene expression. Cell Death Dis. 2018;9(5):500. doi: 10.1038/s41419-018-0524-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chow MT, Luster AD. Chemokines in cancer. Cancer Immunol Res. 2014;2(12):1125–1131. doi: 10.1158/2326-6066.CIR-14-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mollica Poeta V, Massara M, Capucetti A, Bonecchi R. Chemokines and chemokine receptors: new targets for cancer immunotherapy. Front Immunol. 2019;10:379. doi: 10.3389/fimmu.2019.00379. [DOI] [PMC free article] [PubMed] [Google Scholar]