

Abstract

In the present work, 2-imino-1,3-thiazolines featuring highly fluorinated fragments were synthesized through a straightforward cyclization of diversely substituted thioureas with 2-bromo-1-(4-fluorophenyl)ethan-1-one. The target compounds were obtained in good yields, and structures were established by FTIR and 1H- and 13C NMR spectroscopic methods. The in vitro biological assay revealed that all the compounds significantly obstruct the α-glucosidase. Compound 6d (3-fluoro-N-(3-(2-fluorophenyl)-4-(4-fluorophenyl)thiazol-2(3H)-ylidene)benzamide) showed the highest antidiabetic potential with an IC50 value of 1.47 ± 0.05 μM. In addition, computational analysis revealed the binding energy of −11.1 kcal/mol for 6d which was lower than the positive standard, acarbose (−7.9 kcal/mol). Several intermolecular interactions between the active site residues and 6d highlight the significance of 2-imino-1,3-thiazoline core in attaining the potent efficacy and making these compounds a valuable pharmacophore in drug discovery.
1. Introduction
2-Imino-1,3-thiazoline nucleus has emerged as a promising scaffold in medicinal chemistry and drug discovery research considering its broad pharmacological spectrum.1 The literature survey documented that 2-imino-1,3-thiazoline core serves as a diverse pharmacophore acting as platelet GPIIb/IIIa receptor antagonists,2 alkaline phosphatase inhibitors,3 anticancer agents,4 antibacterial agents,5,6 carbonic anhydrase,7 cyclooxygenase (COX) and lipoxygenase (LOX) inhibitors,8 selective cannabinoid receptor type 2 (CB2) agonists,9 p-53 inactivators pifithrin-α,10 insecticidal agents,11 urease inhibitors,12 herbicidal agents,13 glucosidase inhibitors,14 HIV-1 reverse transcriptase inhibitors,15 and fungicides16 (Figure 1). Several synthetic approaches have been reported detailing the enhanced structure–activity relationships to achieve the desired biological efficacy. In a quest to further broaden the biological activity scope of 2-imino-1,3-thiazolines, fluorine substitution has been exploited extensively in drug discovery. Moreover, fluorinated compounds tend to be more resistant to metabolic degradation due to the high bond energies and heat of formation of the H–O and C–O bonds relative to those of the F–O bond.17 Fluorine substitution at a certain position in a drug molecule can influence not only pharmacokinetic properties such as absorption, tissue distribution, secretion, and the route and rate of biotransformation but also the pharmacodynamics and toxicology, thus improving the efficacy of drugs.18 Despite the fact that fluorine is slightly larger than hydrogen, several studies have demonstrated that it is a reasonable hydrogen mimic and is expected to cause minimal steric perturbations with respect to the compound’s mode of binding to a receptor or enzyme.19 Furthermore, the fluorine substitution has also emerged as one of the most prominent atoms in the application of positron emission tomography due to the favorable half-life of the 18F isotope (109.8 min) when compared to 11C (20.4 min) and 124I (4.2 days). Therefore, new synthetic methodologies are considerably making use of this atom, particularly for central nervous system drug discovery.20,21 KHG22394, a fluorinated 2-imino-1,3-thiazoline derivative, significantly inhibits melanin production in a dose-dependent manner, without directly inhibiting tyrosinase, the rate-limiting melanogenic enzyme. It has been reported that the activation of extracellular signal-regulated kinase reduces melanin synthesis by down regulating microphthalmia-associated transcription factor (Mitf). 2-Imino-1,3-thiazoline derivatives with flurbiprofen have been found to act as nonsteroidal anti-inflammatory drugs.22
Figure 1.

Selected examples of biologically active 2-imino-1,3-thiazolines and our target structures.
In this research work, α-glucosidase has been targeted which is produced by salivary and pancreatic glands and metabolizes the starch into small polysaccharides.23 The enzyme is also located in the lining of small intestine where it plays a crucial role in the final stage of glucose metabolism. The enzyme functions by catalyzing the α-glucosidic disaccharide and oligosaccharide bonds owing to the presence of duplicated glycoside hydrolase domains (GH31).24,25 Besides carbohydrate digestion, α-glucosidase enzyme is also essential for numerous biological processes such as degradation of lysosomal glycoconjugates and the modification of post-translational glycoproteins.26 The enzymatic activity of α-glucosidase is significant in the management of type 2 diabetes mellitus (T2DM) as its inhibition retards the rate of breakdown of starch. It helps in regulating the postprandial hyperglycemia.27 Previously, numerous fluorinated compounds have been analyzed to inhibit the α-glucosidase enzyme, and it was observed that the extent of fluorination is directly related to the inhibitory activity.28
Encouraged by the broad-spectrum bioactivity profile of fluorinated compounds, we herein envisioned the synthesis of 2-imino-1,3-thiazoline derivatives by a facile cyclization of diversely substituted thioureas with 2-bromo-1-(4-fluorophenyl)ethan-1-one. In vitro α-glucosidase inhibition potential was examined while in silico computational methods were employed to rationalize the binding mode analysis.
2. Results and Discussion
2.1. Synthesis and Characterization
Due to the influence of the broad spectrum of fluorinated 2-imino-1,3-thiazolines, we have synthesized the targeted compounds by following the literature reported procedures.29,30 The precursor diversely substituted thioureas 4(a–j) were prepared according to the reported procedure involving the reaction of acid chlorides with potassium thiocyanate in anhydrous acetone, followed by the condensation of the resulting isothiocyanate intermediates with substituted anilines 3(a–j).
In the FTIR spectra, the 1-(fluorobenzoyl)-3-(fluorophenyl)thiourea 6d, representative example, was characterized by absorption bands at 3351 and 3200 cm–1 for NH stretching, 1670 cm–1 for carbonyl, and 1240 cm–1 for thiocarbonyl functionalities. 1H NMR analysis indicated two singlets at 9.0 and 12 ppm for NH groups, whereas 13C NMR showed peaks at 170 and 179 ppm for carbonyl and thiocarbonyl groups, respectively. Cyclization of thioureas 4(a–j) with 2-bromo-1-(4-fluorophenyl)ethan-1-one (5) in an alkaline aqueous medium led to the formation of the corresponding series of 2-imino-1,3-thiazolines 6(a–j). The structures were confirmed by a slight shifting of carbonyl absorption bands to 1585–1660 cm–1 and the appearance of a characteristic C=N stretching at 1440–1480 cm–1 in the FTIR spectra, in addition to the absence of thiourea NH absorptions. The emergence of characteristic proton singlet for H-4 at 6.34–6.46 ppm in 1H NMR evidenced the formation of 1,3-thiazoline ring. 13C NMR data revealed the characteristic signals for olefinic carbon at 104.4–105.1 ppm and C=N signals at 154–157 ppm in conjunction with signals for aromatic protons which agreed to the proposed structures. The corresponding 13C–19F coupling is also observed having typically J1 of 152.5 Hz, J2 of 7.5 Hz, and J3 of 3.0 Hz, which varies accordingly in the corresponding structure. The synthesis of the title compounds 6(a–j) is shown in Scheme 1.
Scheme 1. Synthesis of 2-Imino-1,3-thiazoline Derivatives 6(a–j) from Cyclocondensation of 1-Aroyl-3-arylthioureas 4(a–j) and 2-Bromo-1-(4-fluorophenyl)ethan-1-one (5).

2.2. In Vitro Glucosidase Inhibition and Structure–Activity Relationships
The inhibitory potential of 2-imino-1,3-thiazoline derivatives 6(a-j) against α-glucosidase and β-glucosidase was assessed through in vitro inhibition assays, with the aim of determining their respective inhibitory concentration 50 (IC50) values. The results (Table 1) predicted that 2-imino-1,3-thiazoline derivatives exhibit selective inhibitory effect on α-glucosidase as they presented minimal inhibition against β-glucosidase. Additionally, 6d stands out as the most effective inhibitor compared to all other compounds including positive control, acarbose (IC50 = 35.1 ± 0.14 μM) due to its notably low IC50 value (1.47 ± 0.05 μM).
Table 1. Inhibitory Activities of 2-Imino-1,3-thiazoline Derivatives 6(a–j) against α-Glucosidase and β-Glucosidase.
| compound | substituents |
α-glucosidase IC50 ± SEM (μM) | β-glucosidase % age inhibition | |
|---|---|---|---|---|
| R1 | R2 | |||
| 6a | 3,4,5-triOMe | 3-Cl-4-F | 18.2 ± 0.02 | 11.2 |
| 6b | 2-Cl | 2-F | 3.23 ± 0.07 | 24.2 |
| 6c | 2-F | 2-F | 2.98 ± 0.04 | 36.3 |
| 6d | 3-F | 2-F | 1.47 ± 0.05 | 18.6 |
| 6e | 3,4,5-triOMe | 4-F | 2.10 ± 0.06 | 9.33 |
| 6f | 2-Cl-4-NO2 | 4-F | 4.04 ± 0.16 | 19.4 |
| 6g | 4-F | 3-Cl-4-F | 3.16 ± 0.15 | 20.1 |
| 6h | 2-Cl | 4-F | 4.33 ± 0.19 | 17.5 |
| 6i | 3,4,5-triOMe | 3-F | 8.31 ± 0.13 | 15.4 |
| 6j | 2-F | 3-Cl-4-F | 12.5 ± 0.22 | 12.8 |
| acarbosea | 35.1 ± 0.14 | 63.7 | ||
Positive control.
All the newly synthesized compounds are highly fluorinated and are modified at two aryl groups referred as R1 and R2 (Scheme 1). Compounds 6a, 6g, and 6j contain 3-Cl and 4-F substituents as R2, so their degree of α-glucosidase inhibition is dependent on R1 modification. The presence of 4-fluoro (4-F) as R1 in 6g, characterized by its strong electronegativity, has the potential to effectively block the activity of α-glucosidase, displaying an inhibitory effect with an IC50 value of 3.16 ± 0.15 μM. However, the inhibitory activity significantly decreases when F is relocated to 2-position of aromatic ring (6j), resulting in the diminished inhibitory activity with an IC50 value of 12.5 ± 0.22 μM. Likewise, when 3,4,5-trimethoxy group is affixed as R1 (6a), it leads to an adequate inhibition of α-glucosidase with an IC50 value of 18.2 ± 0.02 μM. This is attributed to the electron-donating nature of the methoxy group (Figure 2).
Figure 2.

Structure–activity relationship of compounds 6a, 6g, and 6h.
The inhibitory effect of 2-imino-1,3-thiazoline derivatives containing a 2-F substituent as R2 was also altered when R1 is changed as observed in 6b, 6c, and 6d (Figure 3). In the presence of electron-withdrawing group such as chlorine at the ortho-position of aroyl ring (6b), the inhibitory activity was 3.23 ± 0.07 μM. This α-glucosidase inhibitory potential increases when the Cl is replaced with another highly electronegative atom fluorine (6c; IC50 = 2.98 ± 0.04 μM). However, a sharp rise in the inhibitory potential was observed when fluorine is substituted as R1 at the meta-position of aroyl ring (6d; IC50 = 1.47 ± 0.05 μM).
Figure 3.

Structure–activity relationship of compounds 6b, 6c, and 6d.
When a 4-F substitution occurs as R2, the inhibitory effect of various R1 substituents in compounds 6e, 6f, and 6h demonstrated satisfactory outcomes (Figure 4). Compound 6e, characterized by the presence of a trimethoxy substituent at aroyl ring demonstrated the highest level of α-glucosidase inhibitory activity (IC50 = 2.10 ± 0.06 μM). However, replacing the electron-donating group (OMe) with electron-deficient 2-Cl and 4-NO2 groups (6f), a reduction in the inhibitory activity was observed (IC50 = 4.04 ± 0.16 μM). The ability to inhibit α-glucosidase is further diminished when NO2 is eliminated and just 2-Cl is present (6h; IC50 = 4.33 ± 0.19 μM).
Figure 4.

Structure–activity relationship of compounds 6e, 6f, 6h, and 6i.
Compound 6i shares R1 substitution that is identical to those found in 6e, but the point of distinction lies in the R2 substitution, specifically the presence of 3-F substituent (Figure 4). As a result, a pronounced reduction in its inhibitory activity against α-glucosidase occurred (IC50 = 8.31 ± 0.13 μM). Overall, a distinctive combination of fluoro substituents on three aromatic ring makes compound 6d as the lead inhibitor and deserves further research to be developed as a highly fluorinated α-glucosidase inhibitor.
2.3. Computational Analysis
2.3.1. Prediction and Authentication of Glucosidase Crystal Structures
The prediction of the tertiary structure of α-glucosidase involved the utilization of the Saccharomyces cerevisiae α-glucosidase protein sequence, which comprises 583 amino acids. This sequence was used for the model development using Modeler version 10.3. The protein with a PDB ID of 3AJ7 exhibited a notably high similarity score, prompting its selection as the template for the development of the model. A total of five distinct models were formulated, and the one demonstrating the highest confidence score was selected for subsequent assessment (Figure 5).
Figure 5.

Predicted tertiary model of α-glucosidase formed via modeler version 10.3.
The selected protein model was validated through Ramachandran plot, ERRAT, and ProSA-Web. The results of this analysis indicated that a significant portion, specifically 83.9%, of the total amino acid residues, is situated within the highly favorable region, which is visually represented by the red color on the plot. In contrast, approximately 15.1% of the amino acid residues is positioned within the accepted region on the plot. Nevertheless, only 0.4% of the amino acid residues can be found in the generously accepted region represented by the pale yellow color. Furthermore, a minor fraction of the amino acid composition, specifically 0.6%, corresponding to three residues, occupies the disallowed region highlighted in white (Figure 6A).
Figure 6.

Validation of the tertiary structure of α-glucosidase. (A) Ramachandran plot in which the majority of amino acids (83.9%) are present within the highly favorable region (red color). Nevertheless, a small fraction of amino acids is found in other regions. (B) ProSA-web plot depicts the model quality through z-score, with a higher z-score indicating higher model quality. In the case of predicted α-glucosidase model, the z-score is −10.78. (C) Averaged 3D–1D score of α-glucosidase graphically depicted using VERIFY3D. The amino acid residues of α-glucosidase are denoted by green dots, highlighting their aggregation within the region greater than 0.1. (D) Quality factor illustration of α-glucosidase from ERRAT reveals that the error value peaks are below 95% on an average.
Subsequently, the calculation of the z-score was executed via protein structure analysis (ProSA)-web, providing an assessment of the quality of model, as illustrated in Figure 6B. The model displayed a z-score of −10.78, which closely resembles the z-score of a protein with comparable amino acid composition.31
The VERIFY3D tool assesses the compatibility between amino acid sequence and protein crystal structure.32 The graphical representation indicates that an approximate 97.94% of the residues exhibits a mean 3D-1D score of greater than or equal to 0.1 (Figure 6C). This proportion of favorable score predicts the successful VERIFY3D validation for the α-glucosidase model.
ERRAT is based on the fundamental principle that proteins contain different types of atoms, and these atoms tend to arrange themselves in a nonrandom fashion relative to one another.33 The α-glucosidase model demonstrated a remarkable overall quality factor of 91.115%. It has also been revealed that the protein structure resolution lies within the range of 2.5–3 Å, as visually depicted in Figure 6D.
2.3.2. Molecular Docking
The molecular docking of the most potent inhibitor (6d), the least potent inhibitor (6a), and positive control (acarbose) was performed by SeeSAR version 13.0 (www.biosolveit.de/SeeSAR).34 SeeSAR incorporates FlexX feature, which functions according to incremental algorithm and identifies a limited number of interaction sites within the active pocket to precisely position functional groups of ligands.35 Therefore, subsequent to the molecular docking different poses having varied spatial arrangements and affinity with the active pocket were obtained for 6a, 6d, and acarbose. The docked poses of 6a showed clashes with the amino acid residues in the binding pocket. The binding energy of the favorable spatially arranged pose of 6d and acarbose was −11.1 and −7.8 kcal/mol, respectively. The best spatially arranged pose of 6d in complex with the most druggable active pocket can be visualized in Figure 7.
Figure 7.
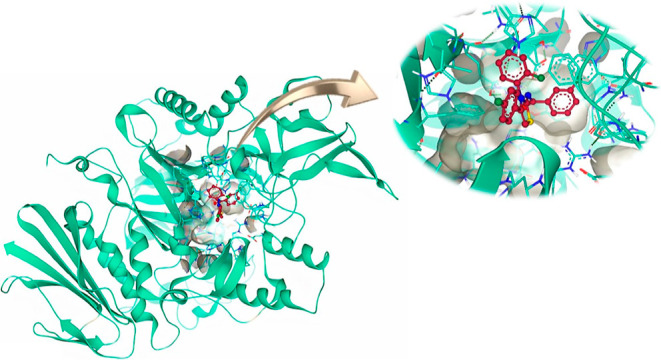
Compound 6d docked in the active pocket of α-glucosidase having DoGSiteScore of 0.38.
2.3.3. ADME Properties
The absorption, distribution, metabolism, and excretion (ADME) properties of 6d were analyzed by SwissADME web tool (http://www.swissadme.ch/index.php). The analysis revealed the druggable properties of 6d. The molecular weight of 6d was below 500 g/mol and contains 29 heavy atoms, 4 rotatable bonds, 5 hydrogen bond acceptors, and none hydrogen bond donor atom. The topological polar surface area, which is an indicator of solubility, was 62.60 Å2. It is highly absorptive via gastrointestinal tract and unable to penetrate the blood–brain barrier. In addition, no alert was observed for pan-assay interference compounds (PAINS) and Brenk depicting the specificity, responsiveness, and safety of 6d. Moreover, the potent compound 6d is unable to cross the epithelium of skin as its log Kp (skin permeation) was −4.78 cm/s.
2.3.4. Protein–Ligand Interactions
In the docking analysis, compound 6a showed unfavorable interactions and does not interact favorably with any residue of the active pocket of α-glucosidase, as shown in Figure 8A. On the contrary, the most active compound 6d interacts favorably with the binding pocket of the target protein by developing hydrogen bonds, π-lone pair, π-anion, π-sulfur, π-alkyl, π–π stacked, and π–π T-shaped bonds. His348 and Arg212 are responsible for developing conventional hydrogen bonds with the O23 of 6d. Similarly, N14 also forms conventional hydrogen bond with Glu267 of the ligand, as shown in Figure 8. F7 and F29 donate their lone pair to the electron-deficient hydrogen atoms of His279 and His111, respectively, to form the hydrogen bond. In addition, Phe177 also develops π-lone pair intermolecular interaction with F29 of 6d. The π-alkyl bonds are attributed to the interaction of Leu218 and Ala278 with the aromatic ring, whereas Asp214 is involved in the formation of π-anion interaction with another aromatic ring of 6d. π–sulfur bond is formed by the Phe300 with S10. Furthermore, Phe300 also forms two π–π T-shaped bonds; one with aromatic ring and the other with thiazole ring of 6d. At last, Tyr71 forms π–π stacked interaction with aromatic ring, as shown in Table 2.
Figure 8.

(A) Docking pose of 6a (yellow sticks) with the active pocket residues of α-glucosidase (green cartoon); (B) elucidation of intermolecular interactions between 6d (reddish brown) and amino acid residues of α-glucosidase. The dotted lines indicate the intermolecular interactions.
Table 2. Intermolecular Interactions, Interacting Atoms, Amino Acid Residues, and Bond Lengths of Each Type of Interaction in Compound 6d.
| compound | binding interactions |
|||
|---|---|---|---|---|
| ligand atoms | receptor residues | interaction types | distance (Å) | |
| 6d | F7 | His279 | H-bond | 2.99 |
| N14 | Glu276 | H-bond | 3.1 | |
| O23 | Arg212 | H-bond | 2.37 | |
| O23 | His348 | H-bond | 2.61 | |
| F29 | His111 | H-bond | 2.34 | |
| F29 | Phe177 | π-lone pair | 2.89 | |
| aromatic ring | Leu218 | π-alkyl | 4.99 | |
| aromatic ring | Ala278 | π-alkyl | 3.82 | |
| aromatic ring | Asp214 | π-anion | 4.10 | |
| aromatic ring | Tyr71 | π–π stacked | 4.07 | |
| aromatic ring | Phe300 | π–π T shaped | 4.90 | |
| thiazole ring | Phe300 | π–π T shaped | 5.04 | |
| thiazole ring | Phe300 | π-sulfur | 5.82 | |
3. Conclusions
In summary, a series of highly fluorinated 2-imino-1,3-thiazolines were designed and synthesized to explore the antidiabetic potential via targeting glucosidase enzymes. An efficient and simple cyclization strategy was employed to connect duly substituted thioureas with 2-bromo-1-(4-fluorophenyl)ethan-1-one. The target compounds were obtained in good yield and their structures were established with FTIR and NMR spectroscopy and mass spectrometry. In vitro biological testing against α-glucosidase and β-glucosidase enzymes revealed the selective inhibition of α-glucosidase over β-glucosidase. Among the screened derivatives, compound 6d, 3-fluoro-N-(3-(2-fluorophenyl)-4-(4-fluorophenyl)thiazol-2(3H)-ylidene)benzamide, exhibited the highest α-glucosidase inhibitory activity with an IC50 value of 1.47 ± 0.05 μM. This inhibitory efficacy profile was ∼24-folds stronger than the standard drug, acarbose (IC50 = 35.1 ± 0.14 μM). Furthermore, computational analysis unveiled that the binding energy of lead inhibitor 6d (−11.1 kcal/mol) exceeded the standard inhibitor, acarbose (−7.9 kcal/mol), thus representing the presence of strong interaction and affinity of 6d with the target. These interactions, as revealed through computational analysis, shed light on the critical role of 2-imino-1,3-thiazoline core within these compounds in facilitating their activity at the molecular level. Finally, the ADME properties acquired by SwissADME web tool revealed that the most potent inhibitor 6d displays druggable properties. Overall, the in vitro data augmented with computational assessment make this class of compounds as efficient alpha-glucosidase inhibitors for the treatment of diabetes mellitus type 2.
4. Experimental Section
4.1. Materials and Methods
Melting points were determined using a Gallenkamp melting point apparatus (MP-D) and are uncorrected. Rf values were determined using aluminum-precoated silica gel plates Kieselgel 60 F254 from Merck (Germany) using solvent system n-hexane/ethyl acetate (4:1). Infrared spectra were recorded using a FTS 3000 MS, Bio-Rad Marlin (Excalibur Model) spectrophotometer. 1H NMR spectra were obtained using a Bruker 300 MHz NMR spectrometer in CDCl3 and (CD3)2CO solvents. Chemical shifts are given in δ-scale (ppm). Abbreviations s, d, dd, and m are used for singlet, doublet, double of doublet, and multiplets, respectively. 13C NMR spectra were measured in CDCl3 and (CD3)2CO solvents at using a Bruker NMR spectrometer at 75 MHz.
4.2. General Procedure for the Synthesis of Thiourea Derivatives 4(a–j)
Substituted benzoic acids (0.01 mol) 1(a–j) were converted to their corresponding acid chlorides 2(a–j) using thionyl chloride which were treated with potassium thiocyanate (0.01 mol) to form corresponding isothiocyanates 3(a–j). In the subsequent step, different substituted anilines (0.01 mol) were reacted with isothiocyanates 3(a–j) to obtain the desired thioureas 4(a–j) in moderate to good yields.
4.2.1. N-((3-Chloro-4-fluorophenyl)carbamothioyl)-3,4,5-trimethoxybenzamide (4a)
The product was obtained as an off-white crystal, mp 112–115 °C, yield = 71%, Rf = 0.54 (n-Hex/EtOAc: 4:1). 1H NMR (300 MHz, CDCl3): δ 8.85 (s, 1H), 7.27–7.14 (m, 4H), 7.04 (t, 1H), 3.84 (s, 9H). 13C NMR (75 MHz, CDCl3): δ 180.93, 166.87, 154.38, 152.28, 149.98, 143.74, 135.19, 128.54, 124.31 (d, J = 7.6 Hz), 121.01, 120.79, 120.24 (d, J = 6.7 Hz), 116.83, 116.62, 108.36, 60.65, 56.78. LCMS (m/z): 398 (M+); Anal. Calcd for C17H16ClFN2O4S: C, 51.19; H, 4.04; N, 7.02; S, 8.04%. Found: C, 51.18; H, 4.03; N, 7.01; S, 8.03%.
4.2.2. 2-Chloro-N-((2-fluorophenyl)carbamothioyl)benzamide (4b)
The product was obtained as a white crystal, mp 142–145 °C, yield = 71%, Rf = 0.53 (n-Hex/EtOAc: 4:1). 1H NMR (300 MHz, CDCl3): δ 8.85 (s, 1H), 7.27–7.14 (m, 4H), 7.04 (t, 1H), 3.84 (s, 9H). 13C NMR (75 MHz, CDCl3): δ 180.59, 167.42, 160.04, 157.94 (d, J = 7.6 Hz), 134.10, 131.67, 130.77, 127.82, 127.61, 127.34, 127.02, 125.72 (d, J = 3.7 Hz), 124.40, 123.93 117.25, 117.04. LCMS (m/z): 308 (M+); Anal. Calcd for C14H10ClFN2OS: C, 54.46; H, 3.26; N, 9.07; S, 10.39%. Found: C, 54.45; H, 3.25; N, 9.08; S, 10.38%.
4.2.3. 2-Fluoro-N-((2-fluorophenyl)carbamothioyl)benzamide (4c)
The product was obtained as a creamy solid, mp 110–112 °C, yield = 68%, Rf = 0.51 (n-Hex/EtOAc: 4:1). 1H NMR (300 MHz, CDCl3): δ 8.51 (s, 1H), 7.92 (m, 1H), 7.51–7.42 (m, 1H), 7.31–6.55 (m, 6H). 13C NMR (75 MHz, CDCl3): δ 180.59, 166.23, 162.09 (d, J = 7.6 Hz), 160.01 (d, J = 5.7 Hz), 157.94, 133.63, 128.21, 127.82, 127.61, 125.72, 124.83, 124.40, 123.93, 121.57, 121.35, 117.25, 117.04, 116.30, 116.07. LCMS (m/z): 292 (M+); Anal. Calcd for C14H10F2N2OS: C, 57.53; H, 3.45; N, 9.58; S, 10.97%. Found: C, 57.52; H, 3.44; N, 9.57; S, 10.96%.
4.2.4. 3-Fluoro-N-((2-fluorophenyl)carbamothioyl)benzamide (4d)
The product was obtained as a white crystal, mp 108–110 °C, yield = 73%, Rf = 0.60 (n-Hex/EtOAc: 4:1). 1H NMR (300 MHz, CDCl3): δ 8.51 (s, 1H), 7.96–7.89 (m, 1H), 7.51–7.42 (m, 1H), 7.31–6.55 (m, 6H). 13C NMR (75 MHz, CDCl3): δ 180.59, 166.23, 162.09, 160.01, 157.94, 153.33 (d, J = 157.5 Hz), 133.63, 128.21, 127.82, 127.61, 125.72, 124.83, 124.40, 123.93, 121.57, 121.35, 117.25, 117.04, 116.30, 116.07. LCMS (m/z): 292 (M+); Anal. Calcd for C17H17FN2O4S: C, 57.53; H, 3.45; N, 9.58; S, 10.97%. Found: C, 57.53; H, 3.44; N, 9.56; S, 10.96%.
4.2.5. N-((4-Fluorophenyl)carbamothioyl)-3,4,5-trimethoxybenzamide (4e)
The product was obtained as a white crystal, mp 118–120 °C, yield = 69%, Rf = 0.50 (n-Hex/EtOAc: 4:1). 1H NMR (300 MHz, CDCl3); δ: 12.54 (s, 1H, NH), 9.10 (s, 1H, NH), 7.67–7.63 (m, 2H, ArH), 7.21–7.08 (m, ArH), 3.95 (s, 9H, OCH3). 13C NMR (75 MHz, CDCl3): δ 180.93, 166.87, 159.48 (d, J = 67.5 Hz), 149.98, 143.74, 135.59 (d, J = 3.7 Hz), 128.54, 122.46 (d, J = 6.7 Hz), 115.45, 115.23, 108.36, 60.65, 56.78. LCMS (m/z): 364 (M+); Anal. Calcd for C17H17FN2O4S: C, 56.03; H, 4.70; N, 7.69; S, 8.80%. Found: C, 56.02; H, 4.69; N, 7.68; S, 8.79%.
4.2.6. 2-Chloro-N-((4-fluorophenyl)carbamothioyl)-4-nitrobenzamide (4f)
The product was obtained as a yellow solid, mp 98–102 °C, yield = 65%, Rf = 0.49 (n-Hex/EtOAc: 4:1). 1H NMR (300 MHz, CDCl3): δ 7.36–7.27 (m, 2H), 7.08 (m, 4H), 3.83 (d, 9H). 13C NMR (75 MHz, CDCl3): δ 180.93, 167.42, 159.48 (d, J = 157.5 Hz), 148.60, 137.60, 135.71, 135.45, 128.08, 125.25, 122.46, 121.41, 115.34 (d, J = 16.5 Hz). LCMS (m/z): 326 (M+); Anal. Calcd for C14H9ClF2N2OS: C, 51.46; H, 2.78; N, 8.57; S, 9.81%. Found: C, 51.44; H, 2.77; N, 8.56; S, 9.80%.
4.2.7. 2-Chloro-N-((4-fluorophenyl)carbamothioyl)benzamide (4h)
The product was obtained as a white solid, mp 125–127 °C, yield = 78%, Rf = 0.52 (n-Hex/EtOAc: 4:1). 1H NMR (300 MHz, CDCl3): δ 9.27 (s, 1H), 7.71 (s, 1H), 7.44 (d, 2H), 7.32 (s, 1H), 7.27–7.23 (m, 2H), 7.11–7.05 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 180.93, 167.42, 159.48 (d, J = 157.5 Hz), 135.59, 134.10, 131.67, 130.77, 127.34, 127.02, 122.46, 115.34 (d, J = 16.5 Hz). LCMS (m/z): 308 (M+); Anal. Calcd for C14H10ClFN2OS: C, 54.46; H, 3.26; N, 9.07; S,10.39%. Found: C, 54.45; H, 3.25; N, 9.06; S, 10.38%.
4.2.8. N-((3-Fluorophenyl)carbamothioyl)-3,4,5-trimethoxybenzamide (4i)
The product was obtained as a white crystal, mp 114–116 °C, yield = 75%, Rf = 0.55 (n-Hex/EtOAc: 4:1). 1H NMR (300 MHz, CDCl3): δ 8.14 (s, 1H), 7.33–7.24 (m, 3H), 7.03 (m, 1H), 6.87 (m, 2H), 3.82 (d, 9H). 13C NMR (75 MHz, CDCl3): δ 180.93, 166.87, 160.08 (d, J = 157.5 Hz), 149.98, 143.74, 140.08 (d, J = 7.6 Hz), 129.54 (d, J = 6.7 Hz), 128.54, 118.44 (d, J = 3.7 Hz), 112.26, 112.05, 111.41, 111.19, 108.36, 60.65, 56.78. LCMS (m/z): 364 (M+); Anal. Calcd for C17H17FN2O4S: C, 56.03; H, 4.70; N, 7.69; S, 8.80%. Found: C, 56.02; H, 4.69; N, 7.68; S, 8.79%.
4.2.9. N-((3-Chloro-4-fluorophenyl)carbamothioyl)-2-fluorobenzamide (4j)
The product was obtained as a white crystal, mp 101–105 °C, yield = 67%, Rf = 0.51 (n-Hex/EtOAc: 4:1). 1H NMR (300 MHz, CDCl3): δ 8.73 (s, 1H), 7.73 (m, 1H), 7.51–7.43 (m, 1H), 7.28 (m, 1H), 7.23–7.08 (m, 3H), 7.04 (t, 1H). 13C NMR (75 MHz, CDCl3): δ 180.93, 166.23, 161.04 (d, J = 157.5 Hz), 154.38, 152.28, 135.19 (d, J = 3.7 Hz), 133.63 (d, J = 6.7 Hz), 128.21 (d, J = 7.6 Hz), 124.83 (d, J = 3.7 Hz), 124.31 (d, J = 7.6 Hz), 121.57, 121.35, 121.01, 120.79, 120.24 (d, J = 6.7 Hz), 116.83, 116.62, 116.30, 116.07. LCMS (m/z): 326 (M+); Anal. Calcd for C14H9ClF2N2OS: C, 51.46; H, 2.78; N, 8.57; S, 9.81%. Found: C, 51.47; H, 2.77; N, 8.56; S, 9.80%.
4.3. General Procedure for the Synthesis of 2-Imino-1,3-thiazoline Derivatives 6(a–j)
To a solution of substituted thiourea (0.01 mol) 4(a–j) in absolute ethanol (10 mL) was added 4-florophenacyl bromide (5) and mixture refluxed for 24 h. The progress of the reaction was monitored through TLC (eluent: 10% ethyl acetate in hexanes). Upon completion of the reaction, the precipitated product 6(a–j) was filtered through Whatman filter paper and washed with water and ethanol. Finally, the compounds 6(a–j) were recrystallized from ethanol.
4.3.1. N-(3-(3-Chloro-4-fluorophenyl)-4-(4-fluorophenyl)thiazol-2(3H)-ylidene)-3,4,5-trimethoxybenzamide (6a)
The product was obtained as a light yellow solid, mp 255–257 °C, yield = 72%, Rf = 0.55 (n-Hex/EtOAc: 4:1). 1H NMR (300 MHz, CDCl3); δ: 7.68 (s, 1H), 7.66 (s, 2H), 7.45–7.12 (m, 3H), 7.06–6.97 (m, 3H), 6.72 (s, 1H), 3.87–3.89 (s, 9H). 13C NMR (75 MHz, CDCl3): δ 173.93, 170.69, 169.60 (d, J = 96.75 Hz), 152.7, 141.15, 137.32, 134.10, 133.80, 131.42 (d, J = 16.5 Hz), 130.88, 130.76, 128.31 (d, J = 7.5 Hz), 126.04, 116.74, 116.44 (d, J = 22.5 Hz), 116.24, 115.95, 107.76, 106.19, 60.92, 55.88. LCMS (m/z): 516 (M+); Anal. Calcd for C25H19ClF2N2O4S: C, 58.09; H, 3.70; N, 5.42; S, 6.20%. Found: C, 58.08; H, 3.69; N, 5.41; S, 6.19%
4.3.2. 2-Chloro-N-(3-(2-fluorophenyl)-4-(4-fluorophenyl)thiazol-2(3H)-ylidene)benzamide (6b)
The product was obtained as a pale yellow solid, mp 264–266 °C, yield = 75%, Rf = 0.51 (n-Hex/EtOAc: 4:1). 1H NMR (300 MHz, CDCl3): δ 7.67 (m, 1H), 7.52–7.42 (m, 2H), 7.40–7.04 (m, 3H), 7.00–6.75 (m, 6H), 6.58 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 174.35, 173.03, 166.06, 165.80, 163.96 (d, J = 138 Hz), 163.70, 136.80 (d, J = 3.7 Hz), 136.09, 134.96, 132.74, 131.86 (d, J = 7.6 Hz), 130.65, 130.19, 128.67, 128.46, 128.05 (d, J = 7.6 Hz), 127.50, 126.61 (d, J = 3.7 Hz), 119.39, 119.16, 116.28, 116.07, 110.91. LCMS (m/z): 426 (M+); Anal. Calcd for C22H13ClF2N2OS: C, 61.90; H, 3.07; N, 6.56; S, 7.51%. Found: C, 61.89; H, 3.05; N, 6.50; S, 7.50%.
4.3.3. 2-Fluoro-N-(3-(2-fluorophenyl)-4-(4-fluorophenyl)thiazol-2(3H)-ylidene)benzamide (6c)
The product was obtained as a yellow solid, mp 204–206 °C, yield = 69%, Rf = 0.55 (n-Hex/EtOAc: 4:1). 1H NMR (300 MHz, CDCl3); δ: 7.95 (s, 1H), 7.44–7.37 (m, 4H), 7.30–7.14 (m, 4H), 7.10–7.02 (m, 2H), 6.99–6.94 (m, 2H), 6.7 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 164.12, 161.39, 160.69, 158.95, 155.60, 137.87, 133.02, 132.90, 132.23, 131.22 (d, J = 7.5 Hz), 130.76, 130.65, 130.26, 126.04 (d, J = 30 Hz), 125.32, 125.15, 124.88, 124.78, 124.60, 124.55, 123.49, 123.43, 116.90, 116.60, 116.49, 115.84, 115.54, 107.55. LCMS (m/z): 410 (M+); Anal. Calcd for C22H13F3N2OS: C, 64.38; H, 3.19; N, 6.83; S, 7.81%. Found: C, 64.37; H, 3.18; N, 6.82; S, 7.80%.
4.3.4. 3-Fluoro-N-(3-(2-fluorophenyl)-4-(4-fluorophenyl) Thiazol-2(3H)-ylidene)benzamide (6d)
The product was obtained as a pale yellow solid, 222–224 °C, yield = 65%, Rf = 0.37 (n-Hex/EtOAc: 4:1). 1H NMR (300 MHz, CDCl3): δ 7.56 (m, 1H), 7.53–7.40 (m, 2H), 7.39–7.30 (m, 2H), 7.30–7.21 (m, 1H), 7.00–6.75 (m, 6H), 6.57 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 173.68, 172.91, 166.06, 165.80, 163.97 (d, J = 152.25 Hz), 163.70, 162.62, 137.43 (d, J = 6.7 Hz), 136.80 (d, J = 3.7 Hz), 131.86 (d, J = 7.6 Hz), 130.52, 130.07, 128.67, 128.46, 128.05, 126.61, 123.96, 119.93, 119.71, 119.39, 119.16, 118.11, 117.89, 116.28, 116.07, 110.91. LCMS (m/z): 410 (M+); Anal. Calcd for C22H13F3N2OS: C, 64.38; H, 3.19; N, 6.83; S, 7.81%. Found: C, 64.36; H, 3.19; N, 6.81; S, 7.80%.
4.3.5. N-(3,4-Bis(4-fluorophenyl)thiazol-2(3H)-ylidene)-3,4,5-trimethoxybenzamide (6e)
The product was obtained as a white solid, mp 212–214 °C, yield = 69%, Rf = 0.74 (n-Hex/EtOAc: 4:1). 1H NMR (300 MHz, CDCl3): δ 12.54 (s, 1H), 9.10 (s, 1H), 7.67–7.63 (m, 2H), 7.28–7.08 (m, 4H), 3.95 (s, 9H). 13C NMR (75 MHz, CDCl3): δ 173.70, 169.50, (d, J = 61.5 Hz), 164.17, 161.26, 160.45, 152.63, 141.07, 137.78, 133.46, 133.42, 130.82, 130.55, 126.29 (d, J = 3 Hz), 116.00, 115.93, 115.71, 115.63, 107.46, 106.28, 60.88, 55.83. LCMS (m/z): 482 (M+); Anal. Calcd for C25HF2N2O4S: C, 62.23; H, 4.18; N, 5.81; S, 6.65%. Found: C, 62.23; H, 4.18; N, 5.81; S, 6.65%.
4.3.6. 2-Chloro-N-(3-(3-chloro-4-fluorophenyl)-4-(4-fluorophenyl)thiazol-2(3H)-ylidene)-6-nitrobenzamide (6f)
The product was obtained as a black solid, mp 240–242 °C, yield = 67%, Rf = 0.44 (n-Hex/EtOAc: 4:1). 1H NMR (300 MHz, CDCl3): δ 7.67 (m, 1H), 7.52–7.42 (m, 2H), 7.40–7.04 (m, 3H), 7.00–6.75 (m, 6H), 6.58 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 174.35, 173.03, 164.75 (d, J = 152.5 Hz), 154.46 (d, J = 156.75 Hz), 136.80, 136.09, 134.96, 132.74, 131.86, 130.65, 130.19, 128.67, 128.46, 128.05, 127.50, 126.61, 119.27 (d, J = 17.25 Hz), 116.28, 116.07, 110.91. LCMS (m/z): 504 (M+); Anal. Calcd for C22H11Cl2F2N3O3S: C, 52.19; H, 2.19; N, 8.30; S, 6.33%. Found: C, 52.18; H, 2.18; N, 8.29; S, 6.32%.
4.3.7. N-(3-(3-Chloro-4-fluorophenyl)-4-(4-fluorophenyl)thiazol-2(3H)-ylidene)-4-fluorobenzamide (6g)
The product was obtained as a white solid, mp 198–201 °C, yield = 66%, Rf = 0.62 (n-Hex/EtOAc: 4:1). 1H NMR (300 MHz, (CD3)2CO): δ 8.09 (s, 2H), 7.78 (s, 1H), 7.77–7.38 (m, 4H), 7.17–7.08 (m, 4H). 13C NMR (75 MHz, (CD3)2CO): δ 172.43, 169.96, 166.58, 164.55 (d, J = 152.3 Hz), 162.27 (d, J = 150 Hz), 159.19, 155.89, 137.60, 134.56 (d, J = 3.75 Hz), 133.35, 133.31, 131.79, 131.68, 131.63, 131.51, 131.40, 129.80, 129.69, 126.80, 126.76, 120.53, 107.82. LCMS (m/z): 444 (M+); Anal. Calcd for C22H12ClF3N2OS: C, 59.40; H, 2.72; N, 6.30; S, 7.21%. Found: C, 59.39; H, 2.70; N, 6.29; S, 7.20%.
4.3.8. N-(3,4-Bis(4-fluorophenyl) Thiazol-2(3H)-ylidene)-2-chlorobenzamide (6h)
The product was obtained as an off-white solid, mp 236–238 °C, yield = 63%, Rf = 0.31 (n-Hex/EtOAc: 4:1). 1H NMR (300 MHz, CDCl3): δ 7.66 (m, 1H), 7.52–7.42 (m, 2H), 7.41–6.92 (m, 5H), 6.86 (t, 2H), 6.78 (m, 2H), 6.59 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 174.35, 170.52, 164.75 (d, J = 157.5 Hz), 161.69 (d, J = 157.5 Hz), 137.22, 136.37 (d, J = 3.7 Hz), 136.09, 134.96, 132.74, 130.37, 130.19, 129.19 (d, J = 6.7 Hz), 127.50, 116.28, 116.09 (d, J = 6.7 Hz), 115.91, 108.90. LCMS (m/z): 426 (M+); Anal. Calcd for C22H13ClF2N2OS: C, 61.90; H, 3.07; N, 6.56; S, 7.51%. Found: C, 61.89; H, 3.06; N, 6.55; S, 7.50%.
4.3.9. N-(3-(3-Fluorophenyl)-4-(4-fluorophenyl)thiazol-2(3H)-ylidene)-3,4,5-trimethoxybenzamide (6i)
The product was obtained as a white solid, mp 249–251 °C, yield = 70%, Rf = 0.53 (n-Hex/EtOAc: 4:1) 1H NMR (300 MHz, (CD3)2CO): δ 7.52–7.42 (m, 2H), 7.41–7.39 (m, 2H), 7.29–7.23 (s, 4H), 7.16–7.10 (m, 4H), 7.05 (s, 1H), 3.77 (s, 9H). 13C NMR (75 MHz, d6-Acetone): δ 172.78, 169.25, 164.50 (d, J = 38.5 Hz), 161.22, 160.74, 160.98 (d, J = 36 Hz), 152.90, 141.29, 139.32 (d, J = 10.5 Hz), 137.49, 132.15, 130.15 (d, J = 9 Hz), 130.09, 127.03 (d, J = 3 Hz), 125.13 (d, J = 3.75 Hz), 116.62 (d, J = 24 Hz), 115.52, 115.23, 107.43, 106.30, 59.65, 55.16. LCMS (m/z): 482 (M+); Anal. Calcd for C25H20F2N2O4S: C, 62.23; H, 4.18; N, 5.81; S, 6.65%. Found: C, 62.22; H, 4.17; N, 5.80; S, 6.64%.
4.3.10. N-(3-(3-Chloro-4-fluorophenyl)-4-(4-fluorophenyl)thiazol-2(3H)-ylidene)-2-fluorobenzamide (6j)
The product was obtained as a gray solid, mp 232–234 °C, yield = 66%, Rf = 0.39 (n-Hex/EtOAc: 4:1). 1H NMR (500 MHz, CDCl3): δ 7.72 (m, 1H), 7.55–7.46 (m, 1H), 7.40–7.31 (m, 2H), 7.25–7.17 (m, 2H), 6.97 (t, 2H), 6.88–6.78 (m, 2H), 6.73 (m, 1H), 6.59 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 170.50, 165.80, 163.70, 162.96, 160.87, 155.50, 153.41, 139.12 (d, J = 3.7 Hz), 137.22, 135.33 (d, J = 7.6 Hz), 130.52 (d, J = 6.7 Hz), 130.07 (d, J = 3.7 Hz), 129.65 (d, J = 7.6 Hz), 126.57 (d, J = 7.6 Hz), 125.91 (d, J = 6.7 Hz), 125.44, 125.34, 119.97, 119.74, 116.10 (d, J = 26.7 Hz), 115.10, 114.88, 108.90. LCMS (m/z): 444 (M+); Anal. Calcd for C22H12ClF3N2OS: C, 59.40; H, 2.72; N, 6.30; S, 7.21%. Found: C, 59.39; H, 2.71; N, 6.29; S, 7.20%.
4.4. Glucosidase Inhibition Assay
The procedure employed for conducting α-glucosidase and β-glucosidase inhibitory assays in 96-well plates was consistent with established methods.14 For these assays, 0.07 M phosphate buffer (pH 6.8) was used to make solutions containing 2.5 U/mL α-glucosidase, 2.0 U/mL β-glucosidase, and the substrate 4-nitrophenyl-β-d-glucopyranoside (p-NPG) at a concentration of 10 mM. Additionally, test compounds were prepared (1 mM solution each) using 10% DMSO, in order to attain a final concentration of 100 μM in every well. To initiate the assay, 10 μL of the respective enzyme was incubated with 10 μL of the test compounds, allowing a 5 min incubation period at 37 °C. Subsequently, 10 μL of p-NPG was introduced into each well, and the mixture was subjected to an incubation period of 30 min at 37 °C. After incubation, 80 μL of a stop solution containing Na2CO3 at a concentration of 200 mM was added to each well. The measurement of absorbance was carried out at 405 nm. The degree of inhibition exhibited by each test compound was quantified using the following equation.
Percentage inhibition = 100 – [slope of test compound/slope of enzyme control] × 100.
Acarbose was used as the reference drug in evaluating the effectiveness of the test compounds against α-glucosidase and β-glucosidase. The determination of the IC50 for each of the test compound was accomplished using GraphPad Prism version 10.0.
4.5. Computational Analysis
4.5.1. Prediction and Authentication of the Glucosidase Crystal Structure
The generation of the three-dimensional structure of α-glucosidase was accomplished using Modeler 10.3 homology modeling software. This process utilized the crystal structure of S. cerevisiae isomaltase (PDB id: 3AJ7) as a reference template. The preparation of the model involved utilizing the FASTA format amino acid sequence of α-glucosidase. This specific sequence was acquired from UniProt under the access code P53341.14 Subsequently, model quality and its stereochemical characteristics were assessed using the Ramachandran plot,36 as sourced from PROCHECK version 3.5 (available at https://saves.mbi.ucla.edu).37 Furthermore, the validation of the anticipated 3D structure of α-glucosidase involved the utilization of Verify3D,38 ERRAT,39 and ProSA-web (accessible at https://prosa.services.came.sbg.ac.at/prosa.php).40
4.5.2. Molecular Docking
Following the prediction of the tertiary structure of the target protein, α-glucosidase, the binding affinity of the most potent inhibitor was assessed using the FlexX functionality within SeeSAR version 13.0, accessible at www.biosolveit.de/SeeSAR.34 The protein was first configured in the protein mode, and subsequently, the binding site was chosen in the binding site mode. Table 3 illustrates the binding sites in α-glucosidase with distinct colors denoting the presence of nine vacant binding sites. Each of these binding sites was assessed individually to investigate their affinity for potent inhibitors. The results revealed that the first binding site, characterized by its yellow hue, emerged as the most favorable and promising druggable binding site.41 Subsequently, the highly potent inhibitor was subjected to standard docking within the chosen binding site. The optimal conformation was determined by assessing the lowest binding energy of the ligand in the docked complex.42
Table 3. Unoccupied Druggable Binding Pockets of α-Glucosidase and Their Properties.

4.5.3. ADME Analysis
SwissADME (http://www.swissadme.ch/index.php) served as the analytical tool for assessing the absorption, distribution, metabolism, and excretion (ADME) properties of the most potent inhibitor.43 The evaluation not only predicts the toxicological attributes but also offers valuable insights into its suitability as a drug candidate and various physicochemical characteristics. The simplified molecular-input line-entry system of the query compound was used as the input to obtain the results.43
4.5.4. Protein–Ligand Interactions
Discovery Studio 2021 molecular visualization software was employed to scrutinize and analyze the favorable interactions occurring between a highly effective inhibitor and the specific amino acid residues located within the binding pocket of the target protein.44 Various favorable interactions between proteins and ligands are represented by dotted lines of distinct colors. These interactions encompass hydrophobic, electrostatic, and hydrophilic interactions. Furthermore, it is also possible to visualize the length of these bonds.44,45
Acknowledgments
The authors extend their appreciation to the Ministry of Education in KSA for funding this research work through the project number KKU-IFP2-DA-8.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c00529.
1H NMR and 13C NMR spectra of synthesized compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Dawane B. S.; Konda S. G.; Kamble V. T.; Chavan S. A.; Bhosale R. B.; M S. B. Multicomponent one-pot synthesis of substituted, Hantzsch thiazole derivatives under solvent free conditions. Eur. J. Chem. 2009, 6 (S1), S358–S362. 10.1155/2009/752580. [DOI] [Google Scholar]
- Manaka A.; Sato M.; Aoki M.; Tanaka M.; Ikeda T.; Toda Y.; Yamane Y.; Nakaike S. 2-Acylimino-3H-thiazoline derivatives: A novel template for platelet GPIIb/IIIa receptor antagonists. Bioorg. Med. Chem. Lett. 2001, 11 (8), 1031–1035. 10.1016/S0960-894X(01)00123-8. [DOI] [PubMed] [Google Scholar]
- Mustafa M. N.; Channar P. A.; Sarfraz M.; Saeed A.; Ejaz S. A.; Aziz M.; Alasmary F. A.; Alsoqair H. Y.; Raza H.; Kim S. J.; Hamad A. Synthesis, kinetic studies and in-silico investigations of novel quinolinyl-iminothiazolines as alkaline phosphatase inhibitors. J. Enzyme Inhib. Med. Chem. 2023, 38 (1), 2163394. 10.1080/14756366.2022.2163394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai R.; Zhu J.; Bai Z.; Mao Q.; Zhang Y.; Hui Z.; Luo X.; Ye X.-Y.; Xie T. Second generation β-elemene nitric oxide derivatives with reasonable linkers: potential hybrids against malignant brain glioma. J. Enzyme Inhib. Med. Chem. 2022, 37 (1), 379–385. 10.1080/14756366.2021.2016734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed A.; Shaheen U.; Hameed A.; Kazmi F. Synthesis and antimicrobial activity of some novel 2-(substituted fluorobenzoylimino)-3-(substituted fluorophenyl)-4-methyl-1,3-thiazolines. J. Fluorine Chem. 2010, 131 (3), 333–339. 10.1016/j.jfluchem.2009.11.005. [DOI] [Google Scholar]
- Saeed A.; Abbas N.; Flörke U. Synthesis and antibacterial activity of some novel 2-aroylimino-3-aryl-thiazolidin-4-ones. J. Braz. Chem. Soc. 2007, 18, 559–565. 10.1590/S0103-50532007000300010. [DOI] [Google Scholar]
- Meleddu R.; Distinto S.; Cottiglia F.; Angius R.; Gaspari M.; Taverna D.; Melis C.; Angeli A.; Bianco G.; Deplano S.; Fois B.; Del Prete S.; Capasso C.; Alcaro S.; Ortuso F.; Yanez M.; Supuran C. T.; Maccioni E. Tuning the dual inhibition of carbonic anhydrase and cyclooxygenase by dihydrothiazole benzensulfonamides. ACS Med. Chem. Lett. 2018, 9 (10), 1045–1050. 10.1021/acsmedchemlett.8b00352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maghraby M. T.; Abou-Ghadir O. M. F.; Abdel-Moty S. G.; Ali A. Y.; Salem O. I. A. Novel class of benzimidazole-thiazole hybrids: The privileged scaffolds of potent anti-inflammatory activity with dual inhibition of cyclooxygenase and 15-lipoxygenase enzymes. Bioorg. Med. Chem. 2020, 28 (7), 115403. 10.1016/j.bmc.2020.115403. [DOI] [PubMed] [Google Scholar]
- Caillé F.; Cacheux F.; Peyronneau M. A.; Jego B.; Jaumain E.; Pottier G.; Ullmer C.; Grether U.; Winkeler A.; Dollé F.; Damont A.; Kuhnast B. From structure-activity relationships on thiazole derivatives to the in vivo evaluation of a new radiotracer for cannabinoid subtype 2 PET imaging. Mol. Pharmaceutics 2017, 14 (11), 4064–4078. 10.1021/acs.molpharmaceut.7b00746. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Singh M.; Selivanova G.; Peuget S. Pifithrin-α alters p53 post-translational modifications pattern and differentially inhibits p53 target genes. Sci. Rep. 2020, 10 (1), 1049. 10.1038/s41598-020-58051-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murru S.; Singh C. B.; Kavala V.; Patel B. K. A convenient one-pot synthesis of thiazol-2-imines: application in the construction of pifithrin analogues. Tetrahedron 2008, 64 (8), 1931–1942. 10.1016/j.tet.2007.11.076. [DOI] [Google Scholar]
- Saeed A.; Mahmood S. U.; Rafiq M.; Ashraf Z.; Jabeen F.; Seo S. Y. Iminothiazoline-sulfonamide hybrids as Jack Bean Urease inhibitors; Synthesis, kinetic mechanism and computational molecular modeling. Chem. Biol. Drug Des. 2016, 87 (3), 434–443. 10.1111/cbdd.12675. [DOI] [PubMed] [Google Scholar]
- Kawamura S.; Izumi K.; Sato J.; Sanemitsu Y.; Hamada T.; Shibata H.; Sato R.. Iminothiazolines, their production and use as herbicides, and intermediates for their production. U.S. Patent 5,244,863, 1993.
- a Kazmi M.; Zaib S.; Amjad S. T.; Khan I.; Ibrar A.; Saeed A.; Iqbal J. Exploration of aroyl/heteroaroyl iminothiazolines featuring 2, 4, 5-trichlorophenyl moiety as a new class of potent, selective, and in vitro efficacious glucosidase inhibitors. Bioorg. Chem. 2017, 74, 134–144. 10.1016/j.bioorg.2017.07.012. [DOI] [PubMed] [Google Scholar]; b Chen J.; Li X.; Liu H.; Zhong D.; Yin K.; Li Y.; Zhu L.; Xu C.; Li M.; Wang C. Bone marrow stromal cell-derived exosomal circular RNA improves diabetic foot ulcer wound healing by activating the nuclear factor erythroid 2-related factor 2 pathway and inhibiting ferroptosis. Diabetic Med. 2023, 40 (7), e15031 10.1111/dme.15031. [DOI] [PubMed] [Google Scholar]
- Masuda N.; Yamamoto O.; Fujii M.; Ohgami T.; Fujiyasu J.; Kontani T.; Moritomo A.; Orita M.; Kurihara H.; Koga H.; Kageyama S.; et al. Studies of non-nucleoside HIV-1 reverse transcriptase inhibitors. Part 2: Synthesis and structure-activity relationships of 2-cyano and 2-hydroxy thiazolidenebenzenesulfonamide derivatives. Bioorg. Med. Chem. 2005, 13 (4), 949–961. 10.1016/j.bmc.2004.11.045. [DOI] [PubMed] [Google Scholar]
- Alkorta I.; Rozas I.; Elguero J. Effects of fluorine substitution on hydrogen bond interactions. J. Fluorine Chem. 2000, 101 (2), 233–238. 10.1016/S0022-1139(99)00164-5. [DOI] [Google Scholar]
- Saeed A.; Shaheen U.; Hameed A.; Naqvi S. H. Synthesis, characterization and antimicrobial activity of some new 1-(fluorobenzoyl)-3-(fluorophenyl) thioureas. J. Fluorine Chem. 2009, 130 (11), 1028–1034. 10.1016/j.jfluchem.2009.09.003. [DOI] [Google Scholar]
- Park B. K.; Kitteringham N. R.; O’Neill P. M. Metabolism of fluorine containing drugs. Annu. Rev. Pharmacol. Toxicol. 2001, 41 (1), 443–470. 10.1146/annurev.pharmtox.41.1.443. [DOI] [PubMed] [Google Scholar]
- Le Bars D. Fluorine-18 and medical imaging: Radiopharmaceuticals for positron emission tomography. J. Fluorine Chem. 2006, 127 (11), 1488–1493. 10.1016/j.jfluchem.2006.09.015. [DOI] [Google Scholar]
- Yang Y. Y.; Shi L. X.; Li J. H.; Yao L. Y.; Xiang D. X. Piperazine ferulate ameliorates the development of diabetic nephropathy by regulating endothelial nitric oxide synthase. Mol. Med. Rep. 2019, 19 (3), 2245–2253. 10.3892/mmr.2019.9875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. S.; Jeong Y. M.; Park I. K.; Hahn H. G.; Lee H. K.; Kwon S. B.; Jeong J. H.; Yang S. J.; Sohn U. D.; Park K. C. A new 2-imino-1, 3-thiazoline derivative, KHG22394, inhibits melanin synthesis in mouse B16 melanoma cells. Biol. Pharm. Bull. 2007, 30 (1), 180–183. 10.1248/bpb.30.180. [DOI] [PubMed] [Google Scholar]
- Aly A. A.; Ahmed E. K.; El-Mokadem K. M. A convenient and efficient method for the synthesis of benzo-and naphthothiazolediones. J. Sulphur Chem. 2006, 27 (5), 419–426. 10.1080/17415990600862960. [DOI] [Google Scholar]
- Mandel A. L.; Breslin P. A. S. High Endogenous Salivary Amylase Activity Is Associated with Improved Glycemic Homeostasis following Starch Ingestion in Adults. J. Nutr. 2012, 142, 853–858. 10.3945/jn.111.156984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B.; Gui M.; An H.; Shen J.; Ye F.; Ni Z.; Zhan H.; Che L.; Lai Z.; Zeng J.; Peng J.; Lin J. Babao Dan alleviates gut immune and microbiota disorders while impacting the TLR4/MyD88/NF-κB pathway to attenuate 5-fluorouracil-induced intestinal injury. Biomed. Pharmacother. 2023, 166, 115387. 10.1016/j.biopha.2023.115387. [DOI] [PubMed] [Google Scholar]
- Lombard V.; Golaconda Ramulu H.; Drula E.; Coutinho P. M.; Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuberi Z.; Sauli E.; Cun L.; Deng J.; Li W.-J.; He X.-L.; Li W. Insulin-delivery methods for children and adolescents with type 1 diabetes. Ther. Adv. Endocrinol. Metab. 2020, 11, 204201882090601. 10.1177/2042018820906016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Bai Y.; Jin Z.; Svensson B. Food-derived non-phenolic α-amylase and α-glucosidase inhibitors for controlling starch digestion rate and guiding diabetes-friendly recipes. LWT-Ed. 2022, 153, 112455. 10.1016/j.lwt.2021.112455. [DOI] [Google Scholar]
- Van P. T. B.; Huy L. T.; Thuy N. T. B.; Nga V. T.; Tam L. M.; Phuong H.; Hao H. M. Solvent-free, microwave-assisted, solid-catalyzed synthesis and α-Glucosidase inhibition of chalcones. Vietnam J. Chem. 2023, 61 (3), 325–332. 10.1002/vjch.202200151. [DOI] [Google Scholar]
- Saeed A.; Zaman S.; Bolte M. Synthesis and crystal structure of some novel 2-aroylimino-3-aryl-4-phenyl-1, 3-thiazolines. Synth. Commun. 2008, 38 (13), 2185–2199. 10.1080/00397910802026212. [DOI] [Google Scholar]
- Saeed A.; Parvez M. The crystal structure of 1-(4-chlorophenyl)-3-(4-methylbenzoyl) thiourea. Cent. Eur. J. Chem. 2005, 3 (4), 780–791. 10.2478/BF02475204. [DOI] [Google Scholar]
- Lee Y.; Kim S.; Kim J. Y.; Arooj M.; Kim S.; Hwang S.; Kim B. W.; Park K. H.; Lee K. W. Binding mode analyses and pharmacophore model development for stilbene derivatives as a novel and competitive class of α-glucosidase inhibitors. PLoS One 2014, 9 (1), e85827 10.1371/journal.pone.0085827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dym O.; Eisenberg D.; Yeates T. O.. VERIFY3D. In International Tables for Crystallography Vol. F: Crystallography of biological macromolecules, 1st ed.; Rossmann M. G.; Arnold E., Eds., 2006; pp 521;. [Google Scholar]; b Su M.; Hu R.; Tang T.; Tang W.; Huang C. Review of the correlation between Chinese medicine and intestinal microbiota on the efficacy of diabetes mellitus. Front. Endocrinol. 2023, 13, 1085092. 10.3389/fendo.2022.1085092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dym O.; Eisenberg D.; Yeates T. O.. ERRAT. In International Tables for Crystallography Vol. F: Crystallography of biological macromolecules, 1st ed.; Arnold E.; Himmel D. M.; Rossmann M. G., Eds., 2012; pp 678–679. [Google Scholar]; b Gan Y.; Xu Y.; Zhang X.; Hu H.; Xiao W.; Yu Z.; Sun T.; Zhang J.; Wen C.; Zheng S. Revisiting Supersaturation of a Biopharmaceutical Classification System IIB Drug: Evaluation via a Multi-Cup Dissolution Approach and Molecular Dynamic Simulation. Molecules 2023, 28, 6962. 10.3390/molecules28196962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SeeSAR version 13.0.1; BioSolveIT GmbH: Sankt Augustin, Germany, 2023. www.biosolveit.de/SeeSAR.
- Schellhammer I.; Rarey M. FlexX-Scan: Fast, structure-based virtual screening. Proteins 2004, 57 (3), 504–517. 10.1002/prot.20217. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; Furnham N.; Thornton J. M.. The Ramachandran plot and protein structure validation. In Biomolecular forms and functions: a celebration of 50 years of the ramachandran map; Bansal M., Srinivasan N., Eds., 2013; pp 62–75. [Google Scholar]
- Hasan M. A.; Mazumder M. H. H.; Chowdhury A. S.; Datta A.; Khan M. A. Molecular-docking study of malaria drug target enzyme transketolase in Plasmodium falciparum 3D7 portends the novel approach to its treatment. Source Code Biol. Med. 2015, 10, 7. 10.1186/s13029-015-0037-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D.; Lüthy R.; Bowie J. U.. VERIFY3D, assessment of protein models with three-dimensional profiles. Methods in enzymology; Academic Press, 1997; Vol. 277, pp 396–404. [DOI] [PubMed] [Google Scholar]
- Hasan M. A.; Khan M. A.; Datta A.; Mazumder M. H. H.; Hossain M. U. A comprehensive immunoinformatics and target site study revealed the corner-stone toward Chikungunya virus treatment. Mol. Immunol. 2015, 65 (1), 189–204. 10.1016/j.molimm.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaib S.; Rana N.; Areeba; Hussain N.; Alrbyawi H.; Dera A. A.; Khan I.; Khalid M.; Khan A.; Al-Harrasi A. Designing multi-epitope monkeypox virus-specific vaccine using immunoinformatics approach. J. Infect. Public Health 2023, 16 (1), 107–116. 10.1016/j.jiph.2022.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaib S.; Rana N.; Hussain N.; Ogaly H. A.; Dera A. A.; Khan I. Identification of Potential Inhibitors for the Treatment of Alkaptonuria Using an Integrated In Silico Computational Strategy. Molecules 2023, 28 (6), 2623. 10.3390/molecules28062623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dera A. A.; Zaib S.; Areeba; Hussain N.; Rana N.; Javed H.; Khan I. Identification of Potent Inhibitors Targeting EGFR and HER3 for Effective Treatment of Chemoresistance in Non-Small Cell Lung Cancer. Molecules 2023, 28 (12), 4850. 10.3390/molecules28124850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S.; Sharma A.; Gupta U. Molecular Docking studies on the Anti-fungal activity of Allium sativum (Garlic) against Mucormycosis (black fungus) by BIOVIA discovery studio visualizer 21.1. 0.0. J. J. Antivirals Antiretrovirals 2021, 5, 028–032. 10.17352/aaa.000013. [DOI] [Google Scholar]
- Baskaran K. P.; Arumugam A.; Kandasamy R.; Alagarsamy S. In silico method for prediction of maximum binding affinity and ligand-protein interaction studies on Alzheimer’s disease. Int. J. Res. Granthaalayah. 2020, 8, 362–370. 10.29121/granthaalayah.v8.i11.2020.2472. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.