

Abstract
A definitive diagnosis of nevus or melanoma is not always possible for histologically ambiguous melanocytic neoplasms. In such cases, ancillary molecular testing can support a diagnosis of melanoma if certain chromosomal aberrations are detected. Current technologies for copy number variation (CNV) detection include chromosomal microarray analysis (CMA) and fluorescence in situ hybridization. Although CMA and fluorescence in situ hybridization are effective, their utilization can be limited by cost, turnaround time, and inaccessibility outside of large reference laboratories. Droplet digital polymerase chain reaction (ddPCR) is a rapid, automated, and relatively inexpensive technology for CNV detection. We investigated the ability of ddPCR to quantify CNV in cyclin-dependent kinase inhibitor 2A (CDKN2A), the most commonly deleted tumor suppressor gene in melanoma. CMA data were used as the gold standard. We analyzed 57 skin samples from 52 patients diagnosed with benign nevi, borderline lesions, primary melanomas, and metastatic melanomas. In a training cohort comprising 29 randomly selected samples, receiver operator characteristic curve analysis revealed an optimal ddPCR cutoff value of 1.73 for calling CDKN2A loss. In a validation cohort comprising the remaining 28 samples, ddPCR detected CDKN2A loss with a sensitivity and specificity of 94% and 90%, respectively. Significantly, ddPCR could also identify whether CDKN2A losses were monoallelic or biallelic. These pilot data suggest that ddPCR can detect CDKN2A deletions in melanocytic tumors with accuracy comparable with CMA. With further validation, ddPCR could provide an additional CNV assay to aid in the diagnosis of challenging melanocytic neoplasms.
Keywords: melanoma, histologically ambiguous melanocytic neoplasms, droplet digital PCR, CDKN2A, copy number variation
INTRODUCTION
Although most melanocytic neoplasms can be classified as either benign or malignant by histopathology, copy number variation (CNV) analysis is an important tool for borderline cases.1 Melanomas, in contrast to nevi, often harbor recurrent copy number alterations.2 Chromosomal microarray analysis (CMA) and fluorescence in situ hybridization (FISH) are the gold standards for CNV detection in melanoma. CMA, which includes both comparative genomic hybridization and single nucleotide polymorphism arrays, is used to detect gene amplifications, deletions, and genome-wide chromosomal aberrations.3,4 FISH panels are used to identify aberrations involving a smaller subset of melanoma-associated genes (RREB1, MYC, MYB, CCND1, and cyclin-dependent kinase inhibitor 2A (CDKN2A)).5,6 Although effective, both CMA and FISH have limitations. For FISH, cutoff values can vary across laboratories, and there may be interobserver disagreement when distinguishing putative melanoma cells from those of an accompanying nevus. By contrast, CMA does not require a trained technician to visualize individual cells, but can produce false-negative results when melanoma cells do not comprise enough of the overall tissue cellularity.7 Next-generation sequencing (NGS) can also provide CNV data, but can be limited by time, expense, and complexity. Currently, NGS is not widely used for this specific application.
Droplet digital polymerase chain reaction (ddPCR) is an emerging technology for CNV analysis.8 ddPCR partitions samples into 20,000 nanoliter-sized droplets, with PCR amplification of template molecules occurring in each droplet. Fluorescently labeled TaqMan probes allow for fluorescence detection when the corresponding amplification is present, and Poisson statistics are used to calculate the concentration of target DNA based on the fraction of positive droplets (ie, droplets containing the target sequence). ddPCR runs can be completed within 8 hours, with 96 reactions in a single run, and delivers a straightforward count of target DNA copies per sample. In addition to its automation and rapidity, ddPCR is inexpensive; in our study, the cost of reagents averaged $7.35 per target gene per sample.
ddPCR technology is growing increasingly relevant to pathology. In research studies, ddPCR accurately quantifies CNV in other oncogenes, including HER2 (invasive mammary carcinoma),9 MYCN (neuroblastoma),10 and MET (gastric cancer and hepatocellular carcinoma).11 It has also been used to evaluate microsatellite instability in breast cancers12 and to detect ESR1 mutations in ovarian tumors.13 Moreover, ddPCR is already used clinically to diagnose hereditary diseases of newborns, such as spinal muscular atrophy.14
In this proof-of-concept study, we evaluated the performance of ddPCR compared with CMA in the quantitation of CDKN2A (9p21.3), one of the most diagnostically important genes in melanoma. Copy number gains of protooncogenes and copy number losses of tumor suppressors typically occur in the later stages of melanomagenesis. Such CNVs can mark key transitions in melanoma progression. Loss of the tumor suppressor CDKN2A is common in melanoma: 40%–70% of primary melanomas15 and more than 75% of metastatic melanomas16 harbor either monoallelic or biallelic CDKN2A deletions. CNV data on CDKN2A can have great diagnostic value in melanocytic lesions with borderline histologic features.
MATERIALS AND METHODS
A total of 57 formalin-fixed paraffin-embedded (FFPE) skin samples were retrieved from the surgical pathology archives at our institution, with material ranging from 2013 to 2022. Inclusion criteria were as follows: (1) Final diagnosis of benign nevus, borderline lesion, primary melanoma, or metastatic melanoma was made by at least 1 board-certified dermatopathologist; (2) CMA-determined copy number status of CDKN2A was known; (3) DNA was extracted within 1 month of biopsy; and (4) initial input DNA concentration was at least 10 ng. For purposes of this study, we defined “borderline lesion” as any melanocytic neoplasm not signed out as benign nevus, primary melanoma, or metastatic melanoma; most were atypical nevi or atypical Spitz nevi. Demographic and clinical details were gathered for each patient, including age at biopsy, sex, lesion size and location, TNM staging and Breslow depth (where appropriate), and final diagnosis.
Chromosomal Microarray Analysis
DNA from the 57 FFPE samples was isolated using the QIAamp FFPE Tissue Kit (Qiagen, Valencia, CA). Samples were subjected to CMA for copy number quantitation of the CDKN2A gene using the OncoScan FFPE Assay Kit (Affymetrix, a Thermo Fisher Scientific company, Santa Clara, CA). DNA quantity was measured using Qubit Fluorometer 3.0 and Qubit dsDNA High-Sensitivity Assay Kit (Thermo Fisher Scientific company, Waltham, MA). For samples with CMA-confirmed CDKN2A loss, the Chromosome Analysis Suite (Thermo Fisher Scientific, Santa Clara, CA) software was used to determine whether losses were monoallelic or biallelic.
Droplet Digital PCR
ddPCR was performed using the Bio-Rad QX200 (Bio-Rad, Hercules, CA). Each reaction was prepared in a 22 μL solution consisting of 11 μL of 2× ddPCR supermix for probes (Bio-Rad, Hercules, CA), 1.1 μL HEX-labeled CDKN2A primer/probes, 1.1 μL FAM-labeled reference primer/probes (LIPI, THNSL2, AGO1, RPPH1, and SLAIN2), 10 ng of FFPE DNA, and nuclease-free water. Approximately 20,000 droplets per reaction were generated on the Bio-Rad QX200 droplet generator, per the manufacturer’s instructions. All emulsified PCR reactions were run in a 96-well plate on the C1000 Touch Thermal Cycler (Bio-Rad), starting with incubation at 95°C for 10 minutes, followed by 40 cycles of 94°C for 30 s, 60°C for 60 s, 10 minutes incubation at 98°C, and a final hold at 4°C for 1 hour. PCR products were read on the Bio-Rad QX200 droplet reader, and copy number data were generated using the QuantaSoft software from Bio-Rad.
Fifty-three FFPE samples (93.0%) were run in duplicate. These samples showed minimal variation across the 2 ddPCR measurements, with an average percent difference of 3.56%. The remaining 4 samples (7.0%) were run only once because of limited amounts of available tissue. Fifty of the 57 FFPE samples (87.7%) were run using all 5 reference genes (LIPI, SLAIN2, AGO1, RPPH1, and THNSL2). In addition, owing to limited amounts of tissue, 6 FFPE samples (10.5%) were run using all reference genes except LIPI and SLAIN2, and 1 (1.8%) was run using all reference genes except RPPH1 (Table 1, see Table 1, Supplemental Digital Content 1, http://links.lww.com/AJDP/A134). The final diagnosis of all cases had already been confirmed by a board-certified dermatopathologist before ddPCR analysis.
TABLE 1.
Summary of ddPCR Data, CMA Data, and Lesion Classification for All Samples
| ID | CDKN2A: LIPI ddPCR Output | CDKN2A: SLAIN2 ddPCR Output | CDKN2A: AGO1 ddPCR Output | CDKN2A: THNSL2 ddPCR Output | CDKN2A: RPPH1 ddPCR Output | Mean ddPCR Output | CDKN2A Loss (ddPCR) | CDKN2A Loss (CMA) | Lesion Classification | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||
| 1 | 1.75 | 1.76 | 2.03 | 2.12 | 2.35 | 2.3 | 2.23 | 2.22 | 1.9 | 1.88 | 2.05 | No | No | Borderline lesion |
| 2 | 2.01 | 2.05 | 1.81 | 1.91 | 2.2 | 2.29 | 2.45 | 2.46 | 2.12 | 2.05 | 2.14 | No | No | Benign nevus |
| 3 | 2.15 | 2.14 | 2.13 | 2.12 | 2.59 | 2.59 | 2.49 | 2.45 | - | - | 2.33 | No | No | Benign nevus |
| 4 | 2.3 | 2.23 | 2.22 | 2.18 | 2.31 | 2.36 | 2.23 | 2.33 | 1.88 | 1.87 | 2.19 | No | No | Borderline lesion |
| 5 | 2.4 | 2.29 | 2.42 | 2.39 | 2.17 | 2.21 | 2.21 | 2.18 | 2.01 | 1.99 | 2.23 | No | No | Primary melanoma |
| 6 | 2.06 | 1.94 | 1.82 | 1.87 | 2.28 | 2.29 | 1.96 | 2.03 | 1.76 | 1.74 | 1.98 | No | No | Borderline lesion |
| 7 | 1.82 | 1.8 | 1.8 | 1.96 | 2.49 | - | 2.32 | 2.64 | 1.93 | 1.89 | 2.07 | No | No | Borderline lesion |
| 8 | 3.07 | 3.29 | 1.38 | 1.48 | 2.05 | 2.2 | 1.11 | 1.16 | 0.62 | 0.61 | 1.70 | Yes (monoallelic) | No | Borderline lesion |
| 9 | 3.41 | 3.16 | 1.6 | 1.57 | 2.3 | 2.17 | 1.25 | 1.28 | 0.61 | 0.7 | 1.81 | No | No | Borderline lesion |
| 10 | - | - | - | - | 2.07 | - | 1.65 | 1.99 | 1.44 | 1.6 | 1.75 | No | No | Benign nevus |
| 11 | - | - | - | - | 1.83 | 2.23 | 2.2 | 1.77 | 2.13 | 2.03 | No | No | Borderline lesion | |
| 12 | 1.88 | 1.9 | 1.82 | 1.9 | 2.13 | 2.08 | 2.13 | 2.15 | 1.92 | 1.98 | 1.99 | No | No | Benign nevus |
| 13 | 2.47 | 2.64 | 2.33 | 2.24 | 2.37 | 2.38 | 2.47 | 2.42 | 1.91 | 1.89 | 2.31 | No | No | Borderline lesion |
| 14 | - | - | - | - | 2.25 | - | 2.15 | 2.16 | 1.78 | 1.75 | 2.02 | No | No | Borderline lesion |
| 15 | - | - | - | - | 2.35 | 2.22 | 2.29 | 2.07 | 1.55 | 1.5 | 2.00 | No | No | Borderline lesion |
| 16 | - | - | - | - | 2.34 | 2.46 | 2.41 | 2.49 | 1.8 | 1.67 | 2.20 | No | No | Benign nevus |
| 17 | - | - | - | - | 2.26 | 2.36 | 2.07 | 2.09 | 1.68 | 1.73 | 2.03 | No | No | Benign nevus |
| 18 | 2.49 | 2.43 | 1.98 | 1.97 | 2.07 | 1.97 | 1.84 | 1.85 | 1.69 | 1.77 | 2.01 | No | No | Borderline lesion |
| 19 | 1.98 | 2.01 | 1.89 | 1.85 | 2.16 | 2.17 | 1.81 | 1.82 | 1.85 | 1.87 | 1.94 | No | No | Benign nevus |
| 20 | 1.94 | 2.03 | 1.96 | 2.02 | 2.36 | 2.38 | 2.13 | 2.23 | 1.79 | 1.75 | 2.06 | No | No | Benign nevus |
| 21 | 1.7 | 1.61 | 2.12 | 2.02 | 2.45 | 2.42 | 2.42 | 2.63 | 1.83 | 1.88 | 2.11 | No | No | Borderline lesion |
| 22 | 1.74 | 1.85 | 1.74 | 1.74 | 2.45 | 2.57 | 2.06 | 2.04 | 1.52 | 1.60 | 1.93 | No | Yes (monoallelic) | Primary melanoma |
| 23 | 1.38 | 1.35 | 1.48 | 1.41 | 1.89 | 1.96 | 1.76 | 1.73 | 1.36 | 1.45 | 1.58 | Yes (monoallelic) | Yes (monoallelic) | Borderline lesion |
| 24 | 1.94 | 1.99 | 1.91 | 1.82 | 1.93 | 2.06 | 1.89 | 1.98 | 1.85 | 1.84 | 1.92 | No | Yes (monoallelic) | Primary melanoma |
| 25 | - | - | - | - | 2.22 | 2.11 | 1.40 | 1.38 | 1.01 | 1.04 | 1.53 | Yes (monoallelic) | Yes (monoallelic) | Borderline lesion |
| 26 | 0.88 | 0.89 | 1.48 | 1.51 | 1.79 | 1.57 | 1.37 | 1.44 | 1.73 | 1.79 | 1.44 | Yes (monoallelic) | Yes (monoallelic) | Borderline lesion |
| 27 | 1.19 | 1.19 | 0.46 | 0.48 | 0.43 | 0.44 | 0.39 | 0.39 | 0.42 | 0.42 | 0.58 | Yes (biallelic) | Yes (biallelic) | Metastatic melanoma |
| 28 | 1.26 | 1.26 | 1.12 | 1.09 | 1.03 | 1.03 | 1.15 | 1.15 | 1.03 | 0.98 | 1.11 | Yes (biallelic) | Yes (biallelic) | Primary melanoma |
| 29 | 1.85 | 2.04 | 1.36 | 1.36 | 1.57 | 1.51 | 1.47 | 1.45 | 1.42 | 1.43 | 1.55 | Yes (monoallelic) | Yes (monoallelic) | Metastatic melanoma |
| 30 | 1.30 | 1.26 | 1.59 | 1.62 | 1.65 | 1.62 | 1.81 | 1.75 | 1.47 | 1.42 | 1.55 | Yes (monoallelic) | Yes (monoallelic) | Metastatic melanoma |
| 31 | 1.21 | 1.22 | 1.50 | 1.54 | 1.56 | 1.49 | 1.58 | 1.59 | 1.30 | 1.23 | 1.42 | Yes (monoallelic) | Yes (monoallelic) | Metastatic melanoma |
| 32 | 1.38 | 1.41 | 1.29 | 1.70 | 1.76 | 1.74 | 1.73 | 1.66 | 1.34 | 1.35 | 1.54 | Yes (monoallelic) | Yes (monoallelic) | Borderline lesion |
| 33 | 1.48 | 1.41 | 1.39 | 1.41 | 1.38 | 1.33 | 1.30 | 1.30 | 2.15 | 2.11 | 1.53 | Yes (monoallelic) | Yes (monoallelic) | Metastatic melanoma |
| 34 | 2.43 | 2.40 | 1.34 | 1.27 | 1.92 | 1.71 | 1.49 | 1.48 | 1.32 | 1.36 | 1.67 | Yes (monoallelic) | Yes (monoallelic) | Metastatic melanoma |
| 35 | 2.57 | 2.69 | 1.36 | 1.27 | 1.67 | 1.82 | 1.29 | 1.32 | 1.28 | 1.21 | 1.65 | Yes (monoallelic) | Yes (monoallelic) | Metastatic melanoma |
| 36 | 0.32 | 0.32 | 0.31 | 0.31 | 0.44 | 0.48 | 0.38 | 0.38 | 0.40 | 0.43 | 0.38 | Yes (biallelic) | Yes (biallelic) | Metastatic melanoma |
| 37 | 0.59 | 0.58 | 0.49 | 0.48 | 0.80 | 0.76 | 0.55 | 0.55 | 0.66 | 0.67 | 0.61 | Yes (biallelic) | Yes (biallelic) | Metastatic melanoma |
| 38 | 0.62 | 0.64 | 0.59 | 0.60 | 0.86 | 0.86 | 0.67 | 0.67 | 0.56 | 0.62 | 0.67 | Yes (biallelic) | Yes (biallelic) | Metastatic melanoma |
| 39 | 1.62 | 1.62 | 1.42 | 1.39 | 1.71 | 1.74 | 1.56 | 1.59 | 1.53 | 1.51 | 1.57 | Yes (monoallelic) | Yes (monoallelic) | Metastatic melanoma |
| 40 | 1.32 | 1.44 | 1.32 | 1.31 | 1.36 | 1.40 | 1.36 | 1.38 | 1.18 | 1.17 | 1.32 | Yes (monoallelic) | Yes (monoallelic) | Primary melanoma |
| 41 | 1.62 | 1.59 | 1.10 | 0.99 | 1.14 | 1.05 | 1.18 | 1.16 | 1.21 | 1.19 | 1.22 | Yes (monoallelic) | Yes (monoallelic) | Primary melanoma |
| 42 | 0.74 | 0.65 | 0.46 | 0.42 | 0.46 | 0.49 | 0.41 | 0.37 | 0.38 | 0.39 | 0.48 | Yes (biallelic) | Yes (biallelic) | Primary melanoma |
| 43 | 0.32 | 0.31 | 0.37 | 0.39 | 0.36 | 0.37 | 0.34 | 0.33 | 0.39 | 0.38 | 0.36 | Yes (biallelic) | Yes (biallelic) | Primary melanoma |
| 44 | 0.47 | 0.45 | 0.82 | 0.79 | 0.79 | 0.83 | 0.60 | 0.61 | 1.00 | 0.95 | 0.73 | Yes (biallelic) | Yes (biallelic) | Primary melanoma |
| 45 | 2.70 | 2.50 | 1.18 | 1.09 | 1.55 | 1.71 | 0.85 | 0.65 | 0.67 | 0.65 | 1.36 | Yes (monoallelic) | Yes (monoallelic) | Primary melanoma |
| 46 | 1.69 | 1.85 | 1.58 | 1.49 | 2.20 | 2.07 | 1.65 | 1.58 | 1.51 | 1.57 | 1.72 | Yes (monoallelic) | Yes (monoallelic) | Primary melanoma |
| 47 | 2.61 | 2.55 | 1.93 | 1.86 | 1.78 | 1.80 | 1.79 | 1.80 | 1.63 | 1.60 | 1.94 | No | Yes (monoallelic) | Primary melanoma |
| 48 | 1.17 | 1.23 | 0.77 | 0.72 | 0.8 | 0.78 | 0.73 | 0.66 | 0.88 | 0.96 | 0.87 | Yes (biallelic) | Yes (biallelic) | Primary melanoma |
| 49 | 2.24 | 2.72 | 2.33 | 2.31 | 1.44 | 1.51 | 1.71 | 1.66 | 1.96 | 1.93 | 1.98 | No | Yes (monoallelic) | Primary melanoma |
| 50 | 1.53 | 1.39 | 1.44 | 1.34 | 1.49 | 1.4 | 1.43 | 1.41 | 1.42 | 1.34 | 1.42 | Yes (monoallelic) | Yes (monoallelic) | Primary melanoma |
| 51 | 0.776 | 0.798 | 0.901 | 0.88 | 0.97 | 0.98 | 0.95 | 0.98 | 1.07 | 1.04 | 0.93 | Yes (biallelic) | Yes (monoallelic) | Primary melanoma |
| 52 | 0.388 | 0.35 | 0.332 | 0.34 | 0.411 | 0.405 | 0.351 | 0.344 | 0.376 | 0.379 | 0.37 | Yes (biallelic) | Yes (biallelic) | Primary melanoma |
| 53 | 0.836 | 0.902 | 1.47 | 1.55 | 1.02 | 0.991 | 1.26 | 1.25 | 1.71 | 1.67 | 1.27 | Yes (monoallelic) | Yes (monoallelic) | Primary melanoma |
| 54 | 1.45 | 1.49 | 1.56 | 1.57 | 1.68 | 1.64 | 1.51 | 1.44 | 1.51 | 1.47 | 1.532 | Yes (monoallelic) | Yes (monoallelic) | Primary melanoma |
| 55 | 0.639 | 0.647 | 0.611 | 0.616 | 0.84 | 0.83 | 0.65 | 0.658 | 0.694 | 0.717 | 0.6902 | Yes (biallelic) | Yes (biallelic) | Primary melanoma |
| 56 | 1.45 | 1.49 | 1.79 | 1.8 | 1.77 | 1.74 | 1.68 | 1.66 | 1.65 | 1.67 | 1.67 | Yes (monoallelic) | Yes (monoallelic) | Primary melanoma |
| 57 | 1.68 | 1.64 | 1.5 | 1.4 | 1.12 | 1.16 | 1.14 | 1.14 | 1.24 | 1.3 | 1.33 | Yes (monoallelic) | Yes (monoallelic) | Primary melanoma |
Note: indicates that the ddPCR run was not performed due to limited amounts of tissue. Cases for which ddPCR and CMA had discordant results are bolded. Each data point represents the CDKN2A copy number output generated from a single ddPCR run. The following mean ddPCR cutoff values were used to generate the “CDKN2A Loss (ddPCR)” column, per receiver operator characteristic (ROC) curve analysis.
Mean ddPCR >1.73 = no. of CDKN2A loss.
1.11 <mean ddPCR ≤1.73 = monoallelic CDKN2A loss.
Mean ddPCR ≤1.11 = biallelic CDKN2A loss.
Statistical Analysis
Descriptive statistics were used to calculate clinicopathologic characteristics for all cases. For each FFPE sample, the average of all ddPCR outputs was recorded (Table 1). These mean ddPCR outputs were used to call CDKN2A loss by ddPCR. The 57 FFPE cases were randomly split into 2 groups: a training cohort (29 cases) and a validation cohort (the remaining 28 cases) (Fig. 1). We used the training cohort to determine the optimal mean ddPCR cutoff for calling CDKN2A loss; we then used the validation cohort to evaluate the performance of this cutoff on new, unseen samples.
FIGURE 1.
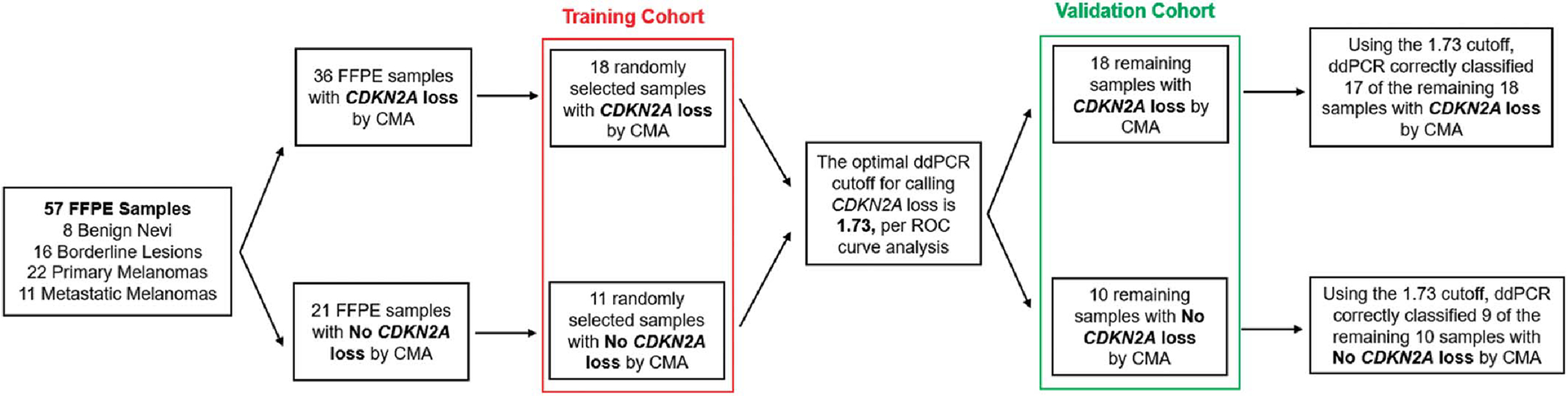
Outline of the study workflow. The 57 FFPE samples were randomly split into 2 groups: a training cohort (to determine the optimal mean ddPCR cutoff for calling CDKN2A loss) and a validation cohort (to test this cutoff on new, unseen samples). Chromosomal microarray analysis (CMA) data were used as the reference standard. Using the ROC-determined optimal cutoff of 1.73, ddPCR correctly classified 26 of the 28 samples in the validation cohort (sensitivity = 94.4%, specificity = 90.0%).
Training Cohort (n = 29)
We plotted sensitivity versus specificity at varying mean ddPCR cutoffs for CDKN2A loss and chose the optimal cutoff of 1.73 because its corresponding sensitivity/specificity pair was closest to the point (1, 1) on the receiver operator characteristic (ROC) curve. Accordingly, samples with mean ddPCR outputs at or below 1.73 were designated as “CDKN2A Loss,” and those with mean ddPCR outputs above 1.73 were designated as “No CDKN2A Loss.”
Validation Cohort (n = 28)
The remaining 28 samples were used to evaluate the performance of this 1.73 cutoff (Fig. 1). Confusion matrices were used to delineate samples for which ddPCR correctly and incorrectly identified CDKN2A copy number status. We assessed the concordance of ddPCR with CMA using sensitivity and specificity. All statistical analyses were performed using R software (R Core Team, Vienna, Austria).
RESULTS
Fifty-seven FFPE samples met inclusion criteria, comprising 8 benign nevi (14.0%), 16 borderline lesions (28.1%), 22 primary melanomas (38.6%), and 11 metastatic melanomas (19.3%). Patient demographic, clinical, and diagnostic information are summarized in Table 2. In total, 36 of the 57 FFPE samples (63.2%) had loss of CDKN2A by CMA, including 4 of the 16 borderline lesions, 21 of the 22 primary melanomas, and all 11 metastatic melanomas (Table 1). A representative case, demonstrating the hematoxylin and eosin (H&E) stain and CMA results of a nevoid melanoma (case ID #42, Table 1), is shown in Figure 2.
TABLE 2.
Summary of Clinicopathologic Characteristics
| Clinical Features | |
|---|---|
|
| |
| Age* (yr), mean ± SD | 55.6 ± 23.9 |
| Female sex,† n (%) | 20 (38.5) |
| Lesion location,‡ n (%) | |
| Trunk | 19 (33.3) |
| Visceral metastases | 6 (10.5) |
| Small bowel | 4 |
| Lung | 2 |
| Head/neck | 15 (26.3) |
| Upper extremity | 9 (15.8) |
| Lower extremity | 8 (14.0) |
| Histopathologic features | |
| Primary lesion size (cm), mean ± SD (range) | 1.4 ± 1.3 (0.3–6.5) |
| Lesion classification | |
| Benign nevus, n (%) | 8 (14.0) |
| Borderline lesion, n (%) | 16 (28.1) |
| Atypical Spitz tumor | 8 |
| Atypical nevus | 6 |
| Combined epithelioid melanocytoma associated with a nevus | 1 |
| Conventional nevus combined with an atypical Spitz nevus | 1 |
| Primary melanoma, n (%) | 22 (38.6) |
| Not otherwise specified | 6 |
| Nodular | 5 |
| Superficial spreading | 4 |
| Nevoid | 2 |
| Spitzoid melanoma in situ | 1 |
| Superficial spreading and spindle cell | 1 |
| Dedifferentiated and spindled | 1 |
| Mixed spindled and desmoplastic | 1 |
| Melanoma in situ | 1 |
| Metastatic melanoma, n (%) | 11 (19.3) |
| Breslow depth§ (mm), mean ± SD (range) | 4.6 ± 4.2 (0.8–21.0) |
At the time of biopsy.
n = 52 patients.
n = 57 lesions.
Of confirmed primary melanomas (n = 22).
FIGURE 2.
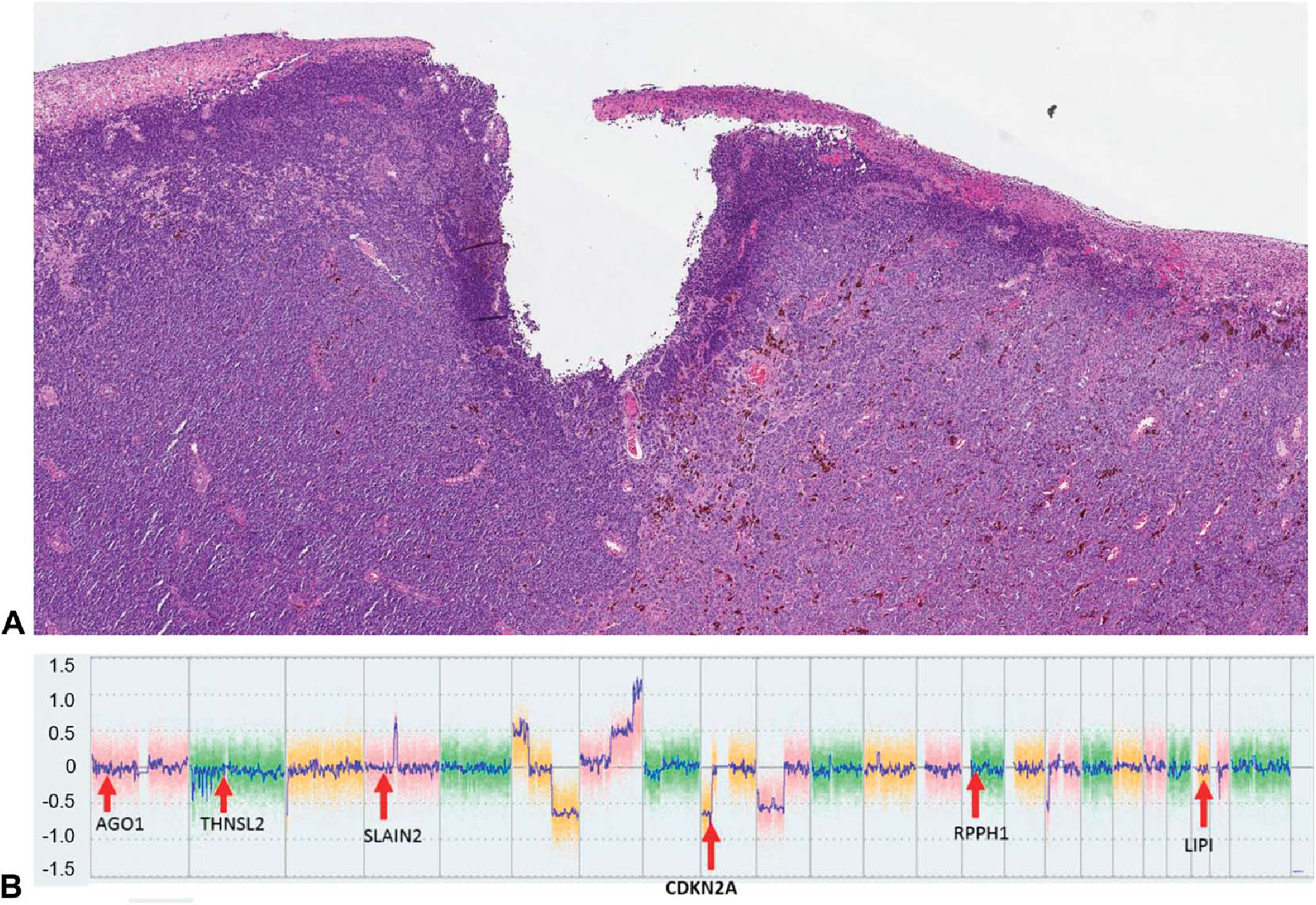
Case 42, from the vulva of an 84-year-old woman. A, (Hematoxylin & Eosin, 2×). A nevoid melanoma, with multiple CMA aberrations. B, CMA output. There is a clear deletion at the CDKN2A locus; all the reference genes are unaffected. This CDKN2A loss was biallelic, per Chromosome Analysis Suite, Thermo Fisher Scientific analysis. The mean ddPCR output for this sample was 0.48, well below the ddPCR cutoff of 1.11 for biallelic CDKN2A loss.
Training Cohort (n = 29)
To create our training cohort, we randomly selected 18 of the 36 samples with CDKN2A loss by CMA and 11 of the 21 samples with no CDKN2A loss by CMA. For these 29 samples, ROC curve analysis revealed an optimal mean ddPCR cutoff of 1.73 for calling CDKN2A loss (mean ddPCR >1.73 = no CDKN2A loss; mean ddPCR ≤1.73 = CDKN2A loss). This cutoff had a sensitivity of 86.1%, a specificity of 100%, and an AUC of 0.98 (95% CI: 0.955–1) for the training cohort (Fig. 3).
FIGURE 3.
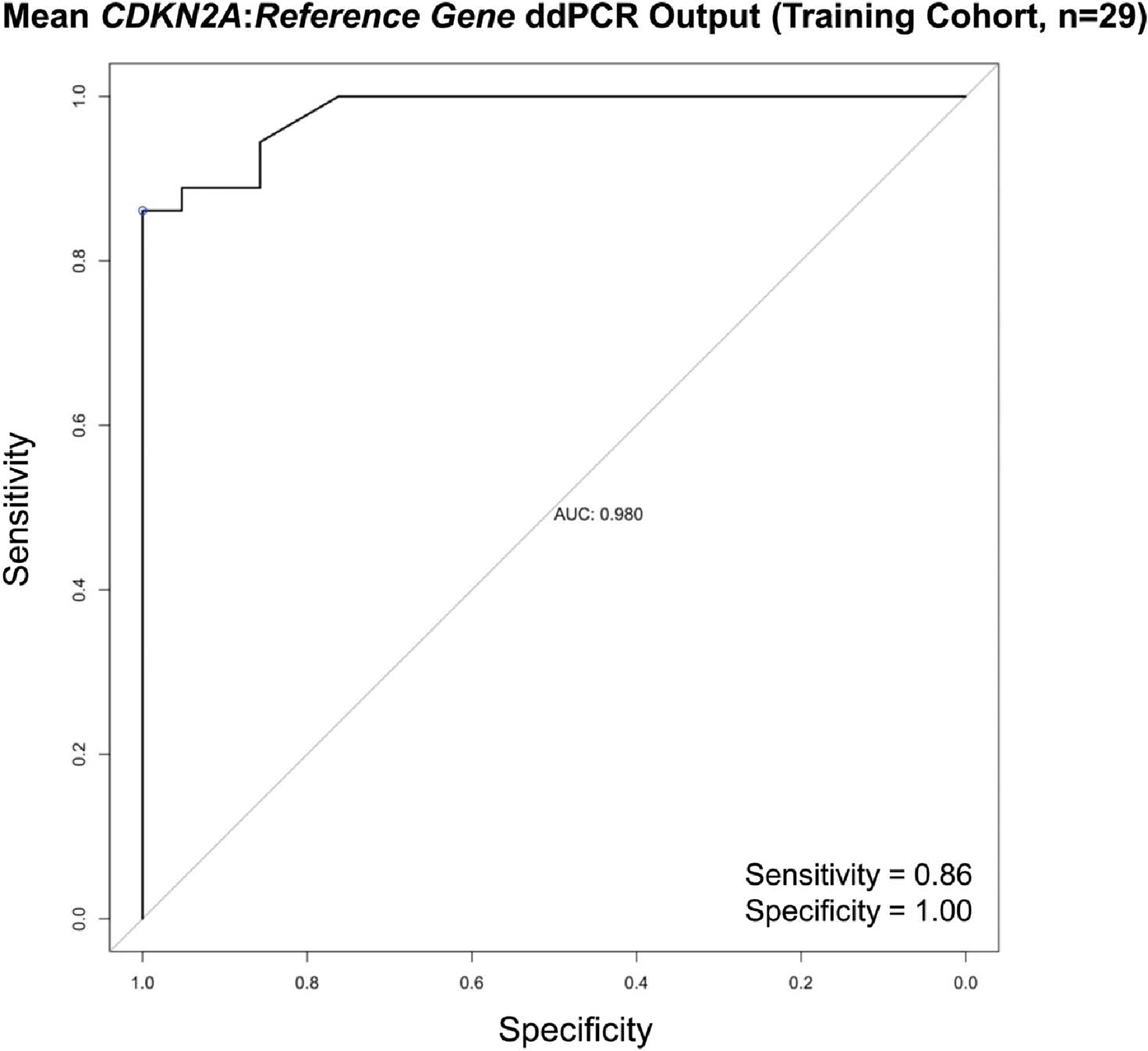
Receiver operator characteristic (ROC) curve for the training cohort (n = 29). We plotted sensitivity versus specificity at various mean ddPCR cutoffs for calling CDKN2A loss. The one closest to the point (1, 1) on the ROC curve was 1.73 (sensitivity = 86%, specificity = 100%, area under the curve 0.98). The performance of this 1.73 cutoff was assessed in the validation cohort (the remaining 28 samples).
Validation Cohort (n = 28)
The remaining 28 samples were used to validate this cutoff. Using the obtained cutoff of 1.73 for CDKN2A loss, ddPCR correctly classified 26 of 28 cases: 17 of the remaining 18 samples with CDKN2A loss and 9 of the remaining 10 samples with no CDKN2A loss (Table 3). Thus, for the 28 validation cohort samples, ddPCR identified CDKN2A copy number status with a sensitivity of 94.4% and a specificity of 90.0% (Table 3).
TABLE 3.
Concordance of ddPCR With CMA for CDKN2A Copy Number Quantitation
| Validation Cohort (n = 28) | CMA |
||||
|---|---|---|---|---|---|
| CDKN2A Loss | No CDKN2A Loss | Total | Sensitivity | Specificity | |
|
| |||||
| ddPCR | |||||
| CDKN2A loss | 17 | 1 | 18 | 94.4% | 90.0% |
| No | 1 | 9 | 10 | ||
| CDKN2A loss | |||||
| Total | 18 | 10 | 28 | ||
Discordant Results
Two cases had ddPCR results that differed from CMA. Case 22 yielded a false-negative result. Its mean ddPCR output was 1.93—above the cutoff of 1.73, implying no CDKN2A loss (Table 1). However, its CMA analysis revealed monoallelic CDKN2A loss. Case 8 yielded a false-positive result, with a mean ddPCR output of 1.70—below the cutoff of 1.73, implying CDKN2A loss (Table 1). However, this sample has no CMA evidence of CDKN2A loss.
DISCUSSION
ddPCR demonstrated high concordance with CMA in distinguishing CDKN2A-positive from CDKN2A-negative samples, detecting CDKN2A loss with a sensitivity and specificity of 94.4% and 90.0%, respectively. Such quantitation has been established to be useful in diagnosis17 and, in some cases, prognostication.18 The causes of the 1 false positive (case 8) and 1 false negative (case 22) were not investigated. Notably, all cases with mean ddPCR values outside of the 1.70–2.00 range were concordant with CMA (Fig. 4). It is possible that cases with mean ddPCR values inside this range may be partially transformed or of intermediate malignant potential. Significantly, the quantitative nature of the ddPCR data lends itself to the possibility of becoming a parameter for staging, index for prognosis or therapeutic response, and/or metastatic potential, in the future.
FIGURE 4.
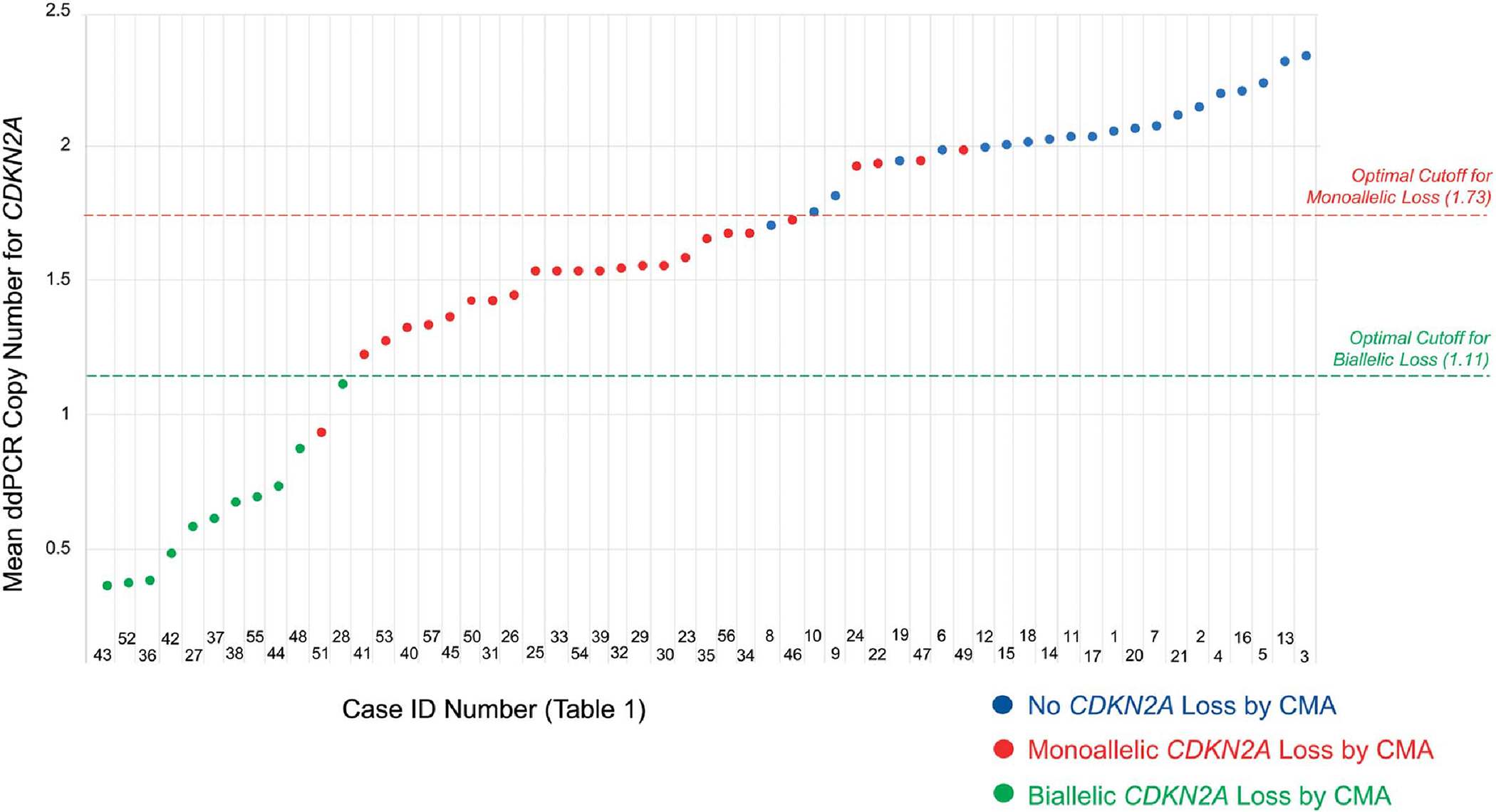
Diagrammatic representation of all cases and mean ddPCR outputs, color-coded by CMA status for CDKN2A. Below a mean ddPCR output of 1.70, all cases have loss of CDKN2A by CMA. Above 2.00, no cases have loss of CDKN2A. Future studies could investigate whether the values in the 1.70–2.00 range reflect lesions with partial malignant transformation. The ROC-determined optimal cutoffs were 1.73 for monoallelic CDKN2A loss and 1.11 for biallelic CDKN2A loss (mean ddPCR >1.73 = no CDKN2A loss; 1.11 < mean ddPCR ≤1.73 = monoallelic CDKN2A loss; mean ddPCR ≤1.11 = biallelic CDKN2A loss).
Clinical Significance of CDKN2A Loss: Monoallelic Versus Biallelic Deletions
Some studies have indicated that CDKN2A deletion is most clinically significant when biallelic.19,20 Of the 36 samples with CMA-confirmed CDKN2A loss, 25 had monoallelic loss (69.4%) and 11 had biallelic loss (30.6%). As shown in Figure 4, the 11 cases with biallelic CDKN2A loss cluster at or below a mean ddPCR copy number of 1.11. Between 1.11 and approximately 1.73, almost all cases have monoallelic loss (Fig. 4). Above 1.73, almost all cases have no CDKN2A loss (Fig. 4). A more rigorous ROC curve analysis confirms that the optimal mean ddPCR cutoff for distinguishing monoallelic from biallelic CDKN2A loss is 1.11, with 1 sample misclassified as biallelic (Fig. 4). Future studies would be needed to validate this cutoff. Notably, ddPCR has been used to distinguish between monoallelic and biallelic CDKN2A loss in other tumors.21
We find it noteworthy that the 1.11 cutoff value for biallelic CDKN2A loss is almost exactly what would be expected from the tumor biology. Theoretically, in a sample composed only of tumor cells with monoallelic CDKN2A loss, the CDKN2A copy number per cell should be 1.0. Thus, for pure samples, the cutoff between monoallelic versus biallelic loss would be 1.0. Certainly, no real patient sample is a pure tumor; both tumor heterogeneity and other nontumor cells are present. One may expect that fact to drastically affect the results, but we found it had a minimal impact, shifting the cutoff value only from 1.00 to 1.11.
Correlation with Histology
The primary purpose of this study was to correlate ddPCR with CMA, not to correlate morphology with ddPCR. Nonetheless, an interesting pattern seems to emerge, which warrants future, more rigorous, study: Our data revealed an inverse relationship between mean CDKN2A copy number and the degree of atypia as assessed by morphology (Fig. 5). This pattern is again consistent with the known tumor biology. Cases signed out as benign nevi had essentially no loss of CDKN2A, while borderline lesions exhibited some loss. Such losses became more dramatic in melanomas and even more so in metastases. Moreover, although our study was not designed to examine this trend, several of the differences were statistically significant (eg, borderline lesions vs. primary melanomas) (Fig. 5).
FIGURE 5.
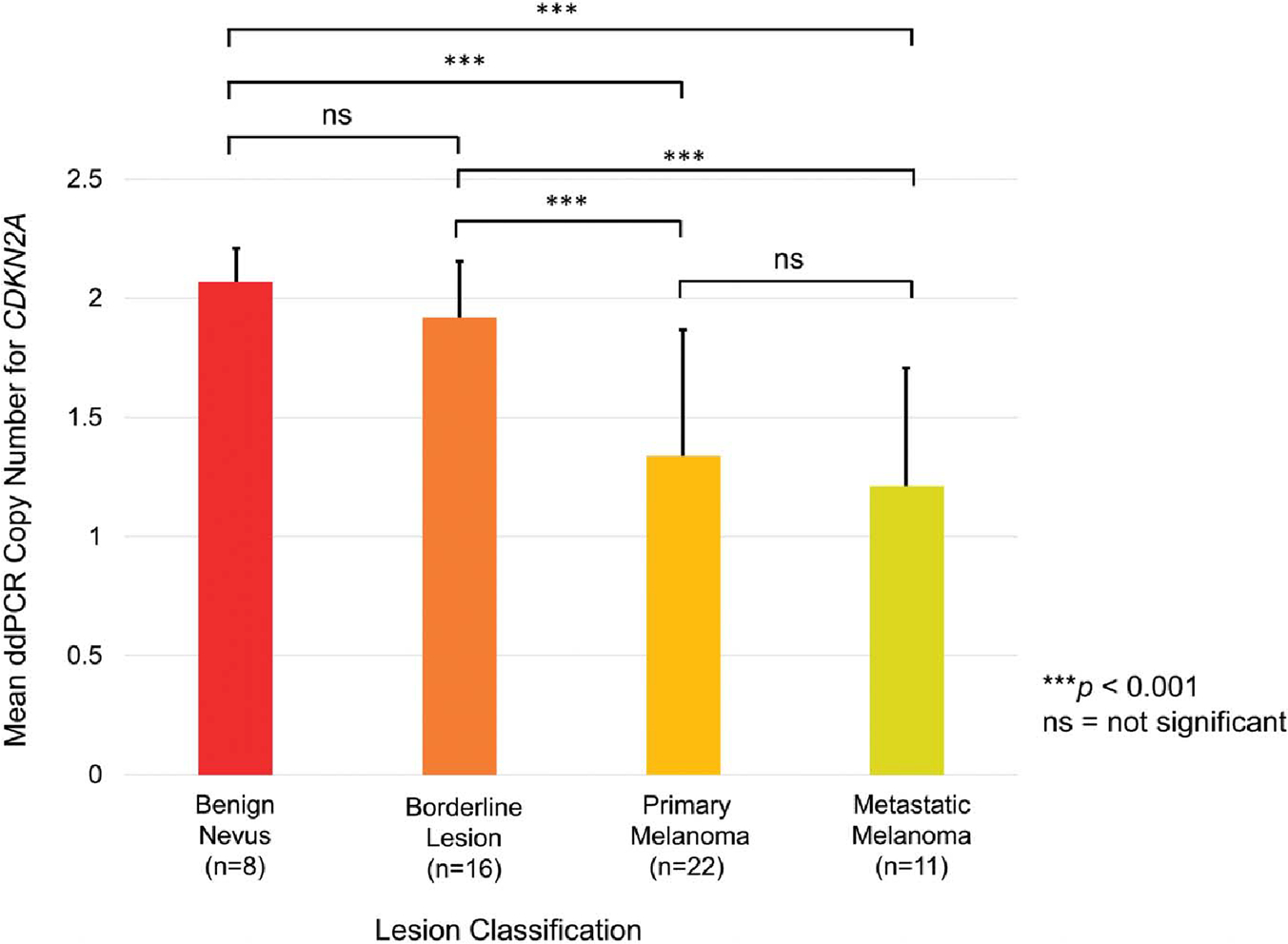
Trends in the data. Error bars represent SD. The average CDKN2A ddPCR output for benign lesions was 2.06 (SD 0.18), followed by 1.90 (SD 0.26) for borderline lesions, 1.29 (SD 0.55) for primary melanomas, and 1.20 (SD 0.52) for metastatic melanomas. Significant differences (2-sample t test) are indicated by asterisks. Notably, the degree of copy number loss increased with increasing atypia. Although this study was not designed to delineate atypia from copy number alone, future work could explore this area further.
Strengths of the Study
Strengths of this study include the broad variety of clinical samples, comprising a broad range of melanocytic neoplasia. The 57 FFPE cases reflected the complexity of clinical care because 16 were borderline lesions that required genomic workup to render a diagnosis in the first place. FFPE samples derived from both pediatric and adult patients were used, with a wide cohort age range of 2–87 years. In addition, to reduce the possibility of deletions or duplications in the reference genes skewing the final CDKN2A:Reference Gene ddPCR ratio, we used a panel of 5 reference genes. These were selected from genomic locations that show relatively infrequent CNVs in dermatological neoplasms.22 Then, instead of measuring CDKN2A copy number against 1 reference gene, we took the average of 5. This averaging dilutes the effect of any 1 spurious reference gene measurement. Although the possibility of such spurious measurements cannot be entirely eliminated, it can be mitigated; in our series, only case 8 seemed to be vulnerable to this effect, whereas 56 of 57 samples (98.2%) were not. We note that, in the literature, FISH and CMA measurements on the same cases have a discordance rate of approximately 10%.23
Limitations of the Study
There were several limitations of this study. First, it was retrospective and limited to a single institution. Our sample size was also constrained by cost and time for CMA analysis. Further research, with larger cohorts across a wide range of laboratories, could prove informative. In addition, ddPCR has its limitations: It can only detect known CNVs, unlike CMA, which can detect previously unknown CNVs. In addition, unlike FISH, ddPCR does not account for tumor heterogeneity. To optimize DNA quality, we only analyzed cases whose DNA was extracted within 1 month of biopsy and in which 10 ng of input DNA could be run per reaction. Future studies could investigate whether these inclusion criteria could be broadened.
Finally, it is worth noting that detection of CDKN2A loss is neither necessary nor sufficient to establish a diagnosis of melanoma. Thus, an important caveat is that CDKN2A loss, even if correctly detected, is not diagnostic of malignancy. Indeed, some benign melanocytic tumors (eg, melanocytomas) can also harbor CDKN2A loss. Conversely, CDKN2A losses are not ubiquitous in melanoma.24 This proof-of-concept study was designed to assess the concordance of ddPCR with CMA. All cases should be evaluated in context of the morphology and traditional parameters.
Future Directions
These preliminary data add to the growing body of literature supporting ddPCR as a robust technology for CNV detection. Future studies could look at further validation, particularly in tissues from different laboratories with different parameters for fixation and embedding. Future studies could also address issues such as whether monoallelic and biallelic CDKN2A deletion, as detected by ddPCR, could correlate with IHC for p16, and how mutations in the gene, detected by NGS, may affect results. Currently, ddPCR’s primary clinical use is for detection of somatic CNV in newborn screening tests. Its utility in oncology is growing and offers some promising future directions for research.
CONCLUSION
The capacity of ddPCR to quantify CDKN2A copy number in FFPE samples with similar sensitivities and specificities to those of CMA and FISH is encouraging. We have begun assembling ddPCR data for other genes that commonly undergo CNV in melanoma. Although not a replacement for CMA and FISH, ddPCR may, with further validation, provide an additional tool in the challenging diagnosis of melanoma.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the members of the Pathology Shared Resource Laboratory, a section of the laboratory for Clinical Genomics and Advanced Technology (CGAT), as well as the Hitchcock Foundation. The data in this study were generated through the CGAT in the Department of Pathology and Laboratory Medicine of the Dartmouth-Hitchcock Medical Center and the Norris Cotton Cancer Center (NCI Cancer Support Grant #5P30CA023108-37).
Supported by a grant from the Hitchcock Foundation.
Footnotes
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Web site (www.amjdermatopathology.com).
REFERENCES
- 1.Scolyer RA, Murali R, McCarthy SW, Thompson JF. Histologically ambiguous (“Borderline”) primary cutaneous melanocytic tumors: approaches to patient management including the roles of molecular testing and sentinel lymph node biopsy. Arch Pathol Lab Med. 2010;134:1770–1777. [DOI] [PubMed] [Google Scholar]
- 2.Shain AH, Yeh I, Kovalyshyn I, et al. The genetic evolution of melanoma from precursor lesions. N Engl J Med. 2015;373:1926–1936. [DOI] [PubMed] [Google Scholar]
- 3.Theisen A. Microarray-based comparative genomic hybridization (aCGH). Nat Educ. 2008;1:45. [Google Scholar]
- 4.Levy B, Burnside RD. Are all chromosome microarrays the same? What clinicians need to know. Prenatal Diagn. 2019;39:157–164. [DOI] [PubMed] [Google Scholar]
- 5.Gerami P, Jewell SS, Morrison LE, et al. Fluorescence in situ hybridization (FISH) as an ancillary diagnostic tool in the diagnosis of melanoma. Am J Surg Pathol. 2009;33:1146–1156. [DOI] [PubMed] [Google Scholar]
- 6.Gerami P, Li G, Pouryazdanparast P, et al. A highly specific and discriminatory fish assay for distinguishing between benign and malignant melanocytic neoplasms. Am J Surg Pathol. 2012;36:808–817. [DOI] [PubMed] [Google Scholar]
- 7.March J, Hand M, Truong A, Grossman D. Practical application of new technologies for melanoma diagnosis. J Am Acad Dermatol. 2015;72:943–958. [DOI] [PubMed] [Google Scholar]
- 8.Bell AD, Usher CL, McCarroll SA. Analyzing copy number variation with droplet digital PCR. In: Karlin-Neumann G, Bizouarn F, eds. Digital PCR. Vol 1768. New York: Springer; 2018:143–160. [DOI] [PubMed] [Google Scholar]
- 9.Belgrader P, Tanner SC, Regan JF, et al. Droplet digital PCR measurement of HER2 copy number alteration in formalin-fixed paraffin-embedded breast carcinoma tissue. Clin Chem. 2013;59:991–994. [DOI] [PubMed] [Google Scholar]
- 10.Somasundaram DB, Aravindan S, Yu Z, et al. Droplet digital PCR as an alternative to FISH for MYCN amplification detection in human neuroblastoma FFPE samples. BMC Cancer. 2019;19:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Tang ET, Du Z. Detection of MET gene copy number in cancer samples using the droplet digital PCR method. PLoS One. 2016;11:e0146784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klouch KZ, Stern MH, Trabelsi-Grati O, et al. Microsatellite instability detection in breast cancer using drop-off droplet digital PCR. Oncogene. 2022;41:5289–5297. [DOI] [PubMed] [Google Scholar]
- 13.Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, et al. Droplet digital polymerase chain reaction assay for screening of ESR1 mutations in 325 breast cancer specimens. Translational Res. 2015;166:540–553.e2. [DOI] [PubMed] [Google Scholar]
- 14.Vidal-Folch N, Gavrilov D, Raymond K, et al. Multiplex droplet digital PCR method applicable to newborn screening, carrier status, and assessment of spinal muscular atrophy. Clin Chem. 2018;64:1753–1761. [DOI] [PubMed] [Google Scholar]
- 15.Ming Z, Lim SY, Rizos H. Genetic alterations in the INK4a/ARF locus: effects on melanoma development and progression. Biomolecules. 2020;10:1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeng H, Jorapur A, Shain AH, et al. Bi-allelic loss of CDKN2A initiates melanoma invasion via BRN2 activation. Cancer Cell. 2018;34:56–68. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Young RJ, Waldeck K, Martin C, et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res. 2014;27:590–600. [DOI] [PubMed] [Google Scholar]
- 18.Helgadottir H, Höiom V, Tuominen R, et al. Germline CDKN2A mutation status and survival in familial melanoma cases. J Natl Cancer Inst. 2016;108:djw135. [DOI] [PubMed] [Google Scholar]
- 19.Yazdan P, Cooper C, Sholl LM, et al. Comparative analysis of atypical spitz tumors with heterozygous versus homozygous 9p21 deletions for clinical outcomes, histomorphology, braf mutation, and p16 expression. Am J Surg Pathol. 2014;38:638–645. [DOI] [PubMed] [Google Scholar]
- 20.Harms PW, Hocker TL, Zhao L, et al. Loss of p16 expression and copy number changes of CDKN2A in a spectrum of spitzoid melanocytic lesions. Hum Pathol. 2016;58:152–160. [DOI] [PubMed] [Google Scholar]
- 21.Wolter M, Felsberg J, Malzkorn B, et al. Droplet digital PCR-based analyses for robust, rapid, and sensitive molecular diagnostics of gliomas. Acta Neuropathologica Commun. 2022;10:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kabbarah O, Nogueira C, Feng B, et al. Integrative genome comparison of primary and metastatic melanomas. PLoS One; 2010. 5:e10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, Rao M, Fang Y, et al. A genome-wide high-resolution array-CGH analysis of cutaneous melanoma and comparison of array-CGH to FISH in diagnostic evaluation. J Mol Diagn. 2013;15:581–591. [DOI] [PubMed] [Google Scholar]
- 24.Sini MC, Manca A, Cossu A, et al. Molecular alterations at chromosome 9p21 in melanocytic naevi and melanoma. Br J Dermatol. 2007;0:243–250. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.