

Abstract
IL12 is a proinflammatory cytokine, that has shown promising antitumor activity in humans by promoting the recruitment and activation of immune cells in tumors. However, the systemic administration of IL12 has been accompanied by considerable toxicity, prompting interest in researching alternatives to drive preferential IL12 bioactivity in the tumor. Here, we have generated XTX301, a tumor-activated IL12 linked to the human Fc protein via a protease cleavable linker that is pharmacologically inactivated by an IL12 receptor subunit beta 2 masking domain. In vitro characterization demonstrates multiple matrix metalloproteases, as well as human primary tumors cultured as cell suspensions, can effectively activate XTX301. Intravenous administration of a mouse surrogate mXTX301 demonstrated significant tumor growth inhibition (TGI) in inflamed and non-inflamed mouse models without causing systemic toxicities. The superiority of mXTX301 in mediating TGI compared with non-activatable control molecules and the greater percentage of active mXTX301 in tumors versus other organs further confirms activation by the tumor microenvironment–associated proteases in vivo. Pharmacodynamic characterization shows tumor selective increases in inflammation and upregulation of immune-related genes involved in IFNγ cell signaling, antigen processing, presentation, and adaptive immune response. XTX301 was tolerated following four repeat doses up to 2.0 mg/kg in a nonhuman primate study; XTX301 exposures were substantially higher than those at the minimally efficacious dose in mice. Thus, XTX301 has the potential to achieve potent antitumor activity while widening the therapeutic index of IL12 treatment and is currently being evaluated in a phase I clinical trial.
Graphical Abstract
Introduction
Cytokines and checkpoint inhibitors represent important modalities in the immunotherapy of cancer (1). However, these agents show limited antitumor activity in immunologically ‘cold’ tumors due to an immunosuppressive tumor microenvironment (TME), characterized by a lack of T-cell infiltration, and the presence of suppressive macrophages and regulatory T cells (2–4). IL12 can induce broad remodeling of the TME and render it to be more immune permissive, effectively inducing a ‘hot’ tumor immune contexture by enhancing immune cell recruitment and effector function (5–7). IL12 naturally forms a heterodimer consisting of IL12 A (p35) and IL12 B (p40) subunits which bind to heterodimeric IL12 receptor subunit beta (IL12Rβ)1/IL12Rβ2 receptor complex leading to STAT4 phosphorylation and downstream effects (8, 9). IL12 binding to its receptors expressed on T cells and NK cells enhances their cytotoxicity predominantly via IFNγ production (10). IL12 facilitates intra-tumoral recruitment of cytotoxic NK cells and T cells by chemokine production (11, 12) and increases antigen presentation by upregulating human leukocyte antigen expression on tumor cells (13). Thus, IL12 is an attractive immunostimulatory agent for the treatment of cancer due to its ability to enhance the immune response in tumors with both ‘hot’ and ‘cold’ immune contexture.
IL12 has demonstrated potent antitumor activity in syngeneic mouse models with cold and hot tumors and promising antitumor activity in clinical trials (6, 14–16). However, the development of IL12 has been limited by severe toxicities including liver damage and multiorgan toxicities attributed to high levels of circulating IFNγ, hence prompting interest in developing alternative therapeutic strategies (16, 17). To localize the effect of IL12 in the TME and overcome systemic toxicities, protein engineering was employed to generate XTX301, a highly potent, half-life extended, and masked IL12. A masking domain based on IL12Rβ2 is designed to pharmacologically inactivate IL12. The masking domain in XTX301 is linked to the human Fc protein for half-life extension to enable less frequent dosing which is ideal for clinical settings. XTX301 includes a protease cleavage site and is designed to be activated by matrix metalloproteases (MMP) that are enriched in the TME (18–20). Thus, XTX301 is intended to have preferential IL12 bioactivity in tumor tissues compared with non-tumor tissues, thereby preserving the antitumor activity of IL12 while reducing its systemic toxicity.
Here we report results from preclinical studies characterizing XTX301. The masking and reactivation of XTX301 were evaluated using in vitro and ex vivo assays. The antitumor activity and mechanism of action were assessed in syngeneic mouse models using a murine surrogate of XTX301, as the human IL12 does not bind to the mouse IL12 receptors (21). A nonhuman primate Good Laboratory Practice (GLP) toxicology study was conducted to determine the tolerability of XTX301. The data from efficacy studies in mice and tolerability studies in nonhuman primates were used to predict the potential therapeutic index of XTX301. In totality, the data suggest that XTX301 has the potential for an improved therapeutic window compared with recombinant IL12.
Methods
Protein expression and purification
XTX301 consists of a human IL12Rβ2 masking moiety, a modified human IL12, and an aglycosylated human IgG1 half-life extension domain. The IL12 in the XTX301 comprises of covalently linked human IL12p35 subunit to a human IL12p40 subunit, both subunits consist of proprietary mutations for optimal pharmacokinetic and manufacturing properties. For additional details on XTX301, murine surrogate of XTX301, unmasked and non-cleavable construct designs, please refer to the patent PCT/US21/25107, available at https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021202678, example 8 and example 11 for XTX301 and murine surrogate respectively (22).
XTX301 was produced from stably transfected Chinese hamster ovary (CHO) cells using the vendor's proprietary CHO-K1 cell line (WuXi Biologics). Cells were passaged 6 times before transfection. The authenticity of the CHO cell line was confirmed using Targeted Ion Torrent Next Generation Sequencing and was determined to be Mycoplasma-negative at the time of transfection. The harvested cell culture fluid contents were purified by Protein A affinity chromatography, anion exchange chromatography, and cation exchange chromatography (CEX). The CEX eluate was buffer exchanged to a final concentration of 6.63 g/L in 20 mmol/L Glutamic Acid-Sodium Hydroxide, 8.8% sucrose, 0.04% polysorbate 80, pH 4.8. All other engineered molecules were produced using transient transfection in a human embryonic kidney cell line Expi293F cell line obtained from ThermoFisher Scientific (Catalog no. A14527, RRID: CVCL_D615). The cells were authenticated using short tandem repeat (STR) profile test by IDEXX Bioanalytical services and passaged less than 25 times prior to transfection. The cells were determined to be Mycoplasma-negative by qPCR (Charles River mycoplasma testing service) at the time of transfection and cultured according to the manufacturer's instructions. Conditioned media was harvested 5 to 7 days after the transfection and purified by affinity chromatography with Protein A beads. Proteins were eluted with a buffer consisting of 25 mmol/L acetate (Sigma, catalog no. A9563–5G) and 100 mmol/L NaCl (pH 3.0; Teknova, catalog no. S5845) and neutralized by adding 5.5% (v/v) 1 mol/L Tris (pH 8.0; Teknova, catalog no. T1085). The proteins were further purified by size exclusion chromatography in a buffer consisting of 20 mmol/L sodium citrate and 150 mmol/L NaCl (pH 5.5; Teknova, catalog no. S5845).
In vitro activation of XTX301 with recombinant enzymes
Recombinant human MMP-1 (Catalog no. 901-MP-010), MMP-2 (Catalog no. 902-MP-010), MMP-7 (Catalog no. 907-MP-010), MMP-8 (Catalog no. 908-MP-010), MMP-9 (Catalog no. 911-MP-010), MMP-10 (Catalog no. 910-MP-010), MMP-14 (Catalog no. 918-MPN-010) were all purchased from R&D Systems. The catalytic efficiency (kcat/Km) of the MMP cleavage of XTX301 was determined by incubating XTX301 with activated MMPs. For experiments in which XTX301+MMP14 was used, MMP14 was activated by incubation with Trypsin-3 (R&D Systems, catalog no. 3714-SE-010) at 37°C for 1 hour followed by the addition of 4-(2-Aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF; R&D Systems, catalog no. 5175/100), at room temperature for 15 minutes without agitation. All the other MMPs were activated by 1 mmol/L 4-aminophenyl-mercuric acetate (Sigma-Aldrich, catalog no. A9563) at 37°C. The incubation time was 1 hour for MMP-2, MMP-7, and MMP-8, 2 hours for MMP-1 and MMP10, and 24 hours for MMP-9. The samples were quenched with 20 mmol/L ethylenediaminetetraacetic acid and analyzed by nonreducing capillary electrophoresis–sodium dodecyl sulfate (CE-SDS) to measure the percentage of cleaved XTX301. Peaks representing intact substrate and cleaved products (IL12 and Fc-mask) were integrated; % cleaved XTX301 was calculated as % cleaved XTX301 = [Product peak area/(Product peak area + Intact XTX301 peak area)]*100. Percent cleaved (aka “Product %”) was plotted against time in GraphPad Prism and the data fit to “One-phase association” model [Y = Y0+(Plateau-Y0)*(1-exp(-K*x))] to calculate the rate constant “K” (in units of min−1). As the concentration of XTX301 <<Km, the exponential constant K was divided by enzyme concentration to yield kcat/Km, as defined by the one-step enzymatic reaction mechanism (23)
Ex vivo cleavage assay for tumor and plasma samples
To determine the cleavage of XTX301 by human tumors, tumor tissues surgically resected from melanoma, colorectal, head and neck (H&N), prostate, lung, ovarian, or renal cell carcinoma (RCC) patients were obtained from the Cooperative Human Tissue Network. Tumor tissues were cut into small pieces (approximately 2 mm) and dissociated by incubating with a cocktail of enzymes (2 mg/mL Collagenase IV (Worthington, catalog no. LS004189) and 5 μg/mL DNAse I (Millipore-Sigma, catalog no. D5025), for 30 minutes at 37°C, with agitation at 200 rotations per minute (rpm). Cells were then filtered using a 70 μmol/L strainer and washed twice in RPMI1640 medium (Gibco, catalog no. 21870–084), with 1X penicillin and streptomycin (Gibco, catalog no. 15070063) and plated at 0.15 × 106 in 100 μL RPMI-50 U/mL penicillin-50 μg/mL streptomycin in a 96 well plate for 24 hours at 37°C. After a 24-hour culture period, cells were incubated with XTX301 (1 μmol/L) for 24 hours at 37°C. To determine the cleavage of XTX301 in the peripheral circulation, frozen plasma samples were obtained from healthy human donors, or RCC, melanoma, H&N, and prostate cancer patients with active disease from BioIVT. Plasma samples were incubated with XTX301 (1 μmol/L) for 24 hours at 37°C and 100 rpm. The % of cleaved XTX301 by either plasma or cells from tumor samples was quantified using fluorescent triplex Western Blot.
Western blot analysis
The human tumor and plasma samples incubated with XTX301, as well as samples from mXTX301 dosed mice, were run on a 4% to 15% Criterion TGX Precast Midi Protein Gel (Bio-Rad, catalog no. 5671085), followed by semi-dry transfer to a nitrocellulose membrane (Bio-Rad, catalog no. 1704159). The membrane was blocked using Intercept TBS blocking buffer (LI-COR, catalog no. 927–60001) for 1 hour at room temperature and 100 rpm, before being incubated overnight at 4°C with the primary antibodies: anti-IL12A mouse monoclonal antibody (clone OTi2A8, Origene, ThermoFisher, catalog no. TA808081), and anti-IL12Rβ2 rabbit monoclonal antibody (ThermoFisher, catalog no. MA5–31162) diluted in Intercept TBS Antibody Diluent (LI-COR, catalog no. 917–65001). The membrane was washed with PBS-T (ThermoFisher, catalog no. 28352) and incubated with: Goat anti-Human IgG Fc Cross-Adsorbed Secondary Antibody, DyLight 488 (Invitrogen, catalog no. SA5–10134, RRID: AB_2556714), IRDye 680RD Goat anti-Mouse IgG Secondary Antibody (LI-COR, catalog no. 926–68070, RRID: AB_10956588) and IRDye 800CW Donkey Anti-Rabbit IgG secondary antibody (LI-COR, catalog no. 926–32213, RRID: AB_621848) for 1 hour at room temperature and 100 rpm. The membrane was washed with PBS-T and imaged using the Bio-Rad Chemidoc MP Imaging system (Catalog no. 12003154). The image analysis was performed using the Bio-Rad Chemidoc MP Imager Software: Image Lab (Ver 6.0.1 build 34 Standard Edition). The intact XTX301 consists of IL12Rβ2, IL12, and Fc, and the cleaved XTX301 consists of IL12Rβ2 and Fc. The anti-IL12Rβ2 fluorescence intensity (FI) was used to quantify the signal from both intact and cleaved bands. The % of cleaved XTX301 was calculated as FI of cleaved /(FI of cleaved + FI of Intact) × 100. The lower limit of quantification of the assay for determining the cleavage is 2.5%.
Surface plasmon resonance analysis
His-tagged Fc-fused human IL12Rβ2 (R&D systems, catalog no. 1959-B2B) and wild-type recombinant human IL12 (rhIL12; R&D Systems, catalog no.219-IL/CF) were used for surface plasmon resonance (SPR) analysis. Anti-His Tag antibody (Cytiva his capture kit, catalog no. 28995056) was diluted to 25 μg/mL in 10 mmol/L sodium acetate pH 4.5 (Cytiva, catalog no. BR100350) and then immobilized onto a CM5 SPR sensor chip (Cytiva, catalog no.BR100530) via amine coupling using EDC/NHS chemistry. His-tagged human IL12Rβ2 was diluted to 2.5 μg/mL in running buffer (1x HBS-EP: 0.01 mol/L HEPES, pH 7.4, 0.15 mol/L NaCl, 0.05% v/v Surfactant P20) supplemented with an additional 0.15 mol/L NaCl) and then was captured onto an anti-his sensor surface. XTX301, proteolytically cleaved XTX301, and rhIL12 were flowed over the chip at concentrations of 3.125 nmol/L to 400 nmol/L with 2-fold dilution. Each concentration of analyte was flowed over the captured ligands for 60 seconds and then a running buffer was flowed for 120 seconds to measure the dissociation of the analyte from the ligands. To remove any residual ligand and/or analyte post-dissociation, the chips were regenerated with 10 mmol/L Glycine pH 1.5 (Cytiva, catalog no. BR100354).
HEK-BLUE IL12 reporter gene assay
HEK Blue IL12 cell line was obtained from InvivoGen (Catalog no. hkb-il12) and incubated in DMEM complete (Gibco, catalog no. 11965118) with 10% (v/v) heat-inactivated FBS (Gibco, catalog no. A38400–01) and 1X Pen-Strep (Corning, catalog no. 30–001-CI). The cells were authenticated using STR profiling service by IDEXX Bioanalytical services and passaged 3 to 6 times prior to in vitro testing. The cells were determined to be Mycoplasma-negative at the time of in vitro testing by qPCR (Charles River mycoplasma testing service). XTX301, proteolytically activated XTX301, or rhIL12 (R&D Systems, catalog no. 219-IL/CF) were serially diluted and added to the cells and incubated at 37°C in 5% CO2 for 24 hours. The cell supernatants were collected and added to Quanti-Blue detection solutions (InvivoGen, catalog no. rep-qbs) and incubated for 2 hours at 37°C. The absorbance was measured at 625 nm using a plate reader (REF NEO2SBioTek Instruments, Inc.).
Phospho STAT4 assay
Primary human peripheral blood mononuclear cells (PBMC; BioIVT) were activated with PMA/Ionomycin (BioLegend, catalog no. 423301) for 4 days at 37°C in RPMI (Gibco, catalog no. 21870–084) with 10% (v/v) FBS (Gibco, catalog no. A38400–01), 1X gentamycin (Gibco, catalog no. 15750–060), 10 mg/L β-mercapto-ethanol (Gibco, catalog no. 21985–023), 1X HEPES (Gibco, catalog no. 15630–080), 1X penicillin/streptomycin (Gibco, catalog no. 15070–063), 1X sodium pyruvate (Gibco, catalog no. 11360–070), 1X MEM (Gibco, catalog no. 11140–050) and 1X l-glutamine (Gibco, catalog no. 25030–081). Activated PBMCs were incubated with XTX301, and proteolytically activated XTX301 using recombinant MMP14 (R&D systems, catalog no. 918-MPN) or rh-IL12 (R&D systems, catalog no. 219-IL-025/CF) at 37°C for 30 minutes. The cells were fixed, permeabilized, and stained for CD8+ T cell and CD4+ T-cell surface markers and for intracellular phosphorylated STAT4 protein according to the manufacturer's instructions. Anti-CD3 (Clone UCHT1, catalog no. 300450, RRID: AB_2563618), anti-CD4 (Clone RPA-T3, catalog no. 300508, RRID: AB_314076), and anti-CD45 (Clone HI30, catalog no. 304028, RRID: AB_893338), anti-CD8 (Clone HIT8a, catalog no. 300934, RRID: AB_2814115) were from BioLegend, anti-phospho STAT4 (Clone 47/STAT4/pY693, catalog no. 558136) was from Becton Dickinson. The cells were analyzed by Celesta flow cytometer (BD Biosciences) and the data were analyzed by FlowJo (Becton Dickinson).
Syngeneic mouse models
All studies were conducted following an approved Institutional Animal Care and Use Committee (IACUC) protocol #XTX-2020–01. The care and use of animals was conducted in accordance with the regulations of the Association for Assessment and Accreditation of Laboratory Animal Care. Female C57BL/6J and BALB/c mice were obtained from the Jackson Laboratories (Jackson Laboratories, catalog no. 000664, RRID: IMSR_JAX:000664 and catalog no. 000651, RRID: IMSR_JAX:000651), at 6 to 8 weeks of age. The MC38 colon adenocarcinoma cell line was obtained from the NIH (RRID:CVCL_B288), CT26 colorectal carcinoma cell line was from ATCC (catalog no. CRL-2638, RRID:CVCL_7254), and B16F10 melanoma cell line was from ATCC (catalog no. CRL-6475, RRID:CVCL_0159). The MC38 and B16F10 cell lines were authenticated using the CellCheck 19 - mouse STR profile test and Interspecies Contamination Test by IDEXX Bioanalytical services. CT26 was authenticated using STR profiling by ATCC. The cell lines were tested by using a PCR based test offered by IDEXX Bioanalytical services and were negative for Mycoplasma. The cell lines were expanded to generate a working bank which was less than 10 passages. The cells for each in vivo experiment were thawed from the working bank and expanded through 2 to 3 passages for in vivo inoculation. The cell lines were cultured according to the manufacturer's recommendations.
Tumors were subcutaneously injected in the right flank of female mice as follows, 1 × 105 CT26 cells into BALB/c mice and 0.5 × 106 MC38 or B16F10 cells into C57BL/6J mice. In efficacy studies with a single intravenous mXTX301 administration, tumor-bearing animals were randomly enrolled into study groups when the mean tumor size reached an average of 60 to 100 mm3. The animals were stratified into treatment groups and dosed with a single intravenous administration of either vehicle control (PBS), mXTX301, or unmasked control. In an efficacy study with repeated intravenous mXTX301 administration, the animals were enrolled when the tumors reached an average size of 360 mm3. The timing and dose information for different experiments can be found in figure legends for individual experiments. Tumor volume, survival, and body weight were measured 2 to 3 times weekly for up to 27 days. Tumor volume was measured using dial calipers and calculated by the formula: Length*(Width2)/2. The percent tumor growth inhibition (% TGI) and survival rate were used as a measure of treatment efficacy. Following the initiation of treatment, the percent change in body weight from baseline was calculated for each mouse as a measure of overall animal health and well-being. The criteria for animal euthanasia were the following: tumor size of more than 2,000 mm3; loss of 20% of their body weight relative to the start of the treatment; cavitated or unhealed tumor ulceration with size greater than 5 mm in diameter; changes in general animal health (hypothermia, progressive dehydration, lethargy, dyspnea or cyanosis, paralysis or ataxia, posturing (including hunched back), severe, intractable diarrhea, skin lesions, anemia, or impaired mobility due to tumors.
In vivo cleavage measurement
To determine the cleavage of mXTX301 in vivo in the tumor, peripheral organs, and plasma, MC38 mice were dosed with 3 mg/kg of mXTX301. After 72 hours, mice were sacrificed and blood, tumor, lung, spleen, kidney, and liver were collected. Tissues were lysed in Pierce IP Lysis buffer (Thermo Fisher, catalog no. 87788) and blood was spun down for plasma preparation. mXTX301 in plasma and tissue lysate was immunoprecipitated using biotinylated-goat anti-Human IgG (Southern Biotech, catalog no. 2049–08) and Pierce Streptavidin Magnetic beads (ThermoFisher, catalog no. 88817). Briefly, the biotinylated- goat anti-human IgG was added to plasma or tissue lysates and incubated at 4°C for 4 hours. The samples were incubated overnight at 4°C with Pierce Streptavidin Magnetic beads according to the manufacturer's instructions. The samples were washed with PBS, eluted with 10 mmol/L glycin (Bio-Rad, catalog no. 161–0718) and neutralized by adding 1 mmol/L Tris (pH 8) (VWR, catalog no. E119). The elution mixture/samples were run on a 4% to 15% Criterion TGX Precast Midi Protein Gel (Bio-Rad, catalog no. 5671085), followed by semi-dry transfer to a nitrocellulose membrane (Bio-Rad, catalog no. 1704159).
The western blot analysis for in vivo cleavage assessment was performed as follows: The membrane was blocked using Intercept TBS blocking buffer (LI-COR, catalog no. 927–60001) for 1 hour at room temperature and 100 rpm, before being incubated overnight at 4°C with the primary antibodies: anti-IL12A mouse monoclonal antibody (clone OTi2A8, Origene, ThermoFisher, catalog no. TA808081), and anti-IL12Rβ2 rabbit monoclonal antibody (ThermoFisher, catalog no. MA5–31162, RRID: AB_2786812) diluted in Intercept TBS Antibody Diluent (LI-COR, catalog no. 917–65001). The membrane was washed with PBS-T (ThermoFisher, catalog no. 28352) and incubated with: Goat anti-Human IgG Fc Cross-Adsorbed Secondary Antibody, DyLight 488 (Invitrogen, catalog no. SA5–10134, RRID: AB_2556714), IRDye 680RD Goat anti-Mouse IgG Secondary Antibody (LI-COR, catalog no. 926–68070, RRID:AB_10956588) and IRDye 800CW Donkey Anti-Rabbit IgG secondary antibody (LI-COR, catalog no. 926–32213, RRID: AB_621848) for 1 hour at room temperature and 100 rpm. The membrane was washed with PBS-T and imaged using the Bio-Rad Chemidoc MP Imaging system (Catalog no. 12003154). The image analysis was performed using the Bio-Rad Chemidoc MP Imager Software: Image Lab (Ver 6.0.1 build 34 Standard Edition). The intact XTX301 consists of IL12Rβ2, IL12, and Fc, and the cleaved XTX301 consists of IL12Rβ2 and Fc. The anti-IgG Fc FI was used to quantify the signal from both intact and cleaved bands. The % of cleaved XTX301 was calculated as FI of cleaved 85 kDa band/(FI of cleaved 85 kDa band + FI of Intact 145 kDa band) × 100.
Pharmacodynamic assessment
Tumors were induced in 6- to 8-week-old female C57BL/6J mice via subcutaneous injection of 0.5 × 106 MC38 tumor cells in the right flank. Tumor-bearing animals were randomly enrolled into study groups (N = 5 animals per treatment group) when the mean tumor size reached approximately 256 mm3. MC38 tumor-bearing mice were intravenously administered a single dose of either vehicle control, mXTX301 (0.13 or 0.39 mg/kg) or unmasked control (0.03 mg/kg). The day of treatment was designated as Day 0. PBS and unmasked control treatment groups were used as negative and positive controls respectively. Tumors and spleens were collected on Day 7, and plasma was collected at 6, 24, 72, 96, and 168-hours post-dose.
IFNγ Meso Scale Discovery
Tumor and spleen IFNγ levels were measured by Meso Scale Discovery (MSD) assay (Catalog no. K152QOD) according to the manufacturer's protocol (Proinflammatory Panel 1 (mouse) kits; www.mesoscale.com), which used anti-mouse IFNγ capture and detection antibodies. Briefly tumor or spleen was homogenized with a Qiagen TissueLyser using ice-cold Pierce IP Lysis Buffer (Fisher Scientific, catalog no. 87788) supplemented with Halt Protease Inhibitor Cocktail (Fisher Scientific; 78438). Lysates were cleared by centrifugation and subjected to MSD assay as per the manufacturer's instructions. IFNγ concentration was determined in comparison with a standard curve using a 1300 MESO QuickPlex SQ120MM MSD plate reader from MSD. The data analysis was performed using MSD Discovery Workbench software.
Flow cytometry
Cell suspensions were prepared from spleens by mechanical disruption followed by lysis of red blood cells using red blood cell lysis buffer (Sigma Aldrich, catalog no. R7757). The tumor tissues were enzymatically digested and mechanically dissociated using Miltenyi Tumor Dissociation Kit (Miltenyi Biotec, catalog no. 130–096–730) and the gentleMACS Dissociator using the manufacturer's program 37C_m_TDK_1. Following tumor digestion, debris was separated by sedimentation, and suspensions were passed through a 40 μmol/L nylon cell strainer. Red blood cells were lysed prior to antibody staining. The optimal concentration for each antibody was predetermined by titration. The following antibodies were used for flow cytometry assessment, anti-mouse CD8 APC (BioLegend, catalog no. 100712, RRID:AB_312751) anti-mouse CD45 BV786 (BD Biosciences, catalog no.564225, RRID:AB_2716861), anti-mouse CD3 APC-Cy7 (BioLegend, catalog no.100248, RRID:AB_2572118), anti-mouse CD4 PerCP-Cy 5.5 (BioLegend, catalog no.100540, RRID:AB_893326), anti-Ki67 FITC (Invitrogen, catalog no. 11–5698–82, RRID:AB_11151330), anti-mouse NKP46 BV650 (BioLegend, catalog no.137635, RRID:AB_2734200), LIVE/DEAD Fixable Aqua Dead Cell Stain (Invitrogen, catalog no. L34957). The live/dead dye staining, cell surface or intracellular staining was performed as per the manufacturer's instructions. The cells were stained and subjected to flow cytometry analysis using FACS Celesta (BD Biosciences), the data was acquired using FACS Diva (BD Biosciences) and subsequently analyzed using FlowJo software.
RNA sequencing
RNA was isolated from tumor samples and subjected to RNA sequencing (RNA-seq) analysis as follows: Library preparation using TrueSeq RNA library prep kit (Illumina) and sequencing performed by Canopy Biosciences (Saint Louis, MO). FastQ files were analyzed by Rosalind Inc. (San Diego, CA). Briefly, reads were trimmed using cutadapt (24). Quality scores were assessed using FastQC (FastQC, RRID:SCR_014583). Reads were aligned to the Mus musculus genome build mm10 using Spliced Transcripts Alignment to a Reference software (25). Gene expression counts in each sample were quantified using high-throughput sequencing (HTSeq) (26). Computing log-transformed Count-Per-Million reads (CPM), filtering, normalization, and differential gene expression analysis was performed in R (version 4.1.0) using the edgeR and the limma libraries. For pathway analysis, cell type-specific gene sets were obtained from the Panglao database (https://panglaodb.se/index.html; ref. 27), while gene ontology (GO) terms were obtained from the GO.db R library (bioconductor). Pathway enrichment analyses were performed using the fisher test function from the R stats package. Relative term expression scores were computed by summing expression z-scores for all genes belonging to a term of interest. Final scores were z-score transformed and used to build boxplots. Figures were prepared using either the pheatmap R library or the ggplot2 R library.
Pharmacokinetic assessment
A single-dose PK study was conducted in mice as follows: 7-week-old female C57BL/6 mice were lightly anesthetized by isoflurane before tumor implantation. Each mouse was subcutaneously injected with MC38 tumor cells (0.5 ×106) in 0.1 mL PBS in the right flank for tumor development. MC38 tumor-bearing mice were administered intravenously a single dose of either vehicle control (PBS) or mXTX301 (0.039, 0.13, 0.39 mg/kg). The day of treatment was designated as Day 0. Blood samples were collected at 5 minutes, 6 hours, 24 hours, 48 hours, 72 hours, 120 hours, and 168 hours post-dose. Plasma mXTX301 concentrations were measured using a MSD assay.
A single-dose PK study was conducted in female cynomolgus monkeys to determine the PK properties of XTX301 after a 60-minute intravenous infusion at doses of 0.1, 0.5, or 2.0 mg/kg. Total measurements were taken to understand the disposition of the drug. Briefly, XTX301 was immunoprecipitated from cynomolgus plasma using biotin-conjugated goat anti-human IgG Fc fragment-specific antibody. Following tryptic digestion, total XTX301 was quantified by LC/MS-MS (Applied Biosystems/MDS Sciex API 6500) using the signature peptide in the Fc domain.
Data representation and statistical analysis
For in vitro activity assays, the background signal was subtracted from the data in Excel (Microsoft Excel, RRID:SCR_016137) and exported to GraphPad Prism 8 (GraphPad Prism, RRID:SCR_002798) for data analyses using a nonlinear sigmoidal, 4PL curve fit model without constraints. All in vitro assays have been repeated at least twice.
For the pSTAT4 assessment, the FlowJo data were imported to GraphPad Prism, where each data series was defined as the percentage of pSTAT4+ cells induced by a single test article in one cell type across multiple doses. Each data series was fit with a curve of nonlinear regression, specifically “[Agonist] vs. response – Variable slope (4 parameters).” The mean EC50 and SD were calculated across 4 assays using Excel.
Tumor growth data were analyzed in GraphPad by plotting tumor growth curves with the time point of each measurement on the X axis and the mean tumor volume of each group on the Y axis; the SEM was represented with error bars. Differences between the treatment groups and the control group were calculated as mean percent (%) TGI using the equation below. Tt and Ct are the mean tumor volumes of treatment and vehicle control groups on the day of the treatment, respectively. T0 and C0 are the mean tumor volumes of treatment and vehicle control groups on Day 0, respectively.
For murine immunophenotyping pharmacodynamic studies, FlowJo V10 software (FlowJo, RRID:SCR_008520) was used to analyze the flow cytometry data prior to exportation for graphing and statistical analysis in GraphPad. Repeated measures two-way ANOVA test with Bonferroni's post hoc pairwise comparison tests were performed to determine the statistical significance of observed differences in tumor volumes. Kaplan–Meier curves were used to estimate the survival function of treated mice. The statistical significance of observed differences between study groups in survival was measured using the log-rank (Mantel–Cox) test with Bonferroni's multiple comparison correction. A one-way ANOVA test with Bonferroni's post hoc pairwise comparison tests was performed to determine the statistical significance of observed differences in immunophenotypic changes. All statistical tests were performed in GraphPad Prism.
Data availability
RNA-seq and gene expression data that support the findings of this study are available on NCBI Gene Expression Omnibus under accession number GSE237982. All other data generated in this study are available within the article and Supplementary Data files.
Supplementary Methods
IFNγ MSD
Plasma IFNγ levels were measured by MSD assay (Catalog no. K152QOD) according to the manufacturer's protocol (Proinflammatory Panel 1 (mouse) kits; www.mesoscale.com), which used anti-mouse IFNγ capture and detection antibodies. Plasma was added to the plate and incubated for 1 hour at room temperature with agitation. IFNγ concentration was determined in comparison with a standard curve using a 1300 MESO QuickPlex SQ120MM MSD plate reader from MSD. The data analysis was performed using MSD Discovery Workbench software.
IFNγ assay
XTX301 and mXTX301 were proteolytically activated using recombinant MMP14 (R&D systems, catalog no. 918-MPN). Murine splenocytes were isolated via mechanical dissociation and pre-activated with PMA/Ionomycin (BioLegend, catalog #423301) for 2 days at 37°C before incubating with varying doses of rm IL12 (R&D Systems, catalog no. 419-ML/CF), murine surrogate mXTX301 or proteolytically activated mXTX301 for 24 hours. Similarly, primary human PBMCs (BioIVT) or cynomolgus PBMCs were thawed and rested overnight at 37°C. Human PBMCs were activated with PMA/Ionomycin for 4 days at 37°C. After 4 days, activated PBMCs were incubated with XTX301, proteolytically activated XTX301, or rh-IL12 (R&D systems, catalog no. 219-IL-025/CF) at 37°C in 5% CO2 for 24 hours. Cyno PBMCs were activated with PMA/Ionomycin, and XTX301 or proteolytically activated XTX301, or rhIL12 was added simultaneously. Cells were incubated for 24 hours and then supernatant was collected. The supernatants were subjected to mouse IFNγ ELISA (BioLegend, catalog no. 430816) human IFNγ ELISA (BioLegend, catalog no. 430101), or cynomolgus IFNγ ELISA (Mabtech, catalog no. 3421M-1H-20). The ELISA assay was performed according to the manufacturer's instructions.
Nonhuman primate GLP toxicology study hematology and clinical chemistry
A GLP toxicology study was conducted following an approved IACUC protocol and in accordance with U.S. Department of Health and Human Services, FDA, United States Code of Federal Regulations, Title 21, Part 58: GLP for Nonclinical Laboratory Studies. Cynomolgus monkeys [3/sex/group (Main Study) and 2/sex/group (Recovery Study)] were administered XTX301 (test article) or Formulation Buffer (control article) via intravenous infusion once weekly for 60 minutes on Days 1, 8, 15, and 22 at 0, 0.05, 0.5, or 2 mg/kg/dose. Hematology and clinical chemistry parameters were evaluated during Week –1 and on Days 1 Predose (hematology only), 3, 6, 10, 13, 17, 20, 24, 27, 35, 42, 49, and 56. Twenty-four animals (12 male and 12 female) were euthanized on Day 30 and the remaining 16 animals were euthanized on Day 57, following a 4-week recovery period.
Results
Multiple MMPs were expressed in both hot and cold tumors and activated XTX301
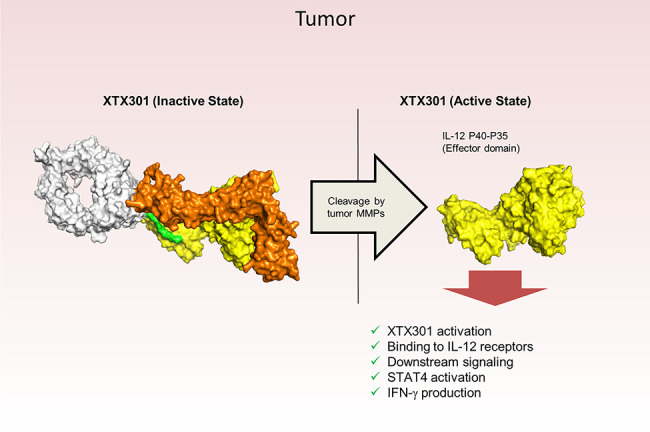
To reduce systemic toxicities by enabling the preferential activity of IL12 in the TME, we have exploited the MMPs biology as they are highly enriched in TME of multiple tumor types. MMPs degrade extracellular matrix proteins and promote tumor growth, metastasis, and angiogenesis (18). Here we have engineered XTX301, a fusion protein of aglycosylated human Fc linked to human IL12 and a masking domain based on IL12Rβ2 designed to pharmacologically inactivate IL12. XTX301 includes a protease cleavage site that is designed to be cleaved by TME-associated MMPs to activate the molecule through the release of IL12 (Fig. 1A). Fig. 1B shows that mRNA expression of multiple MMPs is evident across immunologically cold and hot tumors. The hot, cold, and mixed designations are based on the published literature characterizing the immune contexture, checkpoint inhibitor responsiveness and tumor mutation burden for select indications (4, 7, 28). MMPs from 5 different classes, collagenases (MMP1 and MMP8), gelatinases (MMP2 and MMP9), matrilysin (MMP7), stromelysins (MMP10), and membrane-anchored (MMP14) were selected and evaluated for their ability to cleave XTX301 in vitro (19). Under the conditions tested, recombinant human MMP1, 2, 7, 8, 9, 10, and 14 cleaved XTX301 with catalytic efficiencies ranging from 1.1 × 10−3 to 0.21 (μmol/L−1min−1; Fig. 1C; Supplementary Table S1). The cleavage of XTX301 by human tumors was determined by incubating XTX301 with cells from dissociated primary tumor tissues from melanoma, colorectal, H&N, prostate, lung, ovarian, or RCC cancer patients (Fig. 1D; Supplementary Fig. S1). Samples from all tumor types evaluated in this study were able to activate XTX301, and the percentage of tumors in each tumor type that cleaved XTX301 ranged from 58% to 100%. This study demonstrated that XTX301 was activated by human primary tumors across a broad range of solid tumor types. The cleavage of XTX301 in plasma was determined by incubating XTX301 with plasma from healthy human donors and from RCC, melanoma, H&N or prostate cancer patients with active disease. No significant cleavage was observed in any of the plasma samples tested at 24 hours compared with day 0 (Fig. 1D). Thus, XTX301 was activated by multiple MMPs and cleaved by the majority of human tumor samples tested, but not by human plasma derived from patients with active cancer, suggesting tumor-selective activation.
Figure 1.

Multiple MMPs are expressed in both hot and cold tumors and can activate XTX301. A, A schematic of inactive and active XTX301. B, Heat map showing RNA expression levels of genes encoding for MMPs (MMP-1, -2, -7, -8, -9, -10, and -14) in tumor samples from multiple The Cancer Genome Atlas (TCGA) indications (x-axis). Color intensity indicates median log2-transformed CPM reads for each gene and indication. Column keys (pink, gray, blue) indicate prevalently hot (pink), cold (blue) or mixed tumor indications. Abbreviations: HNSC, head and neck squamous cell carcinoma (n = 522); SKCM, skin cutaneous melanoma (n = 472); KIRC, kidney renal clear cell carcinoma (n = 534); LUAD, lung adenocarcinoma (n = 517); LUSC, lung squamous cell carcinoma (n = 501); COADREAD, colon and rectum adenocarcinoma (n = 382); BRCA, breast invasive carcinoma (n = 1100); PRAD, prostate adenocarcinoma (n = 498); OV, ovarian serous cystadenocarcinoma (n = 307); PAAD, pancreatic adenocarcinoma (n = 179). C, Human recombinant MMPs (1, 2, 7, 8, 9, 10, and 14), were evaluated for their ability to proteolytically cleave XTX301 in vitro. The data points represent the mean of 2 independent experiments analyzed as technical replicates (in duplicate) by CE-SDS; the error bars represent the SEM. D, Heat map showing the percentage of human tumors and plasma able to proteolytically activate XTX301. XTX301 was incubated with cells from dissociated primary tumor tissues from melanoma (n = 8, 100% of samples cleaved), colorectal (n = 11, 91% of samples cleaved), head and neck (H&N) (n = 16, 81% of samples cleaved), prostate (n = 21, 76% of samples cleaved), lung (n = 17, 71% of samples cleaved), ovarian (n = 12, 58% of samples cleaved) or RCC (n = 31, 61% of samples cleaved) cancer patients, or plasma from melanoma (n = 5), H&N (n = 10), prostate (n = 7) and RCC (n = 6) cancer patients. Samples were collected after a 24-hour incubation period, and the percentage of cleaved XTX301 was determined by fluorescent triplex western blot. ND, not determined.
Masking of XTX301 activity and reactivation by MMPs in vitro
The binding of XTX301, proteolytically cleaved XTX301, and rhIL12 to the human IL12 receptor were measured by SPR. The IL12R is composed of 2 different subunits, IL12Rβ1 and IL12Rβ2. IL12 binds to IL12Rβ2 with higher affinity, compared with IL12Rβ1 (29). Only the high-affinity receptor IL12Rβ2 was tested in the SPR experiments. Proteolytically cleaved XTX301 and rhIL12 bound to human IL12Rβ2 with KD values of 7.97 nmol/L and 0.93 nmol/L respectively, while no binding of the intact XTX301 was detected to human IL12Rβ2, demonstrating effective inhibition of IL12Rβ2 binding by the masking domain and restoration of binding upon proteolytic cleavage (Fig. 2A). The binding of IL12 to IL12R results in interactions of the IL12R subunits with JAK2 and TYK2 causing the phosphorylation of tyrosine 693 of STAT4 and subsequent IFNγ production (9). After proteolytic cleavage, XTX301 is designed to engage the IL12R and promote STAT4-mediated transcriptional activity. The potency of XTX301 as an activator of STAT4 was determined with and without proteolytic cleavage in the HEK-Blue IL12 Reporter Gene Assay, which is engineered to express a reporter gene upon STAT4 activation downstream of the IL12R (Fig. 2B; Supplementary Table S2). The half maximal effective concentrations (EC50s) were calculated as described in the statistical analysis section. The results demonstrated that the masking domain of XTX301 inhibited STAT4 activation via the IL12R by 73-fold compared with rhIL12 and 54-fold compared with the unmasked control, indicating effective inhibition of IL12R pathway signaling by the masking domain (Supplementary Table S2). Proteolytic cleavage of the masking domain of XTX301 fully restored STAT4 activation as compared with the unmasked control and rhIL12 (Supplementary Fig. S2B).
Figure 2.

Masking of XTX301 activity and reactivation by MMPs in vitro(A) SPR was used to measure the binding kinetics of XTX301, proteolytically cleaved XTX301 (XTX301 + MMP), and rhIL12 to IL12Rβ2. IL12Rβ2 was immobilized to a sensor chip. Then XTX301, proteolytically cleaved XTX301, and rhIL12 were flowed over at concentrations ranging from 3 nmol/L to 400 nmol/L. The concentrations of each analyte decrease from top to bottom within each panel. XTX301 activity was measured in a reporter cell line and primary human cells. B, IL12 HEK-Blue reporter gene cells were incubated with either rhIL12, unmasked control, or XTX301 at varying doses, and the reporter activity was measured. The data represents one of three independent experiments, data points represent the mean of 2 replicate wells and the error bars represent SD. C, Pre-activated primary human PBMCs were incubated with either rhIL12, unmasked control or XTX301 at varying doses, and STAT4 phosphorylation was assessed in CD8+ T cells via flow cytometry. The data points represent the mean of 2 replicate wells and the error bars represent SD. The data represents one of two independent experiments, each conducted with 2 different PBMC donors.
The ability of XTX301 to induce STAT4 phosphorylation in primary human PBMCs was assessed via flow cytometry with and without proteolytic cleavage by MMP14. The phosphorylation of STAT4 was specifically measured in CD8+ T cells (Fig. 2C) and half maximal effective concentrations (EC50s) were calculated (Supplementary Table S2). The EC50s for XTX301 in CD8+ T cells was 12.3 ± 5.16 nmol/L. After incubation with MMP14, the mean EC50 of proteolytically cleaved XTX301 (0.216 nmol/L) in CD8+ T cells was similar to the unmasked control (0.263 nmol/L). Proteolytically cleaved XTX301 was 26-fold less potent compared with rhIL12. These results demonstrated that the masking domain of XTX301 reduced STAT4 phosphorylation by 47-fold in primary human CD8+ T cells, compared with unmasked control in vitro, indicating effective inhibition of IL12R pathway signaling by the masking domain and restoration of activity by proteolytic cleavage. At concentrations exceeding 10 nmol/L a cleavage-independent activation of STAT4 in response to XTX301 treatment is observed in PBMC. This is consistent with other reports of cleavage-independent activity of masked IL12 and likely result of the non-covalent, reversible nature of the interaction between the mask and IL12 (30, 31).
Human IL12 does not activate the mouse IL12 receptors (21), hence the murine surrogate mXTX301, and unmasked control molecule were created for in vivo pharmacology assessments. We verified the ability of mXTX301 to induce IFNγ production in mouse splenocytes and XTX301 to induce IFNγ production in human and cynomolgus monkey PBMCs (Supplementary Fig. S2). The proteolytic cleavage of the masking domain of mXTX301 or XTX301 fully restored IFNγ production as compared with the unmasked control in all three species (Supplementary Table S3). Thus, we demonstrated that at the dose levels tested, the masking domains of XTX301 and mXTX301 functioned as intended, and that following proteolytic cleavage, full recovery of IL12 signaling activity of XTX301 and mXTX301 in comparison with unmasked control was achieved in primary cells from three different species.
mXTX301 was well tolerated and demonstrated significant TGI across immunologically hot (MC38, CT-26) and immunologically cold (B16F10) syngeneic mouse tumor models
IL12 has demonstrated antitumor effects in a range of malignancies in preclinical syngeneic models (14). The antitumor activity and tolerability of a single intravenous dose of mXTX301 were assessed in MC38, colon adenocarcinoma CT26, colorectal carcinoma, or B16F10, melanoma, tumor-bearing mice, in comparison with the unmasked control. Compared with vehicle control, mXTX301 was effective in inhibiting tumor growth of the MC38 colon tumor model. Significant TGI of 60% to 92% was observed with 0.039 mg/kg to 0.39 mg/kg doses and complete regressions were observed in 2 of 12 and 1 of 12 animals at 0.13 mg/kg and 0.39 mg/kg doses respectively (Fig. 3A; Supplementary Fig. S3A). Compared with the unmasked control, treatment with mXTX301 was well tolerated, with no significant changes in body weight at any of the doses tested. The unmasked control molecule was not well tolerated at a 10-fold lower dose of 0.03 mg/kg and resulted in greater than 20% body weight loss by Day 6 (Fig. 3B). We also assessed the tolerability of repeat dose administration and antitumor activity in mice bearing large MC38 tumors of an average size of 360 mm3. Our data demonstrate that repeated mXTX301 dosing was well tolerated, as demonstrated by no change in body weight (Supplementary Fig. S3), and resulted in dose-dependent TGI in a mouse model bearing large MC38 tumors (Fig. 3C). A significant prolongation of animal survival was also observed in response to treatment with mXTX301 (Supplementary Fig. S3C). In addition, when tested in other models, 0.5 mg/kg mXTX301 mediated significant TGI of 101% in CT26 and 65% in B16F10 mouse tumor models (Fig. 3D and E), and complete regressions were observed in 8 of 10 animals in the CT26 mouse tumor model. Overall, these data demonstrated that mXTX301 exhibited antitumor activity in both immunologically hot and cold tumor-bearing mouse models and was well tolerated upon single or repeat dose administration.
Figure 3.

XTX301 demonstrates antitumor activity in immunologically hot (MC38, CT26) and immunologically cold (B16F10) syngeneic tumor models. C57BL/6J mice were implanted subcutaneously with MC38 tumor cells and received a single intravenous injection of mXTX301, unmasked control, or vehicle (PBS) at indicated dose levels (N = 12). A, Tumor and (B) body weight measurements were taken two/three times a week. The data points represent mean ± SEM. Tumor volume and body weight change were assessed by a two-way ANOVA with Bonferroni's post hoc pairwise comparison test compared with vehicle (PBS) treated animals (**, P < 0.005; ****, P < 0.0001). The tumor growth changes, and body weight data are displayed until Day 14, when 75% of vehicle-treated animals were alive (N = 9). C, TGI was assessed in mice bearing large (∼360 mm3) MC38 tumors. C57BL/6J mice were implanted subcutaneously with MC38 tumor cells and received a total of 3 doses marked by red arrows of mXTX301. Tumor growth data is presented as mean for tumor volume ± SEM. Two-way ANOVA followed by Bonferroni post hoc test (**, P < 0.005; ****, P < 0.0001) (D) Balb/c mice were implanted subcutaneously with CT26 tumor cells and C57BL/6J mice were implanted with (E) B16F10 tumor cells and received a single intravenous injection of mXTX301 at 0.5 mg/kg. Tumor measurements were recorded 2/3 times a week (n = 8–10 per group). Tumor growth data are presented as means for tumor volume ± SEM and were assessed by two-way ANOVA followed by Bonferroni post hoc test (**, P < 0.005; ****, P < 0.0001)
TME-associated protease-dependent activation of mXTX301 in vivo
To assess the protease-dependent activation of mXTX301 in vivo, we engineered a non-activatable control that lacks the MMP cleavage site present in mXTX301. MC38 tumor-bearing mice were administered a single intravenous dose of either vehicle control, non-activatable control, or mXTX301. TGI results demonstrate a significant 72% TGI with mXTX301 treatment compared with 6% TGI in vehicle control. The non-activatable control did not show significant TGI compared with vehicle control (Fig. 4A). In addition, we measured the percentage of cleaved mXTX301 in the tumor, spleen, kidney, lung, liver, and plasma of mXTX301-treated MC38 tumor-bearing mice (Fig. 4B). The results show a significantly higher percentage of cleaved mXTX301 in tumors compared with plasma or any other tissue tested, providing support for preferential activation of mXTX301 in tumors by TME-associated proteases.
Figure 4.

TME-associated protease-dependent activation of mXTX301 in vivo(A) C57BL/6J mice were implanted subcutaneously with MC38 tumor cells and received a single intravenous injection of mXTX301, or non-activatable control lacking protease cleavage site at indicated dose levels. Data represent mean ± SEM. Tumor measurements were assessed by a two-way ANOVA with Bonferroni's post hoc pairwise comparison test compared with vehicle (PBS) treated animals (***, P < 0.0005). The tumor growth changes are displayed until Day 14, when 86% of vehicle-treated animals were alive (N = 6). B, Measurement of percentage of cleaved mXTX301 in the tumor, peripheral organs, and plasma in the MC38 syngeneic tumor model. The percentage of cleaved mXTX301 was quantified by fluorescent triplex WB (LLOQ is represented by the dotted line) The data represents mean ± SEM. Statistical comparisons were performed using Dunnett ordinary one-way ANOVA test versus tumor, where ***, P < 0.001.
mXTX301 demonstrated effective peripheral masking and tumor-selective pharmacodynamic effects in the MC38 syngeneic mouse model
Systemic and intratumoral pharmacodynamic effects of mXTX301 were evaluated in the MC38 mouse model by measuring severity of splenomegaly, IFNγ assessment in plasma, spleen, and tumor, immunophenotyping of lymphocytes in tumor and spleen, and tumor RNA gene expression (Fig. 5). Consistent with effective masking in the periphery, mXTX301 treatment (0.39 mg/kg) resulted in lower plasma IFNγ levels compared with 0.03 mg/kg unmasked control treatment (P < 0.0001; Supplementary Fig. S5). mXTX301 at 0.13 mg/kg demonstrated no significant changes in spleen size on Day 7, compared with vehicle control. A 10-fold lower dose of unmasked control (0.03 mg/kg) increased spleen weight by 1.8-fold (P = 0.0001) on Day 7, compared with mXTX301 (0.39 mg/kg; Fig. 5A). Tumor selective immune cell effector function is evidenced by significant increase in IFNγ in tumor but not in spleen upon mXTX301 treatment (Fig. 5B). In contrast, no tumor selective IFNγ production was observed in the unmasked control treated group evidenced by significant increase in IFNγ production in spleen and tumor (Fig. 5B).
Figure 5.
![Figure 5. mXTX301 demonstrates effective peripheral masking and intratumoral pharmacodynamic effects in MC38 syngeneic tumor model. MC38 tumor-bearing mice were treated with the indicated doses of mXTX301 or unmasked control, (A) The spleen size was assessed by a one-way ANOVA with Bonferroni's post hoc pairwise comparison test compared with vehicle (PBS) treated animals (*, P < 0.05; ***, P < 0.0005; ****, P < 0.0001). The data represents mean ± SEM. B, The animals were sacrificed on day 7 (n = 5 mice per group) spleen and tumor were isolated and IFNγ was assessed by MSD assay. IFNγ measurements were log10-transformed and analyzed by one way ANOVA followed by Dunnett multiple comparisons test (***, P = 0.0001; ****, P < 0.0001). C, Immune cells were characterized via flow cytometry. The data are presented as means ± SEM, spleen (n = 5), tumor-treatment groups (n = 5), tumor-vehicle group (n = 4). Statistical significance was determined by one-way ANOVA test followed by Dunnett multiple comparisons test. #, P = 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.0005; and ****, P < 0.0001 (D) Evaluation of mXTX301 induced gene expression in the MC38 tumors by bulk RNA-seq. Heat map showing relative expression of top mXTX301 differentially expressed genes (N = 10 upregulated and N = 5 downregulated; by P value). Rows (genes) were clustered according to Euclidean distance. Keys above each column (sample)indicate treatment group [vehicle, mXTX301 (0.39 mg/kg), or unmasked control (0.03 mg/kg)]. Box color tracks with z-score-transformed relative expression of each gene across samples (blue, under-expression compared with the mean; red: over-expression compared with the mean) (E) Heat map summarizing results of a pathway enrichment analysis where each box indicates the enrichment significance (-log10 Fisher P value) of a gene set of interest (rows) in the Top 250 most significantly upregulated genes (by P value) computed by comparing tumors after treatment with mXTX301 or unmasked control versus vehicle at Day 7 (columns). Color intensity tracks with significance.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=10993987_421fig5.jpg)
mXTX301 demonstrates effective peripheral masking and intratumoral pharmacodynamic effects in MC38 syngeneic tumor model. MC38 tumor-bearing mice were treated with the indicated doses of mXTX301 or unmasked control, (A) The spleen size was assessed by a one-way ANOVA with Bonferroni's post hoc pairwise comparison test compared with vehicle (PBS) treated animals (*, P < 0.05; ***, P < 0.0005; ****, P < 0.0001). The data represents mean ± SEM. B, The animals were sacrificed on day 7 (n = 5 mice per group) spleen and tumor were isolated and IFNγ was assessed by MSD assay. IFNγ measurements were log10-transformed and analyzed by one way ANOVA followed by Dunnett multiple comparisons test (***, P = 0.0001; ****, P < 0.0001). C, Immune cells were characterized via flow cytometry. The data are presented as means ± SEM, spleen (n = 5), tumor-treatment groups (n = 5), tumor-vehicle group (n = 4). Statistical significance was determined by one-way ANOVA test followed by Dunnett multiple comparisons test. #, P = 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.0005; and ****, P < 0.0001 (D) Evaluation of mXTX301 induced gene expression in the MC38 tumors by bulk RNA-seq. Heat map showing relative expression of top mXTX301 differentially expressed genes (N = 10 upregulated and N = 5 downregulated; by P value). Rows (genes) were clustered according to Euclidean distance. Keys above each column (sample)indicate treatment group [vehicle, mXTX301 (0.39 mg/kg), or unmasked control (0.03 mg/kg)]. Box color tracks with z-score-transformed relative expression of each gene across samples (blue, under-expression compared with the mean; red: over-expression compared with the mean) (E) Heat map summarizing results of a pathway enrichment analysis where each box indicates the enrichment significance (-log10 Fisher P value) of a gene set of interest (rows) in the Top 250 most significantly upregulated genes (by P value) computed by comparing tumors after treatment with mXTX301 or unmasked control versus vehicle at Day 7 (columns). Color intensity tracks with significance.
To investigate the effect of mXTX301 on remodeling of the TME, we assessed the expression of Ki67, a proliferation marker on T and NK cells via flowcytometry. The gating strategy for flow cytometry assessment is described in Supplementary Fig. S4. No changes were observed in the percentage of CD45 and CD3 in tumor in response to mXTX301 treatment. A significant tumor-specific increase in the percentage of CD8+ T cells and CD8+Ki67+ T cells was observed in response to mXTX301 treatment compared with vehicle control (Fig. 5C). A significant decrease in the percentage of CD4+ T cells reflects changes in the CD4/CD8 ratio due to rapid increase in CD8+ T cells. An increase in CD4+Ki67+ T cells and NK+Ki67+ cells in tumor of mXTX301 treated group was also observed, although it did not reach statistical significance. The unmasked control group demonstrated a significant increase in Ki67+ NK cells and T cells in tumors, however, it also induced greater IFNγ production in periphery (Spleen, plasma), caused splenomegaly (Fig. 5A; Supplementary Fig. S5) and induced body weight loss (Fig. 3B). Thus, the flow cytometry data demonstrated that mXTX301 selectively remodeled the TME by modulating T cells and NK cells effector functions.
Transcriptome analysis of tumor samples was performed to characterize the mechanism of action of mXTX301 at 0.39 mg/kg and unmasked control at 0.03 mg/kg. A comprehensive examination of global gene expression revealed a similar pattern of molecular changes for mXTX301 and unmasked control on Day 7 (Fig. 5D), with both molecules inducing the upregulation of several immune-related genes including those encoding for Stat1, IL12Rβ1, Irf1, and Cxcl9.
Gene set enrichment analyses were performed to identify pathways that were consistently upregulated (log2_fold_change ≥ 1.5, FDR-adjusted P < 0.01) by mXTX301 relative to vehicle control. These results demonstrated that similar pathways were induced after treatment with mXTX301 and unmasked control. A significant (Fisher P < 0.01) upregulation of immune-related GO gene sets such as IFNγ cell signaling pathway, antigen processing and presentation, defense response as well as enrichment of marker gene signatures for T cells, NK cells, macrophages, and dendritic cells (Fig. 5E) was observed upon treatment with mXTX301.
We further analyzed the transcriptional activation status of these pathways in each treatment group compared with the vehicle control group on Day 7 (Supplementary Fig. S6). Consistent with previous results, this analysis revealed that samples from mice treated with either mXTX301 or unmasked control showed activation of immune-related transcriptional programs compared with vehicle control and that the extent of such activation was similar for both mXTX301 and unmasked control in all analyzed pathways. Thus, mXTX301 demonstrated pharmacodynamic effects in mouse tumors that are consistent with known IL12 biology in a tumor selective manner (32–34).
XTX301 was tolerated following 4 repeat doses up to 2.0 mg/kg, the highest non-severely toxic dose in a nonhuman primate study
To determine tolerability and the potential for toxicity, XTX301 was administered to cynomolgus monkeys by 1-hour intravenous infusion at 0.05, 0.5, or 2 mg/kg/dose once weekly for a total of 4 doses followed by a four-week recovery period to evaluate the potential reversibility of any findings. Markers of liver toxicity, aspartate aminotransferase (AST) and alanine aminotransferase (ALT), showed minimal to mild increases beginning on Day 6 at ≥0.5 mg/kg/dose to Day 20 (Supplementary Fig. S7A and S7B). However, no corresponding XTX301-related microscopic changes were observed in the liver. There were minimal to moderate decreases in lymphocytes at ≥0.5 mg/kg/dose generally beginning on Day 3 with continued decreases in lymphocytes through Day 10. On Days 10, 13, and/or at subsequent time points to Day 27 at ≥0.5 mg/kg/dose, lymphocytes generally approximated or exceeded pre-study time points (Supplementary Fig. S7C). Finally, albumin was minimally to moderately decreased beginning on Day 6 with persistent decreases to Day 24 or 27 at ≥0.5 mg/kg/dose (Supplementary Fig. S7D). Thus, minimal to moderate changes to AST, ALT, total lymphocytes, and albumin were fully reversible in the main study and recovery animals. Likewise, dose-dependent, and transient increases in plasma IFNγ levels were observed following the first dose of XTX301 and IFNγ levels returned to baseline in all animals following repeated doses. The total XTX301 exposure was relatively dose-proportional across all doses administered. On the basis of the totality of the data the highest non-severely toxic dose (HNSTD) was determined to be 2 mg/kg/dose. At the HNSTD in nonhuman primates (2.0 mg/kg), the terminal half-life of total XTX301 was approximately 97.4 hours, which is 10-fold longer than the published half-life of rhIL12 which is 5.3 to 9.6 hours (16). We expect the half-life of the active XTX301 to be similar to that of the published half-life of rhIL12.
XTX301 (IL12) preclinical data support potential for broad therapeutic index
To estimate the potential therapeutic index of XTX301, the AUC parameter was determined from 0 to 168 hours for concentration versus time profiles from the GLP toxicology study in nonhuman primates and MC38 mouse model pharmacokinetic study. The AUC0–168 (hr*μg/mL) for nonhuman primates dosed at HNSTD of 2.0 mg/kg of XTX301 and the AUC0–168 (hr*μg/mL) of the dose that enabled tumor regressions, 0.13 mg/kg of mXTX301 were 2,540 hr*μg/mL and 37.8 hr*μg/mL respectively (Fig. 6A and B). The potential therapeutic index of 67 was determined to be broad by comparing the systemic exposure of XTX301 at the HNSTD in nonhuman primates to the systemic exposure of mXTX301 associated with TGI in the mouse model (AUCSafety/AUCActivity; Fig. 6C).
Figure 6.

Preclinical data predicts a broad therapeutic index for XTX301. The area-under-the-curve parameter was determined from 0 to 168 hours for (A) concentration versus time profiles from nonhuman primates (N = 5/sex) following the first of the four intravenous doses of the HNSTD of XTX301, 2.0 mg/kg QWx4, and (B) concentration versus time profiles from MC38 tumor-bearing mice (N = 5) following a single intravenous bolus of the efficacious dose of mXTX301, 0.13 mg/kg. C, AUC of the efficacious dose and HNSTD. A large exposure difference in the group mean AUC0–168hr predicts a potential broad therapeutic index for XTX301.
Discussion
IL12 enhances immune cell infiltration and antigen presentation in the TME, T cell and NK cell effector function, and T helper 1 cell polarization. It also reverses immunosuppression and inhibits angiogenesis via IFNγ (35). The ideal cellular targets of IL12 immunotherapy are not lymphocytes in circulation, but rather immune cells within the tumor and nearby lymph nodes, including T cells, NK cells, tumor associated macrophages, and myeloid derived suppressor dendritic cells. Therefore, maximizing the amount of IL12 that reaches the tumor is critical for a robust antitumor response. Recent approaches intended to reduce systemic toxicities associated with IL12 have focused on intratumoral delivery (36–38), attenuated IL12 (29), tumor targeted IL12 (15, 39–41) and masked IL12 (30, 31). While encouraging results have been observed, each strategy has limitations. For example, intra tumoral injection may offer limited benefit to patients with metastasis or inaccessible lesions (14). A tumor targeted IL12 may allow for greater accumulation in tumor but does not completely overcome the issue of systemic lymphocyte activation and consequently the high levels of cytokine production leading to toxicities. Alternatives to overcome systemic toxicities, such as reduced potency IL12 may also limit efficacy (14, 42). More recently, two approaches with masked IL12 were described. The first approach lacks a half-life extension domain and may require frequent dosing in a clinical setting (30), and the second approach uses human serum albumin as a half-life extension domain and reported a half-life of 53 hours in a nonhuman primate study. (43, 44). In addition, the later approach relies on two cleavage events to occur to achieve the release of IL12, which may lead to variability in the activation of the molecule (43). We believe that XTX301, a half-life extended, masked IL12 with tumor selective activation, can overcome some of the challenges highlighted above. XTX301 can be administered intravenously, and the half-life extension domain is designed to reduce the need for frequent dosing, making it highly compatible with clinical use. In addition, to minimize the risk of active IL12 with half-life extension domain leaking into the periphery, XTX301 is designed to enable tumor-specific release of active IL12 with a shorter half-life, compared with the half-life of the intact XTX301.
MMP expression is tightly regulated in the periphery and metalloproteases are aberrantly expressed in the TME by tumor cells, tumor-infiltrating macrophages, and fibroblasts (45, 46). Our data are consistent with the current understanding of MMP biology, as XTX301 can be broadly cleaved by multiple MMPs via incubation with cells derived from hot or cold tumors but not by plasma from patients with cancer; hence, we envision potential broad applicability across multiple cancer indications. In addition, the most efficient recombinant metalloproteases to cleave XTX301 were MMP2 and MMP14, which were also two of the protease-encoding genes expressed at the highest mRNA levels across a range of indications. Also, given the association of MMP expression with tumor progression and metastasis (44, 45), we anticipate that XTX301 will be cleaved in metastatic sites as well as primary tumors. We successfully demonstrated masking of XTX301 activity and reactivation by MMPs using SPR, IL12 reporter gene assays, measuring phosphorylated STAT4 in primary human T cells, and IFNγ assay in primary T cells from multiple species.
The SPR analysis and phosphorylated STAT-4 assessment in primary cells featured a 26-fold reduced potency of cleaved/active XTX301 compared with rhIL12. The reduced potency may be a result of the mutations introduced in the p40 subunit of IL12 for increased stability and favorable pharmacokinetic profile of XTX301 and/or due to linking of p35 and p40 subunits as opposed to rhIL12 which is an unlinked heterodimer. The HEK-Blue reporter gene assays did not show the difference in potency of active XTX301 compared with rhIL12, presumably due to over expression of IL12 receptors in the HEK blue system and/or the longer assay time (overnight vs. 20 min for pSTAT4). Nevertheless, the active XTX301 is still highly potent, and the 26-fold reduced potency can be easily compensated for by increasing the dose of XTX301.
The in vivo proof of concept is supported by significant TGI upon treatment with mXTX301 without the body weight loss in immunologically hot, MC38 and CT26 tumor models, which are historically more responsive to immunotherapy agents and in a cold mouse tumor model, B16-F10, which is less likely to respond to immunotherapy. The non-cleavable form of mXTX301 was less active than mXTX301 demonstrating protease dependent activity of mXTX301 in vivo. Repeated administration of mXTX301 was also well tolerated and was able to control large established tumors. We observed a plateau effect in the survival curve from the repeat dose study, which could be attributed to the rapid proliferation of tumor cells outpacing the activation of cytotoxic immune cells in this large tumor setting or due to the effect of antidrug antibodies as mouse immune cells may produce antibodies to the human Fc present in mXTX301.
The treatment with mXTX301 reduced peripheral IFNγ by 10-fold compared with the unmasked control. In contrast to no increase in IFNγ in spleen, marked increase of up to 10 fold in tumor IFNγ demonstrated mXTX301 mediated immune cell activation and effector function in a tumor selective manner. The upregulation of immune cells and inflammatory gene pathways induced by mXTX301 in the TME are consistent with known IL12 biology (32–34). It has been described that CD8+ T cells play a critical role in mediating the antitumor effect of IL12 in murine tumors (47). Further studies using mice deficient in lymphocyte subsets and/or antibody depletion experiments are needed to confirm the primary cellular drivers of the antitumor effects of mXTX301. Similarly, repeated doses of XTX301 were well tolerated in cynomolgus monkeys with minimal, transient, and reversible changes in AST, ALT, lymphocytes, and serum albumin. The mild transient and reversible changes observed in this study with XTX301 are consistent with published preclinical safety data of rhIL12 but at dose levels that were 500-fold higher than previously assessed (48). The cynomolgus monkeys used in the toxicology study were normal healthy animals without tumors, therefore limited cleavage of XTX301 is expected unlike cleavage mediated by tumors of patients with cancer. The half-life of 97.4 hours and tolerability of multiple dose administration in both mouse model and cynomolgus monkeys are encouraging and relevant to clinical settings as XTX301 is planned to be administered once every 3 weeks (Q3W) in the first-in-human clinical trial. The preclinical results summarized in this paper reveal that systemic administration of a half-life extended tumor activated IL12 molecule, mXTX301, had demonstrated antitumor activity and eradicated established tumors without the toxicity of an unmasked IL12 such as rhIL12. In addition, the minimally active dose exposure in mice was substantially lower than the highest severe non-toxic exposure in cynomolgus monkeys, and thus predicts a potential broad therapeutic index for XTX301 that warrants continued investigation in the clinic.
Overall, our work presents a novel, tumor-activated, engineered IL12 with a potential for clinical translation. The toxicity of a highly potent immunomodulatory treatment such as IL12 makes combination therapy with checkpoint inhibitors or cytokines such as IL2 challenging. On the basis of these preclinical data, XTX301 offers the opportunity for combination and improvement in efficacy of checkpoint inhibitors or cytokines without the associated toxicity of an unmasked IL12. The translatability of the findings from the mouse model efficacy studies and cynomolgus monkey safety studies remains to be determined. A phase I clinical trial evaluating the safety and feasibility of XTX301 in patients with advanced solid tumors is currently ongoing (NCT05684965).
Supplementary Material
Supplemental Figure S1: A representative triplex fluorescent western blot image showing cleavage of XTX301 in primary human tumor specimens. Antibodies against human IL-12 (Green), human IL-12Rβ2 (Red) and human IgG (Blue) were used, a merged image is shown in the figure. The 145 Kda band is the intact XTX301 or non-cleavable construct, the 85Kda band is the cleaved XTX301/unmasked construct, the green arrow indicates free IL-12 released upon cleavage of XTX301. The % of cleavage mXT301 refers to the percentage of cleaved molecule compared to the total (intact and cleaved) molecule detected by western blot using the IL-12Rβ2 signal.
Supplemental Figure S2: XTX301 demonstrates enhanced activity upon proteolytic cleavage in primary cells. (a) Primary mouse splenocytes were pre-activated with PMA & Ionomycin and then incubated with varying doses of rm IL-12 and murine surrogate test articles for 24 hours, and supernatants were assessed by IFN-γ ELISA. (b) Human peripheral blood mononuclear cells (PBMCs) were preactivated with PMA + Ionomycin and incubated with test articles for 4 days and (c) Cynomolgus monkey PBMCs were preactivated with PMA + Ionomycin and incubated with varying doses of rhIL-12 and test articles for 24 hours and evaluated for IFN-γ production by ELISA.
Supplemental Figure S3: Repeated mXTX301 dosing was well tolerated and resulted in dose dependent TGI in mice bearing large (~360mm3) MC38 tumors (a) C57BL/6J mice were implanted subcutaneously with MC38 tumor cells and received a single intravenous injection of mXTX301, unmasked control, or vehicle (PBS) at indicated dose levels (N=12 per group). Tumor measurements were taken two/three times a week. Animals were euthanized due to health issues or body weight loss. The data represent individual survival curves. CR: complete regression, EU: euthanized. (b) C57BL/6J mice were implanted subcutaneously with MC38 tumor cells and received a total of 3 doses marked by red arrows of mXTX301. Body weight was monitored over time (c) Survival prolongation was assessed by Gehan-Breslow-Wilcoxon test comparison at all study time points (until Day 27). ***p = 0.0001; ****p < 0.0001.
Supplemental Figure S4: Gating strategy for multi-color flow cytometry analysis of tumor-infiltrating and splenic immune cell populations.
Supplemental Figure S5: IFN-γ production in plasma: MC38 tumor-bearing mice were treated with the indicated doses of mXT301 or unmasked control. Plasma IFN-γ concentration was measured by MSD assay at indicated time points. The data represent mean ± SEM, N = 3 to 5 mice per group, and IFN-γ level was assessed by a two-way ANOVA with Bonferroni’s post-hoc pairwise comparison test compared to vehicle (PBS) treated animals (*p < 0.05, **p < 0.005, ****p < 0.0001)
Supplemental Figure S6: XTX301 induced cell signature and pathway analysis: single-sample Gene Set Enrichment Analysis (ssGSEA) was performed for all samples and for selected gene sets of interest (see figure 5e). Each ssGSEA score summarizes the overall expression level of genes involved in a certain biological process of interest in a biological sample. For each gene set, ssGSEA scores were analyzed with respect to treatment groups and significant differences compared to the vehicle group were determined by Wilcoxon rank sum test. Boxplots showing ssGSEA scores (y-axis) of all samples for gene sets of interest. Each point is a sample. Samples were grouped by treatment group (x-axis). Colors indicate different treatment groups. * (Wilcoxon test, p< 0.05) ** (Wilcoxon test, p<0.01).
Supplemental Figure S7: XTX301 caused transient and reversible changes to LFTs and other safety parameters in NHP in a repeat dose GLP toxicology study. Male and female NHPs (N=3/sex; 2/sex for recovery animals) were administered XTX301 by intravenous infusion once weekly for a total of four doses, followed by a four-week recovery period. Changes in (a) aspartate aminotransferase, (b) alanine aminotransferase, (c) total lymphocytes, (d) albumin, and (e) IFN-γ were fully reversible in the main study and recovery animals. All values are shown as mean ± SD. The reference ranges, indicated by the grey-shaded area, were derived from the study facility’s historical control values.
Supplementary Table S1: The catalytic efficiency (kcat/Km) of MMP cleavage of XTX301. MMPs from 5 different classes, collagenases (MMP1 and MMP8), gelatinases (MMP2 and MMP9), matrilysin (MMP7), stromelysins (MMP10), and membrane-anchored (MMP14) were selected and evaluated for their ability to cleave XTX301 in vitro. The catalytic efficiency (kcat/Km) of the MMP cleavage of XTX301 was determined by incubating XTX301 with activated MMPs. The data points represent the mean of 2 independent experiments analyzed as technical replicates (in duplicate) by CE-SDS.
Supplementary Table S2: pSTAT4 EC50 in HEK-Blue IL-12 RGA and primary CD8 T cells IL-12 HEK-Blue reporter gene cells were incubated with either rh IL-12, unmasked control, or XTX301 at varying doses, and the reporter activity was measured. The data from three independent experiments, the average of replicates from each experiment are listed in the table. Pre-activated primary human PBMCs were incubated with either rhIL-12, unmasked control or XTX301 at varying doses, and STAT4 phosphorylation was assessed in CD8+ T cells via flow cytometry. The EC50 was calculated as described in the statistical analysis section. The data represents two independent experiments, each conducted with two different PBMC donors.
Supplementary Table S3: IFN-γ production EC50 in mouse, human, and cynomolgus primary cells The ability of mXTX301 to induce IFN-γ production in mouse splenocytes and XTX301 to induce IFN-γ production in human and cynomolgus monkey PBMCs was assessed via ELISA. The EC50 was calculated as described in the statistical analysis section.
This article is featured in Selected Articles from This Issue, p. 405
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
Authors' Disclosures
M. Pederzoli-Ribeil reports a patent for US 2023/0159603-A1 and US-2023-0072822-A1. K.A. Jenkins reports a patent for US 2023/0159603-A and US-2023-0072822-A1. R.K. O'Donnell reports a patent for US 2023/0159603-A1 and US 2023-0072822-A1. P. Johnson reports a patent for US 11,718,655, US 2023/0159603-A1 and US-2023-0072822. D.S. Tomar reports a patent for US 11,718,655, US 2023/0159603-A1, and US-2023-0072822-A1. R. Rozenfeld reports a patent for US 11,718,655, US 2023/0159603-A1, and US-2023-0072822-A1. R.C. O'Hagan reports other support from Xilio Development, Inc. during the conduct of the study, other support from Xilio Development, Inc. outside the submitted work; in addition, R.C. O'Hagan has a patent for US 11,718,655 issued to Xilio Development, Inc. All current employees of Xilio Development, Inc. report that they are equity holders of Xilio Therapeutics, Inc. No disclosures were reported by the other authors.
Authors' Contributions
E. Patel: Conceptualization, data curation, supervision, writing–original draft, project administration, writing–review and editing. N.V. Malkova: Formal analysis, supervision, validation, visualization, methodology, project administration, writing–review and editing. D. Crowe: Formal analysis, supervision, validation, visualization, methodology, project administration, writing–review and editing. M. Pederzoli-Ribeil: Formal analysis, supervision, validation, visualization, methodology, project administration, writing–review and editing. D. Fantini: Resources, data curation, formal analysis, investigation, visualization, methodology, writing–review and editing. M. Fanny: Resources, supervision, investigation, visualization, methodology. H.R. Madala: Resources, formal analysis, investigation, methodology. K.A. Jenkins: Supervision, validation, visualization, project administration, writing–review and editing. O. Yerov: Resources, investigation, visualization, methodology. J. Greene: Resources, investigation, visualization, methodology. W. Guzman: Resources, investigation, methodology. C. O'Toole: Resources, formal analysis, investigation, methodology. J. Taylor: Resources, investigation, methodology. R.K. O'Donnell: Supervision, validation, visualization, methodology, project administration, writing–review and editing. P. Johnson: Resources, visualization, methodology. B.B. Lanter: Resources, investigation, visualization, methodology. B. Ames: Resources, investigation, visualization, methodology. J. Chen: Resources, investigation, visualization. S. Vu: Resources, investigation, methodology. H.-J. Wu: Resources, investigation, methodology. S. Cantin: Resources, investigation, methodology. M. McLaughlin: Methodology. Y.-S.S. Hsiao: Resources, methodology, writing–review and editing. D.S. Tomar: Resources, visualization, methodology. R. Rozenfeld: Conceptualization, supervision, project administration. L. Thiruneelakantapillai: Supervision, project administration. R.C. O'Hagan: Conceptualization, supervision. B. Nicholson: Conceptualization, supervision, project administration, writing–review and editing. J. O'Neil: Conceptualization, supervision, project administration, writing–review and editing. C.U. Bialucha: Conceptualization, supervision, project administration, writing–review and editing.
References
- 1. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med 2016;8:328rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vitale I, Shema E, Loi S, Galluzzi L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat Med 2021;27:212–24. [DOI] [PubMed] [Google Scholar]
- 3. Kalbasi A, Ribas A. Tumor-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol 2020;20:25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maleki Vareki S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J Immunother Cancer 2018;6:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tait Wojno ED, Hunter CA, Stumhofer JS. The immunobiology of the interleukin-12 family: room for discovery. Immunity 2019;50:851–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Langrish CL, McKenzie BS, Wilson NJ, De Waal Malefyt R, Kastelein RA, Cua DJ. IL12 and IL23: master regulators of innate and adaptive immunity. Immunol Rev 2004;202:96–105. [DOI] [PubMed] [Google Scholar]
- 7. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumors with combination immunotherapies. Nat Rev Drug Discov 2019;18:197–218. [DOI] [PubMed] [Google Scholar]
- 8. Presky D H, Yang H, Minetti L J, Chua A O, Nabavi N, Wu C-Y, et al. A functional interleukin 12 receptor complex is composed of two beta-type cytokine receptor subunits. Proc Natl Acad Sci USA 1996;93:14002–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bacon CM, Petricoin EF, Ortaldo JR, Rees RC, Larner AC, Johnston JA, et al. Interleukin 12 induces tyrosine phosphorylation and activation of STAT4 in human lymphocytes. Proc Natl Acad Sci USA 1995;92:7307–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ryffel B. Interleukin-12: role of interferon-gamma in IL12 adverse effects. Clin Immunol Immunopathol 1997;83:18–20. [DOI] [PubMed] [Google Scholar]
- 11. Jenner RG, Townsend MJ, Jackson I, Sun K, Bouwman RD, Young RA, et al. The transcription factors T-bet and GATA-3 control alternative pathways of T-cell differentiation through a shared set of target genes. Proc Natl Acad Sci USA 2009;106:17876–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Allen F, Bobanga ID, Rauhe P, Barkauskas D, Teich N, Tong C, et al. CCL3 augments tumor rejection and enhances CD8(+) T-cell infiltration through NK and CD103+ dendritic cell recruitment via IFNγ. Oncoimmunology 2018;7:e1393598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yun Yue F, Geertsen R, Hemmi S, Burg G, Pavlovic J, Laine E, et al. IL12 directly up-regulates the expression of HLA class I, HLA class II and ICAM-1 on human melanoma cells: a mechanism for its antitumor activity? Eur J Immunol 1999;29:1762–73. [DOI] [PubMed] [Google Scholar]
- 14. Nguyen KG, Vrabel MR, Mantooth SM, Hopkins JJ, Wagner ES, Gabaldon TA, et al. Localized interleukin-12 for cancer immunotherapy. Front Immunol 2020;11:575597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mansurov A, Ishihara J, Hosseinchi P, Potin L, Marchell TM, Ishihara A, et al. Collagen-binding IL12 enhances tumor inflammation and drives the complete remission of established immunologically cold mouse tumors. Nat Biomed Eng 2020;4:531–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Atkins MB, Robertson MJ, Gordon M, Lotze MT, DeCoste M, DuBois JS, et al. Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin Cancer Res 1997;3:409–17. [PubMed] [Google Scholar]
- 17. Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood 1997;90:2541–8. [PubMed] [Google Scholar]
- 18. Isaacson KJ, Martin Jensen M, Subrahmanyam NB, Ghandehari H. Matrix-metalloproteinases as targets for controlled delivery in cancer: an analysis of upregulation and expression. J Control Release 2017;259:62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Murphy G, Nagase H. Progress in matrix metalloproteinase research. Mol Aspects Med 2008;29:290–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Otani Y, et al. Recent progress of matrix metalloproteinase (MMP) research and its clinical application for cancer therapy. Gan To Kagaku Ryoho 1998;25:957–64. [PubMed] [Google Scholar]
- 21. Schoenhaut DS, Chua AO, Wolitzky AG, Quinn PM, Dwyer CM, McComas W, et al. Cloning and expression of murine IL12. J Immunol 1992;148:3433–40. [PubMed] [Google Scholar]
- 22. Rozenfeld R, U.E., Qiu H, Johnson P, Jenkins KA, Pederzoli-Ribeil M, Tomar DS., et al. Masked IL12 Cytokines and Their Cleavage Products. PCT/US21/25107. 2021Available from: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021202678.
- 23. Copeland RA., A practical introduction to structure, mechanism, and data analysis. 2nd Edition ByCopeland R A.. Enzymes. 2000, New York: John Wiley & Sons, Inc. [Google Scholar]
- 24. MARTIN M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011, may v.17:p.10–12. [Google Scholar]
- 25. Dobin A. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Anders S, Pyl PT, Huber W. HTSeq–a python framework to work with high-throughput sequencing data. Bioinformatics 2015;31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Franzen O, Gan LM, Bjorkegren JLM. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019;2019:baz046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wen H, Li F, Bukhari I, Mi Y, Guo C, Liu B, et al. Comprehensive analysis of colorectal cancer immunity and identification of immune-related prognostic targets. Dis Markers 2022;2022:7932655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Glassman CR, Mathiharan YK, Jude KM, Su L, Panova O, Lupardus PJ, et al. Structural basis for IL12 and IL23 receptor sharing reveals a gateway for shaping actions on T versus NK cells. Cell 2021;184:983–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mansurov A, Hosseinchi P, Chang K, Lauterbach AL, Gray LT, Alpar AT, et al. Masking the immunotoxicity of interleukin-12 by fusing it with a domain of its receptor via a tumor-protease-cleavable linker. Nat Biomed Eng 2022;6:819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Skrombolas D, Sullivan M, Frelinger JG. Development of an interleukin-12 fusion protein that is activated by cleavage with matrix metalloproteinase 9. J Interferon Cytokine Res 2019;39:233–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mirlekar B, Pylayeva-Gupta Y. IL12 family cytokines in cancer and immunotherapy. Cancers 2021;13:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shi X, Liu J, Xiang Z, Mitsuhashi M, Wu RS, Ma X. Gene expression analysis in interleukin-12-induced suppression of mouse mammary carcinoma. Int J Cancer 2004;110:570–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li S, Xia X, Mellieon FM, Liu J, Steele S. Candidate genes associated with tumor regression mediated by intratumoral IL12 electroporation gene therapy. Mol Ther 2004;9:347–54. [DOI] [PubMed] [Google Scholar]
- 35. Berraondo P, Etxeberria I, Ponz-Sarvise M, Melero I. Revisiting Interleukin-12 as a cancer immunotherapy agent. Clin Cancer Res 2018;24:2716–8. [DOI] [PubMed] [Google Scholar]
- 36. Li Y, Su Z, Zhao W, Zhang X, Momin N, Zhang C, et al. Multifunctional oncolytic nanoparticles deliver self-replicating IL12 RNA to eliminate established tumors and prime systemic immunity. Nat Cancer 2020;1:882–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hewitt SL, Bailey D, Zielinski J, Apte A, Musenge F, Karp R, et al. Intratumoral IL12 mRNA therapy promotes TH1 transformation of the tumor microenvironment. Clin Cancer Res 2020;26:6284–98. [DOI] [PubMed] [Google Scholar]
- 38. Algazi AP, Twitty CG, Tsai KK, Le M, Pierce R, Browning E, et al. Phase II Trial of IL12 plasmid transfection and PD-1 blockade in immunologically quiescent melanoma. Clin Cancer Res 2020;26:2827–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Puca E, Probst P, Stringhini M, Murer P, Pellegrini G, Cazzamalli S, et al. The antibody-based delivery of interleukin-12 to solid tumors boosts NK and CD8(+) T cell activity and synergizes with immune checkpoint inhibitors. Int J Cancer 2020;146:2518–30. [DOI] [PubMed] [Google Scholar]
- 40. Momin N, Mehta NK, Bennett NR, Ma L, Palmeri JR, Chinn MM, et al. Anchoring of intratumorally administered cytokines to collagen safely potentiates systemic cancer immunotherapy. Sci Transl Med 2019;11:eaaw2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Greiner JW, Morillon YM 2nd, Schlom J. NHS-IL12, a tumor-targeting immunocytokine. Immunotargets Ther 2021;10:155–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lasek W, Zagozdzon R, Jakobisiak M. Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol Immunother 2014;63:419–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nirschl CJ, Brodkin HR, Domonkos C, Dwyer CJ, Hicklin DJ, Ismail N, et al. mWTX-330, an IL12 INDUKINE molecule, activates and reshapes tumor-infiltrating CD8+ T and NK cells to generate antitumor immunity. Cancer Immunol Res 2023;11:962–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Morris K, Brodkin H, Hicklin D, Ismail N, Nirschl C, Salmeron A, et al. WTX-330 is an IL12 pro-drug that is conditionally activated within the tumor microenvironment and induces regressions in mouse tumor models. J Immunother Cancer 2021. [Google Scholar]
- 45. Mason SD, Joyce JA. Proteolytic networks in cancer. Trends Cell Biol 2011;21:228–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2002;2:161–74. [DOI] [PubMed] [Google Scholar]
- 47. Brunda MJ, Luistro L, Warrier RR, Wright RB, Hubbard BR, Murphy M, et al. Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J Exp Med 1993;178:1223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Car BD, Eng VM, Lipman JM, Anderson TD. The toxicology of interleukin-12: a review. Toxicol Pathol 1999;27:58–63. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1: A representative triplex fluorescent western blot image showing cleavage of XTX301 in primary human tumor specimens. Antibodies against human IL-12 (Green), human IL-12Rβ2 (Red) and human IgG (Blue) were used, a merged image is shown in the figure. The 145 Kda band is the intact XTX301 or non-cleavable construct, the 85Kda band is the cleaved XTX301/unmasked construct, the green arrow indicates free IL-12 released upon cleavage of XTX301. The % of cleavage mXT301 refers to the percentage of cleaved molecule compared to the total (intact and cleaved) molecule detected by western blot using the IL-12Rβ2 signal.
Supplemental Figure S2: XTX301 demonstrates enhanced activity upon proteolytic cleavage in primary cells. (a) Primary mouse splenocytes were pre-activated with PMA & Ionomycin and then incubated with varying doses of rm IL-12 and murine surrogate test articles for 24 hours, and supernatants were assessed by IFN-γ ELISA. (b) Human peripheral blood mononuclear cells (PBMCs) were preactivated with PMA + Ionomycin and incubated with test articles for 4 days and (c) Cynomolgus monkey PBMCs were preactivated with PMA + Ionomycin and incubated with varying doses of rhIL-12 and test articles for 24 hours and evaluated for IFN-γ production by ELISA.
Supplemental Figure S3: Repeated mXTX301 dosing was well tolerated and resulted in dose dependent TGI in mice bearing large (~360mm3) MC38 tumors (a) C57BL/6J mice were implanted subcutaneously with MC38 tumor cells and received a single intravenous injection of mXTX301, unmasked control, or vehicle (PBS) at indicated dose levels (N=12 per group). Tumor measurements were taken two/three times a week. Animals were euthanized due to health issues or body weight loss. The data represent individual survival curves. CR: complete regression, EU: euthanized. (b) C57BL/6J mice were implanted subcutaneously with MC38 tumor cells and received a total of 3 doses marked by red arrows of mXTX301. Body weight was monitored over time (c) Survival prolongation was assessed by Gehan-Breslow-Wilcoxon test comparison at all study time points (until Day 27). ***p = 0.0001; ****p < 0.0001.
Supplemental Figure S4: Gating strategy for multi-color flow cytometry analysis of tumor-infiltrating and splenic immune cell populations.
Supplemental Figure S5: IFN-γ production in plasma: MC38 tumor-bearing mice were treated with the indicated doses of mXT301 or unmasked control. Plasma IFN-γ concentration was measured by MSD assay at indicated time points. The data represent mean ± SEM, N = 3 to 5 mice per group, and IFN-γ level was assessed by a two-way ANOVA with Bonferroni’s post-hoc pairwise comparison test compared to vehicle (PBS) treated animals (*p < 0.05, **p < 0.005, ****p < 0.0001)
Supplemental Figure S6: XTX301 induced cell signature and pathway analysis: single-sample Gene Set Enrichment Analysis (ssGSEA) was performed for all samples and for selected gene sets of interest (see figure 5e). Each ssGSEA score summarizes the overall expression level of genes involved in a certain biological process of interest in a biological sample. For each gene set, ssGSEA scores were analyzed with respect to treatment groups and significant differences compared to the vehicle group were determined by Wilcoxon rank sum test. Boxplots showing ssGSEA scores (y-axis) of all samples for gene sets of interest. Each point is a sample. Samples were grouped by treatment group (x-axis). Colors indicate different treatment groups. * (Wilcoxon test, p< 0.05) ** (Wilcoxon test, p<0.01).
Supplemental Figure S7: XTX301 caused transient and reversible changes to LFTs and other safety parameters in NHP in a repeat dose GLP toxicology study. Male and female NHPs (N=3/sex; 2/sex for recovery animals) were administered XTX301 by intravenous infusion once weekly for a total of four doses, followed by a four-week recovery period. Changes in (a) aspartate aminotransferase, (b) alanine aminotransferase, (c) total lymphocytes, (d) albumin, and (e) IFN-γ were fully reversible in the main study and recovery animals. All values are shown as mean ± SD. The reference ranges, indicated by the grey-shaded area, were derived from the study facility’s historical control values.
Supplementary Table S1: The catalytic efficiency (kcat/Km) of MMP cleavage of XTX301. MMPs from 5 different classes, collagenases (MMP1 and MMP8), gelatinases (MMP2 and MMP9), matrilysin (MMP7), stromelysins (MMP10), and membrane-anchored (MMP14) were selected and evaluated for their ability to cleave XTX301 in vitro. The catalytic efficiency (kcat/Km) of the MMP cleavage of XTX301 was determined by incubating XTX301 with activated MMPs. The data points represent the mean of 2 independent experiments analyzed as technical replicates (in duplicate) by CE-SDS.
Supplementary Table S2: pSTAT4 EC50 in HEK-Blue IL-12 RGA and primary CD8 T cells IL-12 HEK-Blue reporter gene cells were incubated with either rh IL-12, unmasked control, or XTX301 at varying doses, and the reporter activity was measured. The data from three independent experiments, the average of replicates from each experiment are listed in the table. Pre-activated primary human PBMCs were incubated with either rhIL-12, unmasked control or XTX301 at varying doses, and STAT4 phosphorylation was assessed in CD8+ T cells via flow cytometry. The EC50 was calculated as described in the statistical analysis section. The data represents two independent experiments, each conducted with two different PBMC donors.
Supplementary Table S3: IFN-γ production EC50 in mouse, human, and cynomolgus primary cells The ability of mXTX301 to induce IFN-γ production in mouse splenocytes and XTX301 to induce IFN-γ production in human and cynomolgus monkey PBMCs was assessed via ELISA. The EC50 was calculated as described in the statistical analysis section.
Data Availability Statement
RNA-seq and gene expression data that support the findings of this study are available on NCBI Gene Expression Omnibus under accession number GSE237982. All other data generated in this study are available within the article and Supplementary Data files.