

Abstract
FmlH, a bacterial adhesin of uropathogenic Escherichia coli (UPEC) has been shown to provide a fitness advantage in colonizing the bladder during chronic urinary tract infections (UTIs). Previously reported ortho-biphenyl glycosides based on βGal and βGalNAc have excellent binding affinity to FmlH and potently block binding to its natural carbohydrate receptor but lack oral bioavailability. In this present paper, we outline studies where we have optimized lead biphenyl galactosides for improved pharmacokinetics leading to the discovery of novel analogs displaying good oral bioavailability. We synthesized galactosides with the anomeric O-linker replaced with more stable S- and C-linked linkers. We also investigated modifications to the GalNAc sugar and modifications to the biphenyl aglycone. We identified GalNAc 69 with an IC50 of 0.19 μM against FmlH and excellent 53% oral bioavailability in mice. Lead compound 69 (AM4085) has potential as a new anti-virulence therapeutic for the treatment of chronic UTIs.
Keywords: glycomimetic, lectin, FmlH, pili, urinary tract infection, glycoside, virulence factor, adhesin, uropathogenic E. coli
Graphical Abstract
INTRODUCTION
Urinary Tract Infections (UTIs) are one of the most common bacterial infections affecting millions of people globally and more than 50% of women will have at least one UTI during their lifetime with almost all caused by uropathogenic strains of the Gram-negative Escherichia coli (UPEC) pathogen. UPEC encode for a variety of virulence factors which are important in facilitating bacterial colonization of and persistence in the urinary tract despite an effectively functioning host defense mechanism1. One of the most important and well-studied determinants of UPEC urovirulence are chaperone-usher pathway (CUP) pili, for which 38 distinct types have been identified thus far. CUP pili are highly extended proteinaceous extracellular fibers expressed by E. coli and other Gram-negative pathogens that are tipped by a two-domain adhesin with a receptor binding domain or lectin domain (LD) for specifically recognizing and binding to carbohydrate receptors.
The most well-known and studied CUP pilus of UPEC is the type 1 pilus which contains the adhesin FimH. FimH recognizes and binds specifically to αMan bearing receptors on bladder and kidney2 epithelial cells. Since 2010, we and others have published extensively on the development of mannoside FimH ligands3–14 as antagonists of bacterial binding in UTIs15 of the bladder (cystitis), and also useful for Crohn’s Disease16–18. As an extension of our work, Fimbrion and GSK have further optimized and developed the orally bioavailable FimH inhibitor clinical candidate GSK3882347, currently in Phase 1b human clinical trials for the treatment of women with uncomplicated UTI. More recently, we have been developing antagonists to the adhesin of the F9/Yde/Fml CUP pilus, FmlH. This pilus has been demonstrated to provide a clear fitness advantage to UPEC in chronic infections of the bladder19 as well as the kidney. The lectin domain of FmLH (FmlHLD) binds Thomsen-Friedenreich (TF) Antigen, Gal(β1–3)GalNAc, on the naïve kidney and chronically infected bladder epithelium. Fml pili enhance UPEC colonization during chronic urinary tract infection (UTI) and vaccination with FmlHLD protects against UTI progression19.
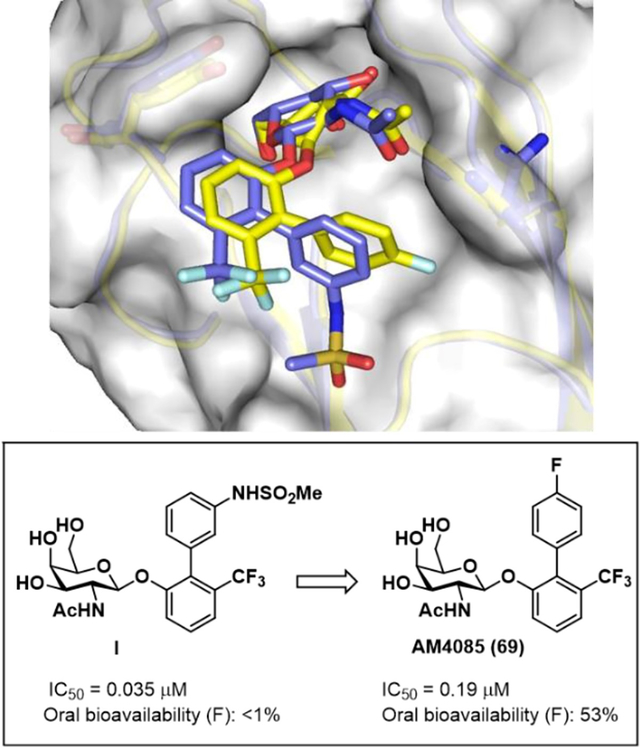
In previous studies, we utilized X-ray crystallography structure-based design (SBDD) combined with directed organic synthesis for developing high-affinity compounds that bind to and competitively inhibit FmlHLD function, relating to adherence to βGal and βGalNAc containing carbohydrate receptors in the bladder and kidney.20 From virtual screening we identified ONPG (ortho-nitrophenyl-β-galactoside) as a FmlH ligand with a KD of 10 μM. Lead identification of ortho-biphenyl galactosides and subsequent medicinal chemistry optimization efforts were rigorously pursued and guided by X-ray structure-based rational design by obtaining multiple co-crystal structures of compounds bound to the lectin domain of FmlH.20, 21 In these studies, we discovered new high-affinity ortho-biphenyl Gal and GalNAc glycomimetic FmlH ligands such as I (Figure 1B) with excellent potency in blocking FmlH adhesion to TF (IC50=34 nM) and good metabolic stability. We also demonstrated that treatment with a FmlH inhibitor reduces bladder and kidney colonization in a mouse model of chronic UTI. Finally, we showed FmlH antagonists effectively block bacterial binding to human kidney tissues20. However, these compounds had poor pharmacokinetics (PK) in rats and mice, including <1% oral bioavailability.
Figure 1.

A) X-ray co-crystal structure of FmlHLD in Complex with I (PDB ID 6MAW)21; B) Summary of strategies followed to modify compound I for improving ADME (Absorption, Distribution, Metabolism, Excretion) properties, including decreased clearance, as well as increased half-life, AUC, and oral bioavailability.
To identify optimized compounds with improved PK including oral bioavailability without foregoing potency, we utilized existing structure activity relationships (SAR) together with X-ray structural information of FmlH ligand binding (Figure 1A) as a guide to rationally design modifications towards this goal.21 To that end, we modified lead GalNAc I using three diverse strategies as shown in Figure 1B to improve ADME (Absorption, Distribution, Metabolism, Excretion) properties and increase the drug-like characteristics of galactosides. First, we explored different anomeric carbon linkers to the biphenyl aglycone as a way to increase half-life and oral absorption. Second, we pursued modifications to the 2-position NHAc group in an attempt to decrease clearance while increasing half-life and oral bioavailability. In another strategy to improve oral absorption, we investigated multiple modifications, including multi-substitution of the biphenyl B-ring of the aglycone, to increase cLogD (a measure of lipophilicity cLogP@pH 7.4) and decrease total polar surface area (tPSA) while retaining optimal ligand binding geometry with key hydrophobic, hydrogen-bonding, and electrostatic interactions with FmlH. In vivo pharmacokinetic (PK) studies established that two new galactosides in mice display good to very good oral bioavailability in mice. We also obtained a high-resolution X-ray co-crystal structure of one the newly designed ligands bound to FmlHLD. Taken together, this work herein provides additional insight into SAR and glycomimetic rational design strategies relevant to lectin antagonist drug discovery and development.
RESULTS AND DISCUSSION
Design and Synthesis of Biphenyl Galactosides: Structural Modifications to Improve Pharmacokinetic (PK) Properties.
In an earlier study we reported on biphenyl-O-GalNAc sulfonamide compound I as a FmlH antagonist21 which has excellent potency and good stability in plasma and simulated gut and intestinal fluids but high clearance, low half-life, and poor oral bioavailability (<1%) in rats. We hypothesized that the highly polar nature of the galactoside itself, combined with the acidic sulfonamide NH might be responsible for the lack of intestinal cell permeability. Thus, we first explored analogs which replaced the secondary meta sulfonamide with tertiary sulfonamides to block the acidic NH. We also incorporated various bioisosteres of the sulfonamide. In a second set of modifications, we changed the O-glycosidic bond (bridging glycosidic oxygen atom to the aglycone) with a carbon (-CH2-) or sulfur (S) linker. We also explored alternative functional groups to the 2-position NHAc group including fluoro and nitro substitutions, as well as varying substituents on the biphenyl B-ring to increase the LogD and decrease the tPSA (total polar surface area) to improve intestinal cell permeability and oral bioavailability.
To introduce the additional substitution on the methyl sulfonamide and to replace the sulfonamide -NH- with other groups, we followed the general synthetic route shown in Scheme 1. The key ortho-bromo-phenyl GalNAc intermediate 2 was synthesized as previously reported21 by glycosylation reaction between 3,4,6-tri-O-acetyl-N-acetyl-D-galactosamine chloride 122 and 2-bromo-3-(trifluoromethyl)phenol using dichloromethane (DCM), 1N aq. sodium hydroxide (NaOH), tetrabutylammonium bromide (TBAB) at room temperature. Next, compound 2 was subjected to a Suzuki cross-coupling reaction with various phenylboronic acids and pinacolate esters, using Pd(PPh3)4, Cs2CO3, and 1,4-dioxane/water (5:1) at 80 °C to generate protected biphenyl intermediates 3–7 which were carried forward without purification and deprotected via standard treatment with 33% methylamine in absolute ethanol solution to give the final target compounds 8–12 in moderate yields.
Scheme 1a.

Synthesis of biphenyl glycoside compounds (8–12).
aReagents and conditions: (a) DCM, 1 N NaOH, TBAB, 2-bromo-3-(trifluoromethyl)phenol, r.t.,1 h; (b) Pd(PPh3)4, Cs2CO3, 1,4-dioxane/water (5:1), 80 °C, 1 h; (c) 33% methylamine in absolute ethanol, r.t., 1h for 8–12; See Table 1 for identity of R1, R2.
Biochemical Activity of ortho-biphenyl GalNAc Compounds 8-12.
The ability of the newly synthesized GalNAc analogs 8–12 to inhibit FmlH binding was assessed using an enzyme-linked immunosorbent assay (ELISA) using TF antigen as a competitive ligand, as we have previously described20. The resultant IC50 values for each compound are shown in Table 1. While still highly active at blocking FmlH, the tertiary sulfonamide compounds 8 (IC50 2.0 μM) and 12 (IC50 0.60 μM), in addition to methylene nitrile (10) had significantly reduced potency relative to parent compound I. Better results were observed with methyl sulfone (9) and the keto sulfonamide (11) analog only losing ~10-fold activity with an IC50 of 0.45 and 0.31 μM, respectively. These results and SAR indicate the importance of the sulfonamide oxygen atoms in making key electrostatic and/or H-bonding binding interactions with FmlH as we previously reported21. Analysis of calculated physical properties (Table 1) suggested that we achieved increases in cLogD and decreased tPSA relative to I with all compounds including 9 except the most potent sulfonyl urea analog 11 which has a much lower cLogD of −1.4 and higher tPSA of 171.5. While these promising results suggested analog 9 would have improved pharmacokinetic properties, the lack of potency towards FmlH precluded us from pursuing further studies.
Table 1.
Biological data for biphenyl galactosaminosides I and 8-12.

| |||||||
|---|---|---|---|---|---|---|---|
| Compound Name | R1 | R2 | IC50 (μM)a | cLogP | cLogD7.4 | CMR (cm3/mol) | tPSA (Å2) |
| I 21 | NHSO2Me | H | 0.035 | 1.9 | 0.7 | 11.92238 | 154.4 |
| 8 | NMeSO2Me | H | 2.0 | 1.4 | 0.7 | 12.5818 | 145.6 |
| 9 | CH2SO2Me | H | 0.45 | 2.1 | 1.2 | 12.06086 | 142.4 |
| 10 | CH2CN | H | 0.80 | 2.2 | 1.5 | 11.26814 | 132 |
| 11 | CONHSO2Me | H | 0.31 | 2.1 | −1.4 | 12.42881 | 171.5 |
| 12 | −N(SO2Me)CH=CH− | 0.60 | 3 | 1.6 | 12.68255 | 147.3 | |
All IC50 values are an average of four replicates
Replacement of O-glycosidic bond and structure-activity relationship (SAR) of its alternatives.
In additional efforts to overcome the rapid clearance, short half-life (t1/2 = 1.16 h and Cl = 57.0 mL/min/kg in rats) and poor oral bioavailability (F <1%) of compound I, we explored a series of direct replacements to the O-glycosidic bond. While the O-glycoside I has good stability in plasma and simulated intestinal and gut fluids21, one cannot rule out action by glycosidases being responsible for the rapid clearance and/or lack of oral bioavailability. Sulfur (S) and carbon (C)-glycosides have increased stability which are not subject to enzymatic hydrolysis by glycosidases. Thus, we constructed three different S-linked analogs (16–18; Figure 2) keeping the meta-substitution as either the sulfonamide of compound I or the carboxyl group of earlier galactosides20, 21. The general synthetic route for the biphenyl S-GalNAc analogs (16-18) is shown in Scheme 2. The key S-GalNAc aryl bromide intermediate 13, was synthesized via thioglycosylation reaction of 122 with 2-bromo-3-methylbenzenethiol. Next, 13 was subjected to a Suzuki cross-coupling reaction with the indicated phenylboronic acids to generate protected biphenyl intermediates 14–15, which were then brought forward to the next step without purification and treated with 33% methylamine in ethanol to give the target compounds sulfonamide 16 and methyl ester 17, with the latter being further converted to carboxylic acid 18 by basic hydrolysis.
Figure 2.

Biological data for biphenyl S-galactosaminosides 16–18.
Scheme 2a.

Synthesis of biphenyl thio GalNAc analogs 16–18.
aReagents and conditions: (a) 2-bromo-3-methylbenzenethiol, DCM, 1 N NaOH, TBAB, r.t., 1 h; (b) Pd(PPh3)4, Cs2CO3,1,4-dioxane/water (5:1), 80 °C, 1 h; (c) 33% methylamine in absolute ethanol, r.t., 1 h; (d) NaOH, methanol/water (1:1), r.t., overnight
We next focused on constructing C-linked galactosides, previously shown to have superior metabolic stability compared to either the O- and S-glycosides in the FimH mannosides3. The synthetic routes to the C-linked galactosides are shown in Schemes 3–5. We first synthesized two analogs of compound I which had the O atom replaced with a methylene CH2 in one analog (27) with also the 2-NHAc (GalNAc) switched to a 2-OH (Gal) while in the other analog it is replaced with a F (28) (Scheme 3). Starting from benzyl protected alkenes 19 or 20 (obtained from Petasis reaction of lactone (3R,4S,5S,6R)-3,4,5-tris(benzyloxy)-6-((benzyloxy)methyl)tetrahydro-2H-pyran-2-one23, X=OBn or (3R,4S,5S,6R)-4,5-bis(benzyloxy)-6-((benzyloxy)methyl)-3-fluorotetrahydro-2H-pyran-2-one, X=F)24, 25, hydroboration of the alkene with 9-borabicyclo[3.3.1]nonane (9-BBN) is followed by carbon-carbon bond formation with 2-iodo-phenol in the precence of PdCl2(dppf) to give 21 and 22. Conversion of the phenol to the triflates 23 and 24 is followed by Suzuki type cross-coupling with (3-(methylsulfonamido)phenyl)boronic acid to give 25 and 26 and then, global debenzylation with boron trichloride yields the target compounds 27 and 28.
Scheme 3a.

Synthesis of biphenyl of C- glycosides 27-28.
aReagents and conditions: (a) i. THF, 9-BBN (0.5M in THF), reflux, 15 h; ii. PdCl2(dppf), 3M K3PO4, DMF; 24 h (b) Tf2O, TEA, DCM, 0 °C, 3 h; (c) Pd(PPh3)4, Cs2CO3, 1,4-dioxane/water (5:1), 80 °C; (d) DCM, BCl3 (1 M in DCM), −78 °C, 3h.
Scheme 5a.

Synthesis of biphenyl glycosides evaluating effect of amide link 41.
aReagents and conditions: (a) MTBE, TMSCN, TBAF (1 M in THF), −78 °C, 2 h; (b) i. THF-Water (1:1), conc. HCl, AcOH, Zn-dust, 0 °C, 2 h; ii. DCM, Ac2O/TEA, DMAP (cat.), r.t., 2 h; (c) dioxane/water (10:1), 4M HCl in dioxane, 60 °C, 2 h; d) 2-bromo aniline, i-butyl chloroformate, NMM, TEA, 0 °C 1 h then r.t., 15 h; e) 3-methylsulfonylamino phenylboronic acid, Pd(PPh3)4, Cs2CO3,1,4-dioxane/water (5:1), 80 °C, 1 h; (f) DCM, BCl3 (1 M in DCM), −78 °C, 3 h.
Synthesis of the biphenyl GalNAc carbon linked analogs 34–35 is shown in Scheme 4. Compound 30 was synthesized by nitro Michael-type addition reaction between tribenzyl nitrogalactal intermediate 2926, 27 and 2-bromobenzylmagnesium bromide in THF at −78 °C. The nitro group of compound 30 was converted reduced to the amine using zinc and acetic acid, which was subsequently acetylated using acetic anhydride and triethylamine (cat. DMAP) at room temperature to give NHAc derivative 31. Suzuki cross-coupling reaction of intermediates 30 and 31 with 3-methylsulfonylamino phenylboronic acid using Pd(PPh3)4, Cs2CO3, and 1,4-dioxane/water (5:1) at 80 °C were employed to generate the benzyl protected biphenyl intermediates 32 and 33, which following treatment with BCl3 at −78 °C gave target compounds 34 and 35.
Scheme 4a.

Synthesis of biphenyl of C- glycosides 34-35.
aReagents and conditions: (a) THF, 2-bromo benzyl magnesium bromide (0.25 M in Et2O), −78 °C, 2 h; (b) i. THF/water (1:1), conc. HCl, AcOH, Zn-dust, 0 °C, 2 h; ii. DCM, Ac2O/TEA, DMAP (cat.), r.t., 2 h; (c) Pd(PPh3)4, Cs2CO3, 1,4-dioxane/water (5:1), 80 °C, 1 h; (d) DCM, BCl3 (1 M in DCM), −78 °C, 3 h.
The general synthetic route for the biphenyl GalNAc amide analog 41 is shown in Scheme 5. Following a similar route to 34 and 35, key β-cyano intermediate 36 was synthesized by nitro Michael-type addition reaction from tribenzyl nitro galactal intermediate 2926, 28 using TMSCN and TBAF in methyl tert-butyl ether (MTBE) at −78 °C. Reduction of the nitro group of compound 36 was achieved once again using zinc and acetic acid and subsequently, the amine was acetylated using acetic anhydride and triethylamine, cat. DMAP at room temperature to yield NHAc compound 37. Next, the cyano group of 37 was converted into the carboxylic acid 38 under strong acid hydrolysis conditions. The amide 39 was synthesized from 38 employing an i-butyl chloroformate derived mixed anhydride activation step followed by reaction with 2-bromo aniline, which was then subjected to a Suzuki cross-coupling reaction with 3-methylsulfonylamino phenylboronic acid utilizing Pd(PPh3)4, Cs2CO3, and 1,4-dioxane/water (5:1) at 80 °C to generate benzyl protected biphenyl intermediate 40. The final target compound 41 was then generated via treatment of the precursor 40 with BCl3 at −78 °C.
Biological activity of glycosidic (anomeric) linker replacement galactosides.
Shown in Figure 2, the FmlH inhibitory activity (ELISA assay) of the S-linked galactosides 17 (IC50 1.3 μM) and 18 (IC50 1.0 μM) were found to be slightly less when compared to the corresponding O-glycoside matched pair compounds (O analog of 18, IC50 = 0.35 μM; O analog of 17, IC50 = 0.63 μM) while 16 (S replaced for the O) lost ~6-fold activity (IC50 0.40 μM) relative to its matched pair (O-analog of 16, IC50 = 0.06 μM)21.
When the C-linked analogs were tested, the methylene (CH2) Gal 27 and GalNAc 35 derivatives also showed weak activity both displaying an IC50 of only 15 μM (Table 2). Replacing the 2-position OH with a F improved the potency 2-fold (IC50 7.3 μM) as seen in compound 28. Interestingly, when the 2-OH was replaced with a nitro group as in 34, all activity against FmlH was eliminated. Finally, the C-linked amide analog 41 has the best activity with an IC50 of 6.9 μM, although this is obvously much weaker when compared to the O-linked parent galactosides. This result is reminiscent of simple C-linked FimH mannoside antagonists which lost activity3. The loss in activity when replacing O with CH2 can be a result of multiple factors including loss of water mediated H-bonding to the anomeric oxygen not possible with CH2 and/or conformational changes of the aglycone itself. Due to the weak activities of the anomeric linker replacements against FmlH, we didn’t pursue any further studies evaluating their pharmacokinetic properties.
Table 2.
Biological data for biphenyl C-galactosides 27–28, 34–35, 41

| |||
|---|---|---|---|
| Compound Name | X | Y | IC50 (μM)a |
| II 20 | NHAc | O | 0.23 |
| III 20 | OH | O | 1.6 |
| 27 | OH | −CH2− | 15 |
| 35 | NHAc | −CH2− | 15 |
| 28 | F | −CH2− | 7.3 |
| 34 | NO2 | −CH2− | >50 |
| 41 | NHAc | −CONH− | 6.9 |
All IC50 values are an average of four replicates
Improving oral bioavailability and half-life through structural simplification of the aglycone.
As discussed above, the activity of all the sulfonamide isosteric replacements and the S-linked and C-linked galactosides which we evaluated was decreased relative to their matched pair compounds. Therefore, in order to develop galactosides with improved ADME properties but that also retain FmlH binding affinity and functional activity, we took an alternate design strategy to investigate a variety of modifications to the aglycone portion of previous lead compound I, specifically to increase LogD and decrease tPSA.
To this end, we replaced the highly polar meta methyl sulfonamide group of the biphenyl B-ring with a variety of other substituents like F, CF3, CN, OMe to decrease polarity while still exploiting the charge-charge and hydrogen bonding interactions with the guanidinium side chain of R142 and K132 (Figure 1). We held the ortho CF3 group constant on the biphenyl A-ring to maximize potency as we discovered in our previous report21. These compounds were synthesized as outlined in Scheme 6 starting with key intermediate 2 which was reacted with various aryl boronic acids in a Suzuki coupling to yield acetyl protected galactosides 42–56. Next, deprotection of the crude intermediates 42–56 was performed using standard conditions to yield the final target compounds 57–71. The ability of the newly synthesized analogs to inhibit FmlH activity was assessed as before with the results shown in Table 3. We found the mono-substituted compounds having a F or other H-bond acceptors in place of the sulfonamide (meta position) lose potency as exemplified by 57–60. The same result was found when this position contained a CF3 group as in 61 and 62. Having said that, it is noteworthy that in all analogs that the R4 (other meta of the B-ring) position also has substitution with either a F, CF3, CN or OMe group (Table 3). In fact, when the R4 position is H as with compound 68, much of the potency returns (IC50 = 480 nM). In contrast, putting a F in the adjacent (para) position retains much of the potency as seen in 69 which has an IC50 of 190 nM. The same we found true for di-F substituted analogs 66 and 67 and tri-F analog 63.
Scheme 6a.

Synthesis of biphenyl glycoside compounds (57–71).
aReagents and conditions: (a) Pd(PPh3)4, Cs2CO3,1,4-dioxane/water (5:1), 80 °C, 1 h; (b) 33% methylamine in absolute ethanol, r.t., 1 h for 57-71; See Table 1 for identity of R1, R2, R3, R4.
Table 3.
Biological data for biphenyl galactosides and galactosaminosides 57−71.
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound Name | R1 | R2 | R3 | R4 | IC50 (μM)a | cLogP | cLogD7.4 | CMR (cm3/mol) | tPSA (Å2) |
| I 21 | H | NHSO2Me | H | H | 0.035 | 1.9 | 0.7 | 11.92238 | 154.4 |
| 57 | H | F | H | F | 6.0 | 3.1 | 2.3 | 10.28273 | 108.2 |
| 58 | H | F | H | CF3 | 9.0 | 4.1 | 2.9 | 10.85846 | 108.2 |
| 59 | H | F | H | CN | 2.0 | 2.6 | 1.7 | 10.83486 | 132 |
| 60 | H | F | H | OMe | 6.0 | 3 | 2.1 | 10.90741 | 117.5 |
| 61 | H | CF3 | H | CN | 6.0 | 2.5 | 2.3 | 11.41059 | 132 |
| 62 | H | CF3 | H | OMe | 18 | 4 | 2.7 | 11.48314 | 117.5 |
| 63 | F | F | F | H | 0.70 | 3.2 | 2.4 | 10.30437 | 108.2 |
| 64 | F | F | H | H | 0.59 | 3 | 2.2 | 10.28273 | 108.2 |
| 65 | F | H | H | H | 0.71 | 2.8 | 1.9 | 10.26109 | 108.2 |
| 66 | F | H | F | H | 0.32 | 3.1 | 2.1 | 10.28273 | 108.2 |
| 67 (AM4075) | H | F | F | H | 0.24 | 2.7 | 2.3 | 10.28273 | 108.2 |
| 68 | H | F | H | H | 0.48 | 2.8 | 2 | 10.26109 | 108.2 |
| 69 (AM4085) | H | H | F | H | 0.19 | 2.9 | 2.1 | 10.26109 | 108.2 |
| 70 | H | NHSO2Me | F | H | 0.89 | 2 | 0.6 | 11.94402 | 154.4 |
| 71 | H | NHSO2Ph | F | H | 0.19 | 3.5 | 1.5 | 13.94431 | 154.4 |
All the IC50 values are an average of four replicates
The ortho-F derivative 65 with an IC50 of 710 nM is less preferred for FmlH than either the meta (68) or the para (69) derivatives. In a final set of compounds, we retained the meta sulfonamide group on the B-ring but added a F to the para position. Surprisingly, the methyl sulfonamide 70 analog lost significant activity relative to I with an IC50 of 890 nM while the phenyl sulfonamide 71 was found to be significantly more active with an IC50 of 190 nM. Compound 71 and para-F galactoside 69 had the best FmlH activity with IC50s of 190 nM, followed closely by meta, para di-F galactoside 67, and ortho, para di-F galactoside 66 with IC50s of 240 nM and 320 nM, respectively. With the exception of sulfonamide 71, the other potent analogs 66, 67, and 69 also show the best improvements in their physiochemical properties (Table 3) with substantial increases in cLogD from 0.7 of I to around 2 and decreases in tPSA from 154 to 108, as well as decreased calculated molar refractivity (CMR) from 11.9 to 10.3. These are three parameters which often influence cellular permeability of small molecules, in some cases directly correlating with oral bioavailability. For example, 67 has a cLogP of 2.7 and a cLogD of 2.3 with a tPSA of 108.2 and a CMR of 10.28 whereas starting compound I has a cLogP of 1.9 and cLogD of 0.7 with a tPSA of 154.4 and CMR of 11.92. GalNAc 69 is similar to 67 with a cLogP of 2.9, cLogD of 2.1, tPSA of 108.2, and CMR of 10.26. These results, combined with their best potency attributes, prompted us to conduct X-ray crystallographic studies with FmlH to understand our SAR, in addition to evaluation of the pharmacokinetic properties of 67 and 69 in mice.
X-ray co-crystal structure of galactoside 69 (AM4085) bound to FmlH.
We obtained a 1.63 Å high-resolution structure of our newly discovered lead GalNAc 69 co-crystallized with the lectin domain of FmlH (Figure 3). The para-fluoro group appears to be within electrostatic interaction distance (3.5 A) with both of the guanidinium nitrogens of R142. Y46 also seems to be in an edge-to-face π-stacking electrostatic interaction with the biphenyl A-ring containing the CF3 group. Shown in Figure 4a, the structure of 69 resembles I (yellow, PDB: 6MAW)21 with respect to the GalNAc sugar binding but both the biaryl A- and B-rings positions are significantly altered and twisted relative to that seen in that of compound I which contains a meta-sulfonamide on the B-ring whereas 69 has a para-F group. The torsion angle (twist) between the two biphenyl rings is much larger in 69, apparently to allow for the F to engage in an electrostatic and hydrophobic interaction with FmlH via R142. However, the interaction with the sulfonamide and FmlH is much stronger as can been deduced from the potency differences between I and 69. Interestingly, when comparing the bound structure with our original hit lead compound IV (FmlH IC50 = 0.64 μM) as shown in Figure 4b (blue, PDB: 6AS8)20, we found that the overlay is better with the sugar and both rings of the biphenyl on 69 closely matching the positions and torsion angles of the GalNAc compound IV having the meta 3-carboxy group on the biphenyl B-ring. It should be noted though that the ortho-CF3 group of I and 69 is not present on the A-ring of this compound. The twisted ring of I could be due to the close proximity of the CF3 and the sulfonamide groups. Alternatively, the twist could be allowing for binding interactions with FmlH in the first place. In either case, we surmise that the loss in binding affinity of 69 relative to I has to do with a loss in entropy upon binding of 69. This premise is partly based on the significant loss in binding affinity seen by compound 70 by simply adding the para-F substituent to compound I.
Figure 3.

Compound 69 (AM4085) bound to the FmlH lectin domain. PDB ID: 8USM (Data collection and refinement in Table S2).
Figure 4.

Overlay of the X-ray structure of 69 (green, PDB: 8USM) with A) I (yellow, PDB: 6MAW)21, B) with I (yellow), and IV (blue, PDB: 6AS8)20.
ADME and in vivo pharmacokinetic (PK) studies.
In previous studies on Gal and GalNAc based FmlH antagonists including compound I, metabolic stability was assessed for the most potent lead compounds in simulated gastric fluid (SGF), simulated intestinal fluid (SIF), mouse liver microsomes, and blood plasma21. All compounds evaluated exhibited a high degree of stability, with some variation seen in the plasma stability. These findings are consistent with our earlier characterization of FimH O-mannoside antagonists but we found the O-galactosides are generally more stable than the latter5, 7. In these current studies, we tested the new most promising compounds 67 and 69 for their PK in mice where we determined the concentration of compounds 67 and 69 in plasma following either a 10 mg/kg oral dose (PO; red triangles) or a 3 mg/kg intravenous dose (IV; blue diamonds) (Figure 5). Analysis of the mouse PK data (Figure 5) revealed that compounds 67 and 69 both had respectable oral bioavailability (% F) of 31% and 53%, respectively. Interestingly, 69 contains only one fluorine on the B-ring while there are two fluorine atoms on the B-ring of 67 resulting in the lower oral bioavailability. In addition to the higher oral bioavailability, the AUC of 69 is almost 4-fold higher than that of 67 and its clearance is also slower going from 28 to 13.6 mL/min/kg (Figure 5), but these differences cannot be explained by their cLogD, CMR, and tPSA, which are all similar for both compounds. A plausible explanation might be gleaned from the Cmax which is three times higher for 69 and could be a result of decreased solubility of 67 relative to 69 leading to decreased absorption. Nonetheless, in an additional PK study we determined the urine concentrations of lead compound 69 over a 24-hour period after a 2 mg/kg and 20 mg/kg PO dose in mice. Shown in Figure 6, urine concentrations of 69 reached and maintained levels many fold above the IC50 for over 12 hours in both the low dose and high dose groups. This data, consisting of good oral bioavailability coupled with its exceptionally high urine levels above the FmlH IC50 strongly supports 69 (AM4085) as a promising oral candidate drug for testing in animal models of chronic urinary tract infections.
Figure 5.

In vivo pharmacokinetics (PK) of 67 (AM4075) and 69 (AM4085) in mice following a 3 mg/kg IV and 10 mg/kg PO dose. Shown is the mean plasma concentration over an 8 h period. Oral bioavailability (%F) of 67 and 69 were calculated to be 31% and 53% respectively.
Figure 6.

Urine concentration of 69 (AM4085) in mice following a 2 mg/kg and 20 mg/kg PO dose. Shown is the mean urine concentration over a 24 h period. 1 μM = 459.39 ng/mL; IC50 = 0.19 μM. Concentrations at the 24 h timepoint are 15.8 and 66.3 ng/mL.
CONCLUSION
The constant rise in antibiotic resistance is a cause for concern especially with most pharmaceutical companies abandoning their drug discovery programs. There is an urgent need to identify new therapeutic approaches to conquer this problem. Targeting virulence mechanisms in bacteria is an attractive approach since it often does not kill the bacteria but does disables primary functions of the bacteria necessary for survival in the infected host. Bacterial adhesins which bind to glycoprotein receptors via their lectin domains are one example of such virulence factors. These lectins are not only necessary for bacterial attachment to target surfaces but also are critical to the formation of protective biofilms to elude the immune system. The most studied lectin is FimH which is important in the pathogenesis of UTIs. We previously developed potent orally bioavailable glycomimetic mannosides targeting FimH via rational drug design, and which has culminated in a clinical candidate drug GSK3882347, now being evaluated in humans. In this paper, we have extended our subsequent work to the rational design of new orally bioavailable galactoside lectin antagonists against FmlH. In UPEC, this factor is important in chronic UTIs where remodeling of the glycoprotein receptors on the bladder epithelium resulting in a significant increase in βGalNAc tipped receptors. Here, we followed a logical three-pronged systematic approach to improve the oral bioavailability of previous lead compounds20, 21. We successfully identified two new lead compounds, 67 (AM4075) which has good 31% intestinal permeability and 69 (AM4085) which has a better 53% oral exposure in mice. This result also translated into achieving remarkably high urine concentrations of 69 (61 μM) over 300-fold higher than the FmlH IC50. We also obtained a co-crystal structure of 69 bound to FmlH which explains how we were able to use fluorine substituents in place of sulfonamides without sacrificing a significant amount of binding affinity to FmlH. Using tPSA to predict both CNS and intestinal oral permeability has been applied successfully to many other small molecule drugs29, 30 including the glycomimetic drugs including the P2Y12 antagonist Ticagrelor31, the Galectin-3 inhibitor Selvigaltin32, and most recently SGLT-2 inhibitors33–35. This learned strategy, to increase LogD and decrease tPSA and/or CMR of the aglycone, in this case by adding aryl fluorine or fluorinated alkyl groups for increasing cellular permeability and oral bioavailability can be applied to other glycoside-based drugs13. In summary, we have developed ortho-biphenyl galactoside 69 (AM4085) as a promising lead candidate worthy of further potency optimization and biological evaluation in advanced preclinical studies in the treatment of chronic UTIs of the bladder and kidney.
EXPERIMENTAL
Materials and Methods.
Starting materials, reagents, and solvents were purchased from trusted commercial vendors unless otherwise noted. In general, anhydrous solvents are used for performing all reactions. 1H and 13C NMR spectra were measured on a Varian 400 MHz NMR spectrometer and the chemical shifts were reported as δ ppm relative to TMS using residual solvent peak as the reference unless otherwise noted. The following abbreviations were used to express the peak multiplicities: s = singlet; d = doublet; t = triplet; q = quartet; m = multiplet; br = broad. Preparatory High-Performance Liquid Chromatography (HPLC) was conducted on a GILSON GX-281 using and Waters Prep C18 5μM, 19*150mm reverse phase column, eluted with a gradient system of 5:95 to 95:5 acetonitrile:water with a buffer consisting of 0.05–0.1% TFA. Mass spectroscopy (MS) was performed on HPLC/MSD Agilent system using a C18 reversed phased Waters C18 5μM, 4.6*50mm column with a solvent gradient of 5:95 to 95:5 acetonitrile:water with a buffer consisting of 0.05–0.1% TFA and electrospray ionization (ESI) for detection. All reactions were monitored by thin layer chromatography (TLC) carried out on either Merck silica gel plates (0.25 mm thick, 60F254) and visualized by using UV (254 nm) or stains such as 5% H2SO4 in ethanol. Silica gel chromatography was carried out on a Teledyne ISCO CombiFlash MPLC purification system using pre-packed silica gel columns (4 g to 80 g sizes). All final compounds were purified by reverse phase preparative HPLC using a C18 column (acetonitrile/water/TFA mobile phase) giving products as solids. All compounds assessed in biological assays are greater than 98% purity based on 1H and 13C NMR, and HPLC/MS as determined by absorbance of peaks at 220 nm and 254 nm wavelengths.
FmlH Protein Expression and Purification.
FmlH protein was expressed and purified as previously described20. Briefly, protein was expressed in the periplasm of E. coli C600 cells containing pTRC99a encoding the first 182 amino acids of the CFT073 FmlH protein (corresponding to the signal sequence and lectin domain) and a C-terminal 6x-his tag. Periplasmic isolates prepared as previously described were washed over a cobalt affinity column (GoldBio) and eluted in 20 mM Tris 8.0 + 250 mM Imidazole. Fractions containing protein of the expected molecular weight were then diluted 5-fold in 20 mM Tris 8.0 to a final concentration of 50 mM Imidazole, washed over an anion exchange column (GE Healthcare Mono Q) with 20 mM Tris 8.0, and eluted in 20 mM Tris 8.0 + 250 mM NaCl. Resulting fractions were pooled and dialyzed in 1 mM HEPES pH 7.5 + 50 mM NaCl and concentrated as needed for further study. Protein used in ELISA assays was biotinylated using an NHS-PEG4-Biotin and Biotinylation Kits (ThermoFisher).
ELISA Assay and Determination of IC50 Values.
Enzyme-linked immunosorbent assays (ELISAs) were used to quantify the IC50 of final galactoside compounds as previously described20. Briefly, 1 μg bovine submaxillary mucin (Sigma) in 100 uL PBS were incubated with Immulon 4HBX 96-well plates overnight prior to treatment with 1 mU Arthrobacter ureafaciens sialidase for 1 hour at 37 °C to remove terminal sialic acid sugars. Wells were then blocked with 200 μL PBS + 1% BSA for 2 hours at room temperature. Biotinylated FmlHLD was diluted to 20 μg/mL in blocking buffer and incubated in the presence or absence of compounds serially diluted 2x down eight rows for 1 hour at room temperature. Wells were washed three times with PBS 0.05% TWEEN-20 then incubated with 100 μL of streptavidin-HRP conjugate (BD Biosciences; 1:2,000 dilution in blocking buffer) for one hour. After three additional PBS +0.05% TWEEN washes, plates were developed with 100 μL of tetramethylbenzidine (BD Biosciences) substrate and quenched with 50 μL of 1 M H2SO4. Total bound portion concentration was measured by the absorbance at 450 nm. IC50s were determined using the GraphPad Prism software.
Crystallography.
The UTI89 FmlH lectin domain, truncated immediately before the penultimate cysteine, with a 6-his tag in 20 mM Tris 8.2 + 20 mM NaCl was co-crystallized with AM4085 dissolved in 100% DMSO via the hanging drop method. 75 uL of non-filtered FmlH lectin domain with a concentration range of 8.118–15.87 mg/mL (measured against H2O) was co-incubated with 2 uL of 200 mM AM4085 in DMSO for approximately 1 hour at room temperature rotating at 75 rpm in the dark. Co-crystallization was achieved over 0.7 M LiSO4 + 2% PEG 8000. 0.15 M Tri-Na-Citrate + 0.1 M HEPES 7.5 + 5% Isopropanol + 20% Glycerol was used as the cryoprotectant solution. Data was collected at ALS Beamline 4.2.2. Data processing was performed in XDS and molecular replacement with 6AOY was performed in Phenix. The SMILES string CC(=O)N[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1OC1=CC=CC(C(F)(F)F)=C1C1=CC=C(F)C=C1 was used to generate Compound 69 (AM4085) in Phenix. Ligand fitting, restraints, and overall refinement was performed in Phenix. Data collection and refinement information was submitted to the Protein Data Bank (PDB ID: 8USM). Authors will release the atomic coordinates upon article publication.
Mouse Pharmacokinetics (Urine Levels).
Compound 69 (AM4085) was administered via oral (PO) dosing at 2 mg/kg and 20 mg/kg to 9-week-old C3H/HeN mice from Envigo (n=5 per group). Compound 69 was prepared in 1% methyl cellulose, vortexed for 30 s, sonicated another 20 min followed by placing in a 50 °C water bath for 10 min and vortexing for a final 30 s. The compound was then immediately administered via oral gavage. Urine was collected at 0.5, 1.5, 3, 6, 9, 12, and 24 h post dosing and analyzed for compound concentrations using standard HPLC/MS/MS techniques.
Supplementary Material
ACKNOWLEDGMENTS
We thank the National Institutes of Health (NIH) for funding awarded by the National Institute of Diabetes and Digestive Diseases (NIDDK), R01 DK10884 and National Institutes of Allergy and Infectious Diseases (NIAID), U19 AI157797 and R37 AI048689. We also thank Jay Nix at ALS 4.2.2 for his help with data processing.
ABBREVIATIONS USED
- ACN
acetonitrile
- aq
aqueous
- DMSO
Dimethyl sulfoxide
- DCM
Dichloromethane
- DMF
dimethylformamide
- °C
degrees Celsius
- MsCl
methanesulfonyl chloride
- μM
micromolar
- PDB
Protein Data Bank
- TEA
Triethyl amine
- TBAF
tetrabutylammonium fluoride
Footnotes
ASSOCIATED CONTENT
Supporting information includes details on the synthesis of compounds, 1H and 13C NMR, HPLC/MS spectral data of all the final tested compounds as well as the X-ray crystallography parameters and animal protocols with raw data for the mouse pharmacokinetic studies performed by WuXi AppTec (Shanghai) Co., Ltd. Molecular formula strings for all final compounds are also provided.
REFERENCES
- (1).Klein RD; Hultgren SJ Urinary tract infections: microbial pathogenesis, host-pathogen interactions and new treatment strategies. Nat Rev Microbiol 2020, 18 (4), 211–226. DOI: 10.1038/s41579-020-0324-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).McLellan LK; McAllaster MR; Kim AS; Tothova L; Olson PD; Pinkner JS; Daugherty AL; Hreha TN; Janetka JW; Fremont DH; et al. A host receptor enables type 1 pilus-mediated pathogenesis of Escherichia coli pyelonephritis. PLoS Pathog 2021, 17 (1), e1009314. DOI: 10.1371/journal.ppat.1009314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Mydock-McGrane L; Cusumano Z; Han Z; Binkley J; Kostakioti M; Hannan T; Pinkner JS; Klein R; Kalas V; Crowley J; et al. Antivirulence C-Mannosides as Antibiotic-Sparing, Oral Therapeutics for Urinary Tract Infections. J Med Chem 2016, 59 (20), 9390–9408. DOI: 10.1021/acs.jmedchem.6b00948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Jarvis C; Han Z; Kalas V; Klein R; Pinkner JS; Ford B; Binkley J; Cusumano CK; Cusumano Z; Mydock-McGrane L; et al. Antivirulence Isoquinolone Mannosides: Optimization of the Biaryl Aglycone for FimH Lectin Binding Affinity and Efficacy in the Treatment of Chronic UTI. ChemMedChem 2016, 11 (4), 367–373. DOI: 10.1002/cmdc.201600006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Han Z; Pinkner JS; Ford B; Chorell E; Crowley JM; Cusumano CK; Campbell S; Henderson JP; Hultgren SJ; Janetka JW Lead optimization studies on FimH antagonists: discovery of potent and orally bioavailable ortho-substituted biphenyl mannosides. J Med Chem 2012, 55 (8), 3945–3959. DOI: 10.1021/jm300165m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cusumano CK; Pinkner JS; Han Z; Greene SE; Ford BA; Crowley JR; Henderson JP; Janetka JW; Hultgren SJ Treatment and prevention of urinary tract infection with orally active FimH inhibitors. Sci Transl Med 2011, 3 (109), 109ra115. DOI: 10.1126/scitranslmed.3003021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Han Z; Pinkner JS; Ford B; Obermann R; Nolan W; Wildman SA; Hobbs D; Ellenberger T; Cusumano CK; Hultgren SJ; et al. Structure-based drug design and optimization of mannoside bacterial FimH antagonists. J Med Chem 2010, 53 (12), 4779–4792. DOI: 10.1021/jm100438s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Klein T; Abgottspon D; Wittwer M; Rabbani S; Herold J; Jiang XH; Kleeb S; Luthi C; Scharenberg M; Bezencon J; et al. FimH Antagonists for the Oral Treatment of Urinary Tract Infections: From Design and Synthesis to in Vitro and in Vivo Evaluation. Journal of Medicinal Chemistry 2010, 53 (24), 8627–8641. DOI: Doi 10.1021/Jm101011y. [DOI] [PubMed] [Google Scholar]
- (9).Schwardt O; Rabbani S; Hartmann M; Abgottspon D; Wittwer M; Kleeb S; Zalewski A; Smiesko M; Cutting B; Ernst B Design, synthesis and biological evaluation of mannosyl triazoles as FimH antagonists. Bioorg Med Chem 2011, 19 (21), 6454–6473. DOI: 10.1016/j.bmc.2011.08.057. [DOI] [PubMed] [Google Scholar]
- (10).Kleeb S; Pang L; Mayer K; Eris D; Sigl A; Preston RC; Zihlmann P; Sharpe T; Jakob RP; Abgottspon D; et al. FimH antagonists: bioisosteres to improve the in vitro and in vivo PK/PD profile. J Med Chem 2015, 58 (5), 2221–2239. DOI: 10.1021/jm501524q. [DOI] [PubMed] [Google Scholar]
- (11).Pang LJ; Kleeb S; Lemme K; Rabbani S; Scharenberg M; Zalewski A; Schadler F; Schwardt O; Ernst B FimH Antagonists: Structure-Activity and Structure-Property Relationships for Biphenyl a-D-Mannopyranosides. Chemmedchem 2012, 7 (8), 1404–1422. DOI: DOI 10.1002/cmdc.201200125. [DOI] [PubMed] [Google Scholar]
- (12).Jiang X; Abgottspon D; Kleeb S; Rabbani S; Scharenberg M; Wittwer M; Haug M; Schwardt O; Ernst B Antiadhesion therapy for urinary tract infections--a balanced PK/PD profile proved to be key for success. Journal of Medicinal Chemistry 2012, 55 (10), 4700–4713, Research Support, Non-U.S. Gov’t. DOI: 10.1021/jm300192x. [DOI] [PubMed] [Google Scholar]
- (13).Schonemann W; Cramer J; Muhlethaler T; Fiege B; Silbermann M; Rabbani S; Datwyler P; Zihlmann P; Jakob RP; Sager CP; et al. Improvement of Aglycone pi-Stacking Yields Nanomolar to Sub-nanomolar FimH Antagonists. ChemMedChem 2019, 14 (7), 749–757. DOI: 10.1002/cmdc.201900051. [DOI] [PubMed] [Google Scholar]
- (14).Spaulding CN; Klein RD; Ruer S; Kau AL; Schreiber HL; Cusumano ZT; Dodson KW; Pinkner JS; Fremont DH; Janetka JW; et al. Selective depletion of uropathogenic E. coli from the gut by a FimH antagonist. Nature 2017, 546 (7659), 528–532. DOI: 10.1038/nature22972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Sarshar M; Behzadi P; Ambrosi C; Zagaglia C; Palamara AT; Scribano D FimH and Anti-Adhesive Therapeutics: A Disarming Strategy Against Uropathogens. Antibiotics (Basel) 2020, 9 (7). DOI: 10.3390/antibiotics9070397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sivignon A; Bouckaert J; Bernard J; Gouin SG; Barnich N The potential of FimH as a novel therapeutic target for the treatment of Crohn’s disease. Expert Opin Ther Targets 2017, 21 (9), 837–847. DOI: 10.1080/14728222.2017.1363184. [DOI] [PubMed] [Google Scholar]
- (17).Mydock-McGrane LK; Hannan TJ; Janetka JW Rational design strategies for FimH antagonists: new drugs on the horizon for urinary tract infection and Crohn’s disease. Expert Opin Drug Discov 2017, 12 (7), 711–731. DOI: 10.1080/17460441.2017.1331216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chalopin T; Alvarez Dorta D; Sivignon A; Caudan M; Dumych TI; Bilyy RO; Deniaud D; Barnich N; Bouckaert J; Gouin SG Second generation of thiazolylmannosides, FimH antagonists for E. coli-induced Crohn’s disease. Org Biomol Chem 2016, 14 (16), 3913–3925. DOI: 10.1039/c6ob00424e. [DOI] [PubMed] [Google Scholar]
- (19).Conover Matt S.; Ruer S; Taganna J; Kalas V; De Greve H; Pinkner Jerome S.; Dodson Karen W.; Remaut H; Hultgren Scott J. Inflammation-Induced Adhesin-Receptor Interaction Provides a Fitness Advantage to Uropathogenic E. coli during Chronic Infection. Cell Host & Microbe 2016, 20 (4), 482–492. DOI: 10.1016/j.chom.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kalas V; Hibbing ME; Maddirala AR; Chugani R; Pinkner JS; Mydock-McGrane LK; Conover MS; Janetka JW; Hultgren SJ Structure-based discovery of glycomimetic FmlH ligands as inhibitors of bacterial adhesion during urinary tract infection. Proc Natl Acad Sci U S A 2018, 115 (12), E2819–E2828. DOI: 10.1073/pnas.1720140115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Maddirala AR; Klein R; Pinkner JS; Kalas V; Hultgren SJ; Janetka JW Biphenyl Gal and GalNAc FmlH Lectin Antagonists of Uropathogenic E. coli (UPEC): Optimization through Iterative Rational Drug Design. J Med Chem 2019, 62 (2), 467–479. DOI: 10.1021/acs.jmedchem.8b01561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Boutureira O; Bernardes GJL; Fernández-González M; Anthony DC; Davis BG Selenenylsulfide-Linked Homogeneous Glycopeptides and Glycoproteins: Synthesis of Human “Hepatic Se Metabolite A”. Angewandte Chemie International Edition 2012, 51 (6), 1432–1436. DOI: doi: 10.1002/anie.201106658. [DOI] [PubMed] [Google Scholar]
- (23).Kuzuhara H; Fletcher HG Jr. Syntheses with partially benzylated sugars. 8. Substitution at C-5 in an aldose. The synthesis of 5-O-methyl-D-glucofuranose derivatives. J Org Chem 1967, 32 (8), 2531–2534. DOI: 10.1021/jo01283a035. [DOI] [PubMed] [Google Scholar]
- (24).Sadurni A; Kehr G; Ahlqvist M; Wernevik J; Sjogren HP; Kankkonen C; Knerr L; Gilmour R Fluorine-Directed Glycosylation Enables the Stereocontrolled Synthesis of Selective SGLT2 Inhibitors for Type II Diabetes. Chemistry 2018, 24 (12), 2832–2836. DOI: 10.1002/chem.201705373. [DOI] [PubMed] [Google Scholar]
- (25).Leshch Y; Waschke D; Thimm J; Thiem J D--hept-2-ulose and Novel Deoxyfluoro Derivatives as Seven-Carbon Analogues of F-Deoxy-D-glucose (FDG). Synthesis-Stuttgart 2011, (23), 3871–3877. DOI: 10.1055/s-0031-1289598. [DOI] [Google Scholar]
- (26).Das J; Schmidt RR Convenient glycoside synthesis of amino sugars: Michael-Type addition to 2-nitro-D-galactal. European Journal of Organic Chemistry 1998, 1998 (8), 1609–1613. [Google Scholar]
- (27).Delaunay T; Poisson T; Jubault P; Pannecoucke X Stereoselective Access to β--Glycosamines by Nitro-Michael Addition of Organolithium Reagents. European Journal of Organic Chemistry 2014, 2014 (16), 3341–3345. DOI: 10.1002/ejoc.201402001. [DOI] [Google Scholar]
- (28).Xue W; Sun J; Yu B An efficient route toward 2-amino-beta-D-galacto- and -glucopyranosides via stereoselective Michael-type addition of 2-nitroglycals. J Org Chem 2009, 74 (14), 5079–5082. DOI: 10.1021/jo900609s. [DOI] [PubMed] [Google Scholar]
- (29).Van de Waterbeemd H 5.28 - In Silico Models to Predict Oral Absorption. In Comprehensive Medicinal Chemistry II, Taylor JB, Triggle DJ Eds.; Elsevier, 2007; pp 669–697. [Google Scholar]
- (30).Ertl P Polar Surface Area. In Molecular Drug Properties, 2007; pp 111–126. [Google Scholar]
- (31).Huber K; Hamad B; Kirkpatrick P Ticagrelor. Nature Reviews Drug Discovery 2011, 10 (4), 255–256. DOI: 10.1038/nrd3418. [DOI] [PubMed] [Google Scholar]
- (32).Aslanis V; Slack RJ; MacKinnon AC; McClinton C; Tantawi S; Gravelle L; Nilsson UJ; Leffler H; Brooks A; Khindri SK; et al. Safety and pharmacokinetics of GB1211, an oral galectin-3 inhibitor: a single- and multiple-dose first-in-human study in healthy participants. Cancer Chemotherapy and Pharmacology 2023, 91 (3), 267–280. DOI: 10.1007/s00280-023-04513-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kshirsagar RP; Kulkarni AA; Chouthe RS; Pathan SK; Une HD; Reddy GB; Diwan PV; Ansari SA; Sangshetti JN SGLT inhibitors as antidiabetic agents: a comprehensive review. RSC Advances 2020, 10 (3), 1733–1756, 10.1039/C9RA08706K. DOI: 10.1039/C9RA08706K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Maccari R; Ottanà R Sodium-Glucose Cotransporter Inhibitors as Antidiabetic Drugs: Current Development and Future Perspectives. Journal of Medicinal Chemistry 2022, 65 (16), 10848–10881. DOI: 10.1021/acs.jmedchem.2c00867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Manoj A; Das S; Kunnath Ramachandran A; Alex AT; Joseph A SGLT2 inhibitors, an accomplished development in field of medicinal chemistry: an extensive review. Future Med Chem 2020, 12 (21), 1961–1990. DOI: 10.4155/fmc-2020-0154. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.