

Abstract
Toxoplasma gondii induces strong IFN-γ-based immunity. Innate lymphoid cells (ILC), in particular ILC1, are an important innate source of this protective cytokine during infection. Our objective was to determine how MyD88-dependent signaling influences ILC function during peroral compared to i. p. infection with T. gondii. MyD88+/+ and MyD88−/− mice were orally inoculated with ME49 cysts, and small intestinal lamina propria ILC were assessed using flow cytometry. We observed T-bet+ ILC1, RORγt+ ILC3 and a population of T-bet+RORγt+ double-positive (DP) ILC. In MyD88−/− mice, IFN-γ-producing T-bet+ ILC1 frequencies were reduced compared to wild-type. Treatment of MyD88−/− mice with an antibiotic cocktail to deplete microflora reduced IFN-γ+ ILC1 frequencies. To examine ILC responses outside of the mucosal immune system, peritoneal exudate cells were collected from wild-type and knockout mice after i. p. inoculation with ME49 cysts. In this compartment ILC were highly polarized to the ILC1 subset that increased significantly and became highly positive for IFN-γ over the course of infection. Increased ILC1 was associated with expression of the Ki67 cell proliferation marker, and the response was driven by IL-12p40. In the absence of MyD88, IFN-γ expression by ILC1 was not maintained but proliferation remained normal. Collectively, these data reveal new aspects of ILC function that are influenced by location of infection and shaped further by MyD88-dependent signaling.
Introduction
Toxoplasma gondii is an opportunistic pathogen well known for its ability to stimulate strong IFN-γ-based immunity. This cytokine is central in providing protection against the parasite, but if not appropriately regulated its over-production can lead to inflammatory host pathology and death (1–4). It was long ago recognized that Th1 and CD8+ lymphocytes are important adaptive immune sources of IFN-γ during T gondii infection (5–7). NK cells and more recently neutrophils have been identified as innate sources of IFN-γ produced early during infection prior to emergence of adaptive immunity (8, 9). The success of Toxoplasma as a globally distributed parasite lies at least in part in its skill at inducing robust IFN-γ responses that prevent uncontrolled infection, instead enabling host survival and establishment of parasite latency.
In recent years innate lymphoid cells (ILC)2 have emerged as a new class of common lymphoid precursor-derived cells that lack antigen-specific receptors associated with adaptive immunity. They are regarded as innate counterparts of T lymphocytes, in particular Th1, Th2 and Th17 subsets of T cells (10, 11). ILC1 are dependent upon transcription factor T-bet, they express IFN-γ and unlike NK cells they have little cytotoxic activity. ILC2 are defined by secretion of Type 2 cytokines IL-4, IL-5 and IL-13. ILC3, like Th17 T cells, are dependent upon RORγt and they produce proinflammatory IL-17 and IL-22 cytokines. Despite these polarized phenotypes, it is also known that ILC subsets display plasticity shaped by environmental cues (12). For example, ILC3 can convert into ILC1-like IFN-γ-producing cells associated with down-regulation of RORγt and upregulation of T-bet (13). It has also been characterized that some subsets of RORγt-positive ILC3 co-express T-bet (14). ILC2 are able to convert to functional ILC1 cells in dependence upon expression of T-bet and the IL-12 receptor. There is also evidence that IL-25 promotes emergence of ILC2 producing both type 2 cytokines and IL-17 (15).
In line with the increasing attention ILC continue to receive, their role during Toxoplasma infection is becoming increasingly appreciated (16, 17). For instance, it is known that ILC1 contribute both IFN-γ and TNF-α following oral inoculation with T. gondii (18). It has also been found that IL-33, in synergy with IL-12, promotes emergence of ILC1 in the peritoneal cavity that promotes innate resistance to the parasite (19). In a similar model, IFN-γ produced by ILC1 together with NK cells plays an essential role in maintenance of IRF8-positive DC that produce IL-12 (20). While ILC1 and NK cells often function together to provide innate resistance, it is notable that Toxoplasma infection can drive conversion of NK cells into ILC1-like cells (21). The role of other ILC during T. gondii infection is less clear, but in the intestine ILC3 cells appear to act in limiting T cell hyperactivation and immune pathology dependent upon expression of the aryl hydrocarbon receptor (22). We recently examined the impact of MyD88 signaling on the activity of ILC1 and ILC3 in the lamina propria and found that the frequency IFN-γ+ ILC1 significantly decreased in the absence of MyD88 (23). While levels were lower compared to ILC1, we nonetheless detected expression of IFN-γ in the ILC3 population. Interestingly, ILC3 expression of IFN-γ occurred independently of MyD88. In the present study we compare ILC1 and ILC3 populations in the intestinal mucosa versus the peritoneal cavity following T. gondii infection. Within the intestinal lamina propria, we determine the influence of intestinal microbiota on ILC1 and ILC3 following oral inoculation with Toxoplasma.
Materials and Methods
Ethics Statement
Animal protocols were approved by the Institutional Animal Care and Use Committee at the University of New Mexico (Animal Welfare Assurance Number A4023–01) and all experiments were performed in strict accordance with recommendations set forth by the National Institutes of Health Guide for the Care and Use of Laboratory Animals (8th Edition). Every effort was made to minimize the number of experimental animals and pain and distress throughout the course of performed studies.
Mice
C57BL/6J (WT, Stock # 000664), B6.129P2(SJL)-Myd88tm1.1Defr/J (MyD88−/−, Stock # 009088), and B6.129P2-Tcrbtm1Mom/J (Tcrb−/−, Stock #002118) mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and maintained in a breeding colony in the Department of Biology Animal Research Facility at the University of New Mexico. 6-week-old female CBA/J (Stock #000656) and J:ARC(S) (Swiss Outbred, Strain #034608) mice were purchased from The Jackson Laboratory for maintenance of T. gondii ME49 cysts.
Parasites and Infections
To establish a chronically infected mouse colony, 8 to 12-week-old Swiss outbred and CBA/J mice were infected intraperitoneally with 10 cysts of the type II T. gondii strain ME49. Chronic infection is established after 2–3 weeks, after which time brains were collected and homogenized to obtain ME49 cysts. Experimental mice were infected with 30–40 cysts by either i. p. injection or peroral gavage in 200 μl PBS.
Isolation of primary leukocytes
Small intestines were collected and processed as previously described to obtain lamina propria cells (23, 24). Briefly, small intestines were rinsed to remove partially digested material, and fat tissue and Peyer’s patches were removed. The intestinal tissue was cut longitudinally and trimmed into ~1 cm pieces. Intestinal tissue fragments were incubated twice for 20 min at 37°C in calcium/magnesium-free Hank’s Balanced Salt Solution (HBSS), 10 mM EDTA (VWR, Radnor, PA, USA), and 1 mM dithiothreitol (Sigma Aldrich, St. Louis, MO, USA) to release intestinal epithelial cells and intraepithelial lymphocytes. To obtain lamina propria cells, samples were subsequently incubated in Dulbecco’s Modified Eagle’s Medium containing collagenase (300 U/ml, Worthington Biochemical, Lakewood, NJ, USA) for one hr at 37°C. A discontinuous Percoll (Sigma Aldrich) gradient (40%/ 80% diluted with DMEM) was then utilized to purify leukocytes in the lamina propria compartment. The cell prep was re-suspended in 40% Percoll, layered over 80% Percoll, and centrifuged for 20 min at 1800 rpm without terminal braking. The cells at the interface were collected, assessed for viability and cell number, and used for downstream applications. Peritoneal exudate cells (PEC) were collected by injecting 10 mL PBS into the peritoneal cavity of mice. The injected fluid was recovered, and cell number and viability were assessed to prepare PEC for downstream applications.
Flow Cytometry
To study ILC-derived IFN-γ production, cells (1.5 million) were stimulated in 1 mL DMEM containing 50 ng/mL PMA (Sigma Aldrich), 5 μg/mL ionomycin (Alfa Aesar, Tewksbury, MA, USA), and 10 μg/mL Brefeldin A (Biolegend, San Diego, CA, USA, Catalog #420601) for 4 hr at 37°C. Cells were subsequently rinsed, and surface staining was performed in FACS buffer (1% bovine growth serum with 30 mM NaN3 in PBS) for 20 min at 4°C. Surface staining primary antibodies included anti-CD45 Brilliant Violet 570 (Biolegend, Catalog #103136), anti-lineage 1 cocktail consisting of PerCP Cy5.5-labelled antibodies (anti-CD8α, anti-CD5, anti-CD3ε, Biolegend, Catalog #100734, 100624, 100328 respectively), anti-lineage 2 cocktail composed of APC eFluor 780-conjugated antibodies (anti-CD11b, anti-CD11c, anti-B220, Invitrogen, Waltham, MA, USA, Catalog #47-0112, 47-0114-82, 47-0452-82 respectively), and anti-CD90-PE (Biolegend, Catalog #140308). After surface staining, cells were rinsed and incubated overnight in 1 mL of Fixation/Permeabilization buffer from the FoxP3/Transcription Factor Staining Kit (eBioscience). The following day, samples were rinsed with 2 mL of permeabilization buffer (Foxp3Transcription Factor Staining Kit) prior to staining intracellular and nuclear proteins in permeabilization buffer (1 hr at 4°C). Intracellular and nuclear markers were labeled using primary antibodies including anti-T-bet Alexa-Fluor 647 (Biolegend, Catalog #644804), anti-RORγt Brilliant Violet 650 (BD Biosciences, San Jose, CA, USA, Catalog #564722), anti-IFN-γ Brilliant Violet 421 (Biolegend, Catalog #505829), anti-IL-17a Alexa-Fluor 488 (Biolegend, Catalog #506910), and anti-Ki67 FITC (Biolegend, Catalog #151212). After 1 hr of staining, samples were washed and re-suspended in 400–500 μl FACS buffer prior to analysis. Samples were run on an Attune NXT, 5 laser flow cytometer (ThermoFisher Scientific) and the data were analyzed using FlowJo v.10.8.1 software (FlowJo).
In vivo antibody depletions
Mice were i. p. injected with 500 μg anti-IL-7Rα mAb (BioXCell, Lebanon, NH, USA, Catalog #BE0065), 500 μg anti-IL-2Rα mAb (BioXCell, Catalog # BE0012), 500 μg anti-common gamma chain mAb (CD132, BioXCell, Catalog #BE0271), 500 μg anti-IL-12 mAb (BioXCell, Catalog # BE0051), or 500 μg rat gamma globulin (Jackson Immunoresearch, West Grove, PA, USA, Catalog #012000002) 2 days prior to infection. On day 0, mice were i. p. infected with 30 ME49 cysts and injected with 500 μg neutralizing and isotype antibody. A subsequent neutralizing and isotype antibody injection occurred on day 2 post-infection. On day 4 post-infection, cells were isolated for flow cytometry analysis.
Intestinal Microbiota Depletion
Mice were administered broad spectrum antibiotic cocktail in their drinking water for 3 wk prior to and during infection (total treatment time of four weeks). The antibiotic cocktail was prepared by dissolving vancomycin hydrochloride (0.5 g/L, VWR, Catalog #97062–554), ampicillin (1g/L, Aldon Corp SE, VWR Catalog #470233–552), metronidazole (1g/L, Acros Organics, VWR Catalog #200013–382), neomycin sulfate (1g/L, Enzo Life Sciences, VWR Catalog #89149–866) and sucrose (10%, VWR, Catalog #BDH9308–500G) in de-ionized water (23, 25, 26). Antibiotic water was filter sterilized, placed in sterile water bottles, and protected from direct light exposure using aluminum foil. Water changes were performed weekly.
Quantification of Fecal CFU
Fresh feces were collected from control and antibiotic-treated mice, weighed, and diluted 1:10 with sterile PBS. Fecal pellets were mashed to create a slurry that was serially diluted with PBS, and 100 μl of various dilutions was plated on LB agar (BD Biosciences, Catalog #240110). Plates were incubated overnight at 37°C and colonies were counted the following day.
Statistical Analysis
Data analyses were performed using GraphPad Prism v.9.0.2 (GraphPad, La Jolla, CA). Two-tailed Student’s t-tests were used to compare two normally distributed data sets. A Student’s t-test with Welch’s correction was also used to compare two normally distributed data sets with unequal variances, and a Mann Whitney test was used to compare two data sets that were not normally distributed. A one-way ANOVA with Tukey multiple comparisons post-test was used to compare three or more normally distributed groups. A two-way ANOVA was used to compare two or more normally distributed groups across time or different treatments. A confidence interval of 95% was used as the cut off to denote significant changes between groups. *p≤0.05, **p≤0.01, ***p≤0.001.
Results
Microbiota affects MyD88-dependent and MyD88-independent ILC populations in the lamina propria
Seven days post-infection, lamina propria cells were isolated from MyD88+/+ and MyD88−/− mice, then ILC identified based upon a previously published lineage-negative gating scheme (27). Briefly, and as shown in Fig. 1A and Fig. 1B, leukocytes were identified based upon CD45 expression then amongst these, lin1− (CD5−, CD8−, CD3−) and lin2− (B220−, CD11c−, CD11b−) cells were selected. Within the double-negative population, ILC were defined as cells expressing CD90, a surface molecule widely associated with this innate lymphoid population (11). This subpopulation was found to comprise approximately 3–5% of CD45+ cells isolated from the lamina propria (LP), regardless of MyD88 genotype. In addition, the ILC population was relatively stable, regardless of T. gondii infection status (Fig. 1B). To deplete microbiota, mice were administered a broad-spectrum antibody cocktail for three weeks prior to oral inoculation with cysts of the low virulence Toxoplasma strain ME49 (Supplemental Fig. 1A) (23, 26). This treatment rapidly reduced levels of culturable bacteria to the detection limit of the assay (Supplemental Fig. 1B).
Figure 1.
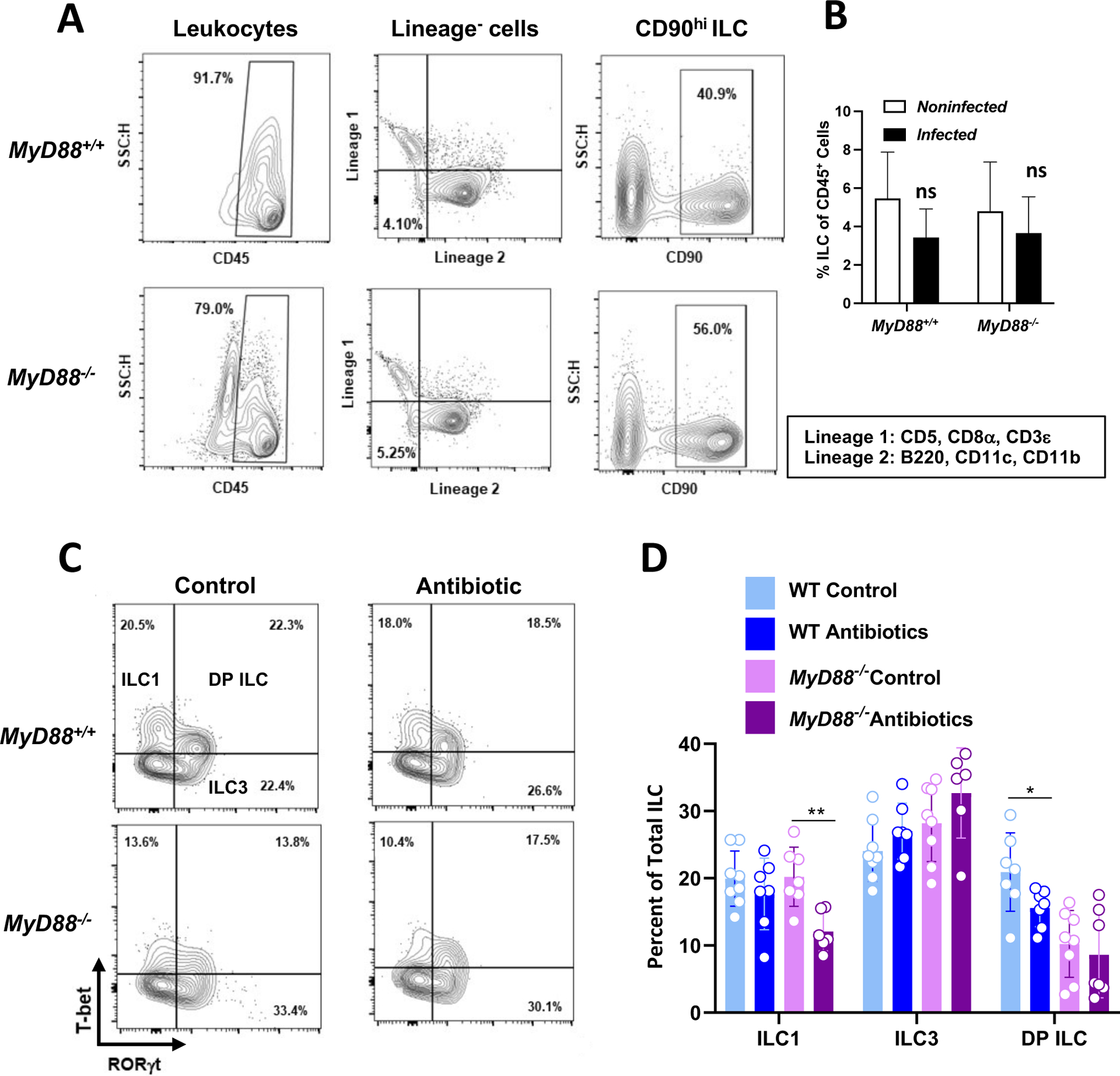
MyD88-dependent and independent signals from the intestinal microbiota regulate T-bet+ ILC1 and RORγt+ T-bet+ ILC in T. gondii infected mice. Lamina propria cells were collected from control and antibiotic treated WT and MyD88 KO mice at day 7 post-infection. (A) Representative flow cytometry plots defining ILC as CD45+, Lineage− (CD5−, CD8α−, CD3ε−, B220−, CD11c−, CD11b−), CD90hi. (B) Average percent ILC over multiple LP isolations. ns, not significant. (C) Subsequent gating defining ILC subsets based on transcription factor expression (ILC1, T-bet+ RORγt−; ILC3, T-bet− RORγt+; DP ILC, T-bet+ RORγt+). (D) Frequencies of ILC1, ILC3, and DP ILC within the total ILC (CD90hi) gate. Values are the mean ± SD of two independent experiments where each symbol represents a single mouse (n=6–8/ group). Unpaired Student’s t test (C) where *p<0.05, **p<0.01.
In the CD90+ population, we identified T-bet+ ILC1 and RORγt+ ILC3 in both WT and KO mice (Fig. 1C). Interestingly, we also identified T-bet+ RORγt+ cells, a population we refer to here as double-positive (DP) ILC. While the ILC1 population in WT mice was unaffected by microbiota depletion, the same treatment resulted in an ILC1 reduction in the KO animals (Fig. 1D). ILC3 populations in both mouse strains were unaffected by antibiotic treatment. However, the DP ILC population was significantly reduced in microbiota-depleted MyD88+/+ mice. Levels of DP cells were lower comparing MyD88+/+ with MyD88−/− mice, and microbiota depletion had no effect on DP ILC in the KO strain (Fig. 1D).
Influence of MyD88 and microbiota on LP ILC IFN-γ expression
Because IFN-γ is the major mediator of defense against Toxoplasma in both acute and chronic infection (8, 28–30), we focused on production of this cytokine by LP ILC after T. gondii inoculation. Representative flow cytometry scatter plots of IFN-γ expression by ILC1, ILC3 and DP ILC are shown in Fig. 2A and cumulative data are displayed in Fig. 2B. As expected, ILC1 represented the most significant pool of IFN-γ amongst ILC. In both WT and KO mice, ILC1 IFN-γ expression significantly decreased in microbiota-depleted animals. In addition, under both microbiota-sufficient and -deficient conditions, the response was partially independent of MyD88. As expected, ILC3 expression of IFN-γ was minimal under each condition. For DP ILC, while some of the cells expressed IFN-γ, there was no significant contribution of either MyD88 or microbiota. We also examined ILC IFN-γ expression in noninfected WT and KO mice. Expression of IFN-γ in ILC1, ILC3 and DP ILC was overall similar to those of infected animals (Supplemental Fig. 2). In this situation, the status of MyD88 expression did not affect IFN-γ levels.
Figure 2.
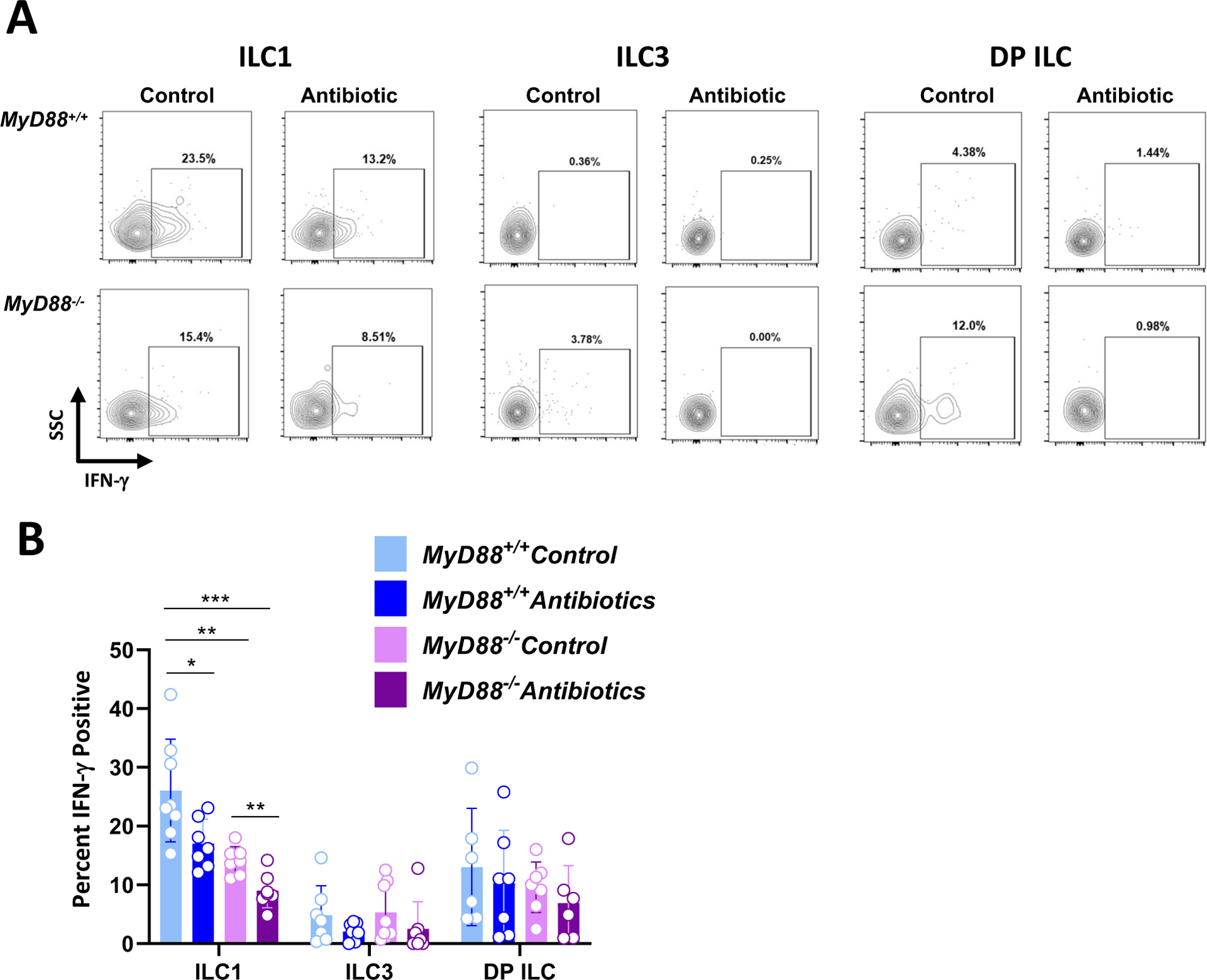
ILC1 function is shaped by the MyD88 signaling cascade and signals from the intestinal microbiota. At day 7 post-infection, lamina propria cells were isolated from control and antibiotic treated MyD88+/+ and MyD88−/− mice. (A) Representative flow plots showing IFN-γ staining within ILC1, ILC3, and DP ILC populations. Numbers indicate the percent of cells falling within each gate (B) IFN-γ+ ILC1, ILC3, and DP ILC frequencies in control and antibiotic treated WT and KO mice. Each symbol represents a single mouse from two independent experiments (n=6–8/group). Two-way ANOVA, where *p<0.05, **p<0.01, ***p<0.001.
Dynamic changes in ILC associated with Toxoplasma infection in the peritoneal cavity
Several investigators have used i. p. infection with Toxoplasma as a model to study ILC responses to the parasite (19–21). We were therefore interested to examine how ILC in the LP compartment compared with responses in the peritoneal cavity following oral versus i. p. ME49 inoculation. Using the same CD45+lin1−lin2−CD90+ gating scheme, we identified ILC in the peritoneal cavity of WT and KO mice (Supplemental Fig. 3A). We found that the proportion of ILC amongst CD45+ cells was approximately 10-fold lower than that occurring in the LP compartment. Despite this, there was a clear increase in the ILC population driven by infection in the peritoneal cavity (Fig. 3A). Using T-bet and RORγt expression, we found a strong skewing to ILC1 in the peritoneal cavity with almost undetectable levels of ILC3 or DP ILC (Supplemental Fig. 3B). Interestingly, we observed a population of T-bet−RORγt− ILC in noninfected mice that decreased over time with infection (Supplemental Fig. 3B). The strong ILC1 bias was preserved over the course of infection (Fig. 3B and Fig. 3C). Indeed, when absolute cell numbers were calculated, we observed that the few ILC3 that were present in noninfected mice rapidly disappeared with infection (Fig. 3D). We found a population of ILC expressing neither T-bet nor RORγt in the uninfected peritoneal cavity, and we note that these cells disappear with infection (Supplemental Fig. 3B). Based upon lack of GATA-3 staining (data not shown), this is not a population of ILC2. A possibility that we are currently examining is that T-bet−RORγt− ILC represent precursor cells that are driven into the ILC1 lineage by Toxoplasma.
Figure 3.
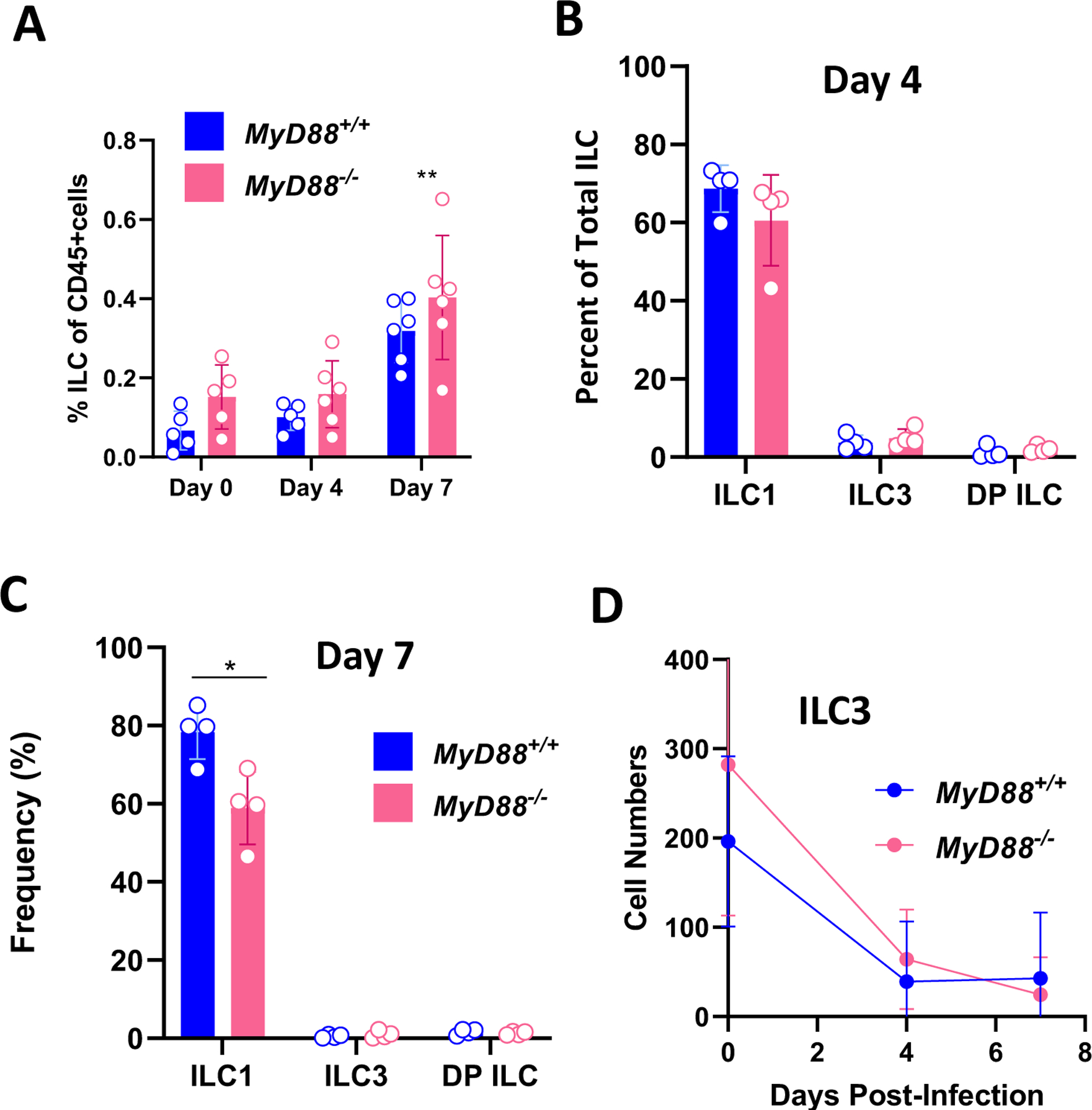
T. gondii infection induces MyD88-independent expansion of ILC1 in the peritoneal cavity. Peritoneal exudate cells (PEC) were collected from WT and MyD88 KO mice at day 0, 4, and 7 post-infection. (A) Percent ILC in the CD45+ cell population in peritoneal exudate from MyD88+/+ and MyD88−/− mice. Frequencies of peritoneal ILC1, ILC3, and DP ILC at day 4 (B) and day 7 (C) post-infection. (D) Declining numbers of ILC3 from day 0 to day 7 post-infection. Each symbol is representative of a single mouse. Two independent experiments were performed with similar results. Two-way ANOVA with Tukey’s multiple comparisons test (A, D), unpaired Student’s t test (B, C), *p<0.05, **p<0.01.
Functional activity of parasite-triggered peritoneal cavity ILC1
We next examined expression of IFN-γ by ILC1 following i. p. infection. In this situation, up to 80–90% of ILC1 expressed IFN-γ following infection in MyD88+/+ mice (Fig. 4A). Regardless of MyD88 status, approximately 20% of ILC1 in noninfected mice expressed IFN-γ. This proportion is similar to that found in the LP compartment (Fig. 4B and Supplemental Fig. 2). In infected MyD88−/− mice, there was a pronounced defect in expression of ILC1 IFN-γ. These cells initially (Day 4) responded with a modest increase in IFN-γ. However, by one week post-infection the response in KO mice had declined to levels detected in noninfected mice while ILC1 IFN-γ expression in WT animals was maintained at high levels (Fig. 4A and B).
Figure 4.
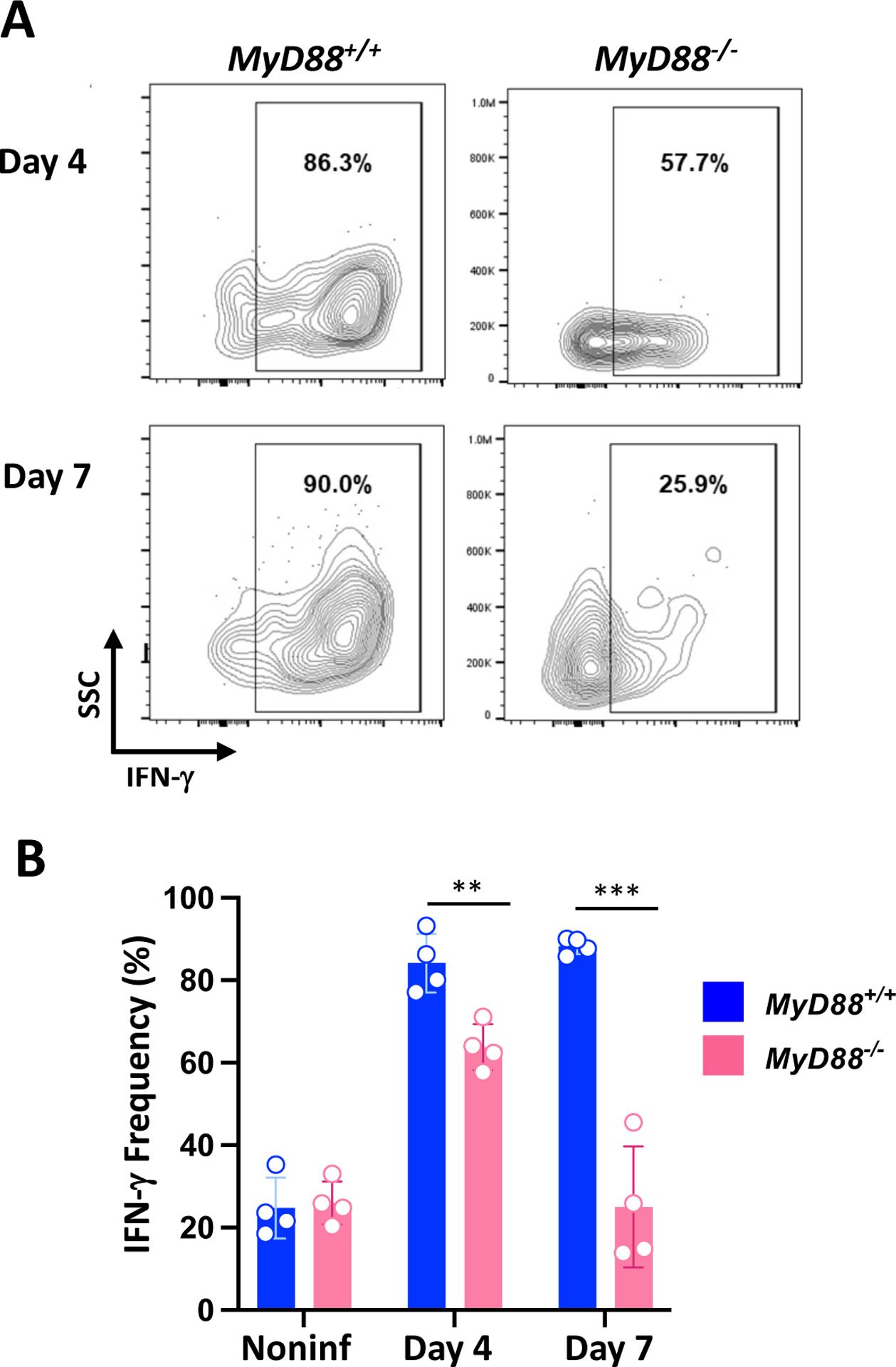
Peritoneal ILC1 require MyD88 to maintain IFN-γ production in response to T. gondii infection. (A) Representative gating showing IFN-γ staining within WT and MyD88 KO ILC1 populations at day 4 and day 7 post-infection. Numbers are the frequency of cells falling within each gate. (B) IFN-γ+ ILC1 frequencies at day 4 and day 7 post-infection. This experiment was repeated twice with similar results. Unpaired Student’s t test (B) where **p<0.01, ***p<0.0001.
The pronounced increase in ILC1 number in the peritoneal cavity following infection could have been due to cell recruitment or proliferation. Accordingly, we examined ILC1 expression of the proliferation marker Ki67. In noninfected mice, <20% ILC1 stained positive for this marker. However, following i. p. infection with ME49 this proportion rapidly increased to approximately 60% (Fig. 5A and B). In accordance with the overall increase in peritoneal cavity ILC induced by infection, both WT and KO cells expressed similar amounts of Ki67 (Fig. 5B). In contrast, Day 7-infected LP ILC expressed only background levels of this proliferation marker, consistent with the relatively stable number of these cells within the intestinal mucosa before and after infection (Fig. 5C, Fig. 1B).
Figure 5.
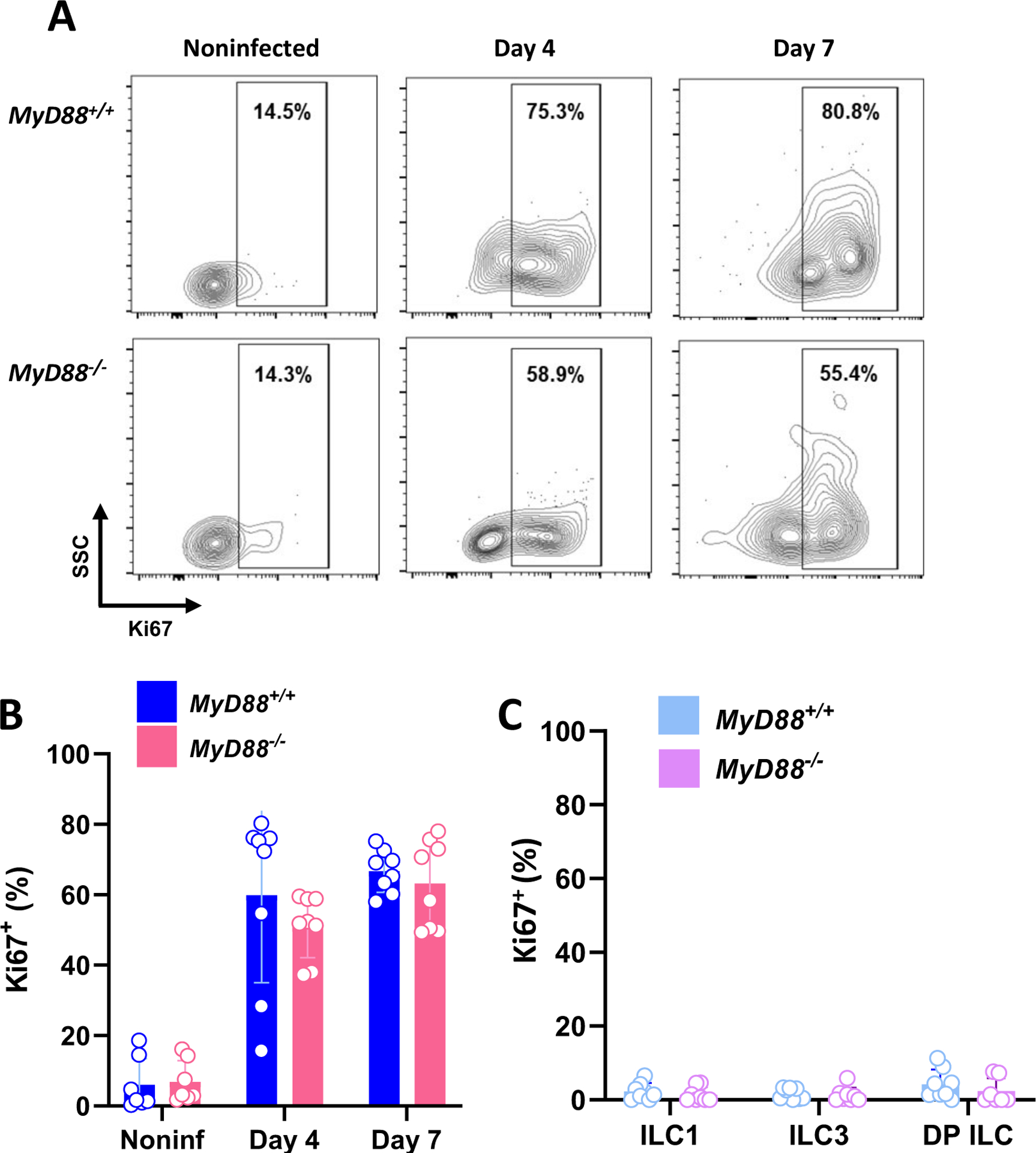
T. gondii infection induces vigorous ILC1 proliferation in the peritoneal cavity. (A) Representative flow gating showing Ki67 staining in MyD88+/+ and MyD88−/− peritoneal ILC1 cells at day 0, 4, and 7 post-infection. (B) Ki67+ ILC1 frequencies in WT and KO peritoneal exudate. (C) Ki67+ frequencies in LP ILC populations at day 7 after oral inoculation. Each symbol is representative of a single mouse (n=7–8 per group at each time point). Values are the mean ± SD of two independent experiments. Unpaired Student’s t test (B), *p<0.05.
We next asked what factors might drive ILC proliferation in the peritoneal cavity. Given the ability of T. gondii to induce robust CD4+ and CD8+ T lymphocyte activation and cytokine production (1), we determined whether T cell factors could drive ILC1 proliferation. As shown in Fig. 6A, induction of ILC1 Ki67 expression was equivalent in WT and T cell KO (Tcrb−/−) mice. Mediators including IL-7, IL-2 and other common γ-chain receptor-dependent cytokines (IL-4, IL-9, IL-15, IL-21) could be involved to varying degrees in ILC generation (31, 32), and we therefore investigated involvement of these factors using depleting mAb. Blocking IL-2Ra, IL-7Ra as well preventing signaling through common γ chain failed to affect ILC1 proliferation, as assessed by Ki67 expression (Fig. 6B). Finally, we determined the role of IL-12 in ILC1 proliferation, since the cytokine is produced in large amount during Toxoplasma infection and is known to display effects on this ILC population (33–36). In this case, administration of depleting anti-IL-12p40 mAb resulted in an approximately 50% decrease in ILC1 proliferation, as measured by Ki67 expression (Fig. 6C and D). In accord with this result, the percent of ILC1 decreased with anti-IL-12 administration, accompanied by a concomitant increase in DN ILC (Fig. 6E and F). We conclude that IL-12 contributes to expansion of ILC1 in the peritoneal cavity of T. gondii infected mice.
Figure 6.
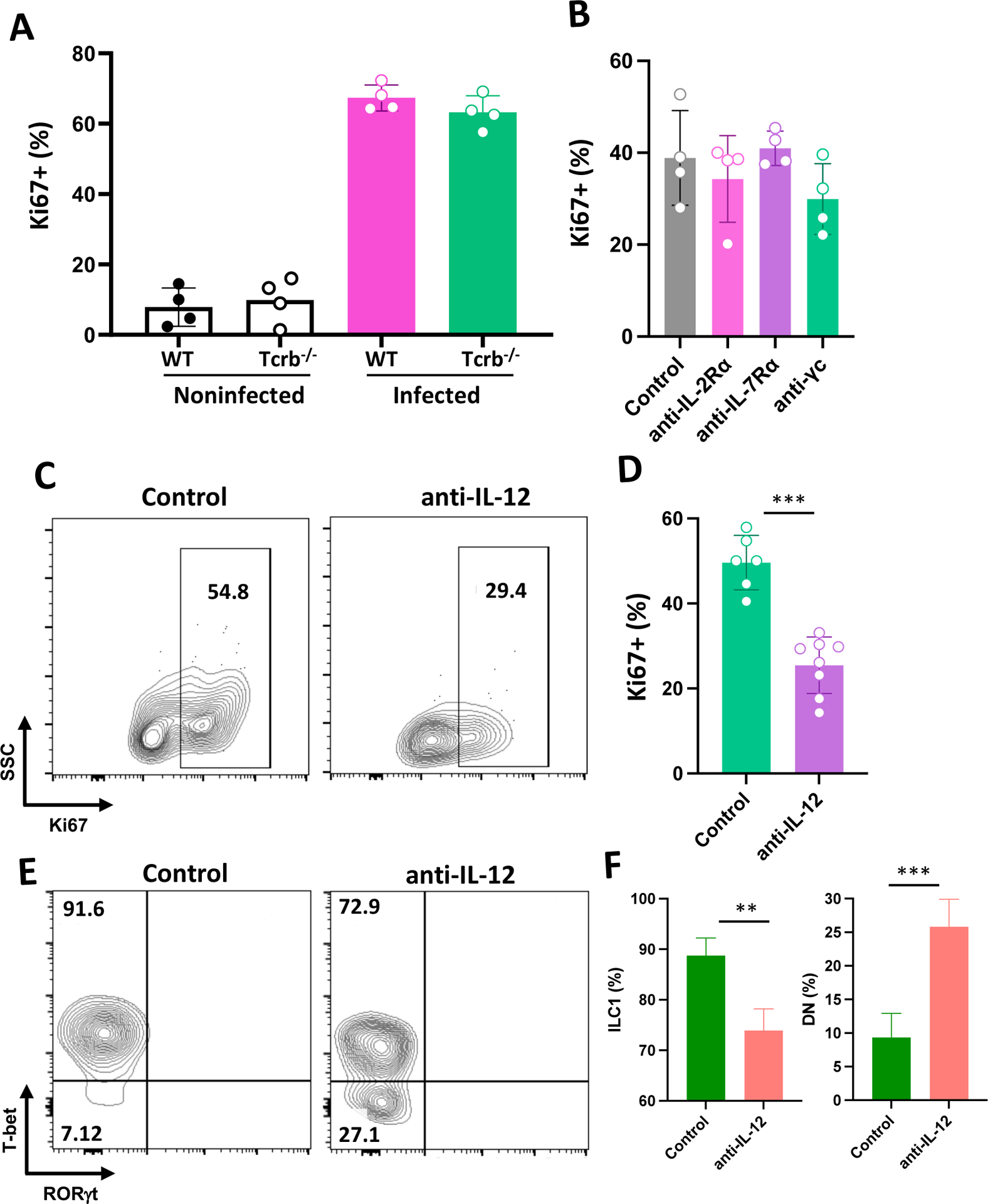
IL-12p40, but not IL-2 or IL-7, drives Toxoplasma-initiated ILC1 proliferation. Mice were infected by i. p. inoculation, then ILC1 were assessed for Ki67 expression 4 days later. (A) Ki67+ ILC1 after infection of WT and Tcrb−/− mice. (B) Peritoneal ILC1 Ki67 expression in WT mice subjected to in vivo mAb mediated blockade of IL-2Ra, IL-7Ra, and common γ chain (γc) receptor. Control, normal rat IgG. (C) Representative flow cytometry plots of Ki67 expression in ILC1 following in vivo isotype and anti-IL-12p40 mAb treatment. (D) Cumulative data showing ILC1 Ki67 expression levels in control and anti-IL-12p40 treated mice. Values are the mean ± SD where each symbol represents of one mouse. (E) Representative flow cytometric plots of ILC1 and DN ILC in mice administered normal rat IgG (Control) and anti-IL-12 mAb. (F) Average ± SD of percent ILC1 and DN relative to ILC in n=4 animals per group. Unpaired Student’s t test (D, E and F), **p<0.01, ***p<0.001. This experiment was repeated three times with the same result.
Discussion
Innate lymphoid cells are now established as an important component of innate immunity with roles in infection, allergy and inflammation (12, 32, 37). Here, we examined ILC populations in the intestinal mucosa and peritoneal cavity following oral and i. p. Toxoplasma infection. A strong Type 1 cytokine response is triggered by T. gondii that is required for protection but that may also reach tissue-damaging levels (38). The immune response is characterized by dendritic cell IL-12 production that stimulates early NK cell IFN-γ, followed by IL-12-dependent emergence of IFN-γ-secreting Th1 cells and CD8+ T lymphocytes with the dual capabilities of IFN-γ production and cytolytic activity. Neutrophils have also been found to be a significant innate source of IFN-γ (9). In accord with this model, we found that ILC1 dominated the overall ILC response to infection in both the LP and peritoneal cavity, results that are in line with those of others (18, 19, 21, 39, 40). Nevertheless, there were notable differences between ILC found in the intestinal LP compared to those occurring in the peritoneal cavity.
ILC levels relative to CD45+ cells in the lamina propria were overall approximately 10-fold higher than that occurring in the peritoneal cavity. IFN-γ expression in LP ILC1 was decreased in infected MyD88 KO mice, and was also diminished after microbiota depletion. In the LP compartment, we detected a significant population of ILC3, as well as T-bet+RORγt+ DP ILC, neither of which were found to any great degree in the peritoneal cavity. While levels were considerably lower, i. p. infection triggered an increase in ILC1 frequency associated with the cell proliferation marker Ki67. These cells were exquisitely responsive to Toxoplasma, insofar as IFN-γ expression increased from 20% of the ILC1 population to up to 90% within 4 days of infection. This response was less robust and was not maintained in the absence of MyD88. Overall, ILC in peritoneal cavity, while lower in number, appear highly dynamic and responsive to infection. In contrast, those in the intestinal LP are more stable without displaying major changes in response to T. gondii, at least within the time-frame of this study.
The nature of the DP ILC we observed in the gut is not clear. Previously, it was found that T-bet-expressing RORγt+ ILC protect the intestinal epithelial layer during Salmonella enterica infection (41). Plasticity within the ILC compartment is a well-known phenomenon (12). For example, it has been shown by several investigators that intestinal ILC3 can deviate into IFN-γ-producing ILC1. This conversion is associated with down regulation of RORγt and concomitant upregulation of T-bet (13, 41–43). It likely involves IL-1β, IL-15 and IL-12, based upon in vitro studies demonstrating ILC3-to-ILC1 conversion in the presence of these cytokines (33, 44). It has also been demonstrated that LP ILC3 are induced to express T-bet in the presence of IL-21, a cytokine previously implicated in optimal T cell and B cell responses in chronic Toxoplasma infection (45, 46).
In our study, we found that the DP ILC population was controlled by signals from both MyD88 and the intestinal microbiota. MyD88 can be regarded as an ancient gatekeeper of immunity, providing the host with an endogenous innate immune system through IL-1 family cytokines and an exogenous innate immune system through the Toll-like receptor (TLR) system (47). For Toxoplasma, parasite profilin was discovered as a classical pathogen-associated molecular pattern (PAMP) molecule recognized by mouse TLR 11/12 (48–50). Expression of MyD88 in CD11c+ DC has been shown to be important in IL-12 production and resistance during T. gondii infection (51). Previous studies have also highlighted the important role of cell intrinsic MyD88 signaling in production of NK cell-derived IFN-γ and protective T cell immunity (52, 53). It is also clear that this signaling adaptor impacts T. gondii infection through its function in signal transduction mediated by IL-1-family cytokines such as IL-1β, IL-18 and IL-33 (19, 54–58). For ILC function, it remains to be determined whether MyD88 is intrinsically involved, and whether its activity is mediated through parasite PAMP molecules, IL-1-family cytokines or a combination of both.
We employed oral dosing of a wide spectrum antibiotic cocktail to determine the influence of the gut microbiota during T. gondii infection. This approach has inherent advantages and disadavantages (59). Although the treatment led to depletion of intestinal bacteria to levels at or below the limit of detection, our assays were based upon evaluation of culturable bacteria only. In addition, it is well recognized that antibiotic administration has an impact on the overall physiology of the intestine that might impact ILC activity. On the other hand, studies using gnotobiotic mice are limited due to the beneficial influence of the gut microbiota on development of the mucosal immune system (60).
In the peritoneal cavity we identified a population of ILC expressing neither T-bet nor RORγt. Strikingly, this population disappeared over the course of infection, a phenomenon that was dependent upon IL-12 (Supplemental Fig 3B; Fig. 6E and F). These cells did not stain for GATA-3 (data not shown), so they are not likely to be ILC2. While ILC are generally thought to differentiate in primary lymphoid tissues, there are multiple lines of evidence for tissue resident precursors that can be driven into specific ILC lineages by environmental cues (61–63). Therefore, it is possible that the T-bet−RORγt− population is composed of ILC precursors such as common helper ILC precursor (CHILP) cells or the more committed innate lymphoid cell precursors (ILCP), both of which express CD90 (18, 64, 65). IL-12, a cytokine induced by T. gondii that is known to be involved in ILC1 effector generation (18, 35, 66), may drive these double-negative cells into the ILC1 lineage. We are currently examining the relationship and function of these cells as related to ILC.
After infection in the peritoneal cavity we observed a pronounced increase in ILC1. These cells are generally not thought to recirculate, in contrast to the related NK cell lineage (67). We found a striking increase in expression of the proliferation marker Ki67 in ILC1 in response to infection, supporting that the cells undergo in situ proliferation rather than recruitment from elsewhere. Amplified Ki67 expression occurred independently of MyD88, in contrast to IFN-γ production by these cells. We did not find evidence for involvement of IL-33 or other MyD88 signaling-dependent cytokines, since cells from both WT and MyD88 KO mice expressed equivalent levels of Ki67 after infection. Furthermore, mAb depletion experiments failed to provide evidence for IL-7 or other common γ-chain receptor cytokines that have been implicated in ILC1 activation (10, 19). Instead, the driving stimulus for ILC1 proliferation was dependent upon signals from IL-12. ILC1 expansion occurred in the absence of MyD88, implicating T. gondii-initiated IL-12 production independent of this TLR/IL-1 signaling adaptor. Indeed, we recently showed that Type II T. gondii induces peritoneal cavity IL-12 in MyD88 KO mice, albeit at lower levels than that occurring in WT animals (68). One candidate parasite driver of this response is dense granule protein GRA24, a molecule that directly activates host p38 MAPK, in turn inducing MyD88-independent IL-12 production (68, 69).
Our studies reveal an important influence of environment on ILC activity during Toxoplasma infection. Those cells in the intestine display an ongoing level of activity that is influenced by T. gondii itself, as well as messaging from the microbiota. In the peritoneal cavity, ILC respond vigorously to infection through a combination of MyD88-dependent and -independent mechanisms that contribute overall to the dominant Type 1 cytokine response associated with infection by this opportunistic pathogen. Our data, and those of others, reinforce a model in which ILC coordinate with other innate and adaptive IFN-γ-producing cellular compartments to provide a robust firewall of protection against Toxoplasma.
Supplementary Material
Footnotes
Supported by PHS grant AI139628
Abbreviations: DN, double-negative; DP, double-positive; ILC, innate lymphoid cell; KO, knockout; LP, lamina propria; WT, wild-type
References
- 1.Denkers EY, and Gazzinelli RT. 1998. Regulation and function of T cell-mediated immunity during Toxoplasma gondii infection. Clin. Microbiol. Rev 11: 569–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dupont CD, Christian DA, and Hunter CA. 2012. Immune response and immunopathology during toxoplasmosis. Semin Immunopathol 34: 793–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaddi PJ, and Yap GS. 2007. Cytokine regulation of immunopathology in toxoplasmosis. Immunol Cell Biol 85: 155–159. [DOI] [PubMed] [Google Scholar]
- 4.Sasai M, and Yamamoto M. 2022. Anti-Toxoplasma host defense systems and the parasitic counterdefense mechanisms. Parasitol Int 89: 102593. [DOI] [PubMed] [Google Scholar]
- 5.Gazzinelli RT, Hakim FT, Hieny S, Shearer GM, and Sher A. 1991. Synergistic role of CD4+ and CD8+ T lymphocytes in IFN-g production and protective immunity induced by an attenuated T. gondii vaccine. J. Immunol 146: 286–292. [PubMed] [Google Scholar]
- 6.Suzuki Y, and Remington JS. 1988. Dual regulation of resistance against Toxoplasma gondii infection by Lyt-2+ and Lyt1+,L3T4+ T cells in mice. J. Immunol 140: 3943–3946. [PubMed] [Google Scholar]
- 7.Wang X, Kang H, Kikuchi T, and Suzuki Y. 2004. Gamma interferon production, but not perforin-mediated cytolytic activity, of T cells is required for prevention of toxoplasmic encephalitis in BALB/c mice genetically resistant to the disease. Infect. Immun 72: 4432–4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gazzinelli RT, Hieny S, Wynn T, Wolf S, and Sher A. 1993. IL-12 is required for the T-cell independent induction of IFN-g by an intracellular parasite and induces resistance in T-cell-deficient hosts. Proc. Natl. Acad. Sci. USA 90: 6115–6119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sturge CR, Benson A, Raetz M, Wilhelm CL, Mirpuri J, Vitetta ES, and Yarovinsky F. 2013. TLR-independent neutrophil-derived IFN-gamma is important for host resistance to intracellular pathogens. Proc Natl Acad Sci U S A 110: 10711–10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eberl G, Colonna M, Di Santo JP, and McKenzie AN. 2015. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science 348: aaa6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie ANJ, Mebius RE, Powrie F, and Spits H. 2018. Innate Lymphoid Cells: 10 Years On. Cell 174: 1054–1066. [DOI] [PubMed] [Google Scholar]
- 12.Bal SM, Golebski K, and Spits H. 2020. Plasticity of innate lymphoid cell subsets. Nat Rev Immunol 20: 552–565. [DOI] [PubMed] [Google Scholar]
- 13.Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, Flach M, Bengsch B, Thimme R, Holscher C, Honig M, Pannicke U, Schwarz K, Ware CF, Finke D, and Diefenbach A. 2010. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity 33: 736–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fiancette R, Finlay CM, Willis C, Bevington SL, Soley J, Ng STH, Baker SM, Andrews S, Hepworth MR, and Withers DR. 2021. Reciprocal transcription factor networks govern tissue-resident ILC3 subset function and identity. Nat Immunol 22: 1245–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang K, Xu X, Pasha MA, Siebel CW, Costello A, Haczku A, MacNamara K, Liang T, Zhu J, Bhandoola A, Maillard I, and Yang Q. 2017. Cutting Edge: Notch Signaling Promotes the Plasticity of Group-2 Innate Lymphoid Cells. J Immunol 198: 1798–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunay IR, and Diefenbach A. 2018. Group 1 innate lymphoid cells in Toxoplasma gondii infection. Parasite Immunol 40. [DOI] [PubMed] [Google Scholar]
- 17.Ivanova DL, Denton SL, Fettel KD, Sondgeroth KS, Munoz Gutierrez J, Bangoura B, Dunay IR, and Gigley JP. 2019. Innate Lymphoid Cells in Protection, Pathology, and Adaptive Immunity During Apicomplexan Infection. Front Immunol 10: 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klose CS, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, Fabiunke C, Pfeifer D, Sexl V, Fonseca-Pereira D, Domingues RG, Veiga-Fernandes H, Arnold SJ, Busslinger M, Dunay IR, Tanriver Y, and Diefenbach A. 2014. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 157: 340–356. [DOI] [PubMed] [Google Scholar]
- 19.Clark JT, Christian DA, Gullicksrud JA, Perry JA, Park J, Jacquet M, Tarrant JC, Radaelli E, Silver J, and Hunter CA. 2021. IL-33 promotes innate lymphoid cell-dependent IFN-gamma production required for innate immunity to Toxoplasma gondii. Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopez-Yglesias AH, Burger E, Camanzo E, Martin AT, Araujo AM, Kwok SF, and Yarovinsky F. 2021. T-bet-dependent ILC1- and NK cell-derived IFN-gamma mediates cDC1-dependent host resistance against Toxoplasma gondii. PLoS Pathog 17: e1008299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park E, Patel S, Wang Q, Andhey P, Zaitsev K, Porter S, Hershey M, Bern M, Plougastel-Douglas B, Collins P, Colonna M, Murphy KM, Oltz E, Artyomov M, Sibley LD, and Yokoyama WM. 2019. Toxoplasma gondii infection drives conversion of NK cells into ILC1-like cells. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagage S, Harms Pritchard G, Dawson L, Buza EL, Sonnenberg GF, and Hunter CA. 2015. The Group 3 Innate Lymphoid Cell Defect in Aryl Hydrocarbon Receptor Deficient Mice Is Associated with T Cell Hyperactivation during Intestinal Infection. PLoS One 10: e0128335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Snyder LM, Doherty CM, Mercer HL, and Denkers EY. 2021. Induction of IL-12p40 and type 1 immunity by Toxoplasma gondii in the absence of the TLR-MyD88 signaling cascade. PLoS Pathog 17: e1009970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lefrancois L, and Lycke N. 2001. Isolation of mouse small intestinal intraepithelial lymphocytes, Peyer’s patch, and lamina propria cells. Curr Protoc Immunol Chapter 3: Unit 3 19. [DOI] [PubMed] [Google Scholar]
- 25.Bora SA, Kennett MJ, Smith PB, Patterson AD, and Cantorna MT. 2018. Regulation of vitamin D metabolism following disruption of the microbiota using broad spectrum antibiotics. J Nutr Biochem 56: 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zarrinpar A, Chaix A, Xu ZZ, Chang MW, Marotz CA, Saghatelian A, Knight R, and Panda S. 2018. Antibiotic-induced microbiome depletion alters metabolic homeostasis by affecting gut signaling and colonic metabolism. Nat Commun 9: 2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou L, Chu C, Teng F, Bessman NJ, Goc J, Santosa EK, Putzel GG, Kabata H, Kelsen JR, Baldassano RN, Shah MA, Sockolow RE, Vivier E, Eberl G, Smith KA, and Sonnenberg GF. 2019. Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature 568: 405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scharton-Kersten TM, Wynn TA, Denkers EY, Bala S, Showe L, Grunvald E, Hieny S, Gazzinelli RT, and Sher A. 1996. In the absence of endogenous IFN-g mice develop unimpaired IL-12 responses to Toxoplasma gondii while failing to control acute infection. J. Immunol 157: 4045–4054. [PubMed] [Google Scholar]
- 29.Suzuki Y, Orellana MA, Schreiber RD, and Remington JS. 1988. Interferon-g: The major mediator of resistance against Toxoplasma gondii. Science 240: 516–518. [DOI] [PubMed] [Google Scholar]
- 30.Yap GS, and Sher A. 1999. Effector cells of both nonhemopoietic and hemopoietic origin are required for interferon (IFN)-g and tumor necrosis factor (TNF)-a-dependent host resistance to the intracellular pathogen, Toxoplasma gondii. J. Exp. Med 189: 1083–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robinette ML, Bando JK, Song W, Ulland TK, Gilfillan S, and Colonna M. 2017. IL-15 sustains IL-7R-independent ILC2 and ILC3 development. Nat Commun 8: 14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zook EC, and Kee BL. 2016. Development of innate lymphoid cells. Nat Immunol 17: 775–782. [DOI] [PubMed] [Google Scholar]
- 33.Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, Munneke JM, Hazenberg MD, Villaudy J, Buskens CJ, Bemelman WA, Diefenbach A, Blom B, and Spits H. 2015. Interleukin-12 and −23 Control Plasticity of CD127(+) Group 1 and Group 3 Innate Lymphoid Cells in the Intestinal Lamina Propria. Immunity 43: 146–160. [DOI] [PubMed] [Google Scholar]
- 34.Frickel EM, and Hunter CA. 2021. Lessons from Toxoplasma: Host responses that mediate parasite control and the microbial effectors that subvert them. J Exp Med 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, Cella M, and Colonna M. 2013. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity 38: 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pifer R, and Yarovinsky F. 2011. Innate responses to Toxoplasma gondii in mice and humans. Trends Parasitol 27: 388–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X, Peng H, and Tian Z. 2019. Innate lymphoid cell memory. Cell Mol Immunol 16: 423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khan IA, and Moretto M. 2022. Immune responses to Toxoplasma gondii. Curr Opin Immunol 77: 102226. [DOI] [PubMed] [Google Scholar]
- 39.Lopez-Yglesias AH, Burger E, Araujo A, Martin AT, and Yarovinsky F. 2018. T-bet-independent Th1 response induces intestinal immunopathology during Toxoplasma gondii infection. Mucosal Immunol 11: 921–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steffen J, Ehrentraut S, Bank U, Biswas A, Figueiredo CA, Holsken O, Dusedau HP, Dovhan V, Knop L, Thode J, Romero-Suarez S, Duarte CI, Gigley J, Romagnani C, Diefenbach A, Klose CSN, Schuler T, and Dunay IR. 2022. Type 1 innate lymphoid cells regulate the onset of Toxoplasma gondii-induced neuroinflammation. Cell Rep 38: 110564. [DOI] [PubMed] [Google Scholar]
- 41.Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y, Goppert N, Croxford AL, Waisman A, Tanriver Y, and Diefenbach A. 2013. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature 494: 261–265. [DOI] [PubMed] [Google Scholar]
- 42.Rankin LC, Groom JR, Chopin M, Herold MJ, Walker JA, Mielke LA, McKenzie AN, Carotta S, Nutt SL, and Belz GT. 2013. The transcription factor T-bet is essential for the development of NKp46+ innate lymphocytes via the Notch pathway. Nat Immunol 14: 389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sciume G, Hirahara K, Takahashi H, Laurence A, Villarino AV, Singleton KL, Spencer SP, Wilhelm C, Poholek AC, Vahedi G, Kanno Y, Belkaid Y, and O’Shea JJ. 2012. Distinct requirements for T-bet in gut innate lymphoid cells. J Exp Med 209: 2331–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cella M, Otero K, and Colonna M. 2010. Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc Natl Acad Sci U S A 107: 10961–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poholek CH, Dulson SJ, Zajac AJ, and Harrington LE. 2019. IL-21 Controls ILC3 Cytokine Production and Promotes a Protective Phenotype in a Mouse Model of Colitis. Immunohorizons 3: 194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stumhofer JS, Silver JS, and Hunter CA. 2013. IL-21 is required for optimal antibody production and T cell responses during chronic Toxoplasma gondii infection. PLoS One 8: e62889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dinarello CA 2018. Introduction to the interleukin-1 family of cytokines and receptors: Drivers of innate inflammation and acquired immunity. Immunol Rev 281: 5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koblansky AA, Jankovic D, Oh H, Hieny S, Sungnak W, Mathur R, Hayden MS, Akira S, Sher A, and Ghosh S. 2012. Recognition of Profilin by Toll-like Receptor 12 Is Critical for Host Resistance to Toxoplasma gondii. Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raetz M, Kibardin A, Sturge CR, Pifer R, Li H, Burstein E, Ozato K, Larin S, and Yarovinsky F. 2013. Cooperation of TLR12 and TLR11 in the IRF8-dependent IL-12 response to Toxoplasma gondii profilin. J Immunol 191: 4818–4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yarovinsky F, Zhang D, Anderson JF, Bannenberg GL, Serhan CN, Hayden MS, Hieny S, Sutterwala FS, Flavell RA, Ghosh S, and Sher A. 2005. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308: 1626–1629. [DOI] [PubMed] [Google Scholar]
- 51.Hou B, Benson A, Kuzmich L, DeFranco AL, and Yarovinsky F. 2011. Critical coordination of innate immune defense against Toxoplasma gondii by dendritic cells responding via their Toll-like receptors. Proc Natl Acad Sci U S A 108: 278–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ge Y, Chen J, Qiu X, Zhang J, Cui L, Qi Y, Liu X, Qiu J, Shi Z, Lun Z, Shen J, and Wang Y. 2014. Natural killer cell intrinsic toll-like receptor MyD88 signaling contributes to IL-12-dependent IFN-gamma production by mice during infection with Toxoplasma gondii. Int J Parasitol 44: 475–484. [DOI] [PubMed] [Google Scholar]
- 53.LaRosa DF, Stumhofer JS, Gelman AE, Rahman AH, Taylor DK, Hunter CA, and Turka LA. 2008. T cell expression of MyD88 is required for resistance to Toxoplasma gondii. Proc Natl Acad Sci U S A 105: 3855–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ryffel B, Huang F, Robinet P, Panek C, Couillin I, Erard F, Piotet J, Le Bert M, Mackowiak C, Torres Arias M, Dimier-Poisson I, and Zheng SG. 2019. Blockade of IL-33R/ST2 Signaling Attenuates Toxoplasma gondii Ileitis Depending on IL-22 Expression. Front Immunol 10: 702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Still KM, Batista SJ, O’Brien CA, Oyesola OO, Fruh SP, Webb LM, Smirnov I, Kovacs MA, Cowan MN, Hayes NW, Thompson JA, Tait Wojno ED, and Harris TH. 2020. Astrocytes promote a protective immune response to brain Toxoplasma gondii infection via IL-33-ST2 signaling. PLoS Pathog 16: e1009027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hunter CA, Chizzonite R, and Remington JS. 1995. IL-1b is required for IL-12 to induce production of IFN-g by NK cells. J. Immunol 155: 4347–4354. [PubMed] [Google Scholar]
- 57.Cai G, Kastelein R, and Hunter CA. 2000. Interleukin-18 (IL-18) enhances innate IL-12-mediated resistance to Toxoplasma gondii. Infect. Immun 68: 6932–6938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Munoz M, Eidenschenk C, Ota N, Wong K, Lohmann U, Kuhl AA, Wang X, Manzanillo P, Li Y, Rutz S, Zheng Y, Diehl L, Kayagaki N, van Lookeren-Campagne M, Liesenfeld O, Heimesaat M, and Ouyang W. 2015. Interleukin-22 Induces Interleukin-18 Expression from Epithelial Cells during Intestinal Infection. Immunity 42: 321–331. [DOI] [PubMed] [Google Scholar]
- 59.Langdon A, Crook N, and Dantas G. 2016. The effects of antibiotics on the microbiome throughout development and alternative approaches for therapeutic modulation. Genome Med 8: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kennedy EA, King KY, and Baldridge MT. 2018. Mouse Microbiota Models: Comparing Germ-Free Mice and Antibiotics Treatment as Tools for Modifying Gut Bacteria. Front Physiol 9: 1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Das A, Harly C, Ding Y, and Bhandoola A. 2022. ILC Differentiation from Progenitors in the Bone Marrow. Adv Exp Med Biol 1365: 7–24. [DOI] [PubMed] [Google Scholar]
- 62.Ghaedi M, Shen ZY, Orangi M, Martinez-Gonzalez I, Wei L, Lu X, Das A, Heravi-Moussavi A, Marra MA, Bhandoola A, and Takei F. 2020. Single-cell analysis of RORalpha tracer mouse lung reveals ILC progenitors and effector ILC2 subsets. J Exp Med 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lim AI, Li Y, Lopez-Lastra S, Stadhouders R, Paul F, Casrouge A, Serafini N, Puel A, Bustamante J, Surace L, Masse-Ranson G, David E, Strick-Marchand H, Le Bourhis L, Cocchi R, Topazio D, Graziano P, Muscarella LA, Rogge L, Norel X, Sallenave JM, Allez M, Graf T, Hendriks RW, Casanova JL, Amit I, Yssel H, and Di Santo JP. 2017. Systemic Human ILC Precursors Provide a Substrate for Tissue ILC Differentiation. Cell 168: 1086–1100 e1010. [DOI] [PubMed] [Google Scholar]
- 64.Constantinides MG, McDonald BD, Verhoef PA, and Bendelac A. 2014. A committed precursor to innate lymphoid cells. Nature 508: 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu W, Cherrier DE, Chea S, Vosshenrich C, Serafini N, Petit M, Liu P, Golub R, and Di Santo JP. 2019. An Id2(RFP)-Reporter Mouse Redefines Innate Lymphoid Cell Precursor Potentials. Immunity 50: 1054–1068 e1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, Hreggvidsdottir HS, Heinsbroek SE, Legrand N, Buskens CJ, Bemelman WA, Mjosberg JM, and Spits H. 2013. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol 14: 221–229. [DOI] [PubMed] [Google Scholar]
- 67.Jiao Y, Huntington ND, Belz GT, and Seillet C. 2016. Type 1 Innate Lymphoid Cell Biology: Lessons Learnt from Natural Killer Cells. Front Immunol 7: 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mercer HL, Snyder LM, Doherty CM, Fox BA, Bzik DJ, and Denkers EY. 2020. Toxoplasma gondii dense granule protein GRA24 drives MyD88-independent p38 MAPK activation, IL-12 production and induction of protective immunity. PLoS Pathog 16: e1008572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mukhopadhyay D, Arranz-Solis D, and Saeij JPJ. 2020. Toxoplasma GRA15 and GRA24 are important activators of the host innate immune response in the absence of TLR11. PLoS Pathog 16: e1008586. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.