

Abstract
Background:
Altered glutamatergic neurotransmission may contribute to impaired default mode network (DMN) function in Alzheimer’s disease (AD). Among the DMN hub regions, frontal cortex (FC) was suggested to undergo a glutamatergic plasticity response in prodromal AD, while the status of glutamatergic synapses in the precuneus (PreC) during clinical-neuropathological AD progression is not known.
Objective:
To quantify vesicular glutamate transporter VGluT1- and VGluT2-containing synaptic terminals in PreC and FC across clinical stages of AD.
Methods:
Unbiased sampling and quantitative confocal immunofluorescence of cortical VGluT1- and VGluT2-immunoreactive profiles and spinophilin-labeled dendritic spines were performed in cases with no cognitive impairment (NCI), mild cognitive impairment (MCI), mild-moderate AD (mAD), or moderate-severe AD (sAD).
Results:
In both regions, loss of VGluT1-positive profile density was seen in sAD compared to NCI, MCI, and mAD. VGluT1-positive profile intensity in PreC did not differ across groups, while in FC it was greater in MCI, mAD, and sAD compared to NCI. VGluT2 measures were stable in PreC while FC had greater VGluT2-positive profile density in MCI compared to sAD, but not NCI or mAD. Spinophilin measures in PreC were lower in mAD and sAD compared to NCI, while in FC they were stable across groups. Lower VGluT1 and spinophilin measures in PreC, but not FC, correlated with greater neuropathology.
Conclusion:
Frank loss of VGluT1 in advanced AD relative to NCI occurs in both DMN regions. In FC, an upregulation of VGluT1 protein content in remaining glutamatergic terminals may contribute to this region’s plasticity response in AD.
Keywords: Alzheimer’s disease, amyloid, glutamate, plasticity, synapse, vesicular glutamate transporter
INTRODUCTION
Alzheimer’s disease (AD) is a chronic neurodegenerative disorder associated with a complex clinical-pathological spectrum [1, 2]. It is characterized neuropathologically by extracellular deposits of fibrillar amyloid-β (Aβ) peptides in the brain parenchyma (plaques) and intracellular neurofibrillary tangles composed of hyperphosphorylated tau proteins. These AD hallmark lesions are accompanied by a variety of coexisting pathologies [3–5] as well as impairments in synaptic structure and loss of synaptic proteins [6–11]. Loss of synapses is a well-established biopathological correlate of impaired cognition and dementia in AD [12–17], while the relative contributions of amyloid and tau lesions to cognitive impairment is less clear. Although there is loss of total synapse number [10, 18–20] and general markers of pre- and post-synaptic elements [11, 21–24] in AD, co-occurring synaptic changes in specific neurotransmitter systems and their association with neuropathology burden and cognitive status during the progression of AD are not well understood.
Impaired synaptic transmission in glutamatergic neuronal circuits is believed to play a key role in the etiology of AD and contribute to the onset of dementia [25–31]. Synaptic availability and release of glutamate, the principal excitatory neurotransmitter in the central nervous system [32] is, in part, regulated by its uptake into presynaptic vesicles by vesicular glutamate transporter (VGluT) proteins [33]. Two major subtypes of VGluT proteins are type 1 (VGluT1) and type 2 (VGluT2) that define glutamatergic presynaptic terminals in cortico-cortical and thalamo-cortical circuitry, respectively [34–42]. Although a reduction of VGluT proteins is postulated as a mechanistic link between glutamate dysfunction and neuropathology in AD [43–45], findings are inconsistent possibly due to differences in brain areas examined, the stages of disease represented in the population, and the method of detection (e.g., protein measurements or bouton counts). For example, biochemical studies have reported a loss of VGluT1 in lateral parietal (BA39) and occipital (BA17) [44] and dorsolateral prefrontal (BA9) cortex [46] in AD, while changes in VGluT2 were more variable. Reduced VGluT2 protein levels have been reported in AD frontal cortex [46] but others have shown stable measures in frontal, temporal, parietal, and occipital cortex [44, 47] in AD. While biochemical analyses of brain tissue homogenates can detect changes in synaptic protein levels, immunohistochemical methods provide the cellular specificity, resolution, and topographic localization needed to analyze disease related changes in synaptology in specific cortical regions and cell layers. In this regard, a light microscopic study found a reduction of VGluT1-immunoreactive profiles in lamina III-IV of parietal and occipital cortex in AD compared to controls [44]. Using quantitative VGluT1 immunohistochemistry, another study reported that VGluT1-immunoreactive presynaptic bouton density in lamina III of the dorsolateral prefrontal cortex (BA 46/9) was lower in clinically mild and severe AD cases and paradoxically higher in prodromal AD (mild cognitive impairment, MCI) [43] compared to cognitively intact subjects, suggesting a transient early upregulation of glutamatergic synapses. The latter finding adds to the wealth of information supporting plasticity within the frontal hub of the default mode network (DMN) connectome that undergoes neuropathologic, metabolic, and connectivity changes early in AD [48–62]. Whether VGluT2 containing presynaptic bouton alterations mimic those related to VGluT1 in the frontal cortex, and the status of VGluT1 and VGluT2 in other hubs of the DMN including the precuneus cortex, remains under-investigated despite showing a high pathology burden early in AD [63–65] and that 60–70% of presynaptic terminals in the precuneus are glutamatergic, with a smaller proportion containing both VGluT1 and VGluT2 [66].
The present study used confocal microscopy combined with an unbiased stereological design to quantify VGluT1- and VGluT2-immunofluorescent profiles, markers of cortico-cortical/intra-cortical and thalamo-cortical presynaptic glutamatergic synapses, respectively, in lamina III of the precuneus cortex. Postmortem tissue was obtained from clinically and neuropathologically well-characterized NCI, MCI, and early AD participants in the Rush Religious Order Study (RROS) [67–70], and severe AD cases from the University of Pittsburgh Alzheimer’s Disease Research Center (ADRC) [71]. We hypothesized that VGluT1-positive terminals in the precuneus cortex are diminished, while VGluT2-positive terminals remain stable across the clinical-pathological stages of AD. In the same diagnostic groups, we also quantified densities of spinophilin-positive postsynaptic dendritic spines in the precuneus and determined the status of VGluT1- and VGluT2-positive terminals and spinophilin-positive postsynaptic dendritic spines in the frontal cortex, the region reported to display an upregulation of VGluT1 in MCI [43].
METHODS
Standard protocol approvals and patient consents
Rush University Medical Center and the University of Pittsburgh’s Committee for Oversight of Research and Clinical Training Involving Decedents approved this study. Written informed consent for research and brain autopsy was obtained for all subjects.
Subjects
A total of 80 cases were examined that included 60 participants from the RROS, a longitudinal clinicopathologic study of aging and AD in retired Catholic clergy [67–70] and 20 cases from the University of Pittsburgh ADRC [71]. The demographic, clinical, and neuropathological characteristics of the cases contributing precuneus and frontal cortex samples are summarized in Tables 1 and 2, respectively. Of the 60 RROS cases, 20 contributed precuneus only, 24 contributed frontal cortex only, and 16 contributed samples from both cortical regions. Of the 20 University of Pittsburgh ADRC cases, 10 cases contributed precuneus and 10 cases contributed frontal cortex tissue. The diagnosis of AD was made using standard diagnostic criteria [72]. The diagnosis of MCI was defined as impairment on neuropsychological testing, but without diagnosis of dementia by the examining neurologist, the criteria also used at other centers [73, 74]. Based on these criteria, and the last clinical evaluation within 12 months before death, the RROS cases were assigned to three clinical diagnostic groups: NCI (Mini-Mental State Exam, MMSE range 25–30), MCI (MMSE range 19–29), and mild to moderate AD (mAD, MMSE range 17–27). The AD cases from the University of Pittsburgh ADRC were clinically classified as moderate to severe AD (sAD, MMSE range 1–18). The neuropathology evaluation included immunohistochemical analyses of Aβ, phosphorylated tau, α-synuclein, and transactive response DNA-binding protein (TDP-43), as well as routine hematoxylin and eosin and Bielschowsky silver staining [75, 76]. Cases with stroke, Parkinson’s disease or hippocampal sclerosis were excluded from the study. Neuropathologic diagnosis was based on the National Institute on Aging (NIA)–Reagan Institute (RI) criteria (NIA-RI) [77], recommendations of the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) [78], and Braak staging of neurofibrillary tangles [79]. Application of the new NIA-Alzheimer’s Association guidelines [80, 81] to the examined cohorts is currently ongoing. All cases were de-identified and randomly assigned a unique 8-digit identifier that was used throughout the study. Investigators were blinded to case demographics and diagnosis throughout the experiment.
Table 1.
Demographic, clinical, and neuropathological characteristics across clinical-pathological diagnostic groups for cases contributing precuneus samples to the study
| Demographic, clinical, and neuropathological characteristics | Clinical-pathological diagnostic
groups and groupwise comparisons |
||||||
|---|---|---|---|---|---|---|---|
| NCI (n = 19) | MCI (n =10) | mAD (n = 7) | sAD (n =10) | Total (n = 46) | p | Groupwise comparisons | |
| Age (y) at death | |||||||
| Mean ± SD | 85.9 ± 5.4 | 87.2 ± 5.4 | 89.1 ± 4.3 | 80.5 ± 7.3 | 85.4 ± 6.3 | 0.03a | sAD<mAD |
| (Range) | (77.3–95.2) | (79.4–94.0) | (83.1–94.5) | (62–90.0) | (62–94.5) | ||
| Education (y) | |||||||
| Mean ± SD | 17.6 ± 3.5 | 17.3 ± 2.1 | 17.7 ± 2.9 | 15.5 ± 3.2 | 17.1 ± 3.1 | 0.36a | ANOVA not significant |
| (Range) | (10–25) | (14–20) | (14–23) | (12–20) | (10–25) | ||
| MMSE | |||||||
| Mean ± SD | 28.6 ± 1.5 | 26.0 ± 3.5 | 22.6 ± 3.2 | 7.5 ± 6.7 | 22.2 ± 9.3 | <0.001a | NCI>mAD; NCI, MCI > sAD |
| (Range) | (25–30) | (19–29) | (17–27) | (1–21) | (1–30) | ||
| PMI (h) | |||||||
| Mean ± SD | 6.3 ± 2.5 | 6.1 ± 2.9 | 5.5 ± 2.5 | 8.2 ± 3.6 | 6.6 ± 2.9 | 0.19a | ANOVA not significant |
| (Range) | (3.1–13) | (2.8–11.5) | (1.5–8.2) | (3–14) | (1.5–14) | ||
| Number (%) males | 8 (42) | 3 (30) | 2 (29) | 6 (60) | 19 (41) | 0.49b | Chi-square not significant |
| APOE ε4 (carrier/noncarrier) | 4/15 | 3/7 | 4/3 | 5/5* | 16/30* | 0.21b | Chi-square not significant |
| CERAD diagnosis | |||||||
| No AD | 5 | 1 | 0 | 0 | 6 | <0.001b | NCI, MCI < sAD |
| Possible | 3 | 1 | 0 | 0 | 4 | ||
| Probable | 10 | 6 | 4 | 0 | 20 | ||
| Definite | 1 | 2 | 3 | 10 | 16 | ||
| Braak stage | |||||||
| 0-II | 7 | 2 | 0 | 0 | 9 | <0.001b | NCI, MCI, mAD < sAD |
| III-IV | 11 | 6 | 5 | 0 | 22 | ||
| V-VI | 1 | 2 | 2 | 10 | 15 | ||
| NIA-RI diagnosis | |||||||
| No AD | 0 | 0 | 0 | 0 | 0 | <0.001b | NCI, MCI < sAD |
| Low | 11 | 3 | 0 | 0 | 14 | ||
| Intermediate | 8 | 6 | 5 | 1 | 20 | ||
| High | 0 | 1 | 2 | 9 | 12 | ||
Kruskal-Wallis one-way analysis of variance
Chi-square analysis.
AD, Alzheimer’s disease; ANOVA, one-way analysis of variance; CERAD, Consortium to Establish a Registry for Alzheimer’s Disease; MCI, mild cognitive impairment; mAD, mild to moderate AD; NIA-RI, National Institute on Aging-Reagan Institute; NCI, no cognitive impairment; PMI, postmortem interval; sAD, moderate to severe AD; SD, standard deviation.
APOE ε4 data not available for one case.
Table 2.
Demographic, clinical, and neuropathological characteristics across clinical-pathological diagnostic groups for cases contributing frontal cortex samples to the study
| Demographic, clinical, and neuropathological characteristics | Clinical-pathological diagnostic
groups and groupwise comparisons |
||||||
|---|---|---|---|---|---|---|---|
| NCI | MCI | mAD | sAD | Total | p | Groupwise comparisons | |
| (n =19) | (n =11) | (n =10) | (n =10) | (n = 50) | |||
| Age (y) at death | |||||||
| Mean ± SD | 85.1 ± 5.2 | 88.0 ± 4.1 | 89.8 ± 4.0 | 76.1 ± 9.2 | 84.8 ± 7.2 | <0.001a | sAD<MCI, mAD |
| (Range) | (76–93) | (81–92) | (83–95) | (59–90) | (59–95) | ||
| Education (y) | |||||||
| Mean ± SD | 17.4 ± 3.5 | 17.8 ± 1.7 | 17.4 ± 2.2 | 14.5 ± 2.5 | 16.9 ± 2.9 | 0.032a | No groupwise differences |
| (Range) | (10–25) | (15–20) | (14–21) | (12–19) | (10–25) | ||
| MMSE | |||||||
| Mean ± SD | 27.8 ± 1.5 | 25.8 ± 2.3 | 22.8 ± 2.0 | 6.8 ± 4.8 | 22.2 ± 8.4 | <0.001a | NCI > mAD, sAD; MCI > sAD |
| (Range) | (26–30) | (22–29) | (20–27) | (1–16) | (1–30) | ||
| PMI (h) | |||||||
| Mean ± SD | 5.0 ± 2.3 | 6.6 ± 3.0 | 5.7 ± 2.7 | 5.6 ± 3.0 | 5.6 ± 2.7 | 0.46a | ANOVA not significant |
| (Range) | (1–10) | (3–14) | (2–11) | (2–10) | (1–14) | ||
| Number (%) males | 7 (37) | 3 (27) | 3 (30) | 3 (30) | 16 (32) | 0.94b | Chi-square not significant |
| APOE ε4 (carrier/noncarrier) | 1/19 | 5/11 | 2/10 | 6/10 | 14/50 | 0.08b | Chi-square not significant |
| CERAD diagnosis | |||||||
| No AD | 6 | 3 | 0 | 0 | 9 | <0.001b | NCI,MCI < sAD; NCI < mAD |
| Possible | 2 | 0 | 0 | 0 | 2 | ||
| Probable | 9 | 6 | 3 | 0 | 18 | ||
| Definite | 2 | 2 | 7 | 10 | 21 | ||
| Braak stage | |||||||
| 0-II | 7 | 0 | 0 | 0 | 7 | <0.001b | NCI, MCI, mAD<sAD |
| III-IV | 11 | 7 | 6 | 0 | 24 | ||
| V-VI | 1 | 4 | 4 | 10 | 19 | ||
| NIA-RI diagnosis | |||||||
| No AD | 0 | 0 | 0 | 0 | 0 | <0.001b | NCI,MCI<sAD; NCI < mAD |
| Low | 9 | 3 | 0 | 0 | 12 | ||
| Intermediate | 10 | 6 | 6 | 0 | 22 | ||
| High | 0 | 2 | 4 | 10 | 16 | ||
Kruskal-Wallis one-way analysis of variance
Chi-square analysis.
AD, Alzheimer’s disease; ANOVA, one-way analysis of variance; CERAD, Consortium to Establish a Registry for Alzheimer’s Disease; MCI, mild cognitive impairment; mAD, mild to moderate AD; NIA-RI, National Institute on Aging-Reagan Institute; NCI, no cognitive impairment; PMI, postmortem interval; sAD, moderate to severe AD; SD, standard deviation.
Tissue preparation
The precuneus (medial BA7) and dorsolateral frontal cortex (BA9) were dissected at autopsy and immersion fixed in 4% paraformaldehyde made in 0.1 M (pH 7.2) phosphate buffer (PB) for 48–72 h at 4°C. Following fixation, samples were cryoprotected in 10% glycerol with 2% dimethyl sulfoxide (DMSO) in 0.1 M phosphate buffer at 4°C for two days, followed by immersion in a solution of 20% glycerol and 2% DMSO. Tissue samples were cut on a freezing sliding microtome at 40 μm thickness and sections were stored in cryoprotectant at −20°C until processed further.
Immunohistochemistry for vesicular glutamate transporters 1 and 2 and spinophilin
To minimize potential variations across multiple sets of tissue sections processed using the immunofluorescence procedure, each of three batches of processed tissue sections included an equal number of cases from each clinical diagnostic group. Sections from select cases were included in all batches to monitor chemical batch-to-batch variation. Immunofluorescence using antibodies generated against VGluT1 or VGluT2 was performed on two sections per case, as described previously [82]. Free-floating tissue sections were washed in PB for 5 min three times to remove the cryoprotection solution. Antigen retrieval was performed by heating the sections in 10 mM sodium citrate buffer (pH 8.6) at 80°C for 75 min. The sections in the antigen retrieval solution were then cooled first at room temperature followed by immersion in an ice bath (approximately 7°C), each for 5 min. After cooling, the sections were removed from the antigen retrieval solution and incubated in the blocking solution “A” (5% goat serum, 5% human serum, 0.5% Triton X-100, 0.1% lysine, and 0.1% glycine made in PB) for 3 h at room temperature. Sections were then rinsed in the blocking solution “B” (1% donkey serum, 1% human serum, 0.3% Triton X-100, 0.1% lysine, and 0.1% glycine made in PB) for 5 min three times. Sections were then incubated in a cocktail of a guinea pig anti-VGluT1 IgG (AB5905, Millipore, Billerica, MA), and a rabbit anti-VGluT2 IgG (V2514, HY-19; Sigma-Aldrich, St. Louis, MO) made in blocking solution B for 96 h. The specificity and labeling patterns of these antibodies have been reported previously [82]. Following primary antibody incubation, sections were rinsed in blocking solution B for 5 min three times and then incubated for 24 h at 4°C in a cocktail of donkey anti-guinea pig Alexa Fluor 594-conjugated (1:500; ThermoFisher, Waltham, MA, #A-11076) and donkey anti-rabbit Alexa Fluor 647-conjugated (1:500; ThermoFisher, #A-31573) secondary antibodies diluted in blocking solution B.
Spinophilin immunofluorescence was performed on two sections of frontal cortex per case, using tissue sections adjacent to those analyzed for VGluT profiles as previously detailed for the precuneus [83]. Briefly, free-floating tissue sections were incubated in a polyclonal rabbit anti-spinophilin antibody (Millipore, Temecula, CA, #AB5669) [83] for 96 h at 4°C. The specificity and selectivity of the spinophilin antibody was described in a previous publication [83]. Following primary antibody incubation, sections were incubated for 24 h at 4°C in biotinylated donkey anti-rabbit secondary antibody (1:200; Jackson, West Grove, PA, #711–066-152), followed by a 24-h incubation at 4°C with a Cy5-streptavidin conjugate (1:500; Jackson, #016–170-084).
A fluorescent counterstain for Nissl substance (Neurotrace, ThermoFisher, #N21480) was used to delineate cortical lamina III [84]. Sections were mounted onto charged slides (Superfrost Plus, ThermoFisher), coverslipped with ProLong™ Diamond Antifade Mountant (ThermoFisher, # P36970) and sealed with Sally Hansen nail hardener.
Cyano-PiB and X-34 histofluorescence procedure and percent area analysis of fibrillar Aβ and tau burden
Tissue sections adjacent to those processed using VGluT1, VGluT2, and spinophilin immunofluorescence were stained separately with cyano-PiB, a marker of fibrillar Aβ in amyloid plaques [85, 86], and the fluorescent Congo red derivative X-34, a pan-amyloid marker of fibrillar Aβ and tau aggregates [87, 88]. For cyano-PiB staining, sections were incubated in 10 mM cyano-PiB for 45 min, dipped three times in potassium phosphate buffer (PBK, 0.1 M, pH 7.4), followed by a 1-min differentiation in PBK, and coverslipped with Fluoromount G (Electron Microscopy Services, Hatfield, PA) [86]. For X-34 staining, sections were washed in PBK three times followed by a 10-min incubation in X-34 (100 μM) solution, then dipped five times in tap water before incubating sections in 0.2% NaOH made in 80% unbuffered ethanol for 2 min. Sections were then rinsed in tap water for 10 min and coverslipped with Fluoromount G [88]. Percent area coverage for cyano-PiB- and X-34-labeled Aβ plaques was determined in three randomly spaced microscopic fields (using a 10 objective) in each of two sections from the same cases used for VGluT1, VGluT2, and spinophilin quantification. Percent area values were obtained by dividing cyano-PiB and X-34 labeled area by total area sampled. To assess the relationship of VGluT1 and VGluT2 immunoreactive profiles with amyloid dye-labeled plaques, a separate set of sections double-immunostained for VGluT1 and VGluT2 were subsequently stained with cyano-PiB.
Confocal sampling and image collection
Image collection was performed as previously described [84]. Briefly, image stacks were acquired using an Olympus BX51WI upright microscope (Olympus, Center Valley, PA) equipped with an Olympus DSU spinning disk confocal, an ORCA-R2 CCD camera (Hamamatsu, Bridgewater, NJ), an MBF CX9000 front-mounted digital camera (MBF Bioscience, Williston, VT) and a BioPrecision2 XYZ motorized stage with linear XYZ encoders (Ludl Electronic Products, Ltd., Hawthorne, NY). The microscope was controlled using Stereo Investigator (MBF Bioscience) and SlideBook software (Intelligent Imaging Innovations, Denver, CO). Using Stereo Investigator software, the borders of cortical lamina III were defined and 14 sites per case were identified within the laminar borders for analysis using systematic uniform random sampling. At each sampling site, tissue thickness was measured. Image stacks were collected using a 60×, N.A. 1.4, super-corrected objective, with a step size of 0.25 μm between Z-axis planes, starting from 4 μm below the tissue surface closest to the cover glass and stepping up until the tissue surface was reached, yielding a 4 μm thick (Z-axis depth) stack comprised of 16 individual two-dimensional planes. Image planes were 512 × 512-pixels (approximately 55 × 55 μm). Exposure time during image stack acquisition was optimized for each site, and differences in exposure were normalized during analysis. Guard zones of 10 pixels were applied around all edges in the X and Y dimensions of each stack, and ten Z-axis planes starting four planes below the cover glass were included in analysis, as antibody penetration was uniform (profile counts and intensities were uniform, not shown) across these Z-axis depths (four planes x 0.25 μm step size = 1 μm disector height). In our quantitative analysis, sampling sites were automatically selected through systematic uniform random sampling using Stereo Investigator and therefore blind to cyano-PiB plaque burden status, preventing the potential bias of selecting only cyano-PiB-positive or only cyano-PiB-negative microscopic fields.
Post-processing and quantification of vesicular glutamate transporters VGluT1 and VGluT2 and spinophilin immunoreactive profiles
Image stacks were post-processed using Slide-Book and Automation Anywhere software (Automation Anywhere, Inc., San Jose, CA) as previously described [82]. Images were then processed using a blind deconvolution algorithm (AutoQuant; Media Cybernetics, Rockville, MD). To improve edge detection during image segmentation, for each image stack two new channels were made by convolving the VGluT1 and VGluT2 channels with a Gaussian function of standard deviations σ =2 and σ = 0.7. The channel transformation with the larger standard deviation (σ = 2) was then subtracted from the one with the smaller standard deviation (σ = 0.7) to make new channels. Intensity segmentation was then coupled with morphological selection using an iterative masking approach [89] where the Otsu algorithm was used to determine the intensity threshold for the first iteration. After each segmentation step, mask objects were size gated to select immunoreactive profiles (0.1 to 2 μm3 for VGluT1, 0.05 to 2 μm3 for VGluT2, and 0.03 to 0.5 μm3 for spinophilin) and merged with the mask generated in the prior segmentation step. Profile mean intensity was extracted from these image stacks through the generated mask to identify objects of interest and normalized to the exposure time for that stack. The density of VGluT1- and VGluT2-immunoreactive boutons as well as spinophilin-immunoreactive dendritic spines was determined by dividing the total number of masked objects counted per each case (Q) by the total volumes, which is the product of the number of sampling sites per case (CF), the area of the counting frame at each site in μm2 (2,823), and the disector height (1 μm) multiplied by the microtome block advance (40 μm) divided by the measured tissue section thickness at each site (tq) to correct for tissue shrinkage [90] as follows:
Cortical thickness estimation
The thickness of precuneus and dorsolateral frontal cortex cortical lamina III in each case was measured on low magnification images (1.25× objective) from sections stained with Neurotrace using the calibrated measuring tool in ImageJ (ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA). Two sections per case were used for quantification.
Statistical analysis
The Kruskall-Wallis test was used to test group differences for cyano-PiB, X-34, protein immunoreactivity measures, and cognitive measures. Chi-square analysis was used to determine if sex, APOE ε4 allele, and pathology group frequencies differed between clinical groups. Spearman correlation was used to assess associations between protein immunoreactivity measures as well as cyano-PiB- and X-34-labeled pathological burden, and cognitive measures. Statistically significant correlations were analyzed using robust regression models that adjusted for age at death, sex, education, and APOE ε4 carrier status. A secondary analysis of VGluT immunoreactivity was performed after dividing the NCI group into Low Pathology (LP-NCI) and High Pathology (HP-NCI) subgroups. LP-NCI cases were defined as having a NIA-RI diagnosis of No AD or Low Likelihood of AD, CERAD rating of No or Possible AD, and Braak stages 0–III for neurofibrillary tangles. HP-NCI cases were defined as having a NIA-RI diagnosis of Moderate or High Likelihood of AD, CERAD rating of Probable or Definite AD, or Braak stage IV–VI for neurofibrillary tangles. False Discovery Rate was used to correct for multiple comparisons among correlations and robust regression models, VGluT1 density, VGluT1 intensity, VGluT2 density, VGluT2 intensity were considered as dependent variables in the analyses. Statistically significance was set at p < 0.05.
RESULTS
Case demographic, clinical, and neuropathological characteristics
Cases contributing precuneus samples
Demographic, clinical, and neuropathological classification data for NCI, MCI, mAD, and sAD groups are shown in Table 1. Among the four groups, years of education, postmortem interval, sex, and APOE ε4 carrier frequency were not significantly different; however, sAD cases were significantly younger than the mAD group (Kruskall-Wallis statistic = 8.776, p = 0.0324; Dunn’s multiple comparisons, p = 0.0346) (Table 1).
There were significant group differences in the MMSE scores (Kruskall-Wallis statistic = 32.87, p < 0.001): the mAD group performed worse than the NCI group (Dunn’s multiple comparisons, p = 0.0148) and the sAD group performed worse than the NCI group and the MCI group (Dunn’s multiple comparisons, p < 0.001 and p = 0.0051, respectively).
The four clinical groups differed significantly by CERAD diagnosis (Kruskall-Wallis statistic = 24.19, p < 0.001; NCI, MCI < sAD, Dunn’s multiple comparisons, p < 0.001 and p = 0.0123, respectively), Braak stage (Kruskall-Wallis statistic = 23.54, p < 0.001; NCI, MCI, mAD < sAD, Dunn’s multiple comparisons, p < 0.001, p = 0.0091, and p = 0.0496, respectively), and likelihood of AD by the NIA-Reagan Institute (NIA-RI) criteria (Kruskall-Wallis statistic = 26.32, p < 0.001; NCI, MCI < sAD, Dunn’s multiple comparisons, p < 0.001 and p = 0.0085, respectively) (Table 1).
Spearman rank order correlation analysis revealed statistically significant associations between lower MMSE scores with higher Aβ pathology burden determined by cyano-PiB staining (rs = −0.6087, p < 0.0001) and with higher total amyloid burden using X-34 staining (rs = −0.6000, p < 0.0001) (both determined in precuneus sections adjacent to those used for VGluT analyses) as well as greater severity of AD neuropathology according to the NIA-RI criteria (rs = −0.6954, p < 0.0001), CERAD diagnosis (rs = −0.6053, p < 0.0001) and Braak stage (rs = −0.7030, p < 0.0001). Robust regression analysis controlling for demographic and biological variables revealed statistically significant correlations between MMSE and Aβ pathology burden quantified by cyano-PiB (β = –1.73, 95% CI (–2.23, –1.23), p < 0.001) and total amyloid burden quantified by X-34 (β = –1.48, 95% CI (–2.10, –0.87), p < 0.001), but not with NIA-RI criteria (β = –2.62, 95% CI (–6.18, 0.93), p = 0.16), CERAD diagnosis (β = –1.48, 95% CI (–3.54, 0.59), p = 0.17), or Braak stage (β = –1.36, 95% CI (–2.81, 0.10), p = 0.08).
Cases contributing frontal cortex tissue
Demographic, clinical, and neuropathological classification data for NCI, MCI, mAD, and sAD groups are shown in Table 2. As observed for the precuneus cases, years of education, postmortem interval, sex, and APOE ε4 carrier frequency were not significantly different among the four clinical diagnostic groups, but the sAD cases were significantly younger than the mAD and MCI cases (Kruskall-Wallis statistic = 17.22, p < 0.001, Dunn’s multiple comparisons, p = 008 and p = <0.001, respectively) (Table 2).
Like the precuneus analysis, clinical and neuropathological variables were significantly different among the four case groups that formed the frontal cortex samples (Table 2). There were significant group differences in the MMSE scores (Kruskall-Wallis statistic = 36.61, p < 0.001): the mAD and sAD groups performed worse than the NCI group (Dunn’s multiple comparisons, p = 0.002 and p < 0.001, respectively) and the sAD group also performed worse than the MCI group (Dunn’s multiple comparisons, p = 0.001). The four clinical groups also differed significantly by CERAD diagnosis (Kruskall-Wallis statistic = 25.36, p < 0.001; NCI, MCI < sAD and NCI < mAD, Dunn’s multiple comparisons, p < 0.001, p = 0.0042 and 0.0059, respectively), Braak stage (Kruskall-Wallis statistic = 33.29, p < 0.001; NCI, MCI, mAD < sAD, Dunn’s multiple comparisons, p < 0.001, p = 0.0061 and p = 0.0317, respectively), and likelihood of AD by the NIA-Reagan Institute criteria (Kruskall-Wallis statistic = 28.36, p < 0.001; NCI, MCI < sAD and NCI < mAD, Dunn’s multiple comparisons, p < 0.001, p = 0.0040 and p = 0.0191 respectively) (Table 2).
Spearman rank order correlation revealed statistically significant associations between MMSE with cyano-PiB-labeled amyloid plaque burden (rs = −0.5467, p < 0.0001) and with X-34-labeled total amyloid burden (rs = −0.5627, p < 0.0001) (both determined in frontal cortex sections adjacent to those used for VGluT and spinophilin analyses) as well as severity of AD neuropathology by NIA-RI criteria (rs = −0.7126, p < 0.0001), CERAD diagnosis (rs = −0.6163, p < 0.0001) and Braak stage (rs = −0.7322, p < 0.0001). Robust regression analysis controlling for demographic and biological factors showed that the associations with MMSE remained significant for the three neuropathological diagnostic measures (NIA-RI criteria, p < 0.001; Braak stage, p < 0.001; CERAD criteria, p = 0.005) and X-34 total amyloid burden (p = 0.03), but not for the cyano-PiB plaque load (p = 0.07) in the frontal cortex.
Precuneus synaptic profile analyses
VGluT protein immunoreactivity in precuneus lamina III
VGluT1 and VGluT2 immunofluorescence appeared as well-defined bright puncta that were co-distributed across the imaged areas within lamina III of the precuneus in the NCI cases (Fig. 1A–C). Similar qualitative patterns of VGluT1 and VGluT2 immunofluorescence were observed in the precuneus from MCI (not shown) and mAD cases (Fig. 1D–F). The sAD group had markedly fewer VGluT1-labeled profiles (Fig. 1G, I), but not VGluT2-labeled profiles (Fig. 1H, I), compared to the other three groups. Both transporter proteins were scarce within cyano-PiB labeled amyloid plaques (Fig. 1J–L). Unbiased quantitative analysis of precuneus VGluT1-immunoreactive profile densities confirmed the lack of significant differences among the NCI, MCI, and mAD groups, while a significantly lower density of VGluT1-positive profiles was detected in sAD compared to the other three groups (Fig. 2A, Table 3). The four groups did not differ statistically when compared for intensity of VGluT1-immunoreactive profiles, a surrogate measure of relative protein level (Fig. 2B, Table 3), or density and intensity of VGluT2-immunoreactive profiles (Fig. 2C, D, Table 3) in the precuneus. The thickness of lamina III of the precuneus was not different among the four clinical groups (not shown).
Fig. 1.
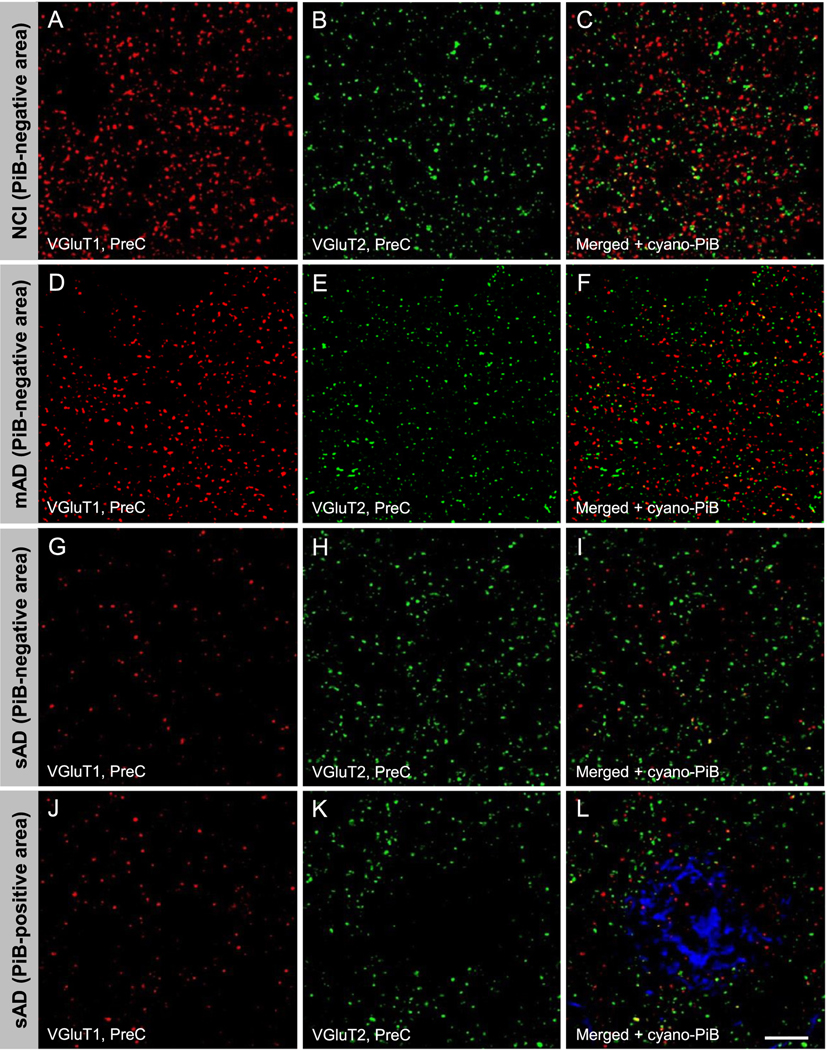
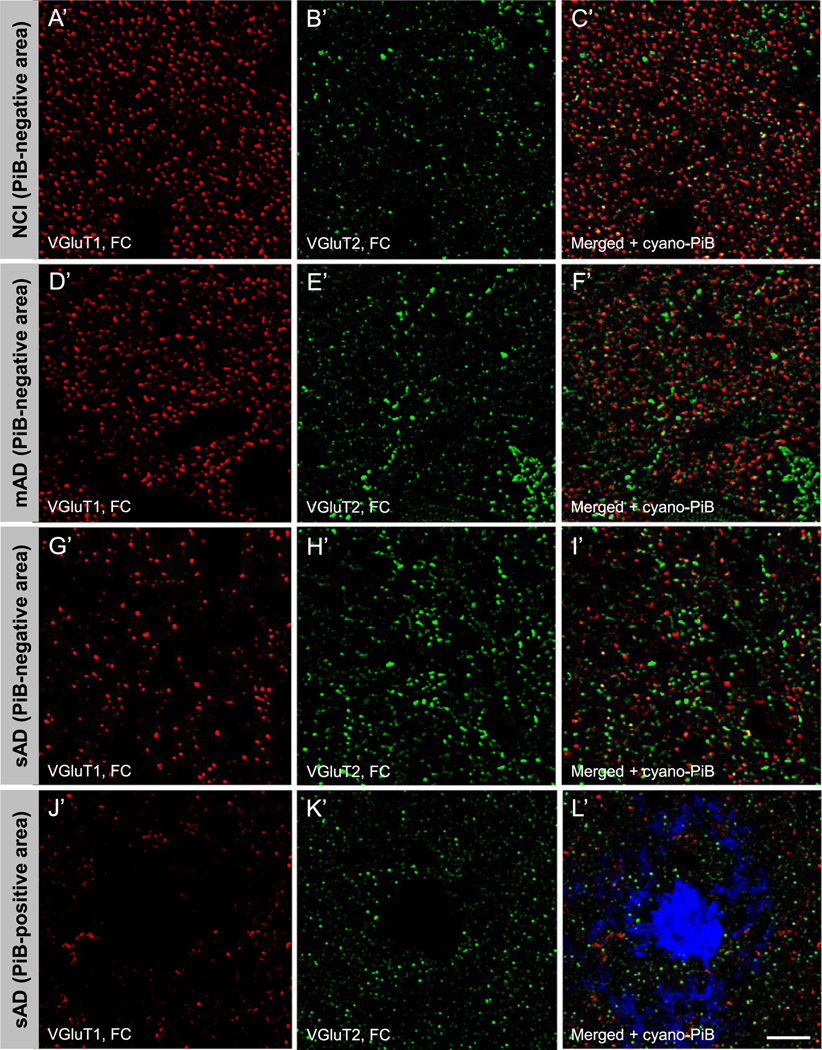
Vesicular glutamate transporter 1 (VGluT1) immunoreactive (red fluorescence, precuneus: A, D, G, J; red fluorescence, frontal cortex: A’, D’, G’, J’) and VGluT2 immunoreactive (green fluorescence, precuneus: B, E, H, K; green fluorescence, frontal cortex: B’, E’, H’, K’) profiles in precuneus lamina III from a case with no cognitive impairment (NCI, precuneus: A, B, C; frontal cortex: A’, B’, C’), a case with mild AD (mAD, precuneus: D, E, F; frontal cortex: D’, E’, F’) and a case with severe Alzheimer’s disease (sAD, precuneus: G, H, I and J, K, L; frontal cortex G’, H’, I’ and J’, K’, L’). Scale bar = 15 μm (A–L); 18 μm (A’–L’).
Fig. 2.

Quantitative measures of vesicular glutamate transporter 1 (VGluT1) immunoreactive profile densities (precuneus, A; frontal cortex, E) and relative protein levels (intensity) (precuneus, B; frontal cortex, F) and vesicular glutamate transporter 2 (VGluT2) immunoreactive profile densities (precuneus, C; frontal cortex, G) and relative protein levels (intensity) (precuneus, D; frontal cortex, H) from cases with no cognitive impairment (NCI), mild cognitive impairment (MCI), mild to moderate Alzheimer’s disease AD (mAD) and moderate to severe AD (sAD). In each clinical diagnostic group, cases with a CERAD diagnosis of No AD/Possible AD (“low pathology”) are identified by shaded symbols and cases with CERAD diagnosis of Probable AD/Definite AD (“high pathology”) are identified by empty symbols. Within the MCI groups, symbols for data points of amnestic cases contain the symbol “×”. The single solid line in each plot indicates the median value. Brackets in panel A indicate statistically significant groupwise differences (p < 0.05) determined by the Kruskall-Wallis analysis of variance and Dunn’s multiple comparisons test.
Table 3.
Clinical group differences for vesicular glutamate transporter and spinophilin immunoreactivity measures obtained in the precuneus cortex
| NCI | MCI | mAD | sAD | p | Groupwise comparison(s) | |
|---|---|---|---|---|---|---|
| VGluTl density | ||||||
| Mean ± SD | 0.045 ± 0.009 | 0.041 ± 0.003 | 0.042 ± 0.003 | 0.030 ± 0.006 | <0.001a | NCI, MCI, mAD>sAD |
| (Median) | (0.0448) | (0.0416) | (0.0426) | (0.0305) | ||
| VGluTl intensity | ||||||
| Mean ± SD | 3583±786 | 3,763 ± 566 | 3,952 ± 619 | 3,762 ± 1,460 | 0.91a | ANOVA not significant |
| (Median) | (3,696) | (3,711) | (3,960) | (4,181) | ||
| VGluT2 density | ||||||
| Mean ± SD | 0.06 ± 0.05 | 0.04 ± 0.02 | 0.06 ± 0.01 | 0.05 ± 0.01 | 0.12a | ANOVA not significant |
| (Median) | (0.0448) | (0.0389) | (0.0556) | (0.0501) | ||
| VGluT2 intensity | ||||||
| Mean ± SD | 477±436 | 737±479 | 456 ± 207 | 427 ± 244 | 0.11a | ANOVA not significant |
| (Median) | (396) | (591) | (377) | (334) | ||
| Spinophilin density* | ||||||
| Mean ± SD | 0.090 ± 0.03 | 0.068 ± 0.030 | 0.042 ± 0.030 | 0.009 ± 0.004 | 0.0003a | NCI, MCI > sAD; NCI > mAD |
| (Median) | (0.086l) | (0.0605) | (0.0344) | (0.0081) | ||
| Spinophilin intensity* | ||||||
| Mean ± SD | 2908 ± 131.6 | 2782 ± 246.6 | 2671 ± 184.8 | 2519 ± 50.86 | 0.0004a | NCI, MCI > mAD, sAD |
| (Median) | (2888) | (2830) | (2636) | (2509) |
Kruskal-Wallis one-way analysis of variance.
These data were published previously [84].
AD, Alzheimer’s disease; ANOVA, one-way analysis of variance; NCI, no cognitive impairment; MCI, mild cognitive impairment; mAD, mild to moderate AD; sAD, moderate to severe AD; SD, standard deviation; VGluT, vesicular glutamate transporter.
Spinophilin protein immunoreactivity in precuneus lamina III
Unbiased quantitative analysis showed statistically lower densities of spinophilin-immunoreactive profiles in the mAD and sAD groups compared to the NCI, and in the sAD group compared to MCI, as well as statistically significantly lower intensity of spinophilin-immunoreactive profiles in the mAD and sAD groups compared to NCI and MCI (Table 3) previously [84].
Frontal cortex synaptic profile analyses
VGluT protein immunoreactivity in frontal cortex lamina III
VGluT1 and VGluT2 immunofluorescent profile appearance and distribution in the dorsolateral frontal cortex lamina III (hereafter referred to as the frontal cortex) was similar in NCI (Fig. 1A’–C’), MCI (not shown), and mAD cases (Fig. 1D’–F’). Cases with sAD had markedly fewer immunoreactive VGluT1 (Fig. 1G’, I’), but not VGluT2 profiles (Fig. 1H’, I’) compared to the other three groups. Both transporter proteins were scarce within cyano-PiB labeled amyloid plaques (Fig. 1J’–L’). Unbiased quantitative analysis of frontal cortex VGluT1-immunoreactive profile densities confirmed the lack of significant differences among the NCI, MCI, and mAD groups, while a statistically significantly lower density of VGluT1-immunoreactive profiles was detected in sAD compared to the other three groups (Fig. 2E, Table 4). In contrast to the data from precuneus, the mean VGluT1-immunoreactive profile intensity (relative VGluT1 protein level) in the frontal cortex was significantly higher in MCI, mAD, and sAD compared to the NCI group (Fig. 2F, Table 4). VGluT2-immunoreactive profile density in the frontal cortex was significantly lower in sAD than MCI (Fig. 2G, Table 4), while no group differences were detected for VGluT2-immunoreactive profile intensity (Fig. 2H, Table 4). The thickness of lamina III of the frontal cortex was not different among the four clinical groups (not shown).
Table 4.
Clinical group differences for vesicular glutamate transporter and spinophilin immunoreactivity measures obtained in the frontal cortex
| NCI | MCI | mAD | sAD | p | Groupwise comparison(s) | |
|---|---|---|---|---|---|---|
| VGluTl density | ||||||
| Mean ± SD | 0.040 ± 0.007 | 0.044 ± 0.007 | 0.043 ± 0.008 | 0.030 ± 0.008 | <0.001a | NCI, MCI, mAD > sAD |
| (Median) | (0.0401) | (0.0451) | (0.0417) | (0.0305) | ||
| VGluTl intensity | ||||||
| Mean ± SD | 3071 ± 680.8 | 3662 ± 786.2 | 3800 ± 501.0 | 4031±1083 | 0.013a | MCI, mAD, sAD>NCI |
| (Median) | (3128) | (3914) | (3951) | (3583) | ||
| VGluT2 density | ||||||
| Mean ± SD | 0.038 ± 0.02 | 0.05 ± 0.01 | 0.05 ± 0.02 | 0.03 ± 0.01 | 0.013a | MCI>sAD |
| (Median) | (0.0331) | (0.0473) | (0.0471) | (0.0284) | ||
| VGluT2 intensity | ||||||
| Mean ± SD | 422 ± 243 | 296 ± 94 | 360±184 | 463 ± 149 | 0.13a | ANOVA not significant |
| (Median) | (347) | (278) | (323) | (494) | ||
| Spinophilin density | ||||||
| Mean ± SD | 0.1301 ± 0.06 | 0.1527 ± 0.03 | 0.1408 ± 0.06 | 0.1031 ± 0.05 | 0.06a | ANOVA not significant |
| (Median) | (0.1099) | (0.1440) | (0.1281) | (0.09) | ||
| Spinophilin intensity | ||||||
| Mean ± SD | 2815 ± 859.7 | 2323 ± 532.4 | 3086 ± 704.5 | 2204 ± 463.8 | 0.09a | ANOVA not significant |
| (Median) | (2604) | (2421) | (3072) | (2247) |
Kruskal-Wallis one-way analysis of variance.
AD, Alzheimer’s disease; ANOVA, one-way analysis of variance; NCI, no cognitive impairment; MCI, mild cognitive impairment; mAD, mild to moderate AD; sAD, moderate to severe AD; SD, standard deviation; VGluT, vesicular glutamate transporter.
Spinophilin protein immunoreactivity in frontal cortex lamina III
The appearance of frontal cortex spinophilin-immunoreactive profiles was like that observed in the precuneus [84]. Unbiased quantitative analysis showed that spinophilin-immunoreactive density or intensity measures did not differ across the four clinical diagnostic groups (Table 4).
Precuneus and the frontal cortex synaptic profiles in NCI cases with low and high neuropathology burden
We divided NCI cases into two subgroups of Low Pathology NCI (LP-NCI) and High Pathology NCI (HP-NCI or “preclinical AD”) cases based on their neuropathology status according to NIA-RI diagnosis (not AD and low likelihood = LP-NCI; moderate and high likelihood of AD = HP-NCI), CERAD diagnosis (CERAD 0, 1 = LP-NCI; CERAD 2, 3 = HP-NCI), or Braak stage for neurofibrillary tangles (Braak 0–III = LP-NCI, Braak IV–VI = HP-NCI). None of these classifications resulted in statistically significant differences in VGluT1, VGLuT2, or spinophilin measures between the LP-NCI and HP-NCI subgroups in either cortical region (not shown).
Correlations between VGluT and spinophilin measures with quantitative amyloid plaque pathology, neuropathology severity, and MMSE
Precuneus cortex
Cyano-PiB-labeled Aβ amyloid burden and X-34-labeled pan-amyloid (Aβ and tau) burden of the precuneus showed statistically significant group differences (Kruskall-Wallis statistic = 19.77 and 18.84, p = 0.0002 and 0.0003, respectively), with sAD displaying a significantly higher amyloid pathology burden than the NCI (Dunn’s multiple comparisons, cyano-PiB: p = 0.0002; X-34: p = 0.0001) and MCI (Dunn’s multiple comparisons, cyano-PiB: p = 0.0024; X-34: p = 0.0178) groups but not the mAD group (Fig. 3A, B). Spearman rank order correlation analysis across all cases showed a statistically significant correlation between greater burden of cyano-PiB positive amyloid plaques and lower density of VGluT1-immunoreactive profiles in the precuneus (rs = −0.4306, p = 0.0028) (Fig. 3C). Lower density of VGluT1 immunoreactive profiles also correlated significantly with greater X-34-labeled total amyloid pathology burden in the precuneus (rs = −0.4216, p = 0.0035) (Supplementary Table 1) and with more severe neuropathology based upon CERAD (rs = −0.5681, p < 0.0001) (Fig. 3D), likelihood of AD by NIA-RI criteria (rs = −0.5018, p = 0.0004) (Supplementary Table 1) or Braak stage (rs = −0.4227, p = 0.0034) (Supplementary Table 1). Robust regression analyses controlling for demographic factors and APOE ε4 allele frequency confirmed that VGluT1 density was associated with quantitative measures of cyano-PiB amyloid plaques (β = −0.001, 95% CI (−0.002, −0.0007), p < 0.001) and X-34 labeled total amyloid pathology (β = −0.0006, 95% CI (−0.0001, −0.0002), p = 0.009), Braak stage (β = −0.002, 95% CI (−0.0003, −0.0009), p < 0.001), NIA-RI criteria (β = −0.0005, 95% CI (−0.0006, −0.003), p < 0.001), but not CERAD diagnosis (β = −0.004, 95% CI (−0.009, 0.001), p = 0.16). Precuneus VGluT1 intensity as well as VGluT2 density and intensity measures did not correlate with neuropathology data.
Fig. 3.

Quantitative measures of cyano-Pittsburgh compound B (cyano-PiB)-labeled amyloid plaque load (precuneus, A; frontal cortex, E) and X-34-labeled total amyloid load (amyloid plaques and neurofibrillary pathology) (precuneus, B; frontal cortex, F) in clinical groups of no cognitive impairment (NCI), mild cognitive impairment (MCI), mild to moderate Alzheimer’s disease (mAD), and moderate to severe AD (sAD). Correlations between VGluT1 immunoreactive profile densities and cyano-PiB (precuneus, C; frontal cortex, G) and VGluT1 immunoreactive profile densities and CERAD diagnosis (precuneus, D; frontal cortex, H). In each group, cases with a CERAD diagnosis of No AD/Possible AD (“low pathology”) are identified by shaded symbols and cases with CERAD diagnosis of Probable AD/Definite AD (“high pathology”) are identified by open symbols. In the clinical group comparison panels, symbols for data points of amnestic MCI cases contain the symbol “x”. The single solid line in each plot indicates the median value. Brackets indicate statistically significant groupwise differences (p < 0.05) determined by the Kruskall-Wallis analysis of variance and Dunn’s multiple comparisons test. Scatter plots illustrating associations of vesicular glutamate transporter 1 (VGluT1) immunoreactive profile densities with cyano-PiB-labeled amyloid plaque pathology burden (precuneus, C; frontal cortex, G) and with CERAD diagnosis (precuneus, D; frontal cortex, H) in clinical diagnostic groups of NCI, MCI, mAD, and sAD.
Spearman rank order correlation revealed a significant association between lower precuneus VGluT1 density and lower MMSE scores (rs = 0.5205, p = 0.0002). However, after controlling for demographic variables the association between VGluT1 density and MMSE was no longer significant (β = 239.02 (95% CI (−48.08, 526.13), p = 0.11). No associations were observed when MMSE scores were plotted against VGluT2 density or with intensity of VGluT1 or VGluT2 in the precuneus (not shown).
Spearman rank order correlation analysis across all cases showed a statistically significant association between greater burden of cyano-PiB positive amyloid plaques and X-34-labeled total amyloid pathology and lower spinophilin density and intensity measures (cyano-PiB and spinophilin density: rs = 0.4980, p < 0.0001; cyano-PiB and spinophilin intensity: rs = −0.5733, p = 0.0072; X-34 and spinophilin density: rs = −0.5710, p = 0.0008; X-34 and spinophilin intensity: rs = −0.4136, p = 0.0207) in the precuneus (Fig. 3E; Supplementary Table 1). Lower precuneus spinophilin-immunoreactive profile densities and intensities were associated with higher Braak stage (Supplementary Table 1) (spinophilin density and Braak: rs = −03768, p = 0.0361; spinophilin intensity and Braak: rs = −0.5353, p = 0.0015) and greater likelihood of AD by NIA-RI criteria (spinophilin density and NIA-RI: rs = −0.4762, p = 0.0015; spinophilin intensity and NIA-RI: rs = −0.5943, p = 0.0015) (Supplementary Table 1). Lower spinophilin-immunoreactive profile density, but not intensity was associated with worse CERAD diagnosis (Fig. 3F) (spinophilin density and CERAD: rs = −0.5561, p = 0.0005).
Spearman rank order correlation analysis showed a statistically significant correlation between higher precuneus spinophilin density or intensity measures and higher MMSE scores (rs = 0.8027, p < 0.0001; rs = 0.6884, p < 0.0001, respectively) as well as with higher precuneus VGluT1-immunoreactive profile density (rs = 0.6466, p < 0.0001; rs = 0.5635, p = 0.0010) and VGluT2-immunoreactive profile intensity (rs = 0.6565, p < 0.0001; rs = 0.4309, p = 0.0174 measures (Supplementary Table 1).
Frontal cortex
Cyano-PiB-labeled Aβ amyloid burden and X-34-labeled pan-amyloid (Aβ and tau) burden within the frontal cortex showed statistically significant group differences (Kruskall-Wallis statistic = 19.75 and 21.47, p = 0.0002 and p < 0.001, respectively), with sAD having statistically significantly higher amyloid burden than NCI (Dunn’s multiple comparisons, cyano-PiB: p = 0.0002; X-34: p < 0.001) and MCI (Dunn’s multiple comparisons, cyano-PiB p = 0.0055; X-34 p = 0.0034) but not mAD (Fig. 3G, Of the VGluT measures in the frontal cortex, lower VGluT1 density was associated with higher Braak stage (rs = −0.2888, p = 0.0419), while no significant associations were detected with other neuropathology measures including cyano-PiB (Fig. 3I) and CERAD diagnosis (Fig. 3J). In contrast, higher VGluT1 intensity was associated with higher cyano-PiB amyloid plaque burden (rs = 0.2815, p = 0.0477), higher likelihood of AD by the NIA-RI criteria (rs = 0.3056, p = 0.0309), and higher Braak stage (rs = 0.3265, p = 0.0206) (Supplementary Table 2). When robust regression analysis controlling for demographic factors and APOE allele frequency was performed, these associations were no longer significant between VGluT1 density and Braak stage (p = 0.43) or between VGluT1 intensity and cyano-PiB plaque burden (p = 0.16), likelihood of AD (p = 0.16), and Braak stage (p = 0.59). VGluT2 density and intensity measures in the frontal cortex did not correlate with neuropathology data. Higher VGluT1 intensity was associated with lower MMSE scores in the frontal cortex (r = −0.43, p = 0.002). This association remained significant after controlling for age, sex, education, and APOE ε4 carrier status (B = −0.002, 95% CI: (−0.004, −0.001), p < 0.001).
Spearman rank order correlation analysis showed that higher spinophilin-immunoreactive density in the frontal cortex was associated with higher VGluT1- and VGluT2-immunoreactive density measures (rs = 0.3130, p = 0.0269; rs = 0.2895, p = 0.0414) (Supplementary Table 2). Spinophilin-immunoreactivity measures were not associated with cyano-PiB- or X-34-labeled amyloid pathology or NIA-RI, CERAD, or Braak stage (Supplementary Table 2). No associations were observed between spinophilin-immunoreactive density or intensity measures and MMSE scores.
DISCUSSION
Glutamatergic neurotransmitter dysfunction occurs in the AD brain [25–29], and recent reports support a role for altered cortical vesicular glutamate transporter (VGluT) proteins in this process [43–47, 66]. However, only a few studies have examined alterations in cortical lamina-specific and cell-compartment localization (i.e., synapses) of VGluT proteins [43, 44] in AD. The present study used an unbiased stereological design combined with high-resolution confocal microscopy to quantify neocortical VGluT1- and VGluT2-containing synaptic profile density and immunosignal intensity (the latter a measure of relative protein level within synaptic profiles) in cases that had a premortem clinical diagnosis of NCI, MCI, mAD, or sAD and received a detailed postmortem neuropathological evaluation. Here, we focused on two hubs of the DMN, the precuneus and the frontal cortex, which display neuropathology early in AD [48–52, 91, 92]. The frontal cortex has been reported to display neuroplastic changes in glutamatergic [43] and cholinergic [93] neuronal circuits.
The main finding of this study is that VGluT1-immunoreactive profile density measures in precuneus and frontal cortex were stable from NCI to MCI to mAD but were significantly lower in sAD cases compared to the other three groups. Our observation of VGluT1-immunoreactive synaptic terminal loss in precuneus is similar to that seen in other areas of cortex examined in AD cases [43, 44]. On the other hand, another study reported an upregulation of VGluT1-containing synaptic terminals in frontal cortex lamina III from MCI compared to NCI, mAD, and sAD cases [43]. This discrepancy could be due to differences in staining methods as we used confocal immunofluorescence microscopy compared to chromogen-based immunohistochemistry [43]. In addition, our NCI cases were selected based upon their last premortem clinical evaluations performed within a year of death, regardless of postmortem neuropathology assessment. Sixty percent of our NCI cases had CERAD diagnosis of probable AD, and one had a CERAD diagnosis of definite AD (see Tables 1 and 2). Similarly, most of our NCI cases were Braak stage III-VI and were classified as having an intermediate likelihood of AD by the NIA-RI criteria. NCI cases were not statistically significantly different from MCI cases either by these neuropathological diagnostic measures (Tables 1 and 2) or by their regional amyloid burden determined by cyano-PiB and X-34 stains (see Fig. 3). In contrast, the other study [43] commented that their NCI cases rarely displayed Aβ plaques and were excluded from analyses of VGluT1 profiles in relation to plaques, but neuropathology data were not provided. NCI cases displaying substantial AD lesions are commonly present in clinical pathological cohorts including the RROS autopsy collection [67, 75, 94–97]. Whether synaptic markers differ in AD pathology-burdened compared to pathology-free NCI cases is an ongoing area of investigation. For example, Perez-Nievas and colleagues [24] reported that general presynaptic (synaptophysin) and postsynaptic (PSD95) measures in the temporal lobe were not significantly different between two subgroups of nondemented cases with low probability or high probability of AD by the NIA-RI criteria (the latter termed “resilient”) [24]. Similarly, our LP-NCI and HP-NCI NCI cases, regardless of the neuropathological criteria used for their designation (i.e., by NIA-RI, CERAD, or Braak stage), did not differ in VGluT terminal measurements in the precuneus or frontal cortex.
Several findings in the current study provide support for the idea that unlike precuneus, the frontal cortex displays glutamatergic synaptic terminal changes suggestive of a plasticity response to the progression of AD neuropathology. In both regions, significantly lower VGluT1 profile density was detected in sAD compared to NCI, but only in the frontal cortex was this loss paralleled by a significantly higher VGluT1 intensity (a measure of relative synaptic protein abundance) in sAD compared to NCI (Fig. 2). Thus, a decrement in cortical glutamatergic input may be associated with higher levels of VGluT1 protein content in remaining synaptic terminals in the frontal cortex but not the precuneus. That the VGluT1 intensity measures in the MCI and mAD cases were higher than in the NCI also suggests possible synaptic changes occurring early in the disease course. We also observed that lower VGluT2 profile densities were associated with higher VGluT2 profile intensities in both regions, suggesting a significant, albeit weaker, compensatory response for VGluT2. Complementary analyses of spinophilin-immunoreactive dendritic spines in lamina III showed that precuneus spinophilin-immunoreactive profile density and intensity were lower in mAD and sAD than NCI and MCI, while in the frontal cortex both measures were stable across clinical groups. Additionally, while lower precuneus spinophilin-immunoreactive profile densities associated with more severe regional and diagnostic neuropathology, similar associations were not observed in the frontal cortex. The strong correlations of VGluT1, but not VGluT2, and spinophilin with neuropathology measures in precuneus, but not frontal cortex, further support the concept of selective regional vulnerability in AD. Perhaps a loss of postsynaptic dendritic spines in the precuneus cortex marks an early stage of AD, when presynaptic VGluT1 terminals are stable, while loss of both postsynaptic dendritic spines and presynaptic cortico-cortical and intra-cortical excitatory VGluT1 terminals in precuneus and frontal cortex designate more advanced AD. However, with clinical AD progression relative protein levels of VGluT1 in synaptic terminals appear to upregulate in the frontal cortex, but not precuneus (see Fig. 2F). Collectively, our data support the concept that compared to the frontal cortex, synaptic profiles in the precuneus cortex are more vulnerable to AD neuropathology, and that this region lacks a sustainable plasticity response.
The degree to which hallmark AD lesions, or the consequences of these lesions, directly affect synapses in the AD brain is not well understood. In the present study, both VGluT1- and VGluT2-immunoreactive profiles were scarce within amyloid plaques in the precuneus and frontal cortex, while amyloid plaque-free neuropil displayed a loss of VGluT1-immunoreactive profiles only in advanced AD. Whether in amyloid plaque-burdened areas VGluT-immunoreactive profiles are affected by fibrillar Aβ, Aβ oligomers, tau-containing dystrophic neurites, or neuroinflammation (e.g., gliosis) associated with amyloid [98–100] remains to be determined.
The present study used a rigorous unbiased quantitative design and statistical analyses. Study limitations include: (i) NCI, MCI, and mAD cases were aging clergy participants in the RROS [67–70], while the sAD group consisted of patients enrolled in the UPitt ADRC. Previous studies have discussed the RROS cohort in comparison to the general population and clinic cohorts [75, 93, 101], and we have reported similar alterations in cortical cholinergic change and changes in neurotrophic factor receptors levels, among others, using tissue from the RROS compared to a general clinic population in AD and individuals with Down syndrome [93, 97, 102–108]. Although the early and advanced AD cases from the RROS and UPitt ADRC, respectively, did not differ by years of education, the RROS early AD subjects were older than the ADRC patients with advanced AD. (ii) The MCI group contributing frontal cortex was 91% (10/11) amnestic, while for the precuneus sample MCI were not as homogeneous, with only 30% (3/10) amnestic. Amnestic MCI (aMCI) is believed more likely to progress to AD than non-aMCI [109, 110], but whether the aMCI subjects in this study would develop AD had they lived longer is an untestable hypothesis. However, it should be noted that VGluT1 densities were clustered tightly in the entire MCI group, and the aMCI cases did not differ appreciably from the non-aMCI cases (see Fig. 2A). (iii) The current analyses focused on cortical lamina III based upon several observations: (a) lamina III of the neocortex displays both VGluT1 and VGluT2 profiles [38], (b) lamina III cortico-cortical neuronal projections are components of higher order cortical circuits that interconnect the precuneus and frontal cortex [111], (c) lamina III projection neurons are selectively vulnerable and develop tau pathology in AD [112–116], and (d) the focus on lamina III also allowed for more relevant comparisons to the study by Bell and colleagues who analyzed the same cortical layer of frontal cortex [43]. Lastly, (iv) due to brain tissue availability overlap of cases was limited which prevented within-case comparisons of VGluT measures between the two brain regions.
In summary, the current study demonstrates differences in VGluT1- and VGluT2-immunoreactive glutamatergic terminal change within layer III of the precuneus and frontal cortex at different clinical-pathological stages of AD. Of particular interest are findings that VGluT1 loss is associated with neuropathology progression in the precuneus, but not frontal cortex, and that a decremental change in frontal cortical VGluT1 terminal density is paralleled by an upregulation of VGluT1 protein content in remaining terminals of frontal but not precuneus cortex. The latter change may signal a preclinical neuroplasticity response. Thus, drugs aimed at preservation of cortical glutamatergic excitatory synaptic function and enhancement of neuroplasticity mechanisms, when combined with other currently available symptomatic and lesion modifying therapies, likely will prove beneficial in treating people who are at the early stages of AD clinical-pathological change.
Supplementary Material
ACKNOWLEDGMENTS
The authors are indebted to the Catholic nuns, priests, and lay brothers who participated in the Rush Religious Orders Study and patients in the University of Pittsburgh’s Alzheimer’s Disease Research Center. We are grateful to Ms. Lan Shao, Ms. Violetta Pivtoraiko, and Dr. Jason T. Newman for expert technical assistance, and to Dr. William Klunk and Dr. Chester Mathis for providing cyano-PiB and X-34. The content of the manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the Department of Veterans Affairs or the United States Government.
FUNDING
This study was supported by grants P01AG014449, P30AG10161, P30AG066468, R01AG052528, and P01AG025204 from the National Institute on Aging, and R01MH116046 and R01MH096985 from the National Institute of Mental Health, National Institutes of Health.
Footnotes
CONFLICT OF INTEREST
ZM, EEA, AYR, MM-A, JKK, KNF, RAS, VLV, and JAS declare no conflict of interest.
EJM has served as consultant to NeuroPhage Pharmaceuticals, Inc., and is an Editorial Board Member of this journal, but was not involved in the peer-review process nor had access to any information regarding its peer-review.
MDI has served as consultant to and received research grants from GE Healthcare
SUPPLEMENTARY MATERIAL
The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-221063.
DATA AVAILABILITY
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- [1].Jellinger KA (2022) Recent update on the heterogeneity of the Alzheimer’s disease spectrum. J Neural Transm (Vienna) 129, 1–24. [DOI] [PubMed] [Google Scholar]
- [2].Mehta RI, Schneider JA (2021) What is ‘Alzheimer’s disease’? The neuropathological heterogeneity of clinically defined Alzheimer’s dementia. Curr Opin Neurol 34, 237–245. [DOI] [PubMed] [Google Scholar]
- [3].DeTure MA, Dickson DW (2019) The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener 14, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Robinson JL, Richardson H, Xie SX, Suh E, Van Deerlin VM, Alfaro B, Loh N, Porras-Paniagua M, Nirschl JJ, Wolk D, Lee VM, Lee EB, Trojanowski JQ (2021) The development and convergence of co-pathologies in Alzheimer’s disease. Brain 144, 953–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schneider JA (2022) Neuropathology of dementia disorders. Continuum (Minneap Minn) 28, 834–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Davies CA, Mann DM, Sumpter PQ, Yates PO (1987) A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J Neurol Sci 78, 151–164. [DOI] [PubMed] [Google Scholar]
- [7].Scheff SW, DeKosky ST, Price DA (1990) Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol Aging 11, 29–37. [DOI] [PubMed] [Google Scholar]
- [8].Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW Jr, Morris JC (2001) Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 56, 127–129. [DOI] [PubMed] [Google Scholar]
- [9].Selkoe DJ (2002) Alzheimer’s disease is a synaptic failure. Science 298, 789–791. [DOI] [PubMed] [Google Scholar]
- [10].Scheff SW, Price DA (2006) Alzheimer’s disease-related alterations in synaptic density: Neocortex and hippocampus. J Alzheimers Dis 9, 101–115. [DOI] [PubMed] [Google Scholar]
- [11].Counts SE, Nadeem M, Lad SP, Wuu J, Mufson EJ (2006) Differential expression of synaptic proteins in the frontal and temporal cortex of elderly subjects with mild cognitive impairment. J Neuropathol Exp Neurol 65, 592–601. [DOI] [PubMed] [Google Scholar]
- [12].DeKosky ST, Scheff SW (1990) Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann Neurol 27, 457–464. [DOI] [PubMed] [Google Scholar]
- [13].Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991) Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann Neurol 30, 572–580. [DOI] [PubMed] [Google Scholar]
- [14].Samuel W, Terry RD, DeTeresa R, Butters N, Masliah E (1994) Clinical correlates of cortical and nucleus basalis pathology in Alzheimer dementia. Arch Neurol 51, 772–778. [DOI] [PubMed] [Google Scholar]
- [15].Masliah E, Mallory M, Hansen L, DeTeresa R, Alford M, Terry R (1994) Synaptic and neuritic alterations during the progression of Alzheimer’s disease. Neurosci Lett 174, 67–72. [DOI] [PubMed] [Google Scholar]
- [16].DeKosky ST, Scheff SW, Styren SD (1996) Structural correlates of cognition in dementia: Quantification and assessment of synapse change. Neurodegeneration 5, 417–421. [DOI] [PubMed] [Google Scholar]
- [17].Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ (1997) Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol 56, 933–944. [DOI] [PubMed] [Google Scholar]
- [18].Scheff SW, Price DA, Schmitt FA, Scheff MA, Mufson EJ (2011) Synaptic loss in the inferior temporal gyrus in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis 24, 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Scheff SW, Price DA, Schmitt FA, Roberts KN, Ikonomovic MD, Mufson EJ (2013) Synapse stability in the precuneus early in the progression of Alzheimer’s disease. J Alzheimers Dis 35, 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Scheff SW, Price DA, Ansari MA, Roberts KN, Schmitt FA, Ikonomovic MD, Mufson EJ (2015) Synaptic change in the posterior cingulate gyrus in the progression of Alzheimer’s disease. J Alzheimers Dis 43, 1073–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Masliah E, Terry RD, DeTeresa RM, Hansen LA (1989) Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci Lett 103, 234–239. [DOI] [PubMed] [Google Scholar]
- [22].Masliah E, Crews L, Hansen L (2006) Synaptic remodeling during aging and in Alzheimer’s disease. J Alzheimers Dis 9, 91–99. [DOI] [PubMed] [Google Scholar]
- [23].Scheff SW, Ansari MA, Mufson EJ (2016) Oxidative stress and hippocampal synaptic protein levels in elderly cognitively intact individuals with Alzheimer’s disease pathology. Neurobiol Aging 42, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Perez-Nievas BG, Stein TD, Tai HC, Dols-Icardo O, Scotton TC, Barroeta-Espar I, Fernandez-Carballo L, de Munain EL, Perez J, Marquie M, Serrano-Pozo A, Frosch MP, Lowe V, Parisi JE, Petersen RC, Ikonomovic MD, Lopez OL, Klunk W, Hyman BT, Gomez-Isla T (2013) Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 136, 2510–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Procter AW, Palmer AM, Francis PT, Lowe SL, Neary D, Murphy E, Doshi R, Bowen DM (1988) Evidence of glutamatergic denervation and possible abnormal metabolism in Alzheimer’s disease. J Neurochem 50, 790–802. [DOI] [PubMed] [Google Scholar]
- [26].Hardy J, Cowburn R, Barton A, Reynolds G, Lofdahl E, O’Carroll AM, Wester P, Winblad B (1987) Region-specific loss of glutamate innervation in Alzheimer’s disease. Neurosci Lett 73, 77–80. [DOI] [PubMed] [Google Scholar]
- [27].Hyman BT, Van Hoesen GW, Damasio AR (1987) Alzheimer’s disease: Glutamate depletion in the hippocampal perforant pathway zone. Ann Neurol 22, 37–40. [DOI] [PubMed] [Google Scholar]
- [28].Greenamyre JT, Maragos WF, Albin RL, Penney JB, Young AB (1988) Glutamate transmission and toxicity in Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry 12, 421–430. [DOI] [PubMed] [Google Scholar]
- [29].Francis PT (2003) Glutamatergic systems in Alzheimer’s disease. Int J Geriatr Psychiatry 18, S15–21. [DOI] [PubMed] [Google Scholar]
- [30].Francis PT (2008) Glutamatergic approaches to the treatment of cognitive and behavioural symptoms of Alzheimer’s disease. Neurodegener Dis 5, 241–243. [DOI] [PubMed] [Google Scholar]
- [31].Francis PT, Ramirez MJ, Lai MK (2010) Neurochemical basis for symptomatic treatment of Alzheimer’s disease. Neuropharmacology 59, 221–229. [DOI] [PubMed] [Google Scholar]
- [32].Fonnum F (1984) Glutamate: A neurotransmitter in mammalian brain. J Neurochem 42, 1–11. [DOI] [PubMed] [Google Scholar]
- [33].Fykse EM, Fonnum F (1996) Amino acid neurotransmission: Dynamics of vesicular uptake. Neurochem Res 21, 1053–1060. [DOI] [PubMed] [Google Scholar]
- [34].Takamori S, Rhee JS, Rosenmund C, Jahn R (2000) Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature 407, 189–194. [DOI] [PubMed] [Google Scholar]
- [35].Takamori S (2006) VGLUTs: ‘Exciting’ times for glutamatergic research? Neurosci Res 55, 343–351. [DOI] [PubMed] [Google Scholar]
- [36].Kaneko T, Fujiyama F (2002) Complementary distribution of vesicular glutamate transporters in the central nervous system. Neurosci Res 42, 243–250. [DOI] [PubMed] [Google Scholar]
- [37].Varoqui H, Schafer MK, Zhu H, Weihe E, Erickson JD (2002) Identification of the differentiation-associated Na+/PI transporter as a novel vesicular glutamate transporter expressed in a distinct set of glutamatergic synapses. J Neurosci 22, 142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].McCullumsmith RE, Meador-Woodruff JH (2003) Expression of transcripts for the vesicular glutamate transporters in the human medial temporal lobe. Ann N Y Acad Sci 1003, 438–442. [DOI] [PubMed] [Google Scholar]
- [39].Fremeau RT Jr., Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA, Edwards RH (2004) Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science 304, 1815–1819. [DOI] [PubMed] [Google Scholar]
- [40].El Mestikawy S, Wallen-Mackenzie A, Fortin GM, Descarries L, Trudeau LE (2011) From glutamate co-release to vesicular synergy: Vesicular glutamate transporters. Nat Rev Neurosci 12, 204–216. [DOI] [PubMed] [Google Scholar]
- [41].Vigneault E, Poirel O, Riad M, Prud’homme J, Dumas S, Turecki G, Fasano C, Mechawar N, El Mestikawy S (2015) Distribution of vesicular glutamate transporters in the human brain. Front Neuroanat 9, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Du X, Li J, Li M, Yang X, Qi Z, Xu B, Liu W, Xu Z, Deng Y (2020) Research progress on the role of type I vesicular glutamate transporter (VGLUT1) in nervous system diseases. Cell Biosci 10, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bell KF, Bennett DA, Cuello AC (2007) Paradoxical upregulation of glutamatergic presynaptic boutons during mild cognitive impairment. J Neurosci 27, 10810–10817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kirvell SL, Esiri M, Francis PT (2006) Down-regulation of vesicular glutamate transporters precedes cell loss and pathology in Alzheimer’s disease. J Neurochem 98, 939–950. [DOI] [PubMed] [Google Scholar]
- [45].Westphalen RI, Scott HL, Dodd PR (2003) Synaptic vesicle transport and synaptic membrane transporter sites in excitatory amino acid nerve terminals in Alzheimer disease. J Neural Transm (Vienna) 110, 1013–1027. [DOI] [PubMed] [Google Scholar]
- [46].Kashani A, Lepicard E, Poirel O, Videau C, David JP, Fallet-Bianco C, Simon A, Delacourte A, Giros B, Epelbaum J, Betancur C, El Mestikawy S (2008) Loss of VGLUT1 and VGLUT2 in the prefrontal cortex is correlated with cognitive decline in Alzheimer disease. Neurobiol Aging 29, 1619–1630. [DOI] [PubMed] [Google Scholar]
- [47].Poirel O, Mella S, Videau C, Ramet L, Davoli MA, Herzog E, Katsel P, Mechawar N, Haroutunian V, Epelbaum J, Daumas S, El Mestikawy S (2018) Moderate decline in select synaptic markers in the prefrontal cortex (BA9) of patients with Alzheimer’s disease at various cognitive stages. Sci Rep 8, 938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Palmqvist S, Scholl M, Strandberg O, Mattsson N, Stomrud E, Zetterberg H, Blennow K, Landau S, Jagust W, Hansson O (2017) Earliest accumulation of beta-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nat Commun 8, 1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sperling RA, Laviolette PS, O’Keefe K, O’Brien J, Rentz DM, Pihlajamaki M, Marshall G, Hyman BT, Selkoe DJ, Hedden T, Buckner RL, Becker JA, Johnson KA (2009) Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 63, 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Drzezga A, Becker JA, Van Dijk KR, Sreenivasan A, Talukdar T, Sullivan C, Schultz AP, Sepulcre J, Putcha D, Greve D, Johnson KA, Sperling RA (2011) Neuronal dysfunction and disconnection of cortical hubs in non-demented subjects with elevated amyloid burden. Brain 134, 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Mormino EC, Smiljic A, Hayenga AO, Onami SH, Greicius MD, Rabinovici GD, Janabi M, Baker SL, Yen IV, Madison CM, Miller BL, Jagust WJ (2011) Relationships between beta-amyloid and functional connectivity in different components of the default mode network in aging. Cereb Cortex 21, 2399–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lim HK, Nebes R, Snitz B, Cohen A, Mathis C, Price J, Weissfeld L, Klunk W, Aizenstein HJ (2014) Regional amyloid burden and intrinsic connectivity networks in cognitively normal elderly subjects. Brain 137, 3327–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Schultz AP, Chhatwal JP, Hedden T, Mormino EC, Hanseeuw BJ, Sepulcre J, Huijbers W, LaPoint M, Buckley RF, Johnson KA, Sperling RA (2017) Phases of hyperconnectivity and hypoconnectivity in the default mode and salience networks track with amyloid and tau in clinically normal individuals. J Neurosci 37, 4323–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sheline YI, Raichle ME, Snyder AZ, Morris JC, Head D, Wang S, Mintun MA (2010) Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol Psychiatry 67, 584–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hedden T, Van Dijk KR, Becker JA, Mehta A, Sperling RA, Johnson KA, Buckner RL (2009) Disruption of functional connectivity in clinically normal older adults harboring amyloid burden. J Neurosci 29, 12686–12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Brier MR, Thomas JB, Ances BM (2014) Network dysfunction in Alzheimer’s disease: Refining the disconnection hypothesis. Brain Connect 4, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sheline YI, Raichle ME (2013) Resting state functional connectivity in preclinical Alzheimer’s disease. Biol Psychiatry 74, 340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chhatwal JP, Schultz AP, Johnson K, Benzinger TL, Jack C Jr., Ances BM, Sullivan CA, Salloway SP, Ringman JM, Koeppe RA, Marcus DS, Thompson P, Saykin AJ, Correia S, Schofield PR, Rowe CC, Fox NC, Brickman AM, Mayeux R, McDade E, Bateman R, Fagan AM, Goate AM, Xiong C, Buckles VD, Morris JC, Sperling RA (2013) Impaired default network functional connectivity in autosomal dominant Alzheimer disease. Neurology 81, 736–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hahn A, Strandberg TO, Stomrud E, Nilsson M, van Westen D, Palmqvist S, Ossenkoppele R, Hansson O (2019) Association between earliest amyloid uptake and functional connectivity in cognitively unimpaired elderly. Cereb Cortex 29, 2173–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Stiernman LJ, Grill F, Hahn A, Rischka L, Lanzenberger R, Panes Lundmark V, Riklund K, Axelsson J, Rieckmann A (2021) Dissociations between glucose metabolism and blood oxygenation in the human default mode network revealed by simultaneous PET-fMRI. Proc Natl Acad Sci USA 118, e2021913118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Dillen KNH, Jacobs HIL, Kukolja J, Richter N, von Reutern B, Onur OA, Langen KJ, Fink GR (2017) Functional disintegration of the default mode network in prodromal Alzheimer’s disease. J Alzheimers Dis 59, 169–187. [DOI] [PubMed] [Google Scholar]
- [62].Badhwar A, Tam A, Dansereau C, Orban P, Hoffstaedter F, Bellec P (2017) Resting-state network dysfunction in Alzheimer’s disease: A systematic review and meta-analysis. Alzheimers Dement (Amst) 8, 73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, Ziolko SK, James JA, Snitz BE, Houck PR, Bi W, Cohen AD, Lopresti BJ, DeKosky ST, Halligan EM, Klunk WE (2008) Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol 65, 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, Klunk WE, Mathis CA, DeKosky ST, Morris JC (2006) [11C]PIB in a nondemented population: Potential antecedent marker of Alzheimer disease. Neurology 67, 446–452. [DOI] [PubMed] [Google Scholar]
- [65].Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW (1991) The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb Cortex 1, 103–116. [DOI] [PubMed] [Google Scholar]
- [66].Sokolow S, Luu SH, Nandy K, Miller CA, Vinters HV, Poon WW, Gylys KH (2012) Preferential accumulation of amyloid-beta in presynaptic glutamatergic terminals (VGluT1 and VGluT2) in Alzheimer’s disease cortex. Neurobiol Dis 45, 381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, Wilson RS (2006) Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 66, 1837–1844. [DOI] [PubMed] [Google Scholar]
- [68].Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE (2004) Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol 61, 378–384. [DOI] [PubMed] [Google Scholar]
- [69].Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT, Barnes LL, Fox JH, Bach J (2002) Natural history of mild cognitive impairment in older persons. Neurology 59, 198–205. [DOI] [PubMed] [Google Scholar]
- [70].Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA (2018) Religious Orders Study and Rush Memory and Aging Project. J Alzheimers Dis 64, S161–S189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Lopez OL, Becker JT, Klunk W, Saxton J, Hamilton RL, Kaufer DI, Sweet RA, Cidis Meltzer C, Wisniewski S, Kamboh MI, DeKosky ST (2000) Research evaluation and diagnosis of probable Alzheimer’s disease over the last two decades: I. Neurology 55, 1854–1862. [DOI] [PubMed] [Google Scholar]
- [72].McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34, 939–944. [DOI] [PubMed] [Google Scholar]
- [73].Morris JC, Price JL (2001) Pathologic correlates of nondemented aging, mild cognitive impairment, and early-stage Alzheimer’s disease. J Mol Neurosci 17, 101–118. [DOI] [PubMed] [Google Scholar]
- [74].Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E (1999) Mild cognitive impairment: Clinical characterization and outcome. Arch Neurol 56, 303–308. [DOI] [PubMed] [Google Scholar]
- [75].Schneider JA, Aggarwal NT, Barnes L, Boyle P, Bennett DA (2009) The neuropathology of older persons with and without dementia from community versus clinic cohorts. J Alzheimers Dis 18, 691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Boyle PA, Yu L, Leurgans SE, Wilson RS, Brookmeyer R, Schneider JA, Bennett DA (2019) Attributable risk of Alzheimer’s dementia attributed to age-related neuropathologies. Ann Neurol 85, 114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Consensus (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol Aging 18, S1–2. [PubMed] [Google Scholar]
- [78].Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L (1991) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–486. [DOI] [PubMed] [Google Scholar]
- [79].Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239–259. [DOI] [PubMed] [Google Scholar]
- [80].Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT, National Institute on Aging; Alzheimer’s Association (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol 123, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Moyer CE, Delevich KM, Fish KN, Asafu-Adjei JK, Sampson AR, Dorph-Petersen KA, Lewis DA, Sweet RA (2013) Intracortical excitatory and thalamocortical boutons are intact in primary auditory cortex in schizophrenia. Schizophr Res 149, 127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Sweet RA, Henteleff RA, Zhang W, Sampson AR, Lewis DA (2009) Reduced dendritic spine density in auditory cortex of subjects with schizophrenia. Neuropsychopharmacology 34, 374–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Mi Z, Abrahamson EE, Ryu AY, Fish KN, Sweet RA, Mufson EJ, Ikonomovic MD (2017) Loss of precuneus dendritic spines immunopositive for spinophilin is related to cognitive impairment in early Alzheimer’s disease. Neurobiol Aging 55, 159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, Lopresti BJ, Ziolko S, Bi W, Paljug WR, Debnath ML, Hope CE, Isanski BA, Hamilton RL, DeKosky ST (2008) Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain 131, 1630–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Ikonomovic MD, Buckley CJ, Abrahamson EE, Kofler JK, Mathis CA, Klunk WE, Farrar G (2020) Post-mortem analyses of PiB and flutemetamol in diffuse and cored amyloid-beta plaques in Alzheimer’s disease. Acta Neuropathol 140, 463–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Styren SD, Hamilton RL, Styren GC, Klunk WE (2000) X-34, a fluorescent derivative of Congo red: A novel histochemical stain for Alzheimer’s disease pathology. J Histochem Cytochem 48, 1223–1232. [DOI] [PubMed] [Google Scholar]
- [88].Ikonomovic MD, Abrahamson EE, Isanski BA, Debnath ML, Mathis CA, Dekosky ST, Klunk WE (2006) X-34 labeling of abnormal protein aggregates during the progression of Alzheimer’s disease. Methods Enzymol 412, 123–144. [DOI] [PubMed] [Google Scholar]
- [89].Fish KN, Sweet RA, Deo AJ, Lewis DA (2008) An automated segmentation methodology for quantifying immunoreactive puncta number and fluorescence intensity in tissue sections. Brain Res 1240, 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Dorph-Petersen KA, Nyengaard JR, Gundersen HJ (2001) Tissue shrinkage and unbiased stereological estimation of particle number and size. J Microsc 204, 232–246. [DOI] [PubMed] [Google Scholar]
- [91].Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Sheline YI, Klunk WE, Mathis CA, Morris JC, Mintun MA (2005) Molecular, structural, and functional characterization of Alzheimer’s disease: Evidence for a relationship between default activity, amyloid, and memory. J Neurosci 25, 7709–7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Jones DT, Machulda MM, Vemuri P, McDade EM, Zeng G, Senjem ML, Gunter JL, Przybelski SA, Avula RT, Knopman DS, Boeve BF, Petersen RC, Jack CR Jr (2011) Age-related changes in the default mode network are more advanced in Alzheimer disease. Neurology 77, 1524–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].DeKosky ST, Ikonomovic MD, Styren SD, Beckett L, Wisniewski S, Bennett DA, Cochran EJ, Kordower JH, Mufson EJ (2002) Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol 51, 145–155. [DOI] [PubMed] [Google Scholar]
- [94].Mufson EJ, Chen EY, Cochran EJ, Beckett LA, Bennett DA, Kordower JH (1999) Entorhinal cortex beta-amyloid load in individuals with mild cognitive impairment. Exp Neurol 158, 469–490. [DOI] [PubMed] [Google Scholar]
- [95].Mufson EJ, Malek-Ahmadi M, Perez SE, Chen K (2016) Braak staging, plaque pathology, and APOE status in elderly persons without cognitive impairment. Neurobiol Aging 37, 147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Ikonomovic MD, Klunk WE, Abrahamson EE, Wuu J, Mathis CA, Scheff SW, Mufson EJ, DeKosky ST (2011) Precuneus amyloid burden is associated with reduced cholinergic activity in Alzheimer disease. Neurology 77, 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Perez SE, He B, Nadeem M, Wuu J, Scheff SW, Abrahamson EE, Ikonomovic MD, Mufson EJ (2015) Resilience of precuneus neurotrophic signaling pathways despite amyloid pathology in prodromal Alzheimer’s disease. Biol Psychiatry 77, 693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Serrano-Pozo A, Muzikansky A, Gomez-Isla T, Growdon JH, Betensky RA, Frosch MP, Hyman BT (2013) Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease. J Neuropathol Exp Neurol 72, 462–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Serrano-Pozo A, Betensky RA, Frosch MP, Hyman BT (2016) Plaque-associated local toxicity increases over the clinical course of Alzheimer disease. Am J Pathol 186, 375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL (2009) Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad SciUSA 106, 4012–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Bennett DA, Schneider JA, Bienias JL, Evans DA, Wilson RS (2005) Mild cognitive impairment is related to Alzheimer disease pathology and cerebral infarctions. Neurology 64, 834–841. [DOI] [PubMed] [Google Scholar]
- [102].Mufson EJ, He B, Nadeem M, Perez SE, Counts SE, Leurgans S, Fritz J, Lah J, Ginsberg SD, Wuu J, Scheff SW (2012) Hippocampal proNGF signaling pathways and beta-amyloid levels in mild cognitive impairment and Alzheimer disease. J Neuropathol Exp Neurol 71, 1018–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Sendera TJ, Ma SY, Jaffar S, Kozlowski PB, Kordower JH, Mawal Y, Saragovi HU, Mufson EJ (2000) Reduction in TrkA-immunoreactive neurons is not associated with an overexpression of galaninergic fibers within the nucleus basalis in Down’s syndrome. J Neurochem 74, 1185–1196. [DOI] [PubMed] [Google Scholar]
- [104].Iulita MF, Do Carmo S, Ower AK, Fortress AM, Flores Aguilar L, Hanna M, Wisniewski T, Granholm AC, Buhusi M, Busciglio J, Cuello AC (2014) Nerve growth factor metabolic dysfunction in Down’s syndrome brains. Brain 137, 860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Davis DG, Schmitt FA, Wekstein DR, Markesbery WR (1999) Alzheimer neuropathologic alterations in aged cognitively normal subjects. J Neuropathol Exp Neurol 58, 376–388. [DOI] [PubMed] [Google Scholar]
- [106].Pivtoraiko VN, Racic T, Abrahamson EE, Villemagne VL, Handen BL, Lott IT, Head E, Ikonomovic MD (2021) Postmortem neocortical (3)H-PiB binding and levels of unmodified and pyroglutamate Abeta in Down syndrome and sporadic Alzheimer’s disease. Front Aging Neurosci 13, 728739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Abrahamson EE, Head E, Lott IT, Handen BL, Mufson EJ, Christian BT, Klunk WE, Ikonomovic MD (2019) Neuropathological correlates of amyloid PET imaging in Down syndrome. Dev Neurobiol 79, 750–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Perez SE, Miguel JC, He B, Malek-Ahmadi M, Abrahamson EE, Ikonomovic MD, Lott I, Doran E, Alldred MJ, Ginsberg SD, Mufson EJ (2019) Frontal cortex and striatal cellular and molecular pathobiology in individuals with Down syndrome with and without dementia. Acta Neuropathol 137, 413–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Johnson JK, Pa J, Boxer AL, Kramer JH, Freeman K, Yaffe K (2010) Baseline predictors of clinical progression among patients with dysexecutive mild cognitive impairment. Dement Geriatr Cogn Disord 30, 344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Petersen RC (2003) Mild cognitive impairment clinical trials. Nat Rev Drug Discov 2, 646–653. [DOI] [PubMed] [Google Scholar]
- [111].Cavanna AE, Trimble MR (2006) The precuneus: A review of its functional anatomy and behavioural correlates. Brain 129, 564–583. [DOI] [PubMed] [Google Scholar]
- [112].Pearson RC, Esiri MM, Hiorns RW, Wilcock GK, Powell TP (1985) Anatomical correlates of the distribution of the pathological changes in the neocortex in Alzheimer disease. Proc Natl Acad SciUSA 82, 4531–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Duyckaerts C, Hauw JJ, Bastenaire F, Piette F, Poulain C, Rainsard V, Javoy-Agid F, Berthaux P (1986) Laminar distribution of neocortical senile plaques in senile dementia of the Alzheimer type. Acta Neuropathol 70, 249–256. [DOI] [PubMed] [Google Scholar]
- [114].Lewis DA, Campbell MJ, Terry RD, Morrison JH (1987) Laminar and regional distributions of neurofibrillary tangles and neuritic plaques in Alzheimer’s disease: A quantitative study of visual and auditory cortices. J Neurosci 7, 1799–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Morrison JH, Hof PR (2002) Selective vulnerability of corticocortical and hippocampal circuits in aging and Alzheimer’s disease. Prog Brain Res 136, 467–486. [DOI] [PubMed] [Google Scholar]
- [116].Bell KF, Ducatenzeiler A, Ribeiro-da-Silva A, Duff K, Bennett DA, Cuello AC (2006) The amyloid pathology progresses in a neurotransmitter-specific manner. Neurobiol Aging 27, 1644–1657. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.