

Abstract
Purpose of review:
New therapies are needed to potentiate the effects of current immunotherapies and overcome resistance. The stimulator of interferon genes (STING)-pathway is an innate immune activating cascade that may enhance current cancer immunotherapies.
Recent findings:
Pre-clinical data has shown that the addition of a STING agonist may enhance the effect of current treatments such as immune checkpoint inhibitor antibodies and radiation therapy. Early phase trials have demonstrated modest efficacy of STING agonists and revealed new mechanistic and technical challenges.
Summary:
STING agonists are a new class of agents that activate the host response immune response to improve tumor control. A wide range of pre-clinical experiments, translational data, and ongoing clinical trials support the therapeutic use of STING agonists in patients. Trials to determine optimal drug combinations and novel delivery mechanisms are continuing in development.
Keywords: STING agonist, anti-tumor immunity, immunotherapy, immune checkpoint inhibitors, tumor microenvironment, innate immune activation
Introduction
Recent advances in cancer immunotherapy including immune checkpoint inhibitors (ICIs), bi-specific T-cell engagers (BiTEs) and chimeric antigen receptor (CAR) T cells have revolutionized the treatment of many cancers. Unfortunately, not all cancers respond to these therapies, and those that do still may develop secondary resistance (1). Resistance may result from features in the tumor microenvironment (TME) preventing or limiting the anti-tumor immune response (2). For example, lack of response may be due to restriction of T cell priming or recruitment, the presence of immune-suppressive cells, secretion of immunosuppressive cytokines, and an overexpression of immune checkpoints by cancer cells(3) The latter problem can be addressed with immune checkpoint therapies, but the other challenges necessitate the development of new therapeutics and methods for administration. The cyclic GMP-AMP synthase (cGAS) - stimulator of interferon genes (STING) pathway emerged as a rational target to improve the rate of response to existing immunotherapies by activation of anti-tumor immune response in the TME. Likewise, the development of STING agonists has coincided with the development of novel therapeutic delivery mechanisms.
cGAS-STING Pathway and its role in the tumor immune microenvironment
The STING pathway is an innate immune activating cascade triggered by cytosolic DNA that potentiates a variety of inflammatory responses that are mediated primarily by type I interferons (4). Cytosolic DNA may be generated from viruses, bacteria, damaged self-DNA, and tumor DNA. Cytosolic DNA is bound by cGAS which catalyzes cyclic GMP-AMP (cGAMP) synthesis (5). cGAMP then binds to the STING receptor located on the endoplasmic reticulum. STING then phosphorylates TBK1 and IRF3, leading to the production of type I interferons which are known to enhance antigen presentation, augment B cell antibody production and amplify the function of dendritic cell and T cell populations (4-6).
Activation of the STING pathway has been shown to be an important mediator of the anti-tumor response of a variety of immune cell types. Prior experiments demonstrated that mice deficient in STING and IRF3 had decreased CD8+ T cell response and impaired control of tumor growth(7). Similarly, the downstream production of type I interferons has been shown to be required for activity of dendritic cells and other tumor-infiltrating antigen presenting cells (APCs)(7, 8). Additional mouse models have also revealed the STING pathway to be vital in the anti-tumor activity of NK cells (9).
Preclinical Studies of STING agonism
Single Agent STING Agonism
Numerous studies have now determined that the stimulation of the STING pathway, through many different approaches, is sufficient to produce immune mediated tumor cell death on its own. Intratumoral injection of a direct sting agonist, 5,6-dimethylxanthenone-4-acetic acid (DMXAA), in q melanoma murine model enhanced the anti-tumor immune response locally, improved control of distal tumors, and invoked T cell memory (10). Other murine models have demonstrated superior immune mediated tumor control by macrophages and CD8+T cells in melanoma, breast cancer, squamous cell cancer and colon cancer injected with cGAMP(11, 12). The synthetic cyclic dinucleotide (CDN) STING agonist ADU-S100 also resulted in enhanced CD8+T cell infiltration and immunogenic tumor control of cervical and pancreatic cancers(13, 14).
STING Agonism with Radiation Therapy
STING agonism may have synergy with a variety of other treatment modalities (Figure 1). The STING pathway has been shown to be activated by irradiated tumors, prompting an innate immune response which can be strengthened with the administration of exogenous cGAMP(15). An inhalable nanoparticle formulation of cGAMP targeting pulmonary APCs (NP-cGAMP) was studied as an adjunct to radiation therapy in mouse models of melanoma and breast cancer. NP-cGAMP was found to induce the STING pathway and create a synergistic effect with radiation therapy that resulted in increased control of lung metastases compared to radiation therapy alone(16). ADU-S100 was also tested alone and in combination with radiation therapy in rats with esophageal cancer. The cohort that received both ADU-S100 and radiation therapy had decreased tumor volume compared to both radiation therapy or ADU-S100 administration alone(17). Similarly, the STING agonist dimeric amidobenzimidazole (diABZI) was shown to sensitize non-small lung cancer (NSCLC) to radiation therapy(18).
Figure 1.
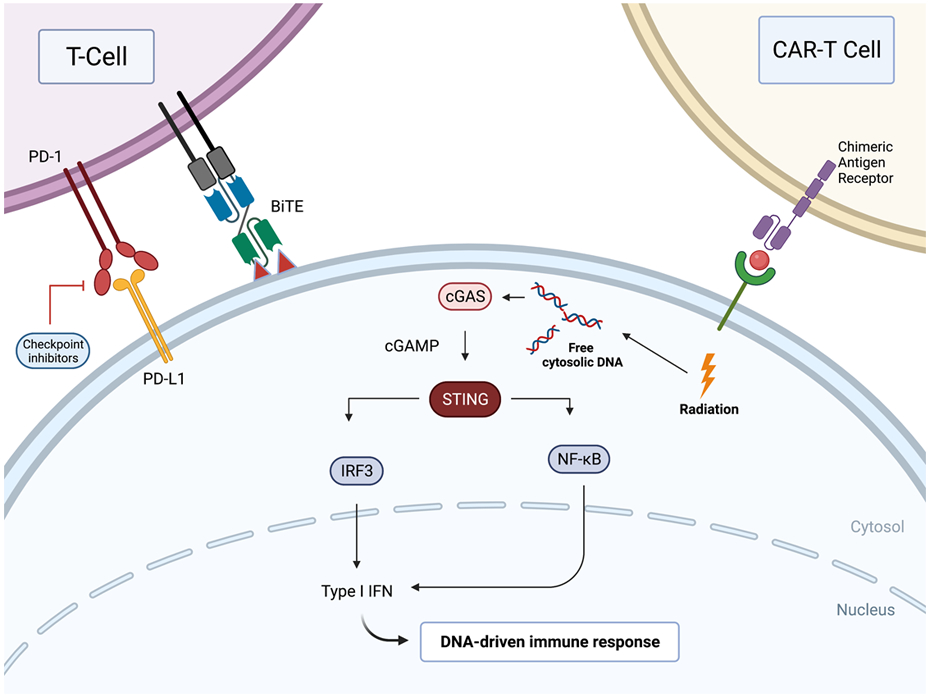
The stimulator of interferon genes (STING) pathway is activated though cytosolic DNA sensors including cyclic GMP-AMP synthase (cGAS). Activation leads to the production of Type I interferons. STING agonists may synergize with various other forms of immunotherapy including CAR-T cells, CPIs, BiTEs, and radiotherapy to improve anti-tumor activity.
STING Agonism with Immune Checkpoint Inhibitors (ICIs)
Several preclinical studies have demonstrated the potential benefit of STING agonists in combination with ICIs. Inhibition of ataxia telangiectasia mutated protein (ATM) activates the STING pathway by promoting the leakage of mitochondrial DNA into the cytosol and was shown in murine models to delay tumor growth and overcome resistance to anti-programmed cell death protein 1 (PD-1) therapy(19). A lipid nano-particle containing a CDN STING agonist (NP-STING) was also used in a murine model to demonstrate a synergistic anti-tumor effect with anti-PD-1 therapy in metastatic melanomas that were previously unresponsive to anti-PD-1 monotherapy (20). A similar study showed that a NP-STING enhanced the response of ICIs in neuroblastoma in vivo(21). In a study of peritoneal carcinomatosis of colon cancer, ADU-S100 in combination with anti-PD-1 therapy greatly reduced tumor burden when compared to either therapy alone (22). Recently, the co-administration of manganese (Mn2+), which has been shown to be an inducer of the STING pathway, and YM101, a TGF-β/ programmed cell death ligand 1 (PD-L1) bispecific antibody, also had a synergistic effect and was able to overcome immunotherapy resistance(23).
STING Agonism with vaccines, cytokines, and CAR-T therapies.
Beyond checkpoint blockade and radiation therapy, STING agonists have been investigated in combination with various other immunotherapeutic agents. Preclinical data have examined the combination of STING agonism with cancer vaccines. CDNs have been observed to improve the effect of peptide-based vaccines in murine melanoma models(24). Additionally, a synergistic effect was demonstrated with the dual use of ADU-S100 and a chimeric peptide vaccine in melanoma cells and lung epithelial cells transfected with HPV16 E6/E7 or c-H-ras oncogenes (25). A comparable study showed the enhanced efficacy of a Listeria monocytogenes-based vaccine with the addition of cyclic diguanylate (c-di-GMP) in metastatic breast cancer cells(26). The coaction of interleukin-15 and an analogue of ADU-S100 had in increased proportion of tumor cell death in an in vitro prostate cancer model when compared to either agent alone(27). Finally, agonism of the STING pathway with DMXAA or cGAMP has been shown to increase the effect of Th/Tc17 CAR T cells in an orthotopic model of breast cancer(28).
STING Agonists in Clinical Trials
Flavonoid small molecule: dimethylxanthone acetic acid (DMXAA, ASA404, vadimezan)
DMXAA was the first STING-targeted therapy to be evaluated in oncologic clinical trials and is the only agent to have reached Phase III (Table 1). During the time of its clinical development by Novartis (2000s through early 2010s), it was primarily thought to act as an anti-angiogenic agent (29). Preclinical work suggested that the drug’s primary utility was as an adjunct to traditional chemotherapy backbones, as its activity as a monotherapy was limited. It was thought to synergize with cytotoxic agents by targeting poorly perfused tumoral regions that might be underexposed to other systemic therapies. It was not until after the publication of Phase III trials that DMXAA’s additional mechanism as a STING agonist was identified (30).
Table 1.
Clinical trials with STING agonists
| STING agonist | Manufacturer | Manufacturer drug status |
NCT number (title, if any) |
Ph. | Indication | RT | Combination (comparator), if any |
NCT trial status |
|---|---|---|---|---|---|---|---|---|
| Vadimezan (DMXAA, ASA404, AS1404) | Novartis, Antisoma | Discontinued (November 2010) | NCT00738387 (ATTRACT-2) | 3 | Advanced NSCLC (stage IIIb/IV), 2nd line | IV | + Docetaxel (vs. placebo + docetaxel) | Terminated |
| NCT00662597 (ATTRACT-1) | 3 | Advanced NSCLC (stage IIIb/IV), 1st line | IV | + PC (vs. placebo + PC) | Terminated | |||
| NCT00111618 | 2 | mHRPC | IV | + Docetaxel | Completed | |||
| NCT01057342 | 2 | Advanced SCLC, 1st line | IV | + PC | Completed | |||
| NCT01071928 (GU09-144) | 2 | Advanced urothelial cancer, 2nd line | IV | + Docetaxel (vs. docetaxel alone) | Withdrawn | |||
| NCT00832494 (AS1404-201) | 1/2 | Advanced NSCLC | IV | + PC | Completed | |||
| NCT00863733 | 1 | Advanced solid tumors | IV | Completed | ||||
| NCT00856336 (DART) | 1 | Advanced solid tumors | IV | Completed | ||||
| NCT01290380 | 1 | Advanced solid tumors | IV | ± either PC or docetaxel | Terminated | |||
| NCT01299415 | 1 | Advanced solid tumors | IV | + Fluvoxamine | Terminated | |||
| NCT01299701 | 1 | Advanced solid tumors | IV | Terminated | ||||
| NCT01278758 | 1 | Advanced solid tumors, impaired renal function | IV | Terminated | ||||
| NCT00674102 | 1 | Advanced NSCLC, 1st line | IV | + PC | Completed | |||
| NCT01278849 | 1 | Advanced solid tumors, impaired liver function | IV | ± either PC or docetaxel | Terminated | |||
| NCT01240642 | 1 | Advanced solid tumors | IV | ± either PC or docetaxel | Terminated | |||
| NCT01285453 | 1 | Advanced solid tumors | IV | + Docetaxel | Completed | |||
| NCT01031212 | 1 | Advanced solid tumors | IV | + PC + cetuximab | Withdrawn | |||
| NCT00003697 | 1 | Advanced solid tumors | IV | Completed | ||||
| ADU-S100) (MIW-815) | Chinook Therapeutics (previously Novartis, Aduro) | Discontinued (November 2020) | NCT03937141 (ADU-CL-20) | 2 | SCCHN (R/M, 1st line, PD-L1 CPS ≥1) | IT | + Pembrolizumab | Terminated |
| NCT03172936 | 1 | Advanced solid tumors or lymphomas | IT | + PDR001 (spartalizumab) | Terminated | |||
| NCT02675439 | 1 | Advanced solid tumors or lymphomas | IT | ± Ipilimumab (terminated prior to accruing any subjects to the ipilimumab arm) | Terminated | |||
| Ulevostinag (MK-1454) | Merck Sharp & Dohme LLC | Not reported | NCT04220866 | 2 | SCCHN (R/M, 1st line, PD-L1 CPS ≥1) | IT | + Pembrolizumab (vs. pembrolizumab alone) | Active, not recruiting |
| NCT03010176 (MK-1454-001) | 1 | Advanced solid tumors or lymphomas | IT | ± Pembrolizumab | Completed | |||
| GSK3745417 | GlaxoSmith-Kline | Ongoing (April 2022) | NCT03843359 | 1/2 | Advanced solid tumors | IV | ± Dostarlimab | Recruiting |
| IMSA101 (GB492) | Immunesensor, Genor Biopharma | Ongoing (March 2022) | NCT04020185 | 1/2 | Advanced solid tumors | IT | ± Geptanolimab (GB226) | Recruiting |
| CDK-002 (exoSTING) | Codiak Biosciences | Ongoing (May 2022) | NCT04592484 | 1/2 | Advanced solid tumors (+ dedicated SCCHN, TNBC, ATC, and cSCC cohorts) | IT | Recruiting | |
| MK-2118 | Merck Sharp & Dohme LLC | Ongoing (May 2022) | NCT03249792 (MK-2118-001) | 1 | Advanced solid tumors or lymphomas | IT or SC | ± Pembrolizumab | Recruiting |
| BMS-986301 | Bristol-Myers Squibb | Ongoing (April 2022) | NCT03956680 | 1 | Advanced solid tumors | IM, IT, or IV | ± Nivolumab / ipilimumab | Recruiting |
| BI 1387446 | Boehringer Ingelheim | Ongoing (March 2022) | NCT04147234 | 1 | Advanced solid tumors | IT | ± Ezabenlimab (BI 754091) ± radiotherapy | Recruiting |
| SB 11285 | F-star Therapeutics | Ongoing (May 2022) | NCT04096638 (SBP-11285-ONC-101) | 1 | Advanced solid tumors (+ dedicated melanoma and SCCHN cohorts) | IV | ± Atezolizumab | Recruiting |
| E7766 | Eisai, H3 | Ongoing (February 2022) | NCT04109092 (INPUT-102) | 1 | Non-muscle invasive urothelial cancer, BCG-unresponsive | I-Ves. | Withdrawn | |
| NCT04144140 (INSTAL-101) | 1 | Advanced solid tumors or lymphomas | IT | Recruiting | ||||
| SNX281 | Stingthera, Merck Sharp & Dohme LLC | Ongoing | NCT04609579 | 1 | Advanced solid tumors or lymphomas | IV | ± Pembrolizumab | Recruiting |
| TAK-676 | Takeda | Ongoing (March 2022) | NCT04420884 | 1 | Advanced solid tumors | IV | ± Pembrolizumab | Recruiting |
| NCT04879849 | 1 | Advanced NSCLC, TNBC, SCCHN | IV | + Pembrolizumab | Recruiting | |||
| SYNB1891 | Synlogic | Ongoing (May 2022) | NCT04167137 | 1 | Advanced solid tumors or lymphomas | IT | ± Atezolizumab | Recruiting |
| STING-dependent activators (STAVs) and stimulated dendritic cells | University of Miami | Ongoing | NCT05321940 | 1 | Aggressive relapsed / refractory leukemias | IV | Not yet recruiting |
Dostarlimab, ezabenlib, geptanolimab, and spartalizumab are programmed cell death protein-1 (PD-1) inhibitors. Parenthetical dates in the “manufacturer drug status” column represent latest company statement of ongoing development, if known. ATC = anaplastic thyroid cancer, BCG = Bacillus Calmete-Guerin, cSCC = cutaneous squamous cell carcinoma, CPS = combined positive scoring, DMXAA = 5,6-dimethylxanthenone-4-acetic acid, IM = intramuscular, IT = intratumoral, IV = intravenous, I-Ves = intravesical, PC = paclitaxel and carboplatin, RT = route, SCCHN = squamous cell carcinoma of the head and neck, TNBC = triple negative breast cancer.
DMXAA’s advancement to Phase III trials in advanced (stage IIIb/IV) NSCLC was based on Phase II data demonstrating improved overall response rates (ORRs) in the first line setting for intravenously administered DMXAA in combination with paclitaxel and carboplatin compared to paclitaxel and carboplatin alone [31% (95% confidence interval (CI) 15.2-47.3) vs 22% (95% CI 6.5-37.9)] (31). Neither median time to progression (TTP) [hazard ratio (HR) 0.94, 95% CI 0.56-1.57, p=0.82] nor median overall survival (OS) (HR 0.86, 95% CI 0.47-1.57, p=0.63) were improved. A subsequent retrospective analysis pooling data from two Phase II trials suggested that patients benefitted regardless of histology, but that DMXAA might offer particular benefit in individuals with squamous tumors, in whom ORR was 40.0% (95% CI 19-64%) vs 14.3% (95% CI 0.4-58%), median TTP was 5.6 months (95% CI 4.1-8.1 months) vs 1.6 months (95% CI 1.3-11.4 months), and median OS was 10.2 months (95% CI 6.0 months-not reached (NR)) vs 5.5 months (95% CI 2.1-12.5 months) (32). The significance of this finding was underscored by the fact that another anti-angiogenic agent, bevacizumab, was known to be associated with diminished activity and increased vascular risk among those with squamous disease compared to those with non-squamous disease (33).
DMXAA was then studied in two large, randomized, placebo-controlled, double-blinded international Phase III trials for individuals with advanced NSCLC. In 1,299 patients treated in the first-line setting, DMXAA combined with paclitaxel/carboplatin failed to demonstrate benefits in response rate, progression-free survival (PFS), or OS over paclitaxel/carboplatin alone (34, 35). Subgroup analyses in patients with non-squamous and squamous diseases demonstrated no benefit in each group. DMXAA was associated with increased rates of grade 4 neutropenia (27% vs 19%) and infusion site pain (10% vs 0.5%) but was not associated with increased rates of dose reductions or delays. The study was discontinued due to futility. The 2nd-line trial compared DMXAA/docetaxel with placebo/docetaxel and also showed no ORR, PFS, or OS benefit, regardless of histology (36). Rates of grade 3/4 adverse events (AEs) were similar by group (80.4% vs 78.0%).
DMXAA also reached Phase II development in metastatic hormone-resistant prostate cancer and extensive-stage small cell lung cancer (SCLC). Among 74 patients in the prostate cancer trial, radiologic ORR was numerically increased for DMXAA/docetaxel vs docetaxel alone (23.1% vs 9.1%), prostate-specific antigen (PSA) response rate (≥50% reduction from baseline) (59.4% vs 36.8%), and median PSA reduction (85% vs 61.9%) without reporting of statistical significance (37). The trial did not demonstrate PFS or OS benefits (2-year OS was with DMXAA/docetaxel was 33.3% vs 22.8% in control arm). Rates of AEs leading to death (6.1% vs 2.6%) and subjects experiencing ≥1 serious AE (SAE) (42.4% vs 34.2%) were numerically higher in the DMXAA/docetaxel group. In the single-arm SCLC trial, 17 patients with previously untreated tumors received DMXAA with carboplatin and docetaxel with an ORR of 94%, disease control rate (DCR) of 100%, median PFS of 7.0 months, and median OS of 14.2 months (median follow-up 17.7 months). In light of the negative Phase III NSCLC trial results, all further development of DMXAA was halted: no further development in mHRPC was pursued, the SCLC was discontinued early (planned accrual was originally n=56), and no data were published on a planned Phase II urothelial cancer trial.
Subsequent translational studies provided some potential explanations for DMXAA’s disappointing results in late-phase trials. First, DMXAA selectively binds and activates mouse STING versus human STING (38). This finding helps explain the discrepancy between DMXAA’s preclinical promise and its lack of clinical activity in NSCLC. Furthermore, the human STING protein was found to be highly polymorphic and DMXAA failed to activate any of the STING isoforms associated with the five main human allele variants (10, 39). Beyond these barriers, DMXAA was mainly tested in combination with cytotoxic chemotherapies (generally taxanes with or without carboplatin) and combinations with newer monoclonal antibody therapies (e.g., immunotherapies) were never studied (a planned combination trial with cetuximab in solid tumors was withdrawn prior to starting accrual). As such, DMXAA’s (and potential derivative compounds’) clinical potential in combination with modern therapies and/or in larger prostate cancer, SCLC, and urothelial cancer trials remain unknown.
Cyclic dinucleotides (CDNs)
In the wake of DMXAA’s disappointing clinical results and the discovery of its STING-targeting mechanism, researchers sought to identify therapeutics that might be active across multiple human STING variants. This led to the development of CDNs, which are thought to agonize STING because of their structural similarity to endogenous cyclic cGAMP (40). These compounds are thought to work best as intratumorally administered therapies, as they are susceptible to destruction by serum phosphatases and hydrolases (41). In 2015 Aduro Biotech developed the CDN ADU-S100, which was shown to activate all five known STING isoforms (10). Aduro clinically developed ADU-S100 jointly with Novartis for oncologic conditions. Intratumorally administered ADU-S100 was tested in Phase I studies as a monotherapy and in combination with immunotherapy agents. ADU-S100 monotherapy phase I data were published in 2021(42). A total of 47 patients were treated with weekly intratumoral injections ranging from 50 to 6,400 micrograms in a weekly. The most frequent treatment-related adverse events (AEs) were noted to be on-target expected toxicities, such as pyrexia and injection site pain. Only one patient discontinued due to AEs. Though the vast majority of injected lesions (94%) were stable or decreased in size, overall efficacy was modest with one confirmed and two unconfirmed PRs. Correlative studies in this trial demonstrated signs of systemic immune activation, including dose-dependent increases in interferon-β levels and clonal T cell proliferation. Interestingly, PK demonstrated rapid absorption into plasma with a half-life was 24 minutes. This suggested that retention of drug in the tumor emerged as an unanticipated challenge to the intratumoral approach. The combination study of ADU-S100 plus spartalizumab concluded and data has not yet been published. A Phase II study was started for first-line recurrent/metastatic squamous cell carcinoma of the head and neck (SCCHN) This study enrolled 16 patients then was terminated after a review of the totality of clinical data with ADU-S100. In June 2020, Aduro merged with Chinook Therapeutics, and given competing priorities the company elected not to further develop ADU-S100(43).
Merck’s intratumorally administered CDN STING agonist MK-1454 (ulevostinag) reported promising Phase I data. The DCR rate was 48% with PRs seen in 6 out of 25 patients (24%) treated in combination with pembrolizumab (3 SCCHN, triple negative breast cancer (TNBC), and 2 anaplastic thyroid cancer (ATC)) (44). Among responders, target lesions (including injected and non-injected lesions) were reduced in size by a median of 83%. The most common AE (experienced by ≥10% of patients) was pruritis and 7% of patients discontinued due to AEs. Ulevostinag advanced to a randomized phase II of development as a combination with pembrolizumab in first-line recurrent/metastatic SCCHN with PD-L1 CPS ≥1. Phase II results have not been reported and the trial was closed with no further development plans reported (45) (46).
Phase I clinical studies are ongoing for multiple other CDN-derivatives including BMS-986301, BI 1387446, and E7766. Bristol-Myers Squibb’s (BMS’s) BMS-986301 is being studied in advanced solid tumors as a single agent and in combination with nivolumab/ipilimumab. Its clinical development program is differentiated from predecessors in its focus on systemic administration such as intramuscular (IM) and intravenous (IV) routes. Intratumorally administered BI 1387446 is being studied with and without Boehringer Ingelheim’s (BI’s) PD-1 inhibitor ezabenlimab and with and without radiotherapy. Eisai and H3 Biomedicine’s E7766 is a macrocycle-bridged CDN that has demonstrated pan-genotypic activity across human STING variants in preclinical studies (47). It is being studied as an intratumorally administered monotherapy for advanced solid tumors (48). A planned study the compound as an intravesical treatment for non-muscle invasive bladder cancer, including those unresponsive to Bacillus Calmette-Guerin (BCG), was withdrawn, given challenges in recruitment (49).
Future Directions:
Other small molecules
Non-CDN small molecule STING agonists have also been developed with different properties The structures of most have not yet been disclosed by manufacturers. In this group, the only product whose structure is publicly known is GlaxoSmithKline’s (GSK’s) GSK3745417, a diABZI. (50). The product was discovered in part with the intention of developing a therapy that could be systemically administered (rather than CDNs, which have largely been studied as intratumorally administered therapies). At the cellular level, the product is thought to work similarly to CDNs (i.e., by agonizing STING at its cGAMP binding pocket). However, its structure is likely more amenable to systemic administration. Like Merck, BMS, and BI, GSK is using the strategy of developing its STING agonist along with its proprietary PD-1 inhibitor (for GSK, the PD-1 monoclonal antibody (mAb) is dostarlimab). Genor Biopharma is also employing this strategy, combining its PD-1 inhibitor geptanolimab with its intratumorally administered STING agonist GB492 (IMSA101), which it is co-developing with ImmuneSensor Therapeutics. Merck has also conducted a phase I study with its STING agonist, MK-2118 (being tested with and without pembrolizumab) and results are pending. Meanwhile several other manufacturers are combining their STING agonists with approved PD-1 inhibitors: Both Stingthera and Takeda are combining their STING agonists (SNX281 and TAK-676, respectively) with pembrolizumab. Stingthera has a formal agreement to co-develop SNX281 with Merck. Finally, F-star Therapeutics is testing its intravenously-administered SB 11285 in collaboration with Roche, combining the novel agent with Roche’s atezolizumab (although it was originally planned as a combination with nivolumab) (51).
Novel delivery systems
To improve the stability of drug after administration another approach has been to devise new ways to “package” traditional CDNs. exoSTING is an intratumorally administered exosome containing Codiak Bioscience’s proprietary CDN STING agonist (52). The exosome not only protects the CDN payload from degradation, but also has surface PTFGRN that is meant to facilitate specific uptake by tumor-resident antigen presenting cells (53). A Phase I/II trial studying exoSTING as a monotherapy in advanced solid tumors is ongoing (54). The trial has dedicated SCCHN, TNBC, ATC, and cutaneous squamous cell carcinoma (cSCC) cohorts. An interim analysis included n=8 subjects with evaluable tumors and showed variable activity from lesion-to-lesion, even within an individual. For example, in a subject receiving exoSTING as a fifth-line agent for parotid gland carcinoma, the injected lesion grew by 50% while a non-injected lesion shrank by 74% (the growth was thought to represent pseudoprogression, or tumor growth due to inflammation rather than due to disease progression). This subject remained on therapy for over seven months. A patient receiving exoSTING as fourth-line therapy for cSCC experienced stable size of an injected lesion and 77% decreased size of a non-injected lesion, while a patient with receiving third-line therapy for chondrosarcoma experienced responsive evaluation criteria in solid tumors (RECIST) SD overall. These data do suggest a meaningful abscopal effect.
Synlogic developed SYNB1891, which is a live Escherichia coli probiotic engineered to produce STING-agonist CDNs in vivo when administered intratumorally with or without systemic atezolizumab (55, 56). A Phase I trial in advanced solid tumors is ongoing. Among 23 patients whose treatment course has been reported, two patients (one with vulvar cancer and one with SCLC, both refractory to immunotherapy) experienced RECIST SD (RECIST −28% and −12%, respectively). Four patients experienced cytokine release syndrome, including one grade 3 event that qualified as a dose-limiting toxicity (DLT). No other SAEs have been reported.
Cell therapy and vaccine
A final STING-targeting approach that has reached clinical development is the administration of University of Miami’s STING-dependent activator (STAV)-loaded autologous leukemic cells combined with a dendritic cell vaccine. A Phase I study in aggressive relapsed/refractory leukemias is planned (57).
In addition to the above immunotherapy combinations, there may be a role for the addition of STING agonists in traditional chemotherapy regimens whose mechanisms have been revealed to involve the STING pathway. Bortezomib, a proteosome inhibitor most used in the treatment of multiple myeloma, has been shown to use a STING mediated pathway to induce an immune response against myeloma cells(58). Multiple studies have also shown that inhibitors of poly(ADP-ribose) polymerase (PARP) promote anti-tumor immunity via activation of the STING pathway(59-61). Likewise, temiposide, a topoisomerase inhibitor, and 5-fluorouracil have been shown to utilize the STIGN pathway for their therapeutic effects (62, 63)
Other mechanisms for delivery of STING agonists are also in development. A recent abstract described the ability of a STING-agonist antibody-drug conjugate (ADC)to induce a complete response and create immunologic memory (64). Given recent growth in effective ADCs for cancer therapy, this strategy may be the most logical to improve intratumoral delivery across multiple tumor sites while pragmatically limiting systemic exposure. Similarly, an oral STING agonist was shown to create sustainable tumor regression and a synergistic effect with ICIs in a murine model(65).
Conclusion:
Resistance to immunotherapies remains a challenge in the growing arsenal of anti-cancer drugs. First generation STING agonist demonstrated modest activity, but a new wave of technologies and combination with other therapies may result in improved outcomes. Many early phase trials and preclinical studies investigating the use of STING agonists and new mechanisms of drug delivery are ongoing and will inform the viability of STING as a successful target for cancer immunotherapy regimens.
References:
- 1.S NL, T MWL, M TSK, S RA. De-novo and acquired resistance to immune checkpoint targeting. The Lancet Oncology. 2017;18(12). doi: 10.1016/S1470-2045(17)30607-1. [DOI] [PubMed] [Google Scholar]
- 2.S P, H-L S, W JA, R A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168(4). doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.B D, Z Z, K B, P-S A, S S, G SH. Resistance to immunotherapy in human malignancies: Mechanisms, research progresses, challenges, and opportunities. Journal of cellular physiology. 2022;237(1). doi: 10.1002/jcp.30575. [DOI] [PubMed] [Google Scholar]
- 4.Hopfner K-P, Hornung V Molecular mechanisms and cellular functions of cGAS–STING signalling. Nature Reviews Molecular Cell Biology. 2020;21(9):501–21. doi: doi: 10.1038/s41580-020-0244-x. [DOI] [PubMed] [Google Scholar]
- 5.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. 2013. doi: 1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Z X, B XC, C ZJ. Structures and Mechanisms in the cGAS-STING Innate Immunity Pathway. Immunity. 2020;53(1). doi: 10.1016/j.immuni.2020.05.013. [DOI] [PubMed] [Google Scholar]
- 7.W SR, F MB, C L, S S, F MJ, L MY, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41(5). doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D MS, K M, M H, M M, D GP, A JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. The Journal of experimental medicine. 2011;208(10). doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.M A, M AJ, L-V M, W L, V RE, R DH. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity. 2018;49(4). doi: 10.1016/j.immuni.2018.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015;11(7):1018–30. Epub 20150507. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohkuri T, Kosaka A, Ishibashi K, Kumai T, Hirata Y, Ohara K, et al. Intratumoral administration of cGAMP transiently accumulates potent macrophages for anti-tumor immunity at a mouse tumor site. Cancer Immunology, Immunotherapy. 2017;66(6):705–16. doi: doi: 10.1007/s00262-017-1975-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D O, DG A, C S, G N, DD J, F L, et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(50). doi: 10.1073/pnas.1512832112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi F, Su J, Wang J, Liu Z, Wang T. Activation of STING inhibits cervical cancer tumor growth through enhancing the anti-tumor immune response. Molecular and Cellular Biochemistry. 2020;476(2):1015–24. doi: doi: 10.1007/s11010-020-03967-5. [DOI] [PubMed] [Google Scholar]
- 14.V EP, B NS, M D, M L, E MA, R MJ, et al. STING Activated Tumor-Intrinsic Type I Interferon Signaling Promotes CXCR3 Dependent Antitumor Immunity in Pancreatic Cancer. Cellular and molecular gastroenterology and hepatology. 2021;12(1). doi: 10.1016/j.jcmgh.2021.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D L, L H, X M, Y X, B B, A A, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity. 2014;41(5). doi: 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Crowe WN, Wang L, Lu Y, Petty WJ, Habib AA, et al. An inhalable nanoparticulate STING agonist synergizes with radiotherapy to confer long-term control of lung metastases. Nature Communications. 2019;10(1):1–15. doi: doi: 10.1038/s41467-019-13094-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zaidi AH, Kelly RJ, Gorbunova A, Omstead AN, Salvitti MS, Zheng P, et al. Intratumoral immunotherapy with STING agonist, ADU-S100, induces CD8+ T-cell mediated anti-tumor immunity in an esophageal adenocarcinoma model. 12. 2021. doi: https://www.oncotarget.com/article/27886/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.X A, S Y, J P, Z S, Z C, H X, et al. Increased activation of cGAS-STING pathway enhances radiosensitivity of non-small cell lung cancer cells. Thoracic cancer. 2022;13(9). doi: 10.1111/1759-7714.14400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.H M, Z M, B X, P D, J M, L X, et al. ATM inhibition enhances cancer immunotherapy by promoting mtDNA leakage and cGAS/STING activation. The Journal of clinical investigation. 2021;131(3). doi: 10.1172/JCI139333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.N T, S T, E R, S S, T N, S Y, et al. STING agonist loaded lipid nanoparticles overcome anti-PD-1 resistance in melanoma lung metastasis via NK cell activation. Journal for immunotherapy of cancer. 2021;9(7). doi: 10.1136/jitc-2021-002852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.W-B L, W M, S D, J J, H BC, G K, et al. Potent STING activation stimulates immunogenic cell death to enhance antitumor immunity in neuroblastoma. Journal for immunotherapy of cancer. 2020;8(1). doi: 10.1136/jitc-2019-000282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.L SJ, Y H, K WR, L YS, L WS, K SJ, et al. STING activation normalizes the intraperitoneal vascular-immune microenvironment and suppresses peritoneal carcinomatosis of colon cancer. Journal for immunotherapy of cancer. 2021;9(6). doi: 10.1136/jitc-2020-002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yi M, Niu M, Zhang J, Li S, Zhu S, Yan Y, et al. Combine and conquer: manganese synergizing anti-TGF-β/PD-L1 bispecific antibody YM101 to overcome immunotherapy resistance in non-inflamed cancers. Journal of Hematology & Oncology. 2021;14(1):1–21. doi: doi: 10.1186/s13045-021-01155-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, Celis E. STING activator c-di-GMP enhances the anti-tumor effects of peptide vaccines in melanoma-bearing mice. Cancer Immunology, Immunotherapy. 2015;64(8):1057–66. doi: doi: 10.1007/s00262-015-1713-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.R M, C S, DB-B W, R E, S-R ML, B E, et al. STING Agonist Combined to a Protein-Based Cancer Vaccine Potentiates Peripheral and Intra-Tumoral T Cell Immunity. Frontiers in immunology. 2021;12. doi: 10.3389/fimmu.2021.695056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.C D, Q-T W, J A, A-A D, R I, S HO, et al. STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer. Cancer immunology research. 2014;2(9). doi: 10.1158/2326-6066.CIR-13-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.E AM, P E, D P, G C. Combination of Interleukin-15 With a STING Agonist, ADU-S100 Analog: A Potential Immunotherapy for Prostate Cancer. Frontiers in oncology. 2021;11. doi: 10.3389/fonc.2021.621550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.X N, P DC, R AC, S P, B H, L SJ, et al. STING agonist promotes CAR T cell trafficking and persistence in breast cancer. The Journal of experimental medicine. 2021;218(2). doi: 10.1084/jem.20200844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siim BG, Lee AE, Shalal-Zwain S, Pruijn FB, McKeage MJ, Wilson WR. Marked potentiation of the antitumour activity of chemotherapeutic drugs by the antivascular agent 5,6-dimethylxanthenone-4-acetic acid (DMXAA). Cancer Chemother Pharmacol. 2003;51(1):43–52. Epub 20021112. doi: 10.1007/s00280-002-0529-0. [DOI] [PubMed] [Google Scholar]
- 30.Prantner D, Perkins DJ, Lai W, Williams MS, Sharma S, Fitzgerald KA, et al. 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)-dependent innate immune pathways and is regulated by mitochondrial membrane potential. J Biol Chem. 2012;287(47):39776–88. Epub 20121001. doi: 10.1074/jbc.M112.382986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKeage MJ, Von Pawel J, Reck M, Jameson MB, Rosenthal MA, Sullivan R, et al. Randomised phase II study of ASA404 combined with carboplatin and paclitaxel in previously untreated advanced non-small cell lung cancer. Br J Cancer. 2008;99(12):2006–12. doi: 10.1038/sj.bjc.6604808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKeage MJ, Jameson MB, Investigators ASSG. Comparative outcomes of squamous and non-squamous non-small cell lung cancer (NSCLC) patients in phase II studies of ASA404 (DMXAA) - retrospective analysis of pooled data. J Thorac Dis. 2010;2(4):199–204. doi: 10.3978/j.issn.2072-1439.2010.02.04.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson DH, Fehrenbacher L, Novotny WF, Herbst RS, Nemunaitis JJ, Jablons DM, et al. Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol. 2004;22(11):2184–91. doi: 10.1200/JCO.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 34.Lara PN Jr., Douillard JY, Nakagawa K, von Pawel J, McKeage MJ, Albert I, et al. Randomized phase III placebo-controlled trial of carboplatin and paclitaxel with or without the vascular disrupting agent vadimezan (ASA404) in advanced non-small-cell lung cancer. J Clin Oncol. 2011;29(22):2965–71. Epub 20110627. doi: 10.1200/JCO.2011.35.0660. [DOI] [PubMed] [Google Scholar]
- 35.A Phase III, randomized, double-blind, placebo-controlled, multi-center study of ASA404 in combination with paclitaxel and carboplatin as first-line treatment for locally advanced or met- astatic (stage IIIb/IV) non-small cell lung cancer (NSCLC). Novartis; 2010. [cited 2022]. Available from: https://www.novctrd.com/ctrdweb/trialresult/trialresults/pdf?trialResultId=4609. [Google Scholar]
- 36.A phase III, randomized, double-blind, placebo-controlled, multi-center study of Vadimezan in combination with docetaxel in second-line treatment of patients with locally advanced or metastatic (stage IIIb/IV) non-small-cell lung cancer (NSCLC) 2011. [cited 2022]. Available from: https://www.novctrd.com/ctrdweb/trialresult/trialresults/pdf?trialResultId=4927. [Google Scholar]
- 37.Pili R, Rosenthal MA, Mainwaring PN, Van Hazel G, Srinivas S, Dreicer R, et al. Phase II study on the addition of ASA404 (vadimezan; 5,6-dimethylxanthenone-4-acetic acid) to docetaxel in CRMPC. Clin Cancer Res. 2010;16(10):2906–14. Epub 20100511. doi: 10.1158/1078-0432.CCR-09-3026. [DOI] [PubMed] [Google Scholar]
- 38.Conlon J, Burdette DL, Sharma S, Bhat N, Thompson M, Jiang Z, et al. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J Immunol. 2013;190(10):5216–25. Epub 20130412. doi: 10.4049/jimmunol.1300097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yi G, Brendel VP, Shu C, Li P, Palanathan S, Cheng Kao C. Single nucleotide polymorphisms of human STING can affect innate immune response to cyclic dinucleotides. PLoS One. 2013;8(10):e77846. Epub 20131021. doi: 10.1371/journal.pone.0077846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, et al. Cyclic [G(2',5')pA(3',5')p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. 2013;153(5):1094–107. Epub 20130503. doi: 10.1016/j.cell.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kato K, Nishimasu H, Oikawa D, Hirano S, Hirano H, Kasuya G, et al. Structural insights into cGAMP degradation by Ecto-nucleotide pyrophosphatase phosphodiesterase 1. Nat Commun. 2018;9(1):4424. Epub 20181024. doi: 10.1038/s41467-018-06922-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meric-Bernstam F, Sweis RF, Hodi FS, Messersmith WA, Andtbacka RHI, Ingham M, et al. Phase I Dose-Escalation Trial of MIW815 (ADU-S100), an Intratumoral STING Agonist, in Patients with Advanced/Metastatic Solid Tumors or Lymphomas. Clin Cancer Res. 2022;28(4):677–88. doi: 10.1158/1078-0432.CCR-21-1963. [DOI] [PubMed] [Google Scholar]
- 43.Chinook Therapeutics. Quarterly report for the quarterly period ended September 30, 2020.
- 44.Harrington K, Brody J, Ingham M, Strauss J, Cemerski S, Wang M, et al. Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Developmental Therapeutics. 2018;29:vii712. [Google Scholar]
- 45.Merck Sharp & Dohme LLC. Study of Intratumoral (IT) Ulevostinag (MK-1454) in Combination With Intravenous (IV) Pembrolizumab (MK-3475) Compared to IV Pembrolizumab Alone as the First Line Treatment of Metastatic or Unresectable, Recurrent Head and Neck Squamous Cell Carcinoma (HNSCC) (MK-1454-002) 2021. [updated October 11, 2021; cited 2022 May 23]. Available from: https://clinicaltrials.gov/ct2/show/NCT04220866.
- 46.Merck Sharp & Dohme LLC. Annual report for the fiscal year ended December 31, 2020.
- 47.Kim DS, Endo A, Fang FG, Huang KC, Bao X, Choi HW, et al. E7766, a Macrocycle-Bridged Stimulator of Interferon Genes (STING) Agonist with Potent Pan-Genotypic Activity. ChemMedChem. 2021;16(11):1740–3. Epub 20210225. doi: 10.1002/cmdc.202100068. [DOI] [PubMed] [Google Scholar]
- 48.Inc. E. Study of Intratumorally Administered Stimulator of Interferon Genes (STING) Agonist E7766 in Participants With Advanced Solid Tumors or Lymphomas - INSTAL-101 2022. [updated April 25, 2022; cited 2022 May 23]. Available from: https://clinicaltrials.gov/ct2/show/NCT04144140.
- 49.Inc. E. A Study of Stimulator of Interferon Genes (STING) Agonist E7766 in Non-muscle Invasive Bladder Cancer (NMIBC) Including Participants Unresponsive to Bacillus Calmette-Guerin (BCG) Therapy, INPUT-102 2020. [updated December 14, 2020; cited 2022 May 23]. Available from: https://clinicaltrials.gov/ct2/show/NCT04109092.
- 50.Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang SY, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature. 2018;564(7736):439–43. Epub 20181107. doi: 10.1038/s41586-018-0705-y. [DOI] [PubMed] [Google Scholar]
- 51.Luke JJ, Janku F, Strauss J, Olszanski AJ, Leach K, Radhakrishnan I, et al. 598TiP A phase I/Ib dose-escalation study of intravenously administered SB 11285 alone and in combination with nivolumab in patients with advanced solid tumours. Annals of Oncology. 2020;31. doi: 10.1016/j.annonc.2020.08.712. [DOI] [Google Scholar]
- 52.EXOSTING FIRST-IN-HUMAN CLINICAL TRIAL UPDATE [Internet]. 2021; November 16, 2021
- 53.Jang SC, Economides KD, Moniz RJ, Sia CL, Lewis N, McCoy C, et al. ExoSTING, an extracellular vesicle loaded with STING agonists, promotes tumor immune surveillance. Commun Biol. 2021;4(1):497. Epub 20210422. doi: 10.1038/s42003-021-02004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Codiak Biosciences. A First-in-Human Study of CDK-002 (exoSTING) in Subjects With Advanced/Metastatic, Recurrent, Injectable Solid Tumors 2022. [updated May 20, 2022; cited 2022 May 29]. Available from: https://clinicaltrials.gov/ct2/show/NCT04592484.
- 55.Riese R, Luke J, Lewis K, Janku F, Piha-Paul S, Verschraegen C, et al. 500 SYNB1891, a bacterium engineered to produce a STING agonist, demonstrates target engagement in humans following intratumoral injection. Journal for ImmunoTherapy of Cancer. 2021;9(Suppl 2):A532–A. doi: 10.1136/jitc-2021-SITC2021.500. [DOI] [Google Scholar]
- 56.Janku F, Luke JJ, Brennan A, Riese R, Varterasian M, Armstrong MB, et al. Abstract CT110: Intratumoral injection of SYNB1891, a synthetic biotic designed to activate the innate immune system, demonstrates target engagement in humans including intratumoral STING activation. Cancer Research. 2021;81(13_Supplement):CT110–CT. doi: 10.1158/1538-7445.Am2021-ct110. [DOI] [Google Scholar]
- 57.Ramos JC. Safety Trial of STING-dependent Activators and Stimulated Dendritic Cells for Aggressive Relapsed/Refractory Leukemias 2022. [updated April 11, 2022; cited 2022 May 29]. Available from: https://clinicaltrials.gov/ct2/show/NCT05321940. [Google Scholar]
- 58.G A, M E, S MK, B C, H T, B G, et al. Bortezomib induces anti-multiple myeloma immune response mediated by cGAS/STING pathway activation. Blood cancer discovery. 2021;2(5). doi: 10.1158/2643-3230.BCD-21-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.P C, S O, DOT M, M AK, K A, W D, et al. PARP Inhibitor Efficacy Depends on CD8 + T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer discovery. 2019;9(6). doi: 10.1158/2159-8290.CD-18-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.S T, R BL, C L, C CMD, M N, F J, et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer discovery. 2019;9(5). doi: 10.1158/2159-8290.CD-18-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.J F, Z F, Z M, L K, X M, L F, et al. Targeting the DNA damage response enhances CD70 CAR-T cell therapy for renal carcinoma by activating the cGAS-STING pathway. Journal of hematology & oncology. 2021;14(1). doi: 10.1186/s13045-021-01168-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.W Z, C J, H J, Z H, X F, H W, et al. cGAS/STING axis mediates a topoisomerase II inhibitor-induced tumor immunogenicity. The Journal of clinical investigation. 2019;129(11). doi: 10.1172/JCI127471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.T J, Z D, K V, W Q, W Y, F D, et al. 5-Fluorouracil efficacy requires anti-tumor immunity triggered by cancer-cell-intrinsic STING. The EMBO journal. 2021;40(7). doi: 10.15252/embj.2020106065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wongthida P, Catcott K, Lancaster K, Bentley K, Dirksen A, Du B, et al. 785 STING-agonist ADCs targeting tumor-associated antigens coordinate immune-mediated killing of antigen-negative cancer cells. Journal for ImmunoTherapy of Cancer. 2021;9(Suppl 2):A820–A. doi: 10.1136/jitc-2021-SITC2021.785. [DOI] [Google Scholar]
- 65.Pan B-S, Perera SA, Piesvaux JA, Presland JP, Schroeder GK, Cumming JN, et al. An orally available non-nucleotide STING agonist with antitumor activity. Science. 2020;369(6506):eaba6098. doi: doi: 10.1126/science.aba6098. [DOI] [PubMed] [Google Scholar]