

Abstract
In addition to genetic variants and copy number alterations, epigenetic deregulation of oncogenes and tumor suppressors is a major contributor in cancer development and propagation. Regulatory elements for gene transcription regulation can be found in promoters which are located in the vicinity of transcription start sites but also at a distance, in enhancer sites, brought to interact with proximal sites when occupied by enhancer protein complexes. These sites provide most of the specific regulatory sequences recognized by transcription factors. A sub-set of enhancers characterized by a longer structure and stronger activity, called super-enhancers, are critical for the expression of specific genes, usually associated with individual cell type identity and function. Super-enhancers show deregulation in cancer, which may have profound repercussions for cancer cell survival and response to therapy. Dysfunction of super-enhancers may result from multiple mechanisms that include changes in their sequence, alterations in the topological neighborhoods where they belong, and alterations in the proteins that mediate their function, such as transcription factors and epigenetic modifiers. These can become potential targets for therapeutic interventions. Genes that are targets of super-enhancers are cell and cancer type specific and could also be of interest for therapeutic targeting. In colorectal cancer, a super-enhancer regulated and over-expressed oncogene is MYC, under the influence of the WNT/β-catenin pathway. Identification and targeting of additional oncogenes regulated by super-enhancers in colorectal cancer may pave the way for combination therapies targeting the super-enhancer machinery and signal transduction pathways that regulate the specific transcription factors operative on them.
Keywords: Epigenetics, gene regulation, histone acetylation, enhancers, promoters
Introduction
Colorectal cancer is a significant cause of cancer-related mortality worldwide. In the United States of America, colorectal cancer contributes to 8% of cancer deaths and constitutes the third most frequent cause of cancer related mortality in both men and women, after lung and prostate cancer in the former and after lung and breast cancer in the latter [1]. Incidence increases with age; about six of every 10,000 persons between age 50 to 54 years-old are diagnosed with the disease, while 20 of every 10,000 persons between age 75 and 79 receive a colorectal cancer diagnosis [2]. Therefore, incidence is expected to increase in the foreseeable future as a result of the aging of the population. Mortality from colorectal cancer results mainly from metastatic disease, which remains mostly incurable, despite prolongation of survival as a result of advancements in systemic treatments [3]. Targeted therapies based on molecular defects, such as microsatellite instability (MSI), BRAF mutations, and HER2 amplifications, which drive colorectal cancer carcinogenesis have been effective for small sub-sets of patients [4-6]. Targetable lesions are present in a minority of colorectal cancers and most metastatic colorectal cancer patients have few effective therapeutic options, besides standard chemotherapy and anti-angiogenic therapies [7]. Great efforts have been invested in the development of targeted therapies that would increase the personalized treatment options for a wider proportion of these patients.
Based on genomic studies, colorectal cancers are categorized into those with high levels of microsatellite instability (MSI-H) and those with chromosomal instability (CIN). A third sub-set includes colorectal cancers that are genomically stable possessing neither MSI nor CIN [8]. Both MSI and CIN result in widespread changes in the DNA of cancer cells not only in gene coding sequences but also in non-coding and regulatory sequences. Consequently, besides alterations in proteins due to mutations in their gene sequences observed in cancer cells, deregulation of expression of various cell components may result from alterations in regulatory elements and alterations in the epigenetic machinery that orchestrates gene expression [9]. Mutations in regulatory elements, such as promoters and enhancers may have significant repercussions in the ability of transcriptions factors to bind and regulate the expression of key cancer proteins, such as oncogenes and tumor suppressors. Super-enhancers are a special type of enhancer sequences, which possess stronger regulatory potency than typical enhancers. Thus their deregulation in cancers may have profound effects [10]. In addition, in cancers that are dependent or addicted to super-enhancer activity, reliable reversal of super-enhancer dysfunction through targeted therapies could provide effective treatment opportunities. This article discusses the role of super-enhancers in colorectal cancer and the arising therapeutic developments in this field.
Definition, molecular structure and physiologic role of super-enhancers
Super-enhancers consist of multiple enhancer DNA sequences that are closely concentrated in the space providing binding sites for enhancer binding proteins [10]. Functionally, genes regulated by super-enhancers are robustly transcribed, since super-enhancers serve as the docking sites for recruitment of an in tandem array of protein complexes required for the assembly of the general transcription machinery [11].
The concept of super-enhancers was first introduced in studies of embryonic stem cells (ESCs) to denote regions bound by stem cell transcription factors Oct4, Sox2 and Nanog, highly bound by the Mediator complex and marked by the chromatin modification H3K27Ac (acetylation of lysine at position 27 of histone 3), which is a marker of active transcription [12]. Since the initial introduction of the concept, super-enhancers have been identified in various cell types and confirmed to be reliably discovered based solely on their high H3K27Ac content [13]. Super-enhancers provide the high transcriptional output required to establish cell identities [14]. Their differentiating properties in comparison to typical enhancers are mostly quantitative, super-enhancers being longer, usually several kilobases in length versus 0.5 to 3 kilobases for typical enhancers, and possessing higher levels of concentrated H3K27Ac [15]. As mentioned above, in ESCs, super-enhancers are regulators of genes that are targets of core stem cell transcription factors. In differentiated cells, super-enhancers regulate genes that are targets of transcription factors associated with the individual cell type identity. For example, super-enhancers of adipocyte differentiation bind transcription factors, including KLF4, KLF5, JunB, FOSL2, nuclear receptors and ATF7, which transcribe adipogenesis promoting genes [16]. In another example, cardiomyocyte differentiation is dependent on super-enhancers bound by transcription factor GATA4, which promotes expression of genes required for contractility and excludes the expression of non-cardiomyocyte related genes [17].
The key epigenetic marker of super-enhancers, H3K27Ac is produced by acetyltransferases of the KAT3 family, KAT3A (also known as CBP, or CREBBP-CREB Binding Protein) and KAT3B (p300) (Figure 1A) [18]. At least in certain tissues, KAT3 acetyltransferases are recruited in super-enhancer sequences marked with methylation of H3K4me1/2 (mono- or dimethylated lysine at position 4 of histone 3) [19]. This epigenetic modification is performed by methyltransferases KMT2C (MLL3) and KMT2B (MLL4). Moreover, demethylation of the repressive dimethylated lysine at position 9 of histone 3 (H3K9me2) by demethylase KDM3 promotes H3K4 methylation in colorectal cancer stem cells [20]. The H3K27Ac histone modification serves as a docking site for protein BRD4 (Bromodomain containing protein 4), which possesses two bromodomains in tandem [21]. In addition, BRD4 recognizes the methylated sub-unit of the SWI-SNF (Switch/Sucrose Non-Fermentable) complex, BAF155 (also called SMARCC1-SWI/SNF-related, Matrix-associated, Actin-dependent Regulator of Chromatin, sub-family C, member 1) [22]. BAF155 methylation is performed by arginine methytransferase CARM1 (Co-activator Associated Arginine Methyltransferase 1, also known as PRMT4), a member of the protein arginine methyltransferase family. The dual acylation on a histone and a SWI/SNF component implies a collaboration of chromatin modifications, performed by distinct methyltransferase and acetyltransferase enzymes, for the optimal recruitment of BRD4. A microRNA, miR-766-5p, which targets both acetyltransferase KAT3A (CBP) and BRD4 leads to reduced H3K27Ac accumulation and interferes with the transcription function of super-enhancers [23]. Moreover, BRD4 interacts with specific transcription factors, including p53, c-Jun, MYC/MAX and TWIST, which may confer specificity of action in certain enhancers, but not in others [24-26]. Interactions of BRD4 with transcription factors occur both in an acetylation dependent manner through the bromodomains of BRD4 or in an acetylation independent manner. BRD4 attracts the Mediator complex and the basic transcription machinery for transcription initiation of super-enhancer target genes (Figure 1A). Moreover, acetylation by p300 and CBP plays a role in pause release of RNA polymerase II and of the general transcription machinery in super-enhancer dependent transcription initiation sites [27].
Figure 1.
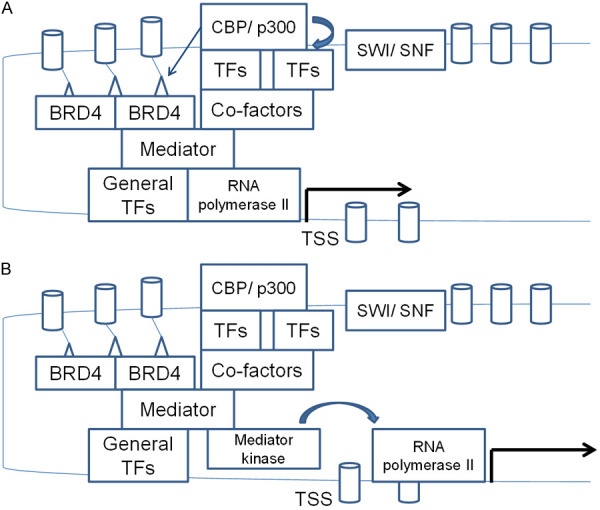
Schematic representation of super-enhancer structure and key components. A. Trithorax component methyltransferases methylate hIstone 3 at lysine 4 (H3K4). The SWI/SNF component of Trithorax displaces or removes histones using energy from ATP hydrolysis, and sequence specific transcription factors bind their cognate sequences. KAT3 acetyltransferases acetylate histone 3 at lysine 27, creating the signal for BRD4 binding. BRD4 contributes in bringing the super-enhancer bound transcription factors in contact with promoter bound Mediator complex and the general transcription factors that recruit RNA polymerase II to initiate DNA transcription from transcription start sites (TSS). B. The Mediator kinase module displaces the rest of Mediator complex from their interaction with RNA polymerase II and allows transcription to proceed to the elongation phase, after an initial pause is released. Both sequence specific transcription factors and Mediator kinases are under the control of up-stream signal transduction pathways. Small triangles represent H3K27 acetylation and cylinders represent the histone octamer.
Another salient epigenetic feature in super-enhancer regulated genes is the acquisition of broad H3K4me3 domains that span the areas up-stream and down-stream of transcription start sites [28]. Broad H3K4me3 domains associated with super-enhancers are aberrantly created at pathologically targeted genes after super-enhancer translocation. This was observed in the classic translocation t(11q13;14q32) that juxtaposes the super-enhancer from the immunoglobulin heavy chain locus at chromosome 14 with the gene CCND1, encoding for cyclin D1, at chromosome 11, and is frequent in mantle cell B cell lymphomas. Cells with this translocation exhibit a broad H3K4me3 domain at CCND1 gene locus, associated with increased expression of cyclin D1 [29]. This suggests that broad H3K4me3 domains are epigenetic features created by the super-enhancer associated modifiers and follow the super-enhancer sequences in their new aberrant location.
The Mediator complex consists of 26 proteins with an aggregate molecular mass of 1.4 MDa and is part of the pre-initiation transcription complex for RNA polymerase II mediated transcription [30]. The Mediator interacts with RNA polymerase II for transcription initiation. A separate module of the Mediator complex called Mediator kinase module consists of Cyclin dependent kinase 8 (CDK8) or CDK19, cyclin C and Mediator units 12 and 13 (MED12 and MED13) or the alternative units MED12L and MED13L, and associates with the rest of Mediator in a RNA polymerase II excluding way. As the association of the Mediator kinase module with the rest of the Mediator complex prevents the association of RNA polymerase II with the complex, the module has been proposed to constitute a negative regulator of polymerase II mediated transcription [31]. However, the kinase module may be required for super-enhancer dependent gene transcription at least in some occasions, a requirement that relates to a positive influence of the module on elongation (Figure 1B) [32]. In this phase of transcription, the kinase module co-operates with the elongation factor PTEFb, a complex of CDK9 kinase and cyclin T [33]. It is also suggested that the complex that includes the kinase module regulates RNA polymerase II dependent gene elongation only in some occasions, while the Mediator unit MED26 associates with the Mediator complex instead of the kinase module for elongation of RNA transcripts in promoters of other genes [33]. CDK8 was shown to participate in the rapid elongation of p53 target genes [34]. MED26, on the other hand, seems to have a key role in the elongation phase of genes transcribed at a steady rate [32]. MED12 and MED13/MED13L play a significant role in super-enhancer mediated transcription of oncogene MYC in colorectal cancer cells, where depletion of the three proteins down-regulated MYC expression, while CDK8 and CDK19 depletion had a lesser effect [35]. In these cells, MED12 and MED13/MED13L directly associate with β-catenin, which is involved in attracting MED12 to the MYC promoter, as witnessed by decreased MED12 binding when β-catenin is depleted.
The two alternative catalytic proteins of the Mediator kinase module, CDK8 and CDK19, are functionally redundant for gene expression [36]. However, double knock out of both kinases results in proliferation reduction and promotion of differentiation and of mucin production in intestinal organoids. Exposure to an inhibitor of the two kinases, SNX-631 recapitulates the knock out phenotype in the same organoid system and in mice in vivo [36]. The effect of CDK8 depletion appears to be content dependent, as witnessed in a conditional knock out Apc (Min) mouse model [37]. In this model, which shows high β-catenin activity, knock out of cdk8 increased tumor growth rate. Intestinal cells with cdk8 deletion displayed loss of the repressive trimethylation at H3K27 and up-regulation of respective genes regulated by this epigenetic modification [37].
SWI/SNF is a multiprotein complex which consists of several sub-units, such as one of the two ATPase sub-units SMARCA2 (SWI/SNF-related, Matrix-associated, Actin-dependent Regulator of Chromatin, sub-family A, member 4, also called BRM - Brahma) or SMARCA4 (also called BRG1 - Brahma related gene 1), SMARCB1 (also known as INI1 - Integrase Interactor 1), SMARCC1 (BAF155 - BRG1/BRM Associated Factor 155) and SMARCC2 (BAF170), that are common for SWI/SNF complexes in all cells and other sub-units that are more cell type specific [38]. SWI/SNF use energy from ATP hydrolysis to remove nucleosomes from chromatin in areas of promoters and enhancers that are associated with H3K27Ac and are devoid of the suppressive methylation at the same lysine, performed by the polycomb repressive complex 2 (PRC2) [39]. Besides this role of nucleosome displacement to facilitate transcription, SWI/SNF complex was shown to directly associate with the Mediator kinase module in intestinal epithelial cells [40]. SWI/SNF sub-units ARID1A, SMARCC2 and PBRM1 are phosphorylated by Mediator kinases, a phosphorylation that regulates SWI/SNF chromatin binding and activity. In intestinal epithelia, loss of CDK8 dependent SWI/SNF phosphorylation results in decrease of cells with the secretory phenotype, through interference with the expression of lineage specification factor ATOH1 [40]. Therefore, SWI/SNF is an integral regulator of super-enhancer function that is required for lineage specification gene expression in the bowel.
Besides transcription, super-enhancer related proteins, such as BRD4 and the SWI/SNF complex proteins are implicated in DNA repair of double strand breaks [41]. Using the BLISS (Breaks Labeling In Situ and Sequencing) method, which can detect genome-wide double strand breaks at a single nucleotide resolution, it was found that super-enhancer sites are enriched in double strand DNA breaks related to the increased transcriptional activity in these sites [42]. As expected from the association of transcriptional activity with double strand breaks production, the map of naturally occurring double breaks across the genome (constituting the “breakome”) is cell type specific [42]. The homologous recombination protein RAD51 is required for repair of these breaks and for continuing transcription from super-enhancers, while RAD51 depletion impairs super-enhancer function [42]. BRD4 is recruited in histone acetylated sites in the vicinity of DNA double breaks and interacts with scaffold factor 53BP1 (p53 Binding Protein 1), which then orchestrates the assembly of the non-homologous end joining machinery for DNA double break repair [43].
Overall, super-enhancers provide the epigenetic framework for the robust expression of genes whose protein or RNA products need to be present for the establishment and maintenance of different cell type identities with diverse cell functions. In this manner, super-enhancers perform a critical function for the creation and maintenance of specialized cells and tissues in multi-cellular organisms. Super-enhancer function is regulated by up-steam signals as well as the 3-dimensional chromatin positioning and the chromatin organization in topologically associated domains (TADs). TADs boundaries are demarcated by insulator protein CTCF and cohesins and are stronger in TADs that contain super-enhancers [44,45].
Super-enhancer hijacking in cancer
In cancer pathogenesis, the importance of super-enhancers is often related to aberrant positioning of pre-existing super-enhancers that were present in non-transformed cells due to translocations [46]. These result in over-production and activation of oncogenes which are not normally targets of the aberrantly positioned super-enhancer. Frequently, the repositioned super-enhancers and target genes that are regulated by them are particularly sensitive to up-stream activated oncogenic pathways [47]. An example in colorectal cancer is oncogene MYC which is a target of the WNT pathway [48]. In addition, existing super-enhancers may become regulators of genes through copy number alterations (amplifications or deletions) that bring remote loci on the same chromosome in physical and functional proximity. For example, a long deletion produced a super-enhancer hijacking event in a colon cancer patient resulting in over-production of TOP2B [49]. After the patient’s genomic lesion was introduced in HCT116 colorectal cancer cell line, the three-dimensional genomic re-arrangement of the patient was reproduced and TOP2B was up-regulated. New super-enhancers can be created in cancer cells by amplifications and juxtaposition in the loci of existing enhancers, enhancing their transcriptional potency [50]. Loss of regulation by normal super-enhancers through similar mechanisms may result in down-regulation of tumor suppressors or identity determining genes, leading to dedifferentiation of transformed cells.
Besides the role of super-enhancers in increasing the transcription of target genes, an additional mechanism of super-enhancer contribution to increased production of proteins from target genes is by increasing target mRNA availability for translation in the cytoplasm [51]. The mechanism involves a physical association of super-enhancer sequences to nuclear pores, which allow newly produced mRNAs from the target gene to be directly transported to the cytoplasm. Given that mRNA decay in the cytoplasm is lower than in the nucleus, direct transport of the nascent mRNA prevents its destruction and results in further increase of protein production from the super-enhancer target gene [52]. An additional role of nuclear pores in super-enhancer function involves the creation of local environments with liquid-liquid phase separation properties [53]. Phase separation promotes the functional interaction of enhancers with promoters and the transcription machinery. Phase separation is favored by intrinsically disordered FG domains with high phenylalanine and glycine content, which are present in several nucleoporins, components of the nuclear pore multiprotein complex [53]. Relevant to colorectal cancer, a super-enhancer upstream of the MYC gene was shown to associate with proteins of the nuclear pore and direct MYC mRNA trafficking in a manner that is dependent on WNT/β-catenin signaling [54]. Inhibition of BRD4 using a novel aminocyclopropenone compound led to down-regulation of nucleoporin NUP210 and interfered with the phase separation micro-environment associated with BRD4 in HCT116 colorectal cancer cells [55]. The drug also showed growth inhibitory effects in this cell line.
Alteration of TADs boundaries is another way of super-enhancer hijacking in cancer without the need for altering the super-enhancer binding sequence or length. TADs boundaries which are created by binding of transcription factor CCTF and cohesins may be lost or, conversely, newly created by point mutations or insertions/deletions altering the binding sequences of TADs boundaries proteins. Altered TADs may deregulate cancer associated genes, by bringing them under the aberrant control of new super-enhancers [44]. Despite resulting in robust modification of expression of cancer associated genes, these types of alterations are difficult to identify even by comprehensive sequencing methods, such as whole exome sequencing, as they occur in non-coding regions of the genome.
Specific types of cancers show distinct super-enhancer aberrations which depend on the transcription factors that are critical for the identity and function of the respective tissues of origin. For example, in prostate cancer, AR is a principal transcription factor and a target of therapeutic interventions. AR regulates the transcriptome of normal prostate tissues and prostate cancer through super-enhancer sequences [56]. Treatment of prostate cancer cells with the anti-androgen darolutamide impaired the transcription output from AR-dependent enhancers and super-enhancers. In colorectal cancer, a new super-enhancer regulating gene POU5F1B, a homologue of ESCs transcription factor OCT4 (also known as POU5F1) is created recurrently and leads to increased expression of POU5F1B, compared with normal colon epithelium [57]. Inhibiting the newly created super-enhancer function through exposure to the BRD4 inhibitor JQ1 reduced the aberrant expression of POU5F1B [57].
Super-enhancer alterations may also contribute to colorectal cancer cases with the MMR phenotype [58]. Loss of a binding sequence for the CCTF factor in a super-enhancer up-stream of the MLH1 gene resulted in impaired expression of MLH1. The MMR phenotype resulting from MLH1 or other MMR gene loss may perturb super-enhancers function across the genome by recurrent mutations in binding sequences [59]. Therefore, colorectal cancers with MMR deficiency may have aberrations in their epigenome added to the widespread alterations created by the MMR defects in coding sequences.
Beyond copy number alterations and structural alterations such as fusions in the super-enhancer sequences, some components of the functional machinery of super-enhancers are frequently altered through point mutations or copy number alterations in their own gene sequences, which provide another mechanism for super-enhancer function deregulation in cancer. For example, component proteins of the SWI/SNF multiprotein complex are variably mutated in a significant minority (up to 25%) of diverse cancer types [60]. The core sub-unit SMARCB1 is mutated in almost all pediatric rhabdoid tumors, aggressive tumors of infancy and early childhood [61]. The ATPase sub-unit SMARCA4 (BRG1) is mutated in the great majority of a rare type of ovarian cancer, small cell carcinoma of the ovary, hypercalcemic type, occurring mostly in pre-menopausal women [62]. In colorectal cancer, mutations of sub-units ARID1A, ARID1B and ARID2 occur in 10.9%, 6.7% and 6.9% of cases, respectively, in the Cancer Genome Atlas (TCGA) cohort [63]. Importantly for colorectal cancer, loss of SMARCB1 results in increased activity of β-catenin/TCF4 pathway [64]. In contrast, SMARCA4 loss decreases WNT signaling through down-regulation of Frizzled receptors, at least in the context of embryonic vascular cells [65]. Mediator kinase module protein MED12 frequently possesses recurrent mutations at exon 2 that are present in uterine fibroids but also in chronic leukemia and colorectal cancer [66]. Mutated MED12 still retains the ability to bind, in conjunction with MED13, to the cyclin C/CDK8 or CDK19 dimer, but is defective in activating the kinase function [66]. Mutations of MED12 and MED13 or the alternative kinase module proteins MED12L and MED13L occur in 6.2% of cases, 6.6% of cases, 4.9% of cases and 3.7% of cases, respectively, in the colorectal cancer cohort of TCGA (Figure 2) [63]. The genes for CDK8, CDK19 and cyclin C are mutated in 2.1%, 1.5% and 0.9% of cases, respectively, in the same cohort. In addition CDK8 locus at chromosome 13q12 is amplified in 4.9% of colorectal cancers. Similar prevalence of alterations is observed in another genomic series of colon cancers reported by the Sidra-LUMC AC-ICAM international collaboration (Figure 2) [67]. Overall, non-overlapping alterations in one of the Mediator kinase module constituent proteins are observed in 20% and 21% of cases in the two series, respectively [63,67].
Figure 2.
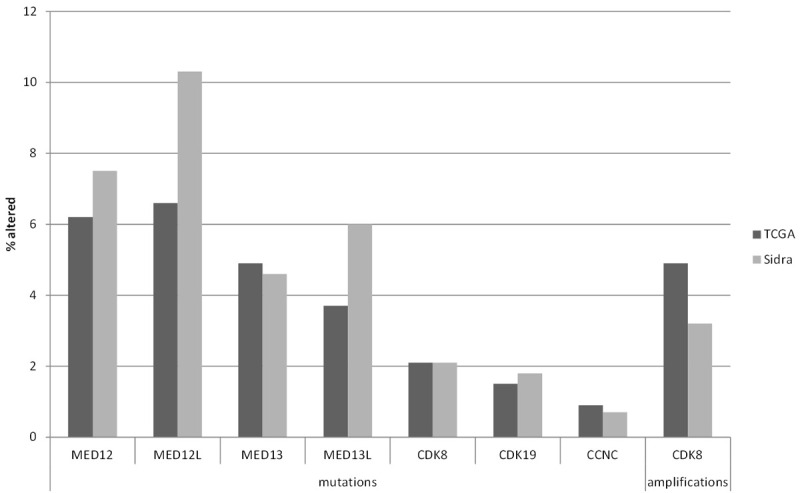
Prevalence of mutations in genes of the Mediator kinase module and amplifications of CDK8 in two colon cancer series, The Cancer Genome Atlas (TCGA) colorectal cancer cohort (black bars) and the Sidra-LUMC AC-ICAM colon cancer cohort (Sidra, grey bars).
Epigenetic deregulation of super-enhancer regulators is also another mechanism that may result in altered expression of these regulators with repercussions for super-enhancer target genes expression. Methylation of CpG islands (CpG Island Methylation Phenotype, CIMP) is observed in a sub-set of colorectal cancers and may affect promoters of super-enhancer component proteins, suppressing their expression. In one study, BRD4 promoter hyper-methylation was present in seven of nine colorectal cancer cell lines and also in colon cancer tumors from patients, compared with adjacent healthy colonic epithelium [68]. Hyper-methylation was associated with the activity of DNA methyltransferases DNMT1 and DNMT3B, while knock out of the two enzymes in HCT116 cells led to BRD4 up-regulation.
Targeting super-enhancers as a colorectal cancer therapy
Super-enhancers act as powerful transcription regulators with critical roles in cell physiology, and are associated with robust target gene deregulation in cancer. Consequently, their targeting has raised interest as a valid cancer therapeutic manipulation [69]. Inhibitors of components of the super-enhancer machinery, such as BET (Bromodomain and Extra-Terminal domain) inhibitors, targeting bromodomain containing proteins, and CDK8/CDK19 inhibitors, or of the signals that are required for their recruitment, such as histone acetyltransferases inhibitors, have been discovered and some are in various phases of clinical development. In colorectal cancer, the MYC gene coding sequence is not commonly altered, but the gene expression is frequently up-regulated under the influence of the oncogenic super-enhancer and the increased WNT/β-catenin pathway activity, in a sub-set of colorectal cases [70]. In the following paragraphs, potential therapeutic interventions targeting super-enhancer structure and function that are in various phases of development and could become clinically relevant in colorectal cancer are discussed.
Target 1: BRD4
The critical role of BRD4 bromodomain containing protein for super-enhancer dependent transcription is suggested by the observation that BET inhibitors inhibit transcription of WNT and super-enhancer regulated MYC oncogene but not other targets of the WNT/β-catenin pathway in HCT116 colorectal cancer cells [71]. Other target genes of BET inhibition included apoptosis regulators XIAP, and FLIP in lung cancer cells [72] and DNA repair proteins BRCA1 and RAD51 in triple negative breast cancer cells [73]. Drugs that bind bromodomains and disrupt the ability of BRD4 to bind acetylated histone tails were sought as a means to interfere with BRD4 function and the transcription of target proteins [74]. The first of such drugs discovered was JQ1, a thieno-triazolo-1,4-diazepine, and I-BET, a triazoyl ring containing benzodiazepine, which bind bromodomains in nanomolar concentrations [75,76]. JQ1 was shown to inhibit preferentially genes regulated by super-enhancers, such as MYC and the immunoglobulin heavy chain IGH, compared to genes regulated by typical enhancers, such as SMARCA4, in multiple myeloma cells [77].
A phase I trial of the oral benzodiazepine BET inhibitor molibresib (GSK525762 or I-BET762) which included patients with Nuclear protein in testis (NUT) carcinomas and other solid tumors established a dose of 75 mg daily as the recommended phase II dose [78]. NUT carcinomas are aggressive cancers that are molecularly defined by fusions of NUTM1 gene at chromosome 15q14 with BRD4 or BRD3 or less commonly other genes [79]. They attracted interest in BET inhibitor trials because of the involvement of BRD homologous genes in the pathogenic rearrangements of these carcinomas which result in deregulation of target genes. In the phase I trial of molibresib, 4 of 14 patients (28.6%) with NUT carcinomas showed a partial response [78]. The trial included 15 evaluable colorectal cancer patients and showed stable disease in 5 patients, but no objective responses were observed. A phase II trial of molibresib with 102 patients with various cancers (12 with NUT carcinomas) included no colorectal cancer patients [80]. Median PFS was 4.7 months and median OS was 6.5 months. Partial responses were observed in 2 patients (one patient with NUT carcinoma and one patient with castration resistant prostate cancer).
Another oral BET inhibitor, birabresib (MK-8628) was investigated in a phase I study that included NUT carcinoma, lung cancer and castration resistant prostate cancer patients [81]. The drug was well tolerated in the recommended dose of 80 mg daily and 3 of 10 patients with NUT carcinomas showed a partial response. A third oral BET inhibitor, trotabresib (CC-90010) is also in clinical development, with the results of an expansion phase Ib study showing stable disease in 63.4% of a cohort of various solid tumors [82]. In 31.7% of these patients stability of the disease lasted for more than 4 months. The oral reversible bivalent BET inhibitor AZD5153 has also completed a phase I trial in relapsed or refractory solid tumors (one participating patient had Non-Hodgkin lymphoma) [83]. The trial also included a cohort of ovarian and pancreatic cancer patients treated with the combination of AZD5153 with the PARP (Poly-ADP Ribose Polymerase) inhibitor olaparib [83]. The recommended phase II dose as monotherapy was 30 mg daily or 15 mg twice daily continuously and the combination was feasible with a reduced AZD5153 dose at 10 mg daily in an intermittent schedule. The monotherapy cohort included 33 evaluable for response patients, among whom there were 4 colorectal cancer patients. Two thirds of patients had received 3 or more lines of previous therapies. No objective responses were observed in the 33 patients, while stability for at least 6 weeks was observed in 48.5% of the patients [83]. In the combination cohort, one patient (6.7%) had a partial response.
Proteolysis targeting chimeras (PROTACs) are constructs that associate a ligand moiety binding a target protein with another ligand for a ubiquitin ligase, an enzyme that catalyzes protein ubiquitination [84]. PROTACs bring in physical proximity the target protein with the ubiquitination machinery and promote ubiquitin attachment to the target which serves as a signal for degradation in the proteasome [85]. A PROTAC targeting BRD4, called A1874, has been developed and tested in primary colorectal cancer cells and the colorectal cancer cell line HCT116 [86,87]. This PROTAC construct consists of a binding moiety for E3 ligase MDM2, idasanutlin, combined through a polyethylene glycol ligand with JQ1 as a BRD4 binder [86]. A1874 showed efficacy in reducing cell proliferation and inducing apoptosis in colorectal cancer cells [87]. Degradation of BRD4 was confirmed, as was the down-regulation of target genes MYC, BCL2 and CCND1. The PROTAC was more effective than BRD4 inhibitors JQ1, CPI203, and I-BET151. Interestingly, activity of A1874 was also observed in colorectal cancer cells with BRD4 knocked-out, suggesting additional mechanisms of cytotoxicity not dependent on BRD4 degradation. Indeed, A1874 showed a stabilizing effect on tumor suppressor p53, which is a target of MDM2 [86]. As a result, expression of cell cycle inhibitor p21, a p53 target gene, was increased after A1874 exposure. Colorectal cancer cell line HCT116 possesses wild type p53, which may increase its sensitivity to A1874, compared with cell line HT29, which possesses an inactivating mutation of p53, or HCT116 cell sub-lines with double p53 knockout [86]. In contrast, another BRD4 targeting PROTAC, A743, employing a different E3 ligase, VHL, was more effective against cells with mutant or deleted p53. No clinical trials with A1874 are currently enlisted in the ClinicalTrials.gov database.
The experience with small molecule BRD4 inhibitors, which suggest low efficacy as monotherapy, as well as with PROTACs, which suggest additional mechanisms of action, besides BRD4 down-regulation, as important contributors of efficacy, pinpoint to combination of BRD4 inhibitors or BRD4 down-regulators with other therapeutic agents as a putative way to improve therapeutic efficacy and to boost the development of useful clinical regimens in colorectal cancer and other cancers. In this respect, it was determined that BRD4 inhibition induces mismatch repair defects and creates therapeutic vulnerability to immune checkpoint inhibitors in MMR proficient cells [88]. BRD4 inhibitors down-regulated MMR-related proteins MSH2, MSH6, MLH1 and PMS2 in colorectal, ovarian, cervical and lung cancer cell lines. BRD4 Inhibitor, JQ1 down-regulated MMR-related proteins in human xenografts, in mice, in vivo [88]. Interestingly, even in cells that had acquired resistance to the growth inhibitory effects of BRD4 inhibitors after prolonged exposure, MMR protein levels remained down-regulated. Cells and tumors with acquired resistance to BRD4 inhibitor AZD5153 displayed sensitivity to an anti-PD-L1 checkpoint inhibitor, as expected in cells with persisting MMR defects, compared to parental cells. Sensitivity was not significantly augmented by continuous exposure to the BRD4 inhibitor [88].
Another pre-clinical study suggested synergy of the combination of BET inhibitors with PARP in homologous recombination proficient cancers [89]. BET inhibitors JQ1, molibresib and OTX015 suppressed the expression of homologous recombination proteins BRCA1 and RAD51 at the transcriptional level and sensitized cells to olaparib. BRCA1 and RAD51 were also down-regulated in BRCA1 and BRCA2 wild-type triple negative breast cancer cells exposed to JQ1 and another BET inhibitor, GSK525762A [73]. This resulted in a homologous recombination defect and sensitization to cisplatin and olaparib. Another study also showed that BRD4 inhibition using various BET inhibitors induced homologous recombination deficiency, in this case through down-regulation of protein CtIP, which co-operates in creation of single strands through end resection for use as DNA templates during the repair process [90]. The produced homologous recombination deficiency sensitizes cells to PARP inhibitors. As mentioned above, a phase I trial with the combination of BET inhibitor AZD5153 and olaparib in pancreatic and ovarian cancer patients showed only a single BRCA proficient pancreatic cancer patient with a partial response (6.7% response rate) lasting for 4 months [83]. Therefore, a better patient selection will be required for translation of the pre-clinical data to successful clinical drug combinations.
Target 2: CDK8 and CDK19
In view of the regulation of the WNT pathway by CDK8 and paralogue kinase CDK19, inhibition of the two kinases with small molecule inhibitors has attracted interest in preclinical models of colorectal cancer [91]. CDK8/CDK19 inhibitors based on diverse chemical scaffolds have been discovered [92]. Inhibition of mediator associated kinases may have pleotropic effects in cancer cells that are tumor promoting or suppressing and are context dependent [93]. Therefore, therapeutic deployment of these inhibitors would also be expected to be successful only in selected cancers where the effects of the target kinases are pro-tumorigenic, but not in other cases where the kinases play a tumor suppressing role. The challenge in development will be to determine the sub-sets of cancers, if any, that are vulnerable to such inhibition. The small molecule selective CDK8/CDK19 inhibitor CCT251545 is a pyridine derivative discovered through a high throughput cell based assay and optimization [94]. CCT251545 alters expression of WNT target genes as well as STAT1 target genes, consistent with its ability to engage and inhibit the target kinases in colorectal cancer cell lines in vitro [95]. Phosphorylation of STAT1 at position S727 was a marker of CDK8 kinase activity in vitro and in vivo in a breast cancer allograft model in mice. Moreover, CCT251545 showed in vivo activity in WNT dependent tumors in the same model [95].
In a study inquiring into biomarkers of efficacy of ATR inhibitors, CDK8 and its partner cyclin C were identified as a requirement for efficacy in cell lines with or without mutations in the related kinase ATM [96]. Loss of CDK8 or Cyclin C suppressed replication stress and produced resistance to ATR inhibitor ceralasertib. This study suggests that pharmacologic inhibition of CDK8 could similarly act antagonistically to ATR inhibition and would not be a good candidate for combination therapy.
Another small molecule CDK8/CDK19 inhibitor, RVU120/SEL120 possesses the ability to down-regulate lineage commitment genes such as KLF1, FLI1, GATA1 and GATA2 [97]. SEL120 has entered phase I/II dose escalation and expansion clinical trials in hematologic malignancies and in solid tumors (NCT05052255). Results are currently not available in the peer reviewed literature. In anticipation of these results, it would be interesting to determine if these inhibitors would prove well tolerated and if any signals of benefit in specific types of cancers or in cancers with specific molecular defects will emerge.
Target 3: CDK9
CDK9 is a serine/threonine kinase, which together with its partner cyclin T constitute the positive transcription elongation factor b (P-TEFb), with a key role in transcription elongation through the release of paused RNA polymerase II [98]. Similar to the Mediator kinases, CDK9 is involved in the regulation of transcription of genes irrespective of the presence of super-enhancers in their regulatory sequences. However, genes with robust transcription activity in given cell types, such as those regulated by super-enhancers are more dependent on the RNA polymerase II transcriptional activity and would be more prone to inhibition of the different phases of this activity [99]. Indeed, the prototypic super-enhancer regulated MYC oncogene is dependent on CDK9 for its transcription [99]. In addition, in cancers with increased MYC activity, such as sub-sets of colorectal cancer, MYC interacts directly with CDK9 in the regulation of target genes [100]. Therefore inhibition of CDK9 provides a rational option for therapeutic targeting of super-enhancer mediated transcription. Concerns regarding adverse effects of CDK9 inhibitors due to the important general physiologic role of the kinase in RNA polymerase II dependent transcription have been partially addressed with the advent of more specific inhibitors that decrease the likelihood of off-target adverse effects, while management of on-target adverse effects are addressed in on-going early phase clinical trials through optimization of doses and schedules [98].
Several CDK9 inhibitors are in clinical development. One inhibitor, the small molecule AZD4573 exhibited selectivity against CDK9 compared to other CDKs and depleted the apoptosis inhibitor MCL1, an anti-apoptotic member of the BCL2 family, inducing cell apoptosis in cell lines in vitro and mouse xenografts in vivo [101]. Hematologic cell lines and patient derived xenografts were more sensitive to the drug than solid tumor cell lines [101]. Cell lines and xenografts that were resistant to AZD4573 were sensitized by the addition of the BH3 mimetic venetoclax, suggesting that inhibition of both MCL1 and BCL2 was required for induction of apoptosis in these resistant cases [101,102]. AZD4573 is currently in phase 2 trials in hematologic malignancies, either as monotherapy or in combination with the Bruton’s tyrosine kinase inhibitor, acalabrutinib [103]. Initial results of the AZD4573/acalabrutinib combination confirmed the feasibility of the regimen and showed an overall response rate of 50% in patients with heavily pretreated relapsed or refractory diffuse large cell lymphoma.
In contrast to AZD4573 which is administered intravenously, another CDK9 small molecule inhibitor, KB-0742 is orally bio-available and has been studied in MYC dependent cancers [104]. In a first in human phase 1 trial that included colon cancer, other solid tumor and lymphoma patients, KB-0742 proved to be well tolerated at a dose of 60 mg daily, which was selected for further study [105]. At this dose two colorectal cancer patients with MYC over-expressing cancers had stable disease and two myxoid liposarcoma patients with transcription factor fusions exhibited partial responses. Overall these early clinical results with selective CDK9 inhibitors are encouraging. Combination therapies in selected patient populations such as those with MYC or MCL1 dependent cancers are the lead candidates for more advanced phase testing.
Target 4: KAT3 inhibitors
The signal for BRD4 recruitment in super-enhancer sequences is provided by the H3K27Ac histone modification which is exceptionally high in these sites. As a result, pharmacologic reversal of these acetylations through histone acetyltransferase inhibitors would be expected to impair the ability of BRD4 to be recruited at super-enhancer sites for triggering the down-stream interactions and transcription initiation and elongation. Inhibitors of p300/CBP acetyltransferases, that perform the H3K27 acetylation, have been discovered and have been confirmed to reduce histone 3 and histone 4 acetylation in leukemia and prostate cancer cells, inducing cell cycle arrest [106,107]. The oxazolidinedione compound A-485 was identified as an acetyl-CoA competitive inhibitor of both KAT3 family acetyltransferases [107]. A-485 is selective for KAT3 acetyltransferases, with negligible activity for other acetyltransferases. The drug inhibited the transcription activity of lineage determining transcription factor MITF (Microphthalmia - associated Transcription Factor) in melanoma cells [108]. This inhibition was associated with decreased histone 3 acetylation but not with displacement of the acetyltransferases from MITF promoters. Acetyltransferase inhibition was mimicked by silencing of EP300, the gene encoding for p300 or silencing of MITF gene. Inhibition of histone acetylation by A-485 was associated with melanoma cell senescence but not with induction of apoptosis [108]. In an attempt to improve the efficacy of A-485 and to induce apoptotic death, the combination of A-485 with the apoptosis inducer TRAIL was evaluated in another preclinical study in lung cancer cells [109]. The combination was found to be more effective than TRAIL alone in apoptosis induction of EGFR inhibitor sensitive and resistant lung cancer cell lines in vitro. Currently, no clinical trials of A-485 or any other KAT3 family acetyltransferase inhibitor have been reported in the peer reviewed literature. It remains to be determined whether the higher order of magnitude of H3K27Ac in super-enhancers will be sufficient to confer selectivity of KAT3 inhibitors for super-enhancer dependent genes, sparing genes not dependent on super-enhancers.
Target 5: Nucleoporins
The arising role of nuclear pores in the facilitation of transcription from super-enhancers, as well as in the prompt export of produced mRNAs of target genes suggests that nuclear pores may be a therapeutic target for interfering with super-enhancer function, although conceivably targeting nuclear pores may have broader implications in cellular functions [53]. An indirect targeting of nuclear pore proteins through decrease of their production by BET inhibitors has also been proposed [55]. An aminocyclopropenone compound inhibiting BRD4 was shown to reduce transcription of the gene NUP210 encoding for a nucleoporin and reducing growth of colorectal cancer cell line HCT116. As expected, the compound also attenuated the expression of BRD4 target MYC. However, knockdown of NUP210 produced similar growth reduction in HCT116 cells with the drug, suggesting that the growth effect was at least in part due to the nucleoporin reduction [55].
Reduced production of nuclear pore components could have global effects on the stoichiometry of the nuclear pore complex and the physiologic function of the multi-protein structure. This could result in triggering of autophagy [110]. Autophagy has a dual effect in cancer and can be both deleterious and protective for cancer cells [111]. Therefore, potential targeting of nuclear pore function in a generic manner may have unanticipated effects. In addition, given the key role of nuclear pores in cell physiology, its dysfunction may not be well tolerated by normal cells, raising concerns regarding the tolerability of compounds that target the nuclear pore. These considerations together with the fact that currently there are no clinical grade compounds targeting nuclear pore components suggest that the nuclear pore may not be the optimal target for super-enhancer inhibition. However, further research may uncover aspects of the nuclear pore complex and its interactions with the super-enhancer machinery that are amenable to specific functional disruption of super-enhancers as machineries for oncogene production.
Target 6: Synthetic lethality
Synthetic lethality refers to the phenomenon where two different molecular lesions are well-tolerated in a cell environment individually, but lead to cell death when they co-exist [112]. The concept was brought in the forefront of cancer therapeutics with the successful treatment of BRCA1 and BRCA2 mutant cancers with PARP inhibitors, which takes advantage of the sensitivity of cells with homologous recombination defects to inhibition of other DNA repair pathways by PARP inhibitors. Related to super-enhancer components, as mentioned previously, BET proteins activity impairment sensitizes colorectal cancer cells to PARP inhibitors [89]. Conversely, BRCA1 deficiency in breast cancer cells sensitized them to BET inhibitors through down-regulation of MYC and alleviating suppression of target gene TXNIP, encoding for a theoredoxin regulator [113]. Moreover, BET inhibitor JQ1 sensitized colorectal cancer cells to topoisomerase I inhibitor camptothecin by preventing recruitment of 53BP1 and MRE11 necessary for the repair of double strand breaks induced by camptothecin [114].
Among components contributing to super-enhancers function, the multi-protein SWI/SNF complex is most frequently mutated in up to a fourth of all cancers. SWI/SNF mutant cancers have been proposed to be sensitive to EZH2 inhibition, based on the fundamentally opposing functions of the PRC2 complex, in which EZH2 belongs, and the trithorax complex, the broader complex encompassing SWI/SNF, in suppressing and promoting transcription, respectively [115,116]. In a phase 1 trial, three of 10 patients with SWI/SNF component SMARCB1 negative malignant rhabdoid tumors and malignant epithelioid sarcomas had a partial response to the EZH2 inhibitor tazemetostat [117]. Two other patients had stable disease and received the drug for over a year. The trial included also two patients with SMARCA4 negative malignant rhabdoid tumors of the ovary (clear cell carcinomas of the ovary of the hypercalcemic type) and a patient with SMARCA4 negative thoracic sarcoma. One of the three patients had a partial response and another patient had stable disease, remaining on treatment for over six months. No objective responses were observed in 30 patients with other solid tumors without SMARCB1 or SMARCA4 negativity and only one patient had a clinical benefit with stable disease lasting for 11 months [117]. In colorectal cancer, three other SWI/SNF components, ARID1A, ARID1B and ARID2 are most frequently mutated than SMARCA4 and SMARCB1. ARID1A mutations are frequently observed in clear cell carcinomas of the ovary and ovarian cancer cells with such mutations were sensitive to EZH2 inhibitor GSK126 [118]. The synthetic lethality was traced in this study to the de-repression of PIK3IP1, a negative regulator of the PI3K/AKT pathway by GSK126 treatment. In contrast, other studies have suggested that ARID1A mutant cell lines do not share the sensitivity of SMARCB1 or SMARCA4 negative cells to tazemetostat [119]. Moreover, in urothelial cancer, ARID1A truncating mutations, which are also the most frequent SWI/SNF sub-unit mutations in this disease, did not confer EZH2 inhibitor sensitivity, implying that the EZH2/SWI/SNF synthetic lethality phenomenon is sub-unit specific and possibly cell context dependent [120].
Deficiency of the mismatch repair protein MSH2 and the related MSH6 protein was associated with super-enhancer dysfunction in gastric cancer cells, independently of the role of the two proteins in mismatch repair [121]. Normal MSH2 in co-operation with MSH6 interacts with SWI/SNF protein SMARCA4 and loss of this interaction due to knockdown of MSH2 results in decreased acetylation of super-enhancer sites with a particular enrichment in super-enhancers of cell adhesion associated genes, such as CLDN4, encoding for tight junction protein Claudin 4, ITGB1, encoding for integrin sub-unit beta 1, and CTNNB1, encoding for β-catenin. Loss of MSH2 sensitized cells to BET inhibitor JQ1. The sensitization was traced to the addiction of cells with MSH2 loss to bromodomain protein BAZ1B, whose inhibition results in arrest of proliferation [121]. Mismatch repair deficiency and microsatellite instability (MSI) may also play a role in the synergism with BET inhibitors as the widespread mutational lesions observed in MSI cells could affect super-enhancer sequences and function. Conceivably, MSI cells that have acquired new super-enhancers and become dependent on them may be sensitive to super-enhancer inhibition.
SMAD4 mutations are frequent in colorectal cancers, occurring in about 10% of cases [63,122]. SMAD4 is a co-factor of the TGFβ cascade and mutations lead to deregulation of the pathway, which has pro-metastatic effects [123]. SMAD4 knockout sensitizes colorectal cancer cells in vitro and xenografts in vivo to BET inhibitors [124]. The specificity for cells without SMAD4 relates to the effect of BET inhibitors in curtailing the function of oncogene MYC which is a target of transcriptional inhibition by SMAD4 and therefore is over-expressed in these cells. Whether BET inhibitors could be effective in colorectal cancer patients with SMAD4 mutations is an interesting hypothesis that may deserve testing.
In conclusion, super-enhancer targeting in colorectal cancer is of clinical interest. The most advanced category of drugs, already in clinical development, are those targeting BET domain proteins. Early clinical results are encouraging so far, but pinpoint also to a need for use of molecular biomarkers to determine populations with the highest probability of deriving clinical benefit. Moreover, it is expected that combination therapies targeting super-enhancers and other vulnerabilities, such as signaling pathways contributing to their activation, will be required to broaden the spectrum of patients effectively targeted. These combinations will require consideration of concomitant oncogene alterations for optimal efficacy. For example, in colorectal cancers with BRAF oncogenic mutations, targeting BRAF with existing inhibitors, such as vemurafenib, and targeting super-enhancer activity with BET inhibitors may provide synergistic results [125]. A putative synergy of targeting the KRAS/BRAF/MEK pathway in combination with the super-enhancer machinery is also suggested by the observation that many super-enhancers across the genome of colorectal cancers are occupied by the transcription factor AP-1 (Activator Protein 1) that is activated downstream of the pathway [126]. The tolerability of targeting super-enhancers, especially in combination with other drugs, is also to be confirmed, although the initial clinical experience with BET inhibitors suggests the feasibility of the approach.
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17–48. doi: 10.3322/caac.21763. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, Cercek A, Smith RA, Jemal A. Colorectal cancer statistics, 2020. CA Cancer J Clin. 2020;70:145–164. doi: 10.3322/caac.21601. [DOI] [PubMed] [Google Scholar]
- 3.Bando H, Ohtsu A, Yoshino T. Therapeutic landscape and future direction of metastatic colorectal cancer. Nat Rev Gastroenterol Hepatol. 2023;20:306–322. doi: 10.1038/s41575-022-00736-1. [DOI] [PubMed] [Google Scholar]
- 4.Yaeger R, Weiss J, Pelster MS, Spira AI, Barve M, Ou SI, Leal TA, Bekaii-Saab TS, Paweletz CP, Heavey GA, Christensen JG, Velastegui K, Kheoh T, Der-Torossian H, Klempner SJ. Adagrasib with or without cetuximab in colorectal cancer with mutated KRAS G12C. N Engl J Med. 2023;388:44–54. doi: 10.1056/NEJMoa2212419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sakamoto Y, Bando H, Nakamura Y, Hasegawa H, Kuwaki T, Okamoto W, Taniguchi H, Aoyagi Y, Miki I, Uchigata H, Kuramoto N, Fuse N, Yoshino T, Ohtsu A. Trajectory for the regulatory approval of a combination of pertuzumab plus trastuzumab for pre-treated HER2-positive metastatic colorectal cancer using real-world data. Clin Colorectal Cancer. 2023;22:45–52. doi: 10.1016/j.clcc.2022.10.003. [DOI] [PubMed] [Google Scholar]
- 6.André T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, Smith D, Garcia-Carbonero R, Benavides M, Gibbs P, de la Fouchardiere C, Rivera F, Elez E, Bendell J, Le DT, Yoshino T, Van Cutsem E, Yang P, Farooqui MZH, Marinello P, Diaz LA Jr KEYNOTE-177 Investigators. Pembrolizumab in microsatellite-instability-high advanced colorectal cancer. N Engl J Med. 2020;383:2207–2218. doi: 10.1056/NEJMoa2017699. [DOI] [PubMed] [Google Scholar]
- 7.Glimelius B, Stintzing S, Marshall J, Yoshino T, de Gramont A. Metastatic colorectal cancer: advances in the folate-fluoropyrimidine chemotherapy backbone. Cancer Treat Rev. 2021;98:102218. doi: 10.1016/j.ctrv.2021.102218. [DOI] [PubMed] [Google Scholar]
- 8.Wang W, Kandimalla R, Huang H, Zhu L, Li Y, Gao F, Goel A, Wang X. Molecular subtyping of colorectal cancer: recent progress, new challenges and emerging opportunities. Semin Cancer Biol. 2019;55:37–52. doi: 10.1016/j.semcancer.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.An X, Lan X, Feng Z, Li X, Su Q. Histone modification: biomarkers and potential therapies in colorectal cancer. Ann Hum Genet. 2023;87:274–284. doi: 10.1111/ahg.12528. [DOI] [PubMed] [Google Scholar]
- 10.Wang M, Chen Q, Wang S, Xie H, Liu J, Huang R, Xiang Y, Jiang Y, Tian D, Bian E. Super-enhancers complexes zoom in transcription in cancer. J Exp Clin Cancer Res. 2023;42:183. doi: 10.1186/s13046-023-02763-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoshino S, Suzuki HI. The molecular understanding of super-enhancer dysregulation in cancer. Nagoya J Med Sci. 2022;84:216–229. doi: 10.18999/nagjms.84.2.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X, Cairns MJ, Yan J. Super-enhancers in transcriptional regulation and genome organization. Nucleic Acids Res. 2019;47:11481–11496. doi: 10.1093/nar/gkz1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kravchuk EV, Ashniev GA, Gladkova MG, Orlov AV, Vasileva AV, Boldyreva AV, Burenin AG, Skirda AM, Nikitin PI, Orlova NN. Experimental validation and prediction of super-enhancers: advances and challenges. Cells. 2023;12:1191. doi: 10.3390/cells12081191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siersbæk R, Rabiee A, Nielsen R, Sidoli S, Traynor S, Loft A, Poulsen LC, Rogowska-Wrzesinska A, Jensen ON, Mandrup S. Transcription factor cooperativity in early adipogenic hotspots and super-enhancers. Cell Rep. 2014;7:1443–1455. doi: 10.1016/j.celrep.2014.04.042. [DOI] [PubMed] [Google Scholar]
- 17.Ang YS, Rivas RN, Ribeiro AJS, Srivas R, Rivera J, Stone NR, Pratt K, Mohamed TMA, Fu JD, Spencer CI, Tippens ND, Li M, Narasimha A, Radzinsky E, Moon-Grady AJ, Yu H, Pruitt BL, Snyder MP, Srivastava D. Disease model of GATA4 mutation reveals transcription factor cooperativity in human cardiogenesis. Cell. 2016;167:1734–1749. e22. doi: 10.1016/j.cell.2016.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Witte S, Bradley A, Enright AJ, Muljo SA. High-density P300 enhancers control cell state transitions. BMC Genomics. 2015;16:903. doi: 10.1186/s12864-015-1905-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lai B, Lee JE, Jang Y, Wang L, Peng W, Ge K. MLL3/MLL4 are required for CBP/p300 binding on enhancers and super-enhancer formation in brown adipogenesis. Nucleic Acids Res. 2017;45:6388–6403. doi: 10.1093/nar/gkx234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Yu B, Deng P, Cheng Y, Yu Y, Kevork K, Ramadoss S, Ding X, Li X, Wang CY. KDM3 epigenetically controls tumorigenic potentials of human colorectal cancer stem cells through Wnt/β-catenin signalling. Nat Commun. 2017;8:15146. doi: 10.1038/ncomms15146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bacabac M, Xu W. Oncogenic super-enhancers in cancer: mechanisms and therapeutic targets. Cancer Metastasis Rev. 2023;42:471–480. doi: 10.1007/s10555-023-10103-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim EJ, Liu P, Zhang S, Donahue K, Wang Y, Schehr JL, Wolfe SK, Dickerson A, Lu L, Rui L, Zhong X, Wisinski KB, Yu M, Suzuki A, Lang JM, Ong IM, Xu W. BAF155 methylation drives metastasis by hijacking super-enhancers and subverting anti-tumor immunity. Nucleic Acids Res. 2021;49:12211–12233. doi: 10.1093/nar/gkab1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gen Y, Muramatsu T, Inoue J, Inazawa J. miR-766-5p targets super-enhancers by downregulating CBP and BRD4. Cancer Res. 2021;81:5190–5201. doi: 10.1158/0008-5472.CAN-21-0649. [DOI] [PubMed] [Google Scholar]
- 24.Wu SY, Lee AY, Lai HT, Zhang H, Chiang CM. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol Cell. 2013;49:843–857. doi: 10.1016/j.molcel.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, Lin Y, Li J, Kang T, Tao M, Rusinova E, Zhang G, Wang C, Zhu H, Yao J, Zeng YX, Evers BM, Zhou MM, Zhou BP. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell. 2014;25:210–225. doi: 10.1016/j.ccr.2014.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54:728–736. doi: 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narita T, Ito S, Higashijima Y, Chu WK, Neumann K, Walter J, Satpathy S, Liebner T, Hamilton WB, Maskey E, Prus G, Shibata M, Iesmantavicius V, Brickman JM, Anastassiadis K, Koseki H, Choudhary C. Enhancers are activated by p300/CBP activity-dependent PIC assembly, RNAPII recruitment, and pause release. Mol Cell. 2021;81:2166–2182. e6. doi: 10.1016/j.molcel.2021.03.008. [DOI] [PubMed] [Google Scholar]
- 28.Kent D, Marchetti L, Mikulasova A, Russell LJ, Rico D. Broad H3K4me3 domains: maintaining cellular identity and their implication in super-enhancer hijacking. Bioessays. 2023;45:e2200239. doi: 10.1002/bies.202200239. [DOI] [PubMed] [Google Scholar]
- 29.Mikulasova A, Kent D, Trevisan-Herraz M, Karataraki N, Fung KTM, Ashby C, Cieslak A, Yaccoby S, van Rhee F, Zangari M, Thanendrarajan S, Schinke C, Morgan GJ, Asnafi V, Spicuglia S, Brackley CA, Corcoran AE, Hambleton S, Walker BA, Rico D, Russell LJ. Epigenomic translocation of H3K4me3 broad domains over oncogenes following hijacking of super-enhancers. Genome Res. 2022;32:1343–1354. doi: 10.1101/gr.276042.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harper TM, Taatjes DJ. The complex structure and function of Mediator. J Biol Chem. 2018;293:13778–13785. doi: 10.1074/jbc.R117.794438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luyties O, Taatjes DJ. The Mediator kinase module: an interface between cell signaling and transcription. Trends Biochem Sci. 2022;47:314–327. doi: 10.1016/j.tibs.2022.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conaway RC, Conaway JW. The Mediator complex and transcription elongation. Biochim Biophys Acta. 2013;1829:69–75. doi: 10.1016/j.bbagrm.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Donner AJ, Ebmeier CC, Taatjes DJ, Espinosa JM. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat Struct Mol Biol. 2010;17:194–201. doi: 10.1038/nsmb.1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Donner AJ, Szostek S, Hoover JM, Espinosa JM. CDK8 is a stimulus-specific positive coregulator of p53 target genes. Mol Cell. 2007;27:121–33. doi: 10.1016/j.molcel.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuuluvainen E, Domènech-Moreno E, Niemelä EH, Mäkelä TP. Depletion of mediator kinase module subunits represses superenhancer-associated genes in colon cancer cells. Mol Cell Biol. 2018;38:e00573-17. doi: 10.1128/MCB.00573-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prieto S, Dubra G, Camasses A, Aznar AB, Begon-Pescia C, Simboeck E, Pirot N, Gerbe F, Angevin L, Jay P, Krasinska L, Fisher D. CDK8 and CDK19 act redundantly to control the CFTR pathway in the intestinal epithelium. EMBO Rep. 2023;24:e54261. doi: 10.15252/embr.202154261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCleland ML, Soukup TM, Liu SD, Esensten JH, de Sousa e Melo F, Yaylaoglu M, Warming S, Roose-Girma M, Firestein R. Cdk8 deletion in the Apc(Min) murine tumour model represses EZH2 activity and accelerates tumourigenesis. J Pathol. 2015;237:508–519. doi: 10.1002/path.4596. [DOI] [PubMed] [Google Scholar]
- 38.Singh A, Modak SB, Chaturvedi MM, Purohit JS. SWI/SNF chromatin remodelers: structural, functional and mechanistic implications. Cell Biochem Biophys. 2023;81:167–187. doi: 10.1007/s12013-023-01140-5. [DOI] [PubMed] [Google Scholar]
- 39.Alver BH, Kim KH, Lu P, Wang X, Manchester HE, Wang W, Haswell JR, Park PJ, Roberts CW. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat Commun. 2017;8:14648. doi: 10.1038/ncomms14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dannappel MV, Zhu D, Sun X, Chua HK, Poppelaars M, Suehiro M, Khadka S, Lim Kam Sian TC, Sooraj D, Loi M, Gao H, Croagh D, Daly RJ, Faridi P, Boyer TG, Firestein R. CDK8 and CDK19 regulate intestinal differentiation and homeostasis via the chromatin remodeling complex SWI/SNF. J Clin Invest. 2022;132:e158593. doi: 10.1172/JCI158593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Donati B, Lorenzini E, Ciarrocchi A. BRD4 and cancer: going beyond transcriptional regulation. Mol Cancer. 2018;17:164. doi: 10.1186/s12943-018-0915-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hazan I, Monin J, Bouwman BAM, Crosetto N, Aqeilan RI. Activation of oncogenic super-enhancers is coupled with DNA repair by RAD51. Cell Rep. 2019;29:560–572. e4. doi: 10.1016/j.celrep.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li X, Baek G, Ramanand SG, Sharp A, Gao Y, Yuan W, Welti J, Rodrigues DN, Dolling D, Figueiredo I, Sumanasuriya S, Crespo M, Aslam A, Li R, Yin Y, Mukherjee B, Kanchwala M, Hughes AM, Halsey WS, Chiang CM, Xing C, Raj GV, Burma S, de Bono J, Mani RS. BRD4 promotes DNA repair and mediates the formation of TMPRSS2-ERG gene rearrangements in prostate cancer. Cell Rep. 2018;22:796–808. doi: 10.1016/j.celrep.2017.12.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Voutsadakis IA. Molecular lesions of insulator CTCF and its paralogue CTCFL (BORIS) in cancer: an analysis from published genomic studies. High Throughput. 2018;7:30. doi: 10.3390/ht7040030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gong Y, Lazaris C, Sakellaropoulos T, Lozano A, Kambadur P, Ntziachristos P, Aifantis I, Tsirigos A. Stratification of TAD boundaries reveals preferential insulation of super-enhancers by strong boundaries. Nat Commun. 2018;9:542. doi: 10.1038/s41467-018-03017-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ottema S, Mulet-Lazaro R, Erpelinck-Verschueren C, van Herk S, Havermans M, Arricibita Varea A, Vermeulen M, Beverloo HB, Gröschel S, Haferlach T, Haferlach C, J Wouters B, Bindels E, Smeenk L, Delwel R. The leukemic oncogene EVI1 hijacks a MYC super-enhancer by CTCF-facilitated loops. Nat Commun. 2021;12:5679. doi: 10.1038/s41467-021-25862-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hnisz D, Schuijers J, Lin CY, Weintraub AS, Abraham BJ, Lee TI, Bradner JE, Young RA. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol Cell. 2015;58:362–370. doi: 10.1016/j.molcel.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yochum GS, Cleland R, Goodman RH. A genome-wide screen for beta-catenin binding sites identifies a downstream enhancer element that controls c-Myc gene expression. Mol Cell Biol. 2008;28:7368–7379. doi: 10.1128/MCB.00744-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim K, Kim M, Lee AJ, Song SH, Kang JK, Eom J, Kang GH, Bae JM, Min S, Kim Y, Lim Y, Kim HS, Kim YJ, Kim TY, Jung I. Spatial and clonality-resolved 3D cancer genome alterations reveal enhancer-hijacking as a potential prognostic marker for colorectal cancer. Cell Rep. 2023;42:112778. doi: 10.1016/j.celrep.2023.112778. [DOI] [PubMed] [Google Scholar]
- 50.Zhou RW, Parsons RE. Etiology of super-enhancer reprogramming and activation in cancer. Epigenetics Chromatin. 2023;16:29. doi: 10.1186/s13072-023-00502-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Göndör A. WNT-mediated gene gating: a novel principle connecting oncogenic super-enhancers with the nuclear pore to drive pathological expression of MYC. Mol Cell Oncol. 2020;7:1710992. doi: 10.1080/23723556.2019.1710992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ben-Yishay R, Shav-Tal Y. The dynamic lifecycle of mRNA in the nucleus. Curr Opin Cell Biol. 2019;58:69–75. doi: 10.1016/j.ceb.2019.02.007. [DOI] [PubMed] [Google Scholar]
- 53.Pascual-Garcia P, Capelson M. The nuclear pore complex and the genome: organizing and regulatory principles. Curr Opin Genet Dev. 2021;67:142–150. doi: 10.1016/j.gde.2021.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chachoua I, Tzelepis I, Dai H, Lim JP, Lewandowska-Ronnegren A, Casagrande FB, Wu S, Vestlund J, Mallet de Lima CD, Bhartiya D, Scholz BA, Martino M, Mehmood R, Göndör A. Canonical WNT signaling-dependent gating of MYC requires a noncanonical CTCF function at a distal binding site. Nat Commun. 2022;13:204. doi: 10.1038/s41467-021-27868-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kondo H, Mishiro K, Iwashima Y, Qiu Y, Kobayashi A, Lim K, Domoto T, Minamoto T, Ogawa K, Kunishima M, Hazawa M, Wong RW. Discovery of a novel aminocyclopropenone compound that inhibits BRD4-driven nucleoporin NUP210 expression and attenuates colorectal cancer growth. Cells. 2022;11:317. doi: 10.3390/cells11030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baumgart SJ, Nevedomskaya E, Lesche R, Newman R, Mumberg D, Haendler B. Darolutamide antagonizes androgen signaling by blocking enhancer and super-enhancer activation. Mol Oncol. 2020;14:2022–2039. doi: 10.1002/1878-0261.12693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tao HC, Wang C, Ma N, Zhu X, Zhou XJ. Recurrent superenhancer of the oncogene POU5F1B in colorectal cancers. Biomed Res Int. 2021;2021:5405060. doi: 10.1155/2021/5405060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Q, Thoms JAI, Nunez AC, Huang Y, Knezevic K, Packham D, Poulos RC, Williams R, Beck D, Hawkins NJ, Ward RL, Wong JWH, Hesson LB, Sloane MA, Pimanda JE. Disruption of a -35 kb enhancer impairs CTCF binding and MLH1 expression in colorectal cells. Clin Cancer Res. 2018;24:4602–4611. doi: 10.1158/1078-0432.CCR-17-3678. [DOI] [PubMed] [Google Scholar]
- 59.Hung S, Saiakhova A, Faber ZJ, Bartels CF, Neu D, Bayles I, Ojo E, Hong ES, Pontius WD, Morton AR, Liu R, Kalady MF, Wald DN, Markowitz S, Scacheri PC. Mismatch repair-signature mutations activate gene enhancers across human colorectal cancer epigenomes. Elife. 2019;8:e40760. doi: 10.7554/eLife.40760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mittal P, Roberts CWM. The SWI/SNF complex in cancer - biology, biomarkers and therapy. Nat Rev Clin Oncol. 2020;17:435–448. doi: 10.1038/s41571-020-0357-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Finetti MA, Grabovska Y, Bailey S, Williamson D. Translational genomics of malignant rhabdoid tumours: current impact and future possibilities. Semin Cancer Biol. 2020;61:30–41. doi: 10.1016/j.semcancer.2019.12.017. [DOI] [PubMed] [Google Scholar]
- 62.Lu B, Shi H. An in-depth look at small cell carcinoma of the ovary, hypercalcemic type (SCCOHT): clinical implications from recent molecular findings. J Cancer. 2019;10:223–237. doi: 10.7150/jca.26978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mora-Blanco EL, Mishina Y, Tillman EJ, Cho YJ, Thom CS, Pomeroy SL, Shao W, Roberts CW. Activation of β-catenin/TCF targets following loss of the tumor suppressor SNF5. Oncogene. 2014;33:933–938. doi: 10.1038/onc.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Griffin CT, Curtis CD, Davis RB, Muthukumar V, Magnuson T. The chromatin-remodeling enzyme BRG1 modulates vascular Wnt signaling at two levels. Proc Natl Acad Sci U S A. 2011;108:2282–2287. doi: 10.1073/pnas.1013751108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Park MJ, Shen H, Spaeth JM, Tolvanen JH, Failor C, Knudtson JF, McLaughlin J, Halder SK, Yang Q, Bulun SE, Al-Hendy A, Schenken RS, Aaltonen LA, Boyer TG. Oncogenic exon 2 mutations in Mediator subunit MED12 disrupt allosteric activation of cyclin C-CDK8/19. J Biol Chem. 2018;293:4870–4882. doi: 10.1074/jbc.RA118.001725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roelands J, Kuppen PJK, Ahmed EI, Mall R, Masoodi T, Singh P, Monaco G, Raynaud C, de Miranda NFCC, Ferraro L, Carneiro-Lobo TC, Syed N, Rawat A, Awad A, Decock J, Mifsud W, Miller LD, Sherif S, Mohamed MG, Rinchai D, Van den Eynde M, Sayaman RW, Ziv E, Bertucci F, Petkar MA, Lorenz S, Mathew LS, Wang K, Murugesan S, Chaussabel D, Vahrmeijer AL, Wang E, Ceccarelli A, Fakhro KA, Zoppoli G, Ballestrero A, Tollenaar RAEM, Marincola FM, Galon J, Khodor SA, Ceccarelli M, Hendrickx W, Bedognetti D. An integrated tumor, immune and microbiome atlas of colon cancer. Nat Med. 2023;29:1273–1286. doi: 10.1038/s41591-023-02324-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rodriguez RM, Huidobro C, Urdinguio RG, Mangas C, Soldevilla B, Domínguez G, Bonilla F, Fernandez AF, Fraga MF. Aberrant epigenetic regulation of bromodomain BRD4 in human colon cancer. J Mol Med (Berl) 2012;90:587–595. doi: 10.1007/s00109-011-0837-0. [DOI] [PubMed] [Google Scholar]
- 69.Shin HY. Targeting super-enhancers for disease treatment and diagnosis. Mol Cells. 2018;41:506–514. doi: 10.14348/molcells.2018.2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, Bot BM, Morris JS, Simon IM, Gerster S, Fessler E, De Sousa E Melo F, Missiaglia E, Ramay H, Barras D, Homicsko K, Maru D, Manyam GC, Broom B, Boige V, Perez-Villamil B, Laderas T, Salazar R, Gray JW, Hanahan D, Tabernero J, Bernards R, Friend SH, Laurent-Puig P, Medema JP, Sadanandam A, Wessels L, Delorenzi M, Kopetz S, Vermeulen L, Tejpar S. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–1356. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tögel L, Nightingale R, Chueh AC, Jayachandran A, Tran H, Phesse T, Wu R, Sieber OM, Arango D, Dhillon AS, Dawson MA, Diez-Dacal B, Gahman TC, Filippakopoulos P, Shiau AK, Mariadason JM. Dual targeting of bromodomain and extraterminal domain proteins, and WNT or MAPK signaling, inhibits c-MYC expression and proliferation of colorectal cancer cells. Mol Cancer Ther. 2016;15:1217–1226. doi: 10.1158/1535-7163.MCT-15-0724. [DOI] [PubMed] [Google Scholar]
- 72.Klingbeil O, Lesche R, Gelato KA, Haendler B, Lejeune P. Inhibition of BET bromodomain-dependent XIAP and FLIP expression sensitizes KRAS-mutated NSCLC to pro-apoptotic agents. Cell Death Dis. 2016;7:e2365. doi: 10.1038/cddis.2016.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mio C, Gerratana L, Bolis M, Caponnetto F, Zanello A, Barbina M, Di Loreto C, Garattini E, Damante G, Puglisi F. BET proteins regulate homologous recombination-mediated DNA repair: BRCAness and implications for cancer therapy. Int J Cancer. 2019;144:755–766. doi: 10.1002/ijc.31898. [DOI] [PubMed] [Google Scholar]
- 74.Oliver SS, Denu JM. Disrupting the reader of histone language. Angew Chem Int Ed Engl. 2011;50:5801–5803. doi: 10.1002/anie.201101414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, White J, Kirilovsky J, Rice CM, Lora JM, Prinjha RK, Lee K, Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Piha-Paul SA, Hann CL, French CA, Cousin S, Braña I, Cassier PA, Moreno V, de Bono JS, Harward SD, Ferron-Brady G, Barbash O, Wyce A, Wu Y, Horner T, Annan M, Parr NJ, Prinjha RK, Carpenter CL, Hilton J, Hong DS, Haas NB, Markowski MC, Dhar A, O’Dwyer PJ, Shapiro GI. Phase 1 study of molibresib (GSK525762), a bromodomain and extra-terminal domain protein inhibitor, in NUT carcinoma and other solid tumors. JNCI Cancer Spectr. 2019;4:pkz093. doi: 10.1093/jncics/pkz093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kaplan HG, Subramaniam S, Vallières E, Barnett T. Prolonged survival of NUT midline carcinoma and current approaches to treatment. Oncologist. 2023;28:765–770. doi: 10.1093/oncolo/oyad177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cousin S, Blay JY, Garcia IB, de Bono JS, Le Tourneau C, Moreno V, Trigo J, Hann CL, Azad AA, Im SA, Cassier PA, French CA, Italiano A, Keedy VL, Plummer R, Sablin MP, Hemming ML, Ferron-Brady G, Wyce A, Khaled A, Datta A, Foley SW, McCabe MT, Wu Y, Horner T, Kremer BE, Dhar A, O’Dwyer PJ, Shapiro GI, Piha-Paul SA. Safety, pharmacokinetic, pharmacodynamic and clinical activity of molibresib for the treatment of nuclear protein in testis carcinoma and other cancers: results of a phase I/II open-label, dose escalation study. Int J Cancer. 2022;150:993–1006. doi: 10.1002/ijc.33861. [DOI] [PubMed] [Google Scholar]
- 81.Lewin J, Soria JC, Stathis A, Delord JP, Peters S, Awada A, Aftimos PG, Bekradda M, Rezai K, Zeng Z, Hussain A, Perez S, Siu LL, Massard C. Phase Ib trial with birabresib, a small-molecule inhibitor of bromodomain and extraterminal proteins, in patients with selected advanced solid tumors. J. Clin. Oncol. 2018;36:3007–3014. doi: 10.1200/JCO.2018.78.2292. [DOI] [PubMed] [Google Scholar]
- 82.Moreno V, Vieito M, Sepulveda JM, Galvao V, Hernández-Guerrero T, Doger B, Saavedra O, Carlo-Stella C, Michot JM, Italiano A, Magagnoli M, Carpio C, Pinto A, Sarmiento R, Amoroso B, Aronchik I, Filvaroff E, Hanna B, Wei X, Nikolova Z, Braña I. BET inhibitor trotabresib in heavily pretreated patients with solid tumors and diffuse large B-cell lymphomas. Nat Commun. 2023;14:1359. doi: 10.1038/s41467-023-36976-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hamilton EP, Wang JS, Oza AM, Patel MR, Ulahannan SV, Bauer T, Karlix JL, Zeron-Medina J, Fabbri G, Marco-Casanova P, Moorthy G, Hattersley MM, Littlewood GM, Mitchell P, Saeh J, Pouliot GP, Moore KN. First-in-human study of AZD5153, a small-molecule inhibitor of bromodomain protein 4, in patients with relapsed/refractory malignant solid tumors and lymphoma. Mol Cancer Ther. 2023;22:1154–1165. doi: 10.1158/1535-7163.MCT-23-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Graham H. The mechanism of action and clinical value of PROTACs: a graphical review. Cell Signal. 2022;99:110446. doi: 10.1016/j.cellsig.2022.110446. [DOI] [PubMed] [Google Scholar]
- 85.Voutsadakis IA. The ubiquitin-proteasome system in colorectal cancer. Biochim Biophys Acta. 2008;1782:800–808. doi: 10.1016/j.bbadis.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 86.Hines J, Lartigue S, Dong H, Qian Y, Crews CM. MDM2-recruiting PROTAC offers superior, synergistic antiproliferative activity via simultaneous degradation of BRD4 and stabilization of p53. Cancer Res. 2019;79:251–262. doi: 10.1158/0008-5472.CAN-18-2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Qin AC, Jin H, Song Y, Gao Y, Chen YF, Zhou LN, Wang SS, Lu XS. The therapeutic effect of the BRD4-degrading PROTAC A1874 in human colon cancer cells. Cell Death Dis. 2020;11:805. doi: 10.1038/s41419-020-03015-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fu Y, Yang B, Cui Y, Hu X, Li X, Lu F, Qin T, Zhang L, Hu Z, Guo E, Fan J, Xiao R, Li W, Qin X, Hu D, Peng W, Liu J, Wang B, Mills GB, Chen G, Sun C. BRD4 inhibition impairs DNA mismatch repair, induces mismatch repair mutation signatures and creates therapeutic vulnerability to immune checkpoint blockade in MMR-proficient tumors. J Immunother Cancer. 2023;11:e006070. doi: 10.1136/jitc-2022-006070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang L, Zhang Y, Shan W, Hu Z, Yuan J, Pi J, Wang Y, Fan L, Tang Z, Li C, Hu X, Tanyi JL, Fan Y, Huang Q, Montone K, Dang CV, Zhang L. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci Transl Med. 2017;9:eaal1645. doi: 10.1126/scitranslmed.aal1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sun C, Yin J, Fang Y, Chen J, Jeong KJ, Chen X, Vellano CP, Ju Z, Zhao W, Zhang D, Lu Y, Meric-Bernstam F, Yap TA, Hattersley M, O’Connor MJ, Chen H, Fawell S, Lin SY, Peng G, Mills GB. BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell. 2018;33:401–416. e8. doi: 10.1016/j.ccell.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Seo JO, Han SI, Lim SC. Role of CDK8 and beta-catenin in colorectal adenocarcinoma. Oncol Rep. 2010;24:285–291. [PubMed] [Google Scholar]
- 92.Xi M, Chen T, Wu C, Gao X, Wu Y, Luo X, Du K, Yu L, Cai T, Shen R, Sun H. CDK8 as a therapeutic target for cancers and recent developments in discovery of CDK8 inhibitors. Eur J Med Chem. 2019;164:77–91. doi: 10.1016/j.ejmech.2018.11.076. [DOI] [PubMed] [Google Scholar]
- 93.Clarke PA, Ortiz-Ruiz MJ, TePoele R, Adeniji-Popoola O, Box G, Court W, Czasch S, El Bawab S, Esdar C, Ewan K, Gowan S, De Haven Brandon A, Hewitt P, Hobbs SM, Kaufmann W, Mallinger A, Raynaud F, Roe T, Rohdich F, Schiemann K, Simon S, Schneider R, Valenti M, Weigt S, Blagg J, Blaukat A, Dale TC, Eccles SA, Hecht S, Urbahns K, Workman P, Wienke D. Assessing the mechanism and therapeutic potential of modulators of the human Mediator complex-associated protein kinases. Elife. 2016;5:e20722. doi: 10.7554/eLife.20722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mallinger A, Crumpler S, Pichowicz M, Waalboer D, Stubbs M, Adeniji-Popoola O, Wood B, Smith E, Thai C, Henley AT, Georgi K, Court W, Hobbs S, Box G, Ortiz-Ruiz MJ, Valenti M, De Haven Brandon A, TePoele R, Leuthner B, Workman P, Aherne W, Poeschke O, Dale T, Wienke D, Esdar C, Rohdich F, Raynaud F, Clarke PA, Eccles SA, Stieber F, Schiemann K, Blagg J. Discovery of potent, orally bioavailable, small-molecule inhibitors of WNT signaling from a cell-based pathway screen. J Med Chem. 2015;58:1717–1735. doi: 10.1021/jm501436m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dale T, Clarke PA, Esdar C, Waalboer D, Adeniji-Popoola O, Ortiz-Ruiz MJ, Mallinger A, Samant RS, Czodrowski P, Musil D, Schwarz D, Schneider K, Stubbs M, Ewan K, Fraser E, TePoele R, Court W, Box G, Valenti M, de Haven Brandon A, Gowan S, Rohdich F, Raynaud F, Schneider R, Poeschke O, Blaukat A, Workman P, Schiemann K, Eccles SA, Wienke D, Blagg J. A selective chemical probe for exploring the role of CDK8 and CDK19 in human disease. Nat Chem Biol. 2015;11:973–980. doi: 10.1038/nchembio.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lloyd RL, Urban V, Muñoz-Martínez F, Ayestaran I, Thomas JC, de Renty C, O’Connor MJ, Forment JV, Galanty Y, Jackson SP. Loss of Cyclin C or CDK8 provides ATR inhibitor resistance by suppressing transcription-associated replication stress. Nucleic Acids Res. 2021;49:8665–8683. doi: 10.1093/nar/gkab628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rzymski T, Mikula M, Żyłkiewicz E, Dreas A, Wiklik K, Gołas A, Wójcik K, Masiejczyk M, Wróbel A, Dolata I, Kitlińska A, Statkiewicz M, Kuklinska U, Goryca K, Sapała Ł, Grochowska A, Cabaj A, Szajewska-Skuta M, Gabor-Worwa E, Kucwaj K, Białas A, Radzimierski A, Combik M, Woyciechowski J, Mikulski M, Windak R, Ostrowski J, Brzózka K. SEL120-34A is a novel CDK8 inhibitor active in AML cells with high levels of serine phosphorylation of STAT1 and STAT5 transactivation domains. Oncotarget. 2017;8:33779–33795. doi: 10.18632/oncotarget.16810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Toure MA, Koehler AN. Addressing transcriptional dysregulation in cancer through CDK9 inhibition. Biochemistry. 2023;62:1114–1123. doi: 10.1021/acs.biochem.2c00609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Morales F, Giordano A. Overview of CDK9 as a target in cancer research. Cell Cycle. 2016;15:519–527. doi: 10.1080/15384101.2016.1138186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Blake DR, Vaseva AV, Hodge RG, Kline MP, Gilbert TSK, Tyagi V, Huang D, Whiten GC, Larson JE, Wang X, Pearce KH, Herring LE, Graves LM, Frye SV, Emanuele MJ, Cox AD, Der CJ. Application of a MYC degradation screen identifies sensitivity to CDK9 inhibitors in KRAS-mutant pancreatic cancer. Sci Signal. 2019;12:eaav7259. doi: 10.1126/scisignal.aav7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cidado J, Boiko S, Proia T, Ferguson D, Criscione SW, San Martin M, Pop-Damkov P, Su N, Roamio Franklin VN, Sekhar Reddy Chilamakuri C, D’Santos CS, Shao W, Saeh JC, Koch R, Weinstock DM, Zinda M, Fawell SE, Drew L. AZD4573 is a highly selective CDK9 inhibitor that suppresses MCL-1 and induces apoptosis in hematologic cancer cells. Clin Cancer Res. 2020;26:922–934. doi: 10.1158/1078-0432.CCR-19-1853. [DOI] [PubMed] [Google Scholar]
- 102.Alcon C, Manzano-Muñoz A, Montero J. A new CDK9 inhibitor on the block to treat hematologic malignancies. Clin Cancer Res. 2020;26:761–763. doi: 10.1158/1078-0432.CCR-19-3670. [DOI] [PubMed] [Google Scholar]
- 103.Strati P, Morschhauser F, Danilov A, Cheah C, Shah H, Jurczak W, Olabode D, Meyer S, Lim Yoon J, Arduini S, Saeh J, Olsson R, Gregory G. P1126: phase 1B/2A study of AZD4573 (CDK9I) and acalabrutinib in patients (PTS) with relapsed/refractory diffuse large B-cell lymphoma (R/R DLBCL) Hemasphere. 2023;7:e47925e9. [Google Scholar]
- 104.Freeman DB, Hopkins TD, Mikochik PJ, Vacca JP, Gao H, Naylor-Olsen A, Rudra S, Li H, Pop MS, Villagomez RA, Lee C, Li H, Zhou M, Saffran DC, Rioux N, Hood TR, Day MAL, McKeown MR, Lin CY, Bischofberger N, Trotter BW. Discovery of KB-0742, a potent, selective, orally bioavailable small molecule inhibitor of CDK9 for MYC-dependent cancers. J Med Chem. 2023;66:15629–15647. doi: 10.1021/acs.jmedchem.3c01233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Villalona-Calero M, Mita M, Mita A, Federman N, Rasco D, Spigel D, Luo J, Hanna GJ, Cote GM, Cutler RE, Kumar P, MacKenzie C, Lin C, DiMartino JF, Olek EA, Van Tine B. A first-in-human study of CDK9 inhibitor KB-0742 demonstrates evidence of tolerability and clinical activity. Mol Cancer Ther. 2023;22:B159. [Google Scholar]
- 106.Milite C, Feoli A, Sasaki K, La Pietra V, Balzano AL, Marinelli L, Mai A, Novellino E, Castellano S, Tosco A, Sbardella G. A novel cell-permeable, selective, and noncompetitive inhibitor of KAT3 histone acetyltransferases from a combined molecular pruning/classical isosterism approach. J Med Chem. 2015;58:2779–2798. doi: 10.1021/jm5019687. [DOI] [PubMed] [Google Scholar]
- 107.Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL, Hansen TM, Risi RM, Frey R, Manaves V, Shaw B, Algire M, Hessler P, Lam LT, Uziel T, Faivre E, Ferguson D, Buchanan FG, Martin RL, Torrent M, Chiang GG, Karukurichi K, Langston JW, Weinert BT, Choudhary C, de Vries P, Van Drie JH, McElligott D, Kesicki E, Marmorstein R, Sun C, Cole PA, Rosenberg SH, Michaelides MR, Lai A, Bromberg KD. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature. 2017;550:128–132. doi: 10.1038/nature24028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang R, He Y, Robinson V, Yang Z, Hessler P, Lasko LM, Lu X, Bhathena A, Lai A, Uziel T, Lam LT. Targeting lineage-specific MITF pathway in human melanoma cell lines by A-485, the selective small-molecule inhibitor of p300/CBP. Mol Cancer Ther. 2018;17:2543–2550. doi: 10.1158/1535-7163.MCT-18-0511. [DOI] [PubMed] [Google Scholar]
- 109.Zhang B, Chen D, Liu B, Dekker FJ, Quax WJ. A novel histone acetyltransferase inhibitor A485 improves sensitivity of non-small-cell lung carcinoma cells to TRAIL. Biochem Pharmacol. 2020;175:113914. doi: 10.1016/j.bcp.2020.113914. [DOI] [PubMed] [Google Scholar]
- 110.Xie J, Yuan Y, Yao G, Chen Z, Yu W, Zhu Q. Nucleoporin 160 (NUP160) inhibition alleviates diabetic nephropathy by activating autophagy. Bioengineered. 2021;12:6390–6402. doi: 10.1080/21655979.2021.1968777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hippert MM, O’Toole PS, Thorburn A. Autophagy in cancer: good, bad, or both? Cancer Res. 2006;66:9349–9351. doi: 10.1158/0008-5472.CAN-06-1597. [DOI] [PubMed] [Google Scholar]
- 112.Tutuncuoglu B, Krogan NJ. Mapping genetic interactions in cancer: a road to rational combination therapies. Genome Med. 2019;11:62. doi: 10.1186/s13073-019-0680-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang B, Lyu J, Liu Y, Wu C, Yang EJ, Pardeshi L, Tan K, Wong KH, Chen Q, Xu X, Deng CX, Shim JS. BRCA1 deficiency sensitizes breast cancer cells to bromodomain and extra-terminal domain (BET) inhibition. Oncogene. 2018;37:6341–6356. doi: 10.1038/s41388-018-0408-8. [DOI] [PubMed] [Google Scholar]
- 114.Lei L, Xie X, He L, Chen K, Lv Z, Zhou B, Li Y, Hu W, Zhou Z. The bromodomain and extra-terminal domain inhibitor JQ1 synergistically sensitizes human colorectal cancer cells to topoisomerase I inhibitors through repression of Mre11-mediated DNA repair pathway. Invest New Drugs. 2021;39:362–376. doi: 10.1007/s10637-020-01014-0. [DOI] [PubMed] [Google Scholar]
- 115.Pyziak K, Sroka-Porada A, Rzymski T, Dulak J, Łoboda A. Potential of enhancer of zeste homolog 2 inhibitors for the treatment of SWI/SNF mutant cancers and tumor microenvironment modulation. Drug Dev Res. 2021;82:730–753. doi: 10.1002/ddr.21796. [DOI] [PubMed] [Google Scholar]
- 116.Schuettengruber B, Bourbon HM, Di Croce L, Cavalli G. Genome regulation by polycomb and trithorax: 70 years and counting. Cell. 2017;171:34–57. doi: 10.1016/j.cell.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 117.Italiano A, Soria JC, Toulmonde M, Michot JM, Lucchesi C, Varga A, Coindre JM, Blakemore SJ, Clawson A, Suttle B, McDonald AA, Woodruff M, Ribich S, Hedrick E, Keilhack H, Thomson B, Owa T, Copeland RA, Ho PTC, Ribrag V. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol. 2018;19:649–659. doi: 10.1016/S1470-2045(18)30145-1. [DOI] [PubMed] [Google Scholar]
- 118.Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, Schultz DC, Liu Q, Shih IeM, Conejo-Garcia JR, Speicher DW, Zhang R. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med. 2015;21:231–238. doi: 10.1038/nm.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chan-Penebre E, Armstrong K, Drew A, Grassian AR, Feldman I, Knutson SK, Kuplast-Barr K, Roche M, Campbell J, Ho P, Copeland RA, Chesworth R, Smith JJ, Keilhack H, Ribich SA. Selective killing of SMARCA2- and SMARCA4-deficient small cell carcinoma of the ovary, hypercalcemic type cells by inhibition of EZH2: in vitro and in vivo preclinical models. Mol Cancer Ther. 2017;16:850–860. doi: 10.1158/1535-7163.MCT-16-0678. [DOI] [PubMed] [Google Scholar]
- 120.Garczyk S, Schneider U, Lurje I, Becker K, Vögeli TA, Gaisa NT, Knüchel R. ARID1A-deficiency in urothelial bladder cancer: no predictive biomarker for EZH2-inhibitor treatment response? PLoS One. 2018;13:e0202965. doi: 10.1371/journal.pone.0202965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nargund AM, Xu C, Mandoli A, Okabe A, Chen GB, Huang KK, Sheng T, Yao X, Teo JMN, Sundar R, Kok YJ, See YX, Xing M, Li Z, Yong CH, Anand A, A I ZF, Poon LF, Ng MSW, Koh JYP, Ooi WF, Tay ST, Ong X, Tan ALK, Grabsch HI, Fullwood MJ, Teh TB, Bi X, Kaneda A, Li S, Tan P. Chromatin rewiring by mismatch repair protein MSH2 alters cell adhesion pathways and sensitivity to BET inhibition in gastric cancer. Cancer Res. 2022;82:2538–2551. doi: 10.1158/0008-5472.CAN-21-2072. [DOI] [PubMed] [Google Scholar]
- 122.Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, Bahl S, Cao Y, Amin-Mansour A, Yamauchi M, Sukawa Y, Stewart C, Rosenberg M, Mima K, Inamura K, Nosho K, Nowak JA, Lawrence MS, Giovannucci EL, Chan AT, Ng K, Meyerhardt JA, Van Allen EM, Getz G, Gabriel SB, Lander ES, Wu CJ, Fuchs CS, Ogino S, Garraway LA. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016;15:857–865. doi: 10.1016/j.celrep.2016.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 124.Shi C, Yang EJ, Liu Y, Mou PK, Ren G, Shim JS. Bromodomain and extra-terminal motif (BET) inhibition is synthetic lethal with loss of SMAD4 in colorectal cancer cells via restoring the loss of MYC repression. Oncogene. 2021;40:937–950. doi: 10.1038/s41388-020-01580-w. [DOI] [PubMed] [Google Scholar]
- 125.Nakamura Y, Hattori N, Iida N, Yamashita S, Mori A, Kimura K, Yoshino T, Ushijima T. Targeting of super-enhancers and mutant BRAF can suppress growth of BRAF-mutant colon cancer cells via repression of MAPK signaling pathway. Cancer Lett. 2017;402:100–109. doi: 10.1016/j.canlet.2017.05.017. [DOI] [PubMed] [Google Scholar]
- 126.Cohen AJ, Saiakhova A, Corradin O, Luppino JM, Lovrenert K, Bartels CF, Morrow JJ, Mack SC, Dhillon G, Beard L, Myeroff L, Kalady MF, Willis J, Bradner JE, Keri RA, Berger NA, Pruett-Miller SM, Markowitz SD, Scacheri PC. Hotspots of aberrant enhancer activity punctuate the colorectal cancer epigenome. Nat Commun. 2017;8:14400. doi: 10.1038/ncomms14400. [DOI] [PMC free article] [PubMed] [Google Scholar]