

Abstract
Introduction
The emerging field of the disc microbiome challenges traditional views of disc sterility, which opens new avenues for novel clinical insights. However, the lack of methodological consensus in disc microbiome studies introduces discrepancies. The aims of this study were to (1) compare the disc microbiome of non‐Modic (nonMC), Modic type 1 change (MC1), and MC2 discs to findings from prior disc microbiome studies, and (2) investigate if discrepancies to prior studies can be explained with bioinformatic variations.
Methods
Sequencing of 16S rRNA in 70 discs (24 nonMC, 25 MC1, and 21 MC2) for microbiome profiling. The experimental setup included buffer contamination controls and was performed under aseptic conditions. Methodology and results were contrasted with previous disc microbiome studies. Critical bioinformatic steps that were different in our best‐practice approach and previous disc microbiome studies (taxonomic lineage assignment, prevalence cut‐off) were varied and their effect on results were compared.
Results
There was limited overlap of results with a previous study on MC disc microbiome. No bacterial genera were shared using the same bioinformatic parameters. Taxonomic lineage assignment using “amplicon sequencing variants” was more sensitive and detected 48 genera compared to 22 with “operational taxonomic units” (previous study). Increasing filter cut‐off from 4% to 50% (previous study) reduced genera from 48 to 4 genera. Despite these differences, both studies observed dysbiosis with an increased abundance of gram‐negative bacteria in MC discs as well as a lower beta‐diversity. Cutibacterium was persistently detected in all groups independent of the bioinformatic approach, emphasizing its prevalence.
Conclusion
There is dysbiosis in MC discs. Bioinformatic parameters impact results yet cannot explain the different findings from this and a previous study. Therefore, discrepancies are likely caused by different sample preparations or true biologic differences. Harmonized protocols are required to advance understanding of the disc microbiome and its clinical implications.
Keywords: Cutibacterium acnes, metagenomics, microbiome, Modic changes
The disc microbiome challenges traditional views of sterility, and this study compares microbiomes in different disc types, aiming to explain discrepancies in prior research. Bioinformatic variations were found to influence outcomes, but dysbiosis in Modic type discs was consistently observed. The study emphasizes the need for standardized protocols in disc microbiome research for a more cohesive understanding.

1. INTRODUCTION
The microbiome of intervertebral discs (IVDs) has become a focal point of intense debates within the spine research community because it challenges the longstanding paradigm of the disc's sterility and because its clinical relevance is unclear. Particularly in the context of Modic changes (MC), the presence of bacteria, specifically Cutibacterium acnes (C. acnes), within the disc has been a long‐debated topic. 1 , 2 , 3 , 4 , 5 Reports of a disc microbiome challenge the paradigm of the sterile disc and raise the questions, of what are commensals and what are pathogenic bacteria. 6 , 7
Rapid technological advancements have revolutionized our ability to explore the microbiome in low‐biomass samples with next‐generation sequencing (NGS). This innovation allows us to get insight into the complete microbial DNA present within a sample, which marks a significant leap forward from the traditional approach of culturing bacteria in vitro. In particular, the significant constraint of selecting cultural conditions that favor the proliferation of specific bacteria was overcome with NGS. 8 In addition, certain bacteria have very slow growth or remain completely unculturable and have therefore never been considered as part of the disc's microbiome. While DNA sequencing is highly sensitive and comprehensive, it cannot differentiate between live and deceased bacteria. Skeptics often focus on this aspect, suggesting that the identified bacterial DNA could potentially result from contamination or dead bacteria rather than from live resident bacteria with the potential to have a functional impact on the disc.
Rajasekaran et al. and Astur et al. were the first to perform in‐depth analysis of the disc's microbiome with NGS. 6 , 9 However, the overlap between the detected bacteria was very small. The potential causes for this disparity could be attributed to one or a combination of the following factors: First, True biologic difference, for example, geographic and ethnic differences in patients, Second, differences in sample preparation and the presence of differing contaminating bacteria in reagents, 10 which mask the number of true bacteria present. Third, the bioinformatic analysis. Since they used different methodologies (points 2 and 3), true biologic differences and their clinical relevance (point 1) cannot be answered, emphasizing the need to harmonize the procedures. For example, both studies handle the control for contamination differently, which greatly affects the amount and speciation of detected bacteria. Consequently, a consensus is needed on the sample preparation, bioinformatic pipeline, and the use of contamination controls to ensure the comparability, reliability, and reproducibility of the results.
Despite the lack of methodological consensus, bacteria and particularly C. acnes have been found in discs using different methods. 7 , 9 , 11 , 12 , 13 The discovery of a vast number of bacteria present within the disc questioned the importance of C. acnes and raised the possibility that other bacteria or a state of a dysbiosis may be clinically more relevant. 6 , 7 , 9 In prior NGS studies of IVDs, Astur et al. 6 did not detect C. acnes in any disc. 6 In contrast, Rajasekaran identified C. acnes, however it was not among the most abundant bacterial species nor was it different between MC and non‐MC discs. Hence, the new paradigm of a disc microbiome challenges the conventional notion of disc sterility and emphasizes the urgent necessity for a harmonized methodology for disc microbiome analysis. Moreover, in the context of MCs, it is essential to determine whether C. acnes should persist as the predominant pathogen under investigation or if a critical reassessment is warranted to encompass a broader spectrum of bacterial species.
The study aimed: (1) to compare our methods and results with previous disc microbiome studies and to identify the differences in the bioinformatic pipeline, (2) to demonstrate, based on our data, how variations in the previously identified bioinformatic parameters can yield distinct outcomes; and (3) to gain insights into the differences in microbiome profiles among non‐Modic change (nonMC) discs, Modic type 1 change (MC1) discs, and Modic type 2 change (MC2) discs.
2. METHODS
2.1. Disc collection
Twenty four nonMC, 25 MC1, and 21 MC2 IVDs were collected aseptically from patients undergoing lumbar spinal fusion surgery at the Balgrist University Hospital Zurich between May 25, 2021 and October 6, 2022. The research followed the principles outlined in the Declaration of Helsinki and discs were collected with informed consent and with approval from the local ethics commission. Exclusion criteria were previous lumbar spinal fusions and current or chronic systemic inflammatory or infectious diseases.
A board‐certified radiologist specialized in musculoskeletal conditions graded disc degeneration according to Pfirrmann, 14 and classified adjacent bone marrow changes according to Modic 15 based on pre‐operative magnetic resonance imaging not older than 3 months. General demographic metrics such as age, gender and BMI were collected, and patients filled out the visual analog score (VAS) for back pain and the Oswestry disability index (ODI) before surgery. Friedman's test was used to determine significant differences in age, body mass index (BMI), ODI, and VAS back pain score among the three groups. Wilcoxon test was used to test differences in Pfirrmann grade between the groups and the gender distribution differences between the three groups were examined with the use of a Fisher's exact test.
Once the disc was removed, it was immediately placed in sterile tubes. All subsequent steps were also conducted in a sterile environment. The entire procedure also involved an additional 10 contamination control samples, which included all the buffers but did not contain any tissue. Each disc was minced into small pieces and mixed. Finally, the discs were snap‐frozen and stored at −80°C until all samples were collected and processed for genomic DNA extraction, 16S rRNA DNA amplification, and amplicon sequencing.
2.2. DNA isolation
All discs underwent genomic DNA extraction using the Qiagen Pathogen Kit, following the manufacturer's protocol with an initial overnight incubation step with Proteinase K.
2.3. 16S rRNA PCR
The 16s rRNA V3‐V4 region was amplified using primers with MiSeq‐overhang adapters (Fwd: 5′TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG and Rev: 5′GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC). The PCR reaction consisting of 7 μL forward/reverse Primer Mix (2 μM), 12.5 μL 2× KAPA Hifi HotStart Ready Mix (Roche, Basel, Switzerland) and 5.5 μL DNA template was performed using the following cycle conditions: initial denaturation at 94°C for 2 min, 35 cycles of denaturation at 94°C for 20 s, annealing at 64°C for 30 s, elongation at 68°C for 30 s, final elongation step at 72°C for 5 min. PCR products were verified with an agarose gel.
2.4. Library preparation and sequencing
A 2‐step PCR approach was used to generate libraries; the first PCR amplifies the specific region (Forward primer: CCTACGGGNGGCWGCAG, Reverse primer: GACTACHVGGGTATCTAATCC) and the second PCR adds Illumina adapters and 8 bp barcodes to the amplicons. This approach employs Truseq tag sequences. The PCR products generated from the second PCR were purified with magnetic beads. The quality and quantity of the libraries were validated using the Agilent 4200 TapeStation system and the GloMax® Explorer System, Promega. After library quantification, libraries were normalized to 10 nM in Tris‐Cl 10 mM, pH 8.5 with 0.1% Tween 20 and pooled equimolarly.
The Miseq Sequencing Systems (Illumina, Inc, California, USA) was used for cluster generation and sequencing according to standard protocol. Sequencing was performed with a run configuration of pair end 250 bp. The sequencing raw data were processed with Trimmomatic (0.39) by trimming the Illumina‐specific adapters, removing reads below average quality of Q20 and shorter than 30 bp. The reads were then mapped to GRCh38.p13 human reference genome using Bowtie2 (2.4.2) with a standard set of parameters, to check for contamination resulting from off‐target amplification. 16 The reads that mapped to the human genome were filtered out from both the forward and the reverse reads using Seqtk (1.3).
2.5. 16 s rRNA data analysis
The described bioinformatic workflow will be referred to as our best practice approach throughout the paper. Data analysis of the pre‐processed 16S rRNA reads was performed with QIIME 2 next‐generation microbiome bioinformatics pipeline (v2022.2). Raw reads were transformed into a QIIME 2 artifact format (.qza) and the amplicon sequencing variants (ASVs) were extracted from the data using Divisive Amplicon Denoising Algorithm 2 (DADA2) implemented in QIIME 2 as a plugin. DADA2 corrected amplicon errors, dereplicated and denoised the sequences, identified and removed chimeras and merged the paired end reads. 17 The extracted representative sequences were assigned a taxonomic lineage using a sklearn‐based Naive Bayes classifier trained on the SILVA v138 99% 16S database narrowed down to the V3‐V4 region. The taxonomic classification was compared with a classifier trained on the Greengenes v138 99% database. SILVA database was chosen as the preferred choice due to the close correspondence between the two but an outdated classification for a small set of bacterial genomes by the Greengenes database. 18 , 19 The phylogeny plugin in QIIME 2, which utilizes MAFFT and FastTree for alignment, was also used to estimate the rooted and the unrooted tree. The data were subsequently processed in Rstudio (version 4.3). First, bacterial contaminants in the experimental samples were identified and removed from the samples with Decontam (1.20.0), using the prevalence method, comparing the composition of the positive samples to the negative controls (threshold = 0.5). Additionally, ASVs present in less than 4% of the samples and having a count of less than or equal to 1 were filtered out. The gram stain of each genus was determined using the AMR R package (v2.1.0). Phyloseq (1.44.0) was used to measure a set of standard alpha diversity metrics (Shannon, Simpson, Observed, Chao1, ACE and Pielou). The significance of the difference in Shannon diversity index between samples was tested using the all‐group and pairwise Kruskal–Wallis test (a non‐parametric version of ANOVA). Beta diversity (Jaccard, Bray‐Curtis and weighted Unifrac) was also estimated with the phyloseq package and it was performed on transformed counts (counts per taxon normalized by the total sum of counts per sample).
2.6. Comparison to other disc microbiome studies
Our study's methods and bioinformatic pipeline were compared to the ones used by Astur et al. 6 and Rajasekaran et al. 9 who previously used metagenomics to investigate the disc microbiome. For comparison of the microbiome disc results only the study of Rajasekaran et al.'s study 9 was used, given that Astur et al.'s 6 research focused on herniated discs. The main difference between the groups of our study to that of Rajasekaran et al. 9 lies in our study's additional division of the MC group into MC1 and MC2.
2.7. Comparison of different bioinformatic approaches
Based on the comparison to Rajasekaran et al.'s study the most critical steps in the bioinformatic processing pipeline were identified and the data of this study were used to compare the results when applying the methodologies from Rajasekaran's study. The main result that was compared was the extracted bacterial genera. The bioinformatic methodologies compared were (1) taxonomic lineage assignment: ASV versus operational taxonomic unit (OTU) and (2) different prevalence cut‐offs: 4% versus 50%. When the prevalence cut‐off is mentioned, it consistently corresponds to the minimum percentage of samples in which a particular ASV or OTU, depending on the analysis, was detected.
OTU table was computed from the ASV table with the tip glom function from the phyloseq R package using agglomerative hierarchical clustering (agnes). The data was then subsequently agglomerated at a taxonomic level of interest (genus). For comparison of OTUs and ASVs, the analysis involved the assessment of the detected genera, their distribution within each group, and the median abundance of all ASVs/OTUs across the three MC groups, both overall as well as split into gram positive and negative genera. The significance of the difference in median abundance among the groups was evaluated with use of the Friedman's test, followed by multiple comparisons adjusted for false positives through Dunn's statistical hypothesis testing.
The different filter methods were based on the number of samples in which a specific OTU or ASV was detected. Three distinct cut‐off criteria were assessed: the requirement for the ASV to be present in at least 4% of all patients, the presence of the ASV in at least 50% of patients within at least one group, or the presence of the ASV in at least 50% of all patients. Pie charts showing the genera distribution within each group based on their prevalence were used for comparison of the different filters.
Invariant results from the bioinformatic variations were compared between nonMC, MC1 and MC2 and compared to Rajasekaran et al.'s 9 MC microbiome study.
3. RESULTS
3.1. Patient demographics
Age and gender did not differ between groups (Table 1). Disc degeneration was not significantly different between the groups (p = 0.448). The nonMC group had higher BMI compared to the MC1 group (p = 0.019) (Table 1).
TABLE 1.
Patient demographics of the groups divided into no Modic change (nonMC), Modic type 1 change (MC1), and Modic type 2 change (MC2) discs. Age, body mass index (BMI), Oswestry disability index (ODI), and visual analog scale (VAS) are indicated as mean ± standard deviation (SD). Gender distribution is described as percentage of females per group and Pfirrmann grade is indicated as the median and the interquartile ratio (IQR). Significant p‐values are highlighted in bold.
| nonMC (n = 24) | MC1 (n = 25) | MC2 (n = 21) | p‐value | |
|---|---|---|---|---|
| Pfirrmann Grade (Median [IQR]) | 4 [2, 4] | 4 [3, 4] | 4 [4, 4.5] | 0.267 |
| Age (Average ± SD) | 63.5 ± 17.1 | 62.0 ± 15.6 | 64.2 ± 10.2 | 0.781 |
| BMI (Average ± SD) | 30.5 ± 6.2 | 26.3 ± 4.2 | 28.2 ± 4.2 | 0.019 |
| Female (Percentage) | 42% | 56% | 29% | 0.197 |
| ODI (Average ± SD) | 43.3 ± 17.8 | 42.9 ± 19.7 | 36.4 ± 15.4 | 0.848 |
| VAS back pain (Average ± SD) | 6.6 ± 2.2 | 6.1 ± 2.7 | 6.7 ± 1.9 | 0.708 |
3.2. Comparison to previous disc microbiome studies
The comparison of the sample preparation and bioinformatic methods of the three disc metagenomic studies is shown in Table 2. All three studies used the same variable 16 s rRNA region, and the same sequencing machine. DNA extraction was the same for Astur et al. and Rajasekaran et al. For contamination controls, our study included 10 tubes with only the reagents. The other two studies did not mention any control samples.
TABLE 2.
The methods for metagenomic sequencing and analysis of discs of this study were compared to two prior disc metagenomic studies of Rajasekaran et al. 9 and Astur et al. 6 study.
| Our study | Rajasekaran et al. 9 | Astur et al. 6 | |
|---|---|---|---|
| Patients | 70 (24 nonMC, 25 MC1, 21 MC2) | 40 (20 non‐Modic, 20 Modic, 20 Control) | 17 (herniated discs) |
| Patient average age | 63 (From 13 to 89 years) | 47 (From 19 to 70 years) | 42.8 years (From 31 to 59 years) |
| Variable region used | V3‐V4 (341F and 806R) | V3‐V4 (341F and 806R) | V3‐V4 |
| Filtered for human genome content | Yes, with Bowtie2 (reference genome GRCh38p13) | No | No |
| DNA extraction | QIAmp UCP Pathogen Mini Kit | QIAmp DNA Microbiome Kit | QIAmp DNA Microbiome Kit |
| Contamination controls | 10 tubes included | N/A | N/A |
| Sequencing platform | Illumina | Illumia NovoSeq 6000 | Illumina |
| Decontamination strategy | Decontam R package | N/A | N/A |
| Database | SILVA | Inhouse database consisting of: Greengenes, SILVA and 16s core bacterial database | RDP tools version 2.12 |
| Filtering |
ASVs present at count <1 Comparison of multiple cutoffs:
|
Presence in at least 70% of samples & >100 OTUs | Minimum of 20 sequence reads |
| Bioinformatic Pipeline | QIIME2 | QIIME2 | Geneious Prime Softwares (Geneious Prime 2020.1.2) |
| Taxonomic lineage assignment | ASVs | OTUs | Neither |
Abbreviations: ASV, amplicon sequencing variant; OTU, operational taxonomic unit.
For sequencing analysis, the three studies all used very different approaches. Differences were seen in taxonomic lineage assignment, filtering strategies, and the database used to annotate the bacteria. No information was provided for bioinformatic identification of potential contaminant reads in the other two studies.
A step‐by‐step description of our methodology is depicted in Figure 1, highlighting different methodologies compared to Rajasekaran's study. Initial taxonomic lineage assignment assigned 2415 ASVs (Figure 1, step 8). Decontam eliminated 120 ASVs (step 9) mainly affecting ASVs belonging to the phylum Proteobacteria, Actinobacteria, and Firmicutes and on genus level this mainly affected ASVs belonging to Escheria‐Shigella, Staphylococcus, and Pelomonas. The second filtration step eliminated ASVs found in fewer than either 4% or 50% of samples with counts greater than 1 (step 10). This resulted in the removal of over 2000 ASVs, which leaves a final count of 180 and 26 ASVs, respectively.
FIGURE 1.
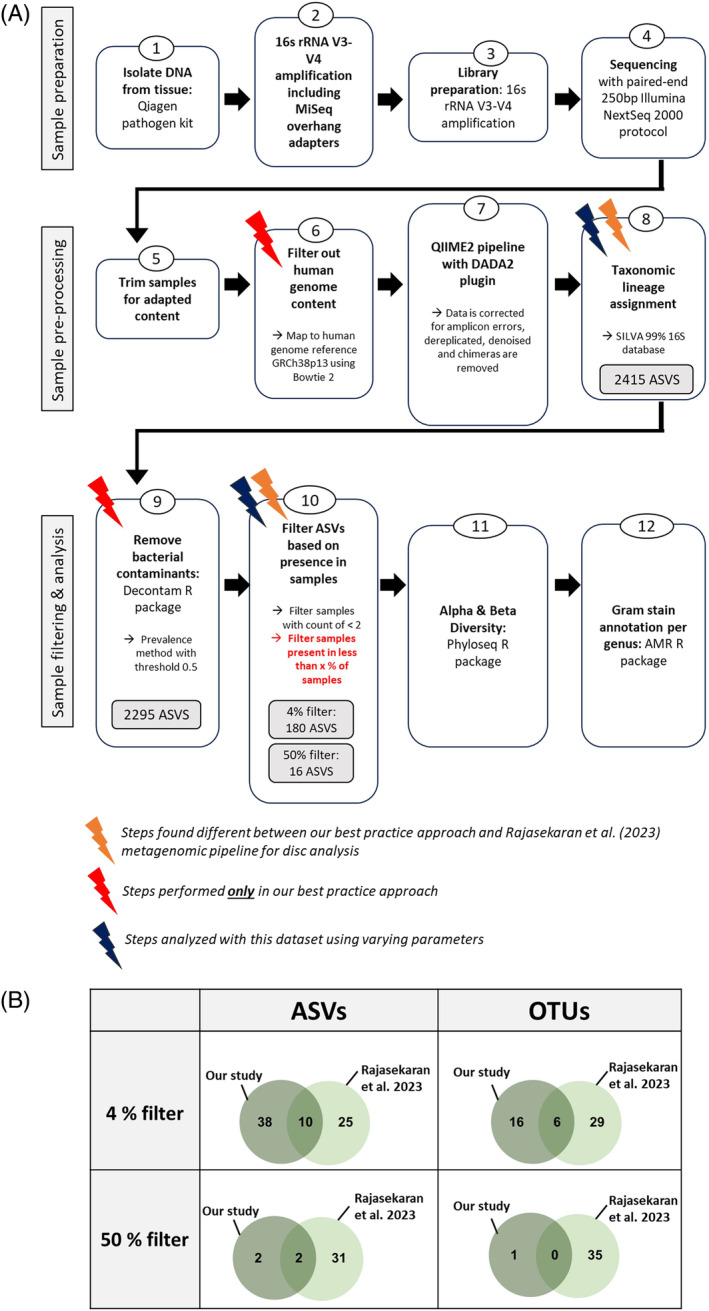
(A) The workflow used to perform our study in a best practice approach. Orange lightning bolts indicate steps found to differ from the prior investigation of the MC metagenome by Rajasekaran et al., 9 while red indicates steps done only in this workflow and not in prior MC microbiome studies. Blue lightning bolts indicate the steps which were further investigated with the use of the dataset from this study. (B) Direct comparison of the number of genera detected in our study compared to the number presented by Rajasekaran et al. 9 Different parameters were used for the analysis of our samples including the use of either ASVs or OTUs as well as the application of two different prevalence cut‐off filters for the number of samples indicating the presence of these genera. ASV, amplicon sequencing variant; OTU, operational taxonomic unit.
Both Rajasekaran et al.'s and our study aimed to identify differences in disc microbiome in MC discs. Therefore, we compared in‐depth the two methodologies. The discrepancies consist of two steps that had different parameters (steps 8 & 10), two steps which were only implemented in this microbiome study (steps 6 & 9) (Figure 1). The two steps that differed between our and Rajasekaran's study were selected for further comparison. We found that different filters had a large effect and that ASVs detected more genera than OTU (Figure 1B). All possible combinations of filters and taxonomic lineage assignment failed to converge our results with Rajasekaran's result. No overlaps in genera were found when using the same parameters as in Rajasekaran's study, indicating that the observed differences are not due to filtering thresholds or taxonomic lineage assignment. Using a setup similar to Rajasekaran's setup (OTU, 50% filtering), Methylobacterium‐Methylorubrum was the sole remaining genera in our dataset, a bacterium that Rajasekaran did not detect. They reported as the top four bacteria Psuedomonas, Acinetobacter, Prevotella, and Orchobactrum. In contrast, under our preferred parameters for exploratory screening (ASVs, 4% filtering) as well as with our most stringent filter (ASVs, 50% filtering), the top four genera were Pelmonas, Sphingomonsa, Methylobacterium‐Methylorubrum, and Cutibacterium.
3.3. Effect of ASVs versus OTUs with respect to MC microbiome
A decrease in bacterial diversity in MC groups was observed, which was not influenced by using ASVs or OTUs. Alpha diversity was similar in all groups (Supplementary Figure 1). Beta diversity was lowest for MC1 compared to all other groups and highest for nonMC compared to all other groups (Figure 2A).
FIGURE 2.
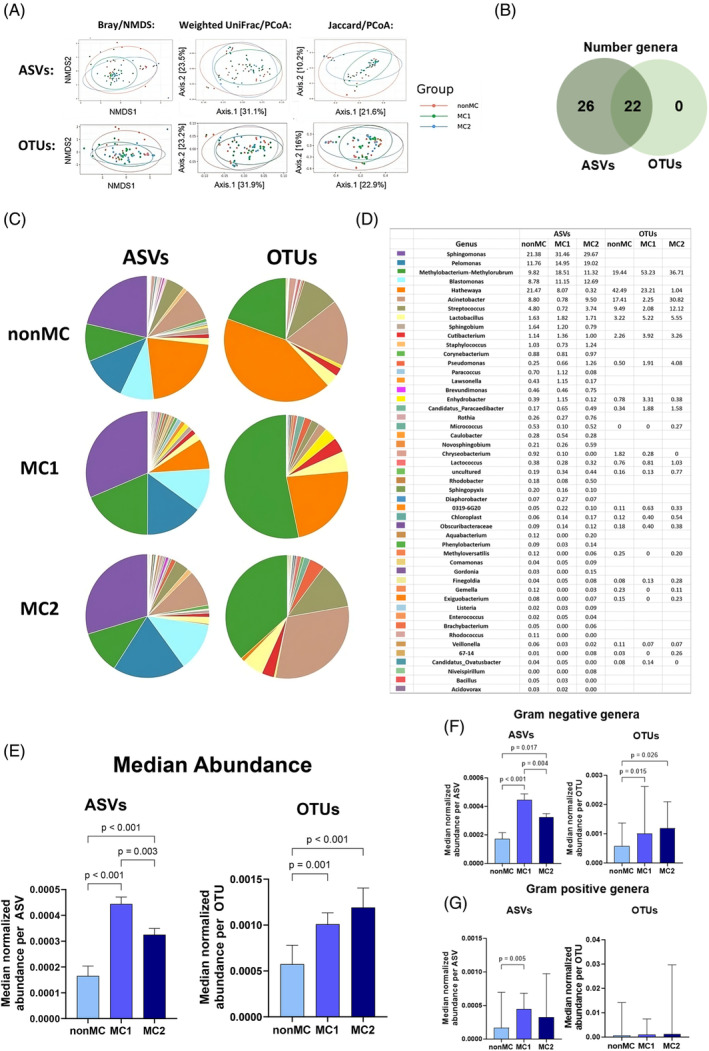
ASV compared to OTU annotation of genera found in at least 4% of all samples. (A) Beta‐diversity calculated with Bray/NMDS, Weighted UniFrac/PCoA and Jaccard/PCoA with either ASV annotation or OTU annotation. (B) The overlap of genera detected with ASVs compared to OTUs. (C) The microbial distribution of ASVs compared to OTU on the genera level with a corresponding (D) table indicating the percentage of each genus based on the median normalized abundance. (E) The median abundance of genera extracted with ASVs or OTUs compared between nonMC, MC1, and MC2. (F) Gram‐negative or (G) gram—positive extracted ASVs and OTUs compared between nonMC, MC1, and MC2. Significance tested with Friedman's test and corrected for multiple comparisons with Dunn's statistical hypothesis testing. ASV, amplicon sequencing variant; OTU, operational taxonomic unit.
The number of genera detected with ASV and OTU were largely different. OTU annotation identified 22, ASV annotation 48 different genera with use of a 4% prevalence cut‐off. All genera identified by OTUs were also identified with ASVs (Figure 2B), indicating that ASVs provided better resolution. The distribution of genera was also strongly affected by taxonomic lineage assignment. ASVs revealed four prominent genera, namely Sphingomonas, Pelmonas, Methylobacterium‐Methylorubrum, and Blastomonas while OTUs were characterized mainly by Methylobacterium‐Methylorubrum and Hathewaya in nonMC and MC1 and by Methylobacterium‐Methylorurum together with Acinetobacter in MC2 (Figure 2C,D). Interestingly, two out of the four dominant genera that contributed significantly to the microbiome with the use of ASVs were absent in the OTU data, which resulted in the pronounced overrepresentation of the previously named bacteria evident in the pie charts of the OTUs (Figure 2C,D).
The analysis of median abundance for all ASVs across the groups resulted in a significantly lower median ASV abundance in nonMC compared to both MC1 (p < 0.001) and MC2 (p < 0.001). Additionally, MC1 displayed a higher overall median ASV abundance compared to MC2 (p = 0.003) (Figure 2E). Gram‐negative bacteria significantly increased in MC1 compared to nonMC for ASVs and OTUs, with an additional increase in MC1 compared to MC2 in ASVs (Figure 2F). For gram‐positive bacteria, ASVs but not OTU revealed a significantly greater abundance in MC1 compared to nonMC (Figure 2G).
Consistent findings independent of the use of ASV or OTU were that MC1 had lower beta diversity, yet more bacteria, in particular gram‐negative genera (Figure 2A,E,F). In addition, similar patterns can be observed when looking at Hathewaya which was found to make up a large part of the microbiome in nonMC and MC1 but not in MC2, while Acinetobacter took its place in MC2.
3.4. Comparison of different filter cut‐offs
Using different filter cut‐offs strongly affected the number and type of detected ASVs and genera (Figure 3). While a 4% cut‐off leaves 180 ASVs and 48 genera, a 50% prevalence cut‐off leaves only the genera Pelmonsa, Methylobacterium‐Methyloruburm, Sphingomonas, and Cutibacterium. However, when a group specific 50% prevalence cut‐off was applied, four additional genera make the cut, as they are predominantly found in nonMC discs. These include Hathewaya, Lactobacillus, Corynebacterium, and Staphylococcus (Figure 3B,C).
FIGURE 3.
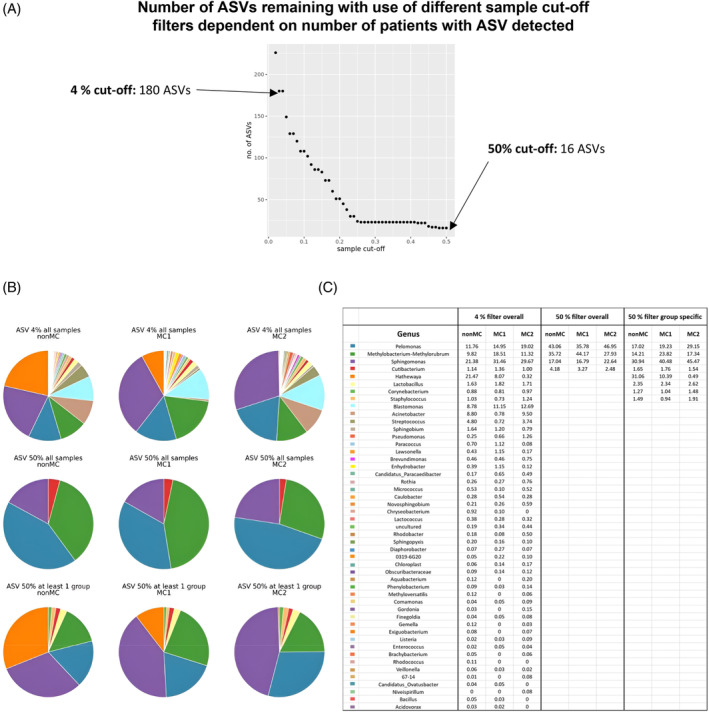
Comparison of different filter cut‐offs. (A) The graph shows the number of ASVs left when the prevalence filter cut‐off is set at different percentages. (B) Genera distribution based on the median abundance per genera with three different filters: genera detected in more than 4% of patients overall (top row), genera detected in more than 50% of patients in at least one group (middle row) and genera detected in overall more than 50% of patients (bottom row). (C) The table depicts the information from the pie charts in percentages based on the median abundance per genus. ASV, amplicon sequencing variant.
3.5. Cutibacterium in the spotlight
Two ASVs were extracted which belong to the genus Cutibacterium. One was detected in 43 samples, the other in 44 out of the 70 samples, with all of them overlapping and the 44 just having one additional patient. Cutibacterium ASVs were present in 71% of nonMC patients, 56% of MC1, and 62% of MC2 discs without statistical difference. This high rate of occurrence led to the genera Cutibacterium being one of four bacteria that was retained throughout all bioinformatic approaches tested.
4. DISCUSSION
The microbiome of the disc challenges the paradigm of a sterile disc and represents a novel and relatively unexplored domain that is currently receiving careful and critical consideration in the field. Disc dysbiosis has previously been reported for MC discs by Rajasekaran et al. and with this study we provide further evidence that the disc harbors its own microbiome. In this study, consistent with Rajasekaran et al., reduced bacterial diversity in MC discs was found, indicating a state of dysbiosis. In addition, absolute number of bacteria was higher in MC1 discs, mainly of gram‐negative bacteria. This suggests that they either infiltrated MC1 discs or that the environment of MC1 discs favored their proliferation. However, large differences in the number and speciation of bacteria were found compared to Rajasekaran's study. There are three possible explanations for the observed differences: (1) true biological differences due to, for example, geographic and ethnic differences, (2) differences in sample preparation, for example, DNA isolation, contaminations in buffers, (3) differences in bioinformatic analysis. Point 1 is the most clinically relevant, yet it cannot be addressed until the source of data variance originating from points 2 and 3 is understood and minimized using a harmonized protocol. With this goal in mind, we used our dataset to test if the bioinformatic discrepancies between Rajasekaran's and our study could account for the observed differences in results. We compared the results generated by our dataset when using: (1) different taxonomic assignments, that is, ASVs versus OTUs and (2) three different filter cut‐offs. We found that changing taxonomic assignment and filter cut‐offs largely affects results but cannot explain the discrepancies to Rajasekaran's study. They reported 35 different genera using OTU and a 50% filter—parameters that, as evidenced by our dataset, lead to a reduction in the number of identified bacteria. In this study, these settings resulted in only one genus. To match the high number of genera detected in their study, a less restrictive 4% filter had to be used. Yet, the majority of genera detected remained different from the ones reported by Rajasekaran et al. This suggests that either their patients had a much richer and different microbiome (biologic variance), or that more and different bacteria were introduced during their sample preparation (e.g., different buffer solutions).
It is known that different taxonomic assignments affect the results of microbiome analysis. 20 OTU uses a 97% sequence similarity to assign taxonomy. This causes grouping of similar sequences. In contrast, ASVs capture single nucleotide differences, and hence provide higher resolution and specificity than OTU. It has been suggested to use ASVs as standard due to their comprehensiveness and easier reproducibility. 20 , 21 , 22 In this study, using ASV instead of OTU almost doubled the number of genera found and mainly increased the abundance of the bacteria found in MC2 discs. Yet, both ASV and OTU consistently showed reduced bacterial diversity in MC discs and more gram‐negative bacteria in MC1 discs potentially making these robust results.
Filtering is an important step to reduce noise and errors from sequencing, mitigate sample contamination, to focus on dominant bacteria, to avoid statistical zero‐inflation, and enhance statistical power. However, in low microbiome biomass tissue like the disc, stringent filtering can remove potentially important bacteria species that are only present in a subset of samples. In this study we found that a stringent filter, which only retained ASVs present in over 50% of patients, excluded group specific ASVs. For example, Hathewaya, Lactobacillus, Corynebacterium, and Staphylococcus were filtered out, because they were predominantly found in nonMC samples. To avoid this risk, the 50% cut‐off filter was applied to each group rather than all samples. This filtering strategy overcomes the mentioned drawbacks by emphasizing the bacteria most relevant for the majority of patients, potentially identifying those crucial for clinical treatments. However, when the aim of the study is to explore the diversity of the microbiome in a low biomass sample, it is not advisable to use such stringent filtering and for this case we propose the use of the 4% cut‐off. To address the risk of contamination reads when using a 4% cut‐off filter, 10 contamination control samples (not tissue) were used along with the decontamination algorithm “Decontam” in R. Decontam leverages the inverse relationship between contaminant‐derived sequences and total DNA concentration for effective decontamination without data loss in low‐biomass metagenomic data. 23 It has previously been suggested to use ligamentum flavum or surrounding tissues as decontamination controls. 6 However, considering them as ‘negative controls’ may be inappropriate because soft tissue may also harbor a microbiome. 24 In addition, the exclusion of discs with microbial presence in the surrounding tissues could eliminate critical samples if the assumption is that pathogenic bacteria enter the disc through surrounding tissue. Besides bioinformatic variations, physical sample processing before sequencing is a likely source of variance. 25 , 26 Therefore, we suggest that a harmonized protocol for disc microbiome analysis should encompass bioinformatic analysis and protocols to isolate bacterial DNA from disc tissue.
Lastly, this dataset adds further evidence for the presence of Cutibacterium in the disc. Notably, the genus Cutibacterium persisted among the five genera even under stringent filter methods using ASVs, being detected in over 50% of samples. Our data are in agreement with Rajasekaran et al. 9 who also found Cutibacterium in discs, not as one of the most abundant genera, and also without a clear association with MC discs. However, the complexity of factors that influence bacterial pathogenicity beyond absolute abundance, underscores the need to further investigate the potential pathogenic role of Cutibacterium in MC. 27 , 28
This study has some limitations. First, multiple surgeons and technical operation assistants were involved in collecting the tissue. Different harvesting techniques may have different risks for contamination. There was no assurance that fresh surgical tools were used to collect the disc into sterile containers. While this could have affected the observed microbiome, this random effect does not affect the observed dysbiosis in MC nor does it restrict the notion that bioinformatic processing has a large impact. Second, the classification into nonMC, MC1, and MC2 does not consider the diverse sub‐ and endo‐phenotypes identified within MCs, nor was endplate damage or potential herniations included in the grouping of the patients. This could also largely impact the composition of the microbiome in each of the groups. 29 In addition to this, variations in MRI machines and sequences used among different centers can also influence the classification. Despite this limitation, it can be said that the overall bacteria detected in the disc through our approach was largely different from the bacteria detected by previous authors investigating the microbiome of the disc, independent of MC classification.
In conclusion, changing key bioinformatic parameters, that is, taxonomic lineage assignment and filtering cut‐offs had a large impact on the resulting microbiome. However, using similar parameters as a prior study investigating the MC microbiome did not converge results. Therefore, the observed discrepancies were either introduced during sample processing or are true biologic differences. Before any clinically relevant conclusions about the role of bacteria in MC and disc degeneration can be drawn, the source of variance needs to be identified and understood, and harmonized protocols for sample processing and bioinformatic analysis are required. Ultimately, the availability of a robust and reproducible methodology will allow the exploration of this untapped metagenomic landscape as a new source of biomarkers and potential treatment targets.
AUTHOR CONTRIBUTIONS
Tamara Mengis: Data curation; investigation; methodology; writing—original draft; visualization. Natalia Zajac: Bioinformatic data analysis; writing—original draft; visualization. Laura Bernhard: Methodology; data curation; writing—review. Irina Heggli: Methodology; writing—review and editing. Nick Herger: Writing—review and editing; methodology. Jan Devan: Writing—review and editing. Roy Marcus: Data curation; writing—review. Florian Brunner: Writing—review and editing. Christoph Laux: Data curation; writing—review. Mazda Farshad: Writing—review and editing. Oliver Distler: Funding acquisition; writing—review and editing. Stefan Dudli: Funding acquisition; writing—review and editing.
FUNDING INFORMATION
This study was supported by a career research grant from the Foundation for Research in Rheumatology (FOREUM) (SD and OD), by a FOREUM Research Voucher (SD), by the ISSLS Basic Science Research Grant (SD) and by a grant from the Swiss National Fond (SD, Grant No. 207989).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
Supporting information
SUPPLEMENTARY FIGURE 1. Alpha diversity of nonMC, MC1, and MC2 with use of OTUs.
ACKNOWLEDGMENTS
We would also like to thank the Swiss Center for Musculoskeletal Biobanking (SCMB), which worked together with us to provide the samples in the shortest time possible for further processing. Lastly, we would like to thank the Functional Genomic Center Zurich staff for giving their inputs in the project planning and performing the sequencing.
Mengis T, Zajac N, Bernhard L, et al. Intervertebral disc microbiome in Modic changes: Lack of result replication underscores the need for a consensus in low‐biomass microbiome analysis. JOR Spine. 2024;7(2):e1330. doi: 10.1002/jsp2.1330
REFERENCES
- 1. Dudli S, Liebenberg E, Magnitsky S, Miller S, Demir‐Deviren S, Lotz JC. Propionibacterium acnes infected intervertebral discs cause vertebral bone marrow lesions consistent with Modic changes. J Orthop Res. 2016;34(8):1447‐1455. doi: 10.1002/jor.23265 [DOI] [PubMed] [Google Scholar]
- 2. Heggli I, Mengis T, Laux CJ, et al. Low back pain patients with Modic type 1 changes exhibit distinct bacterial and non‐bacterial subtypes. Osteoarthr Cartil Open. 2024;6(1):100434. doi: 10.1016/j.ocarto.2024.100434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Manniche C, O'Neill S. New insights link low‐virulent disc infections to the etiology of severe disc degeneration and Modic changes. Future Sci OA. 2019;5(5):FSO389. 10.2144/fsoa-2019-0022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Perry A, Lambert P. Propionibacterium acnes: infection beyond the skin. Expert Rev Anti‐Infect Ther. 2011;9:1149‐1156. doi: 10.1586/eri.11.137 [DOI] [PubMed] [Google Scholar]
- 5. Stirling A, Worthington T, Rafiq M, Lambert PA, Elliott TS. Association between sciatica and Propionibacterium acnes . Lancet. 2001;357(9273):2024‐2025. doi: 10.1016/S0140-6736(00)05109-6 [DOI] [PubMed] [Google Scholar]
- 6. Astur N, Maciel BFB, Doi AM, et al. Next‐generation sequencing (NGS) to determine microbiome of herniated intervertebral disc. Spine J. 2022;22(3):389‐398. doi: 10.1016/j.spinee.2021.09.005 [DOI] [PubMed] [Google Scholar]
- 7. Rajasekaran S, Soundararajan DCR, Tangavel C, et al. Human intervertebral discs harbour a unique microbiome and dysbiosis determines health and disease. Eur Spine J. 2020;29(7):1621‐1640. doi: 10.1007/s00586-020-06446-z [DOI] [PubMed] [Google Scholar]
- 8. Chen P, Sun W, He Y. Comparison of the next‐generation sequencing (NGS) technology with culture methods in the diagnosis of bacterial and fungal infections. J Thorac Dis. 2020;12(9):4924‐4929. doi: 10.21037/jtd-20-930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rajasekaran S, Vasudevan G, Easwaran M, et al. ‘Are we barking up the wrong tree? Too much emphasis on Cutibacterium acnes and ignoring other pathogens’—a study based on next‐generation sequencing of normal and diseased discs. Spine J. 2023;23:1414‐1426. doi: 10.1016/j.spinee.2023.06.396 [DOI] [PubMed] [Google Scholar]
- 10. Mühl H, Kochem AJ, Disqué C, Sakka SG. Activity and DNA contamination of commercial polymerase chain reaction reagents for the universal 16S RDNA real‐time polymerase chain reaction detection of bacterial pathogens in blood. Diagn Microbiol Infect Dis. 2010;66(1):41‐49. doi: 10.1016/j.diagmicrobio.2008.07.011 [DOI] [PubMed] [Google Scholar]
- 11. Agarwal V, Golish SR, Alamin TF. Bacteriologic culture of excised intervertebral disc from immunocompetent patients undergoing single level primary lumbar microdiscectomy. J Spinal Disord Tech. 2011;24(6):397‐400. doi: 10.1097/BSD.0b013e3182019f3a [DOI] [PubMed] [Google Scholar]
- 12. Albert HB, Lambert P, Rollason J, et al. Does nuclear tissue infected with bacteria following disc herniations lead to Modic changes in the adjacent vertebrae? Eur Spine J. 2013;22(4):690‐696. doi: 10.1007/s00586-013-2674-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Capoor MN, Ruzicka F, Machackova T, et al. Prevalence of Propionibacterium acnes in intervertebral discs of patients undergoing lumbar microdiscectomy: a prospective cross‐sectional study. PLoS One. 2016;11(8):e0161676. doi: 10.1371/journal.pone.0161676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pfirrmann CWA, Metzdorf A, Zanetti M, Hodler J, Boos N. Magnetic resonance classification of lumbar intervertebral disc degeneration. Spine (Phila Pa 1976). 2001;26(17):1873‐1878. doi: 10.1097/00007632-200109010-00011 [DOI] [PubMed] [Google Scholar]
- 15. Modic MT, Steinberg PM, Ross JS, Masaryk TJ, Carter JR. Degenerative disk disease: assessment of changes in vertebral body marrow with MR imaging. Radiology. 1988;166(1):193‐199. doi: 10.1148/radiology.166.1.3336678 [DOI] [PubMed] [Google Scholar]
- 16. Walker SP, Barrett M, Hogan G, Bueso YF, Claesson MJ, Tangney M. Non‐specific amplification of human DNA is a major challenge for 16S RRNA gene sequence analysis. Sci Rep. 2020;10(1):16356. doi: 10.1038/s41598-020-73403-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Prodan A, Tremaroli V, Brolin H, Zwinderman AH, Nieuwdorp M, Levin E. Comparing bioinformatic pipelines for microbial 16S RRNA amplicon sequencing. PLoS One. 2020;15(1):e0227434. doi: 10.1371/journal.pone.0227434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. DeSantis TZ, Hugenholtz P, Larsen N, et al. Greengenes, a chimera‐checked 16S RRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069‐5072. doi: 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pruesse E, Quast C, Knittel K, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35(21):7188‐7196. doi: 10.1093/nar/gkm864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chiarello M, McCauley M, Villéger S, Jackson CR. Ranking the biases: the choice of OTUs vs. ASVs in 16S RRNA amplicon data analysis has stronger effects on diversity measures than rarefaction and OTU identity threshold. PLoS One. 2022;17(2):e0264443. doi: 10.1371/journal.pone.0264443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Callahan BJ, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker‐gene data analysis. ISME J. 2017;11(12):2639‐2643. doi: 10.1038/ismej.2017.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Inglis C, Rahmani D, Yeung D, Mun D. A comparison of metabarcoding analysis between ASVs and OTUs‐using data regarding the effects of chronic radiation on the Bank vole gut microbiota. Undergrad J Exp Microbiol Immunol. 2022;27:1–14. https://jemi.microbiology.ubc.ca/ [Google Scholar]
- 23.Davis, NM, Proctor DM, Holmes SP, et al. Simple statistical identification and removal of contaminant sequences in marker‐gene and metagenomics data. Microbiome. 2018;6:226. doi: 10.1186/s40168-018-0605-2 [DOI] [PMC free article] [PubMed]
- 24.Cheng HS, Tan SP, Wong DMK, Koo WLY, Wong SH, Tan NS. The Blood Microbiome and Health: Current Evidence, Controversies, and Challenges. Int J Mol Sci. 2023;24(6):5633. doi: 10.3390/ijms24065633 [DOI] [PMC free article] [PubMed]
- 25. Brooks JP, Edwards DJ, Harwich MD, et al. The truth about metagenomics: quantifying and counteracting bias in 16S RRNA studies. BMC Microbiol. 2015;15(1):66. doi: 10.1186/s12866-015-0351-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hallmaier‐Wacker LK, Lueert S, Roos C, Knauf S. The impact of storage buffer, DNA extraction method, and polymerase on microbial analysis. Sci Rep. 2018;8(1):6292. doi: 10.1038/s41598-018-24573-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Capoor MN, Konieczna A, McDowell A, et al. Pro‐inflammatory and neurotrophic factor responses of cells derived from degenerative human intervertebral discs to the opportunistic pathogen Cutibacterium acnes . Int J Mol Sci. 2021;22(5):1‐16. doi: 10.3390/ijms22052347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dudli S, Miller S, Demir‐Deviren S, Lotz JC. Inflammatory response of disc cells against Propionibacterium acnes depends on the presence of lumbar Modic changes. Eur Spine J. 2018;27(5):1013‐1020. doi: 10.1007/s00586-017-5291-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Määttä JH, Karppinen J, Paananen M, et al. Refined phenotyping of Modic changes. Medicine (United States). 2016;95(22):e3495. doi: 10.1097/MD.0000000000003495 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY FIGURE 1. Alpha diversity of nonMC, MC1, and MC2 with use of OTUs.