

Abstract
Background
Medulloblastoma (MB) is the most common malignant brain tumor in children and requires intensive multimodal therapy. Long-term survival is still dissatisfying and, most importantly, survivors frequently suffer from severe treatment-associated morbidities. The sonic hedgehog pathway (SHH) in SHH MB provides a promising target for specific therapeutic agents. The small molecule Vismodegib allosterically inhibits SMO, the main upstream activator of SHH. Vismodegib has proven effective in the treatment of MB in mice and in clinical studies. However, due to irreversible premature epiphyseal growth plate fusions after systemic application to infant mice and children, its implementation to pediatric patients has been limited. Intraventricular Vismodegib application might provide a promising novel treatment strategy for pediatric medulloblastoma patients.
Methods
Infant medulloblastoma-bearing Math1-cre::Ptch1Fl/Fl mice were treated with intraventricular Vismodegib in order to evaluate efficacy on tumor growth and systemic side effects.
Results
We show that intraventricular Vismodegib treatment of Math1-cre::Ptch1Fl/Fl mice leads to complete or partial tumor remission only 2 days after completed treatment. Intraventricular treatment also significantly improved symptom-free survival in a dose-dependent manner. At the same time, intraventricular application prevented systemic side effects in the form of anatomical or histological bone deformities.
Conclusions
We conclude that intraventricular application of a SHH pathway inhibitor combines the advantages of a specific treatment agent with precise drug delivery and might evolve as a promising new way of targeted treatment for SHH MB patients.
Keywords: medulloblastoma, targeted treatment, intraventricular therapy, Sonic Hedgehog Inhibition
Key Points.
Intraventricular SHH inhibition improves survival of infant SHH medulloblastoma mice and prevents systemic side effects on bone growth.
Intraventricular SHH inhibition constitutes a promising new way of targeted treatment for pediatric SHH MB patients.
Importance of the Study.
Treatment of childhood medulloblastoma requires intensive multimodal therapy, commonly leading to severe treatment related morbidity. Targeted therapy with SHH inhibitor Vismodegib previously proved not feasible in prepubertal pediatric patients because of severe side effects on bone growth upon systemic application. We investigated intraventricular application of Vismodegib in a mouse model of infant SHH medulloblastoma. We observed that intraventricular Vismodegib prevented systemic side effects on bone growth and at the same time significantly improved survival compared to placebo treatment. Intraventricular application of chemotherapeutical agents via a subcutaneous reservoir is already well established in pediatric medulloblastoma therapy. Therefore, we propose that addition of SHH pathway inhibitors to a multimodal treatment regimen would be feasible and might significantly improve treatment response. At the same time, it could reduce the need of other, highly toxic treatment elements like craniospinal irradiation.
Medulloblastome (MB) is a highly malignant childhood brain tumor of the posterior fossa. So far, the prognosis is dismal for many patients. Treatment options of childhood MB comprise maximal surgical resection, polychemotherapy, and craniospinal irradiation (CSI). Because of frequent dissemination via the cerebrospinal fluid, CSI is a central element of this multimodal treatment. However, the immature brain of young children is especially susceptible to radiotherapy-induced long-term side effects including endocrine abnormalities, impaired spinal growth, and neuropsychological dysfunction.1–3 Therefore, there is an imperative to avoid or at least delay CSI especially in children under the age of 3 years and to find alternative strategies to treat and prevent metastasis of childhood MB.4 These alternative strategies so far comprise applying high-dose chemotherapy, intraventricular chemotherapy, for example, with methotrexate (MTX), or a combination with local irradiation.5 Intrathecal chemotherapy has been an integral part of therapy regimens in leukemia and lymphoma for decades.6 In bypassing the blood–brain barrier, intrathecally applied drugs exert their maximal effect in the CSF, while keeping systemic toxicities at a minimal level.7 Prospective studies have provided evidence that intraventricular application of MTX leads to a favorable outcome and can decrease the need for CSI in specific risk groups.3,4,8 Neuropsychological deficits after chemotherapy including intraventricular MTX were less pronounced compared to children receiving CSI.4,9 Hence, intraventricular MTX treatment of pediatric MB is routinely applied and recommended by the HIT MED Guidance in Germany, Switzerland, and Austria. With the method of intraventricular application already established for MB patients in the clinical setting, it is both feasible and urgent to use this treatment modality for a more targeted and less cytotoxic therapy.
MB has been extensively profiled on the molecular level. This led to the definition of four consensus molecular subgroups (Wingless (WNT), Sonic-hedgehog (SHH), Group 3, and Group 4), exhibiting distinctive transcriptional and epigenetic signatures and defining clinically relevant patient subsets.10,11 The SHH subgroup (SHH-MB) accounts for approximately 30% of MBs and is most common in infants under the age of 3 years and in adults.10 The small molecule Vismodegib allosterically inhibits Smoothened (SMO), the main upstream activator of SHH. Vismodegib is an established therapy for basal cell carcinoma harboring SHH pathway mutations and showed significant antitumor effects in the treatment of SHH MB in mice and in clinical studies.12–14 However, a preclinical study showed that oral Vismodegib treatment of skeletally immature mice causes irreversible premature epiphyseal growth plate fusions. In a phase II study, three children developed short stature, skeletal deformities, and disproportionate growth after oral treatment.15,16
These severe side effects prevent the systemic application of Vismodegib in children. We propose that targeting the overactivated SHH pathway in SHH MB with Vismodegib by intraventricular delivery might enable reduction of current intensive chemotherapy and irradiation regimens and thereby contribute to an increased long-term survival with an improved quality of life.
To investigate this, we used a well-established SHH MB mouse model: Math1-cre::Ptch1Fl/Fl mice show a loss of both alleles of Ptch1 in Math1-positive cells, resulting in a constitutively activated SHH pathway in cerebellar granule cell precursors—the cells of origin of SHH MB.17 This leads to the development of SHH MB in early postnatal days. These tumors share histologic, genetic, and transcriptional characteristics of their human SHH MB counterparts.18 We treated this animal model with intraventricular Vismodegib to test both antitumor efficacy and avoidance of premature epiphyseal growth plate fusions, aiming for a targeted therapy strategy for childhood SHH MB.
Materials and Methods
Animals and treatments
Ptch1 Fl/Fl (B6N.129-Ptch1tm1Hahn/J, Jax stock 12457) and Math1-cre (B6.Cg-Tg(Atoh1-cre)1Bfri/J, Jax stock 11104) mice were obtained from The Jackson Laboratory (Bar Harbour, ME, USA). Ptch1Fl/Fl and Math1-cre mice were crossed to generate Math1-cre::Ptch1Fl/Fl mice. All animal procedures were performed in accordance with the German Animal Welfare Act and approved by the Government of Hamburg, Germany (Reference N2018/36). Mice were kept in conventional type II long cages on a constant light–dark rhythm of 12/12 hours. Water and food were given ad libitum. Genotyping of tail tip biopsies at postnatal day (P) 1 was performed by polymerase chain reaction with the primer pairs forward TCCGGGCTGCCACGACCAA, reverse GGCGCGGCAACACCATTTT for Cre and forward TTCATTGAACCTTGGGGAAC, reverse AGTGCGTGACACAGATCAGC for Ptch1. For treatment, Math1-cre::Ptch1Fl/Fl mice were randomized into four treatment groups (male and female mice equally distributed in each group). Treatment was given daily for three consecutive days P11–P13. Mice from the treatment groups OralS1 and OralS2 received 200 mg/kg/d Vismodegib by oral gavage either in solvent 1 (“S1,” 0.5% methyl cellulose/0.2% Tween80) or solvent 2 (“S2,” 80% PEG300). Treatment groups CSF-PlaceboS1 and CSF-PlaceboS2 received S1 or S2 as intraventricular injection into the CSF by stereotactical application. CSF-lowS1-treated mice received 0.2 mg/kg/d Vismodegib in S1 as intraventricular injection. CSF-highS2-treated mice received 1.6 mg/kg/d Vismodegib in S2 as intraventricular injection. The dosage of 0.2 mg/kg/day was approximated from the dosage of intraventricular Methotrexate recommended by the SIOP PNET 5 MB Study (NCT02066220). The stereotactic operation was performed under anesthesia with intranasal isoflurane. Mice were fixated in the stereotactic setup with noninvasive earbars. After disinfection, a sagittal skin incision of approximately 0.5 cm was performed, and the bregma was located. The coordinates for injection were Y: −0.22 mm, X: 0.8 mm, and Z: −1.5 mm from bregma. The injection was performed with a 33 Gauge Hamilton syringe at 40 nl/min. Oral gavage was performed without anesthesia using sterile polypropylene, 22 ga × 25 mm feeding tubes. All mice received analgesia in form of subcutaneous 10 µl/g carprofen (50 mg/mL stock diluted 1:100 in 0.9 % NaCl) at P11–14. Starting at P10, neurological symptoms and weight were noted daily and tibia bone length was measured using a micrometer. Mice were sacrificed at occurrence of neurological symptoms (ataxia, hydrocephalus), or at end of observation either at P15 for evaluation of early treatment effects or at P50 for evaluation of long-term treatment effects.
For investigations of systemic side effects in untreated wild-type (WT) siblings, Ptch1Fl/Fl and Ptch1Fl/Wt mice were treated and observed according to the procedures above. Animals were sacrificed at P15 or P50.
CT imaging
CT images were acquired using the IVIS Spectrum CT. Mice were sacrificed and directly imaged at standard resolution. CT images were edited with the Living Image Software 4.7.3 to create maximum intensity projections.
Chemicals
Vismodegib (synonym: GDC-0449, CAS No.: 879085-55-9) was obtained from MedChemExpress. It was prepared either in solvent 1 (S1): 0.5% methyl cellulose/0.2% Tween80 (Sigma–Aldrich, CAS No.: 9004-67-5 and 9005-65-6) or solvent 2 (S2): 80% PEG300 (Carl Roth GmbH, CAS No: 25322-68-3) for oral and intraventricular dosing after warming and sonication.
X-ray
After sacrifice, the bones of the hindlimbs were prepared by dislocation of the acetabulofemoral joint and removal of surrounding soft tissue. The bones were fixed in 4% PFA overnight. Subsequently, X-rays of the fixed bones were taken by contact radiography (35 kV, 2s exposure; Faxitron XRay Corp., USA)
Histology and immunohistochemistry
Brain tissue was fixed in 4% paraformaldehyde (PFA) for at least 24 h and embedded in paraffin. Sections of 2 µm were stained in hematoxylin and eosin (H&E) according to standard protocols. Immunohistochemical staining of paraffin sections was performed on an automated Ventana staining instrument using the following antibodies: Caspase 3 (Asp175), Cell Signaling, #9664, RRID:AB_2070042, 1:100; CD3, Abcam, ab16669, RRID:AB_443425, 1:100; Cre, Biolegend, #908001, RRID:AB_2565079, 1:75; GFAP, DAKO, M0761, RRID:AB_2109952, 1:200; IBA1, Wako, 019-19741, RRID:AB_839504, 1:1000; iNOS, Antikörper online, ABIN 373696, 1:10; Ki67, Abcam, Ab15580, RRID:AB_443209, 1:100; Neu-N, Sigma–Aldrich, MAB377, RRID:AB_2814948, 1:25; N-Myc, Cell Signaling Technology, 51705S, RRID:AB_2799400, 1:600; OLIG2, Millipore, AB9610, RRID:AB_570666, 1:200; PHH3, Cell Signaling Technology, 9706, RRID:AB_331748, 1:200; SOX2, Abcam, ab97959, RRID:AB_2341193,1:200.
For skeletal phenotyping, dissected skeletons were fixed in 4% PFA for 24 h before they were stored in 80% ethanol. Tibiae and femora were dehydrated in ascending alcohol concentrations and then embedded in methylmethacrylate for undecalcified histology. Histological sections of 4 μm thickness from the longitudinal (tibiae) or frontal (femora) plane were stained by H&E and von Kossa staining procedures as described previously.19,20
For immunohistochemical analysis of collagen type X, dissected skeletons were fixed in 4% PFA and decalcified in Osteosoft (Sigma–Aldrich, Ref 1.01728.1000) before paraffin embedding. Sections of 2 µm were stained with anti-Collagen Type X (Quartett, Cat. No.: 1-CO097-05, 1:200) using the DCS SuperVision2 System (DCS Innovative diagnostic systems, Cat. No.: PD000KIT). For antigen retrieval, the slides were treated 15 min at 37°C with Proteinase K (Abcam, Cat. No.: ab64220).
Image acquisition
H&E and immunohistochemical stainings were imaged with the upright BX43 microscope (Olympus) with a WHN10x-H/22 ocular and Plan Achromat C objective with 20× magnification (“high power field”) or 10× magnification. Pictures were acquired using a SC50 microscope camera (Olympus) and Olympus cellSens Entity 1.15 software.
Overview pictures of brains and bones were acquired using an inverted Research Microscope ECLIPSE Ti2-N (Nikon, Tokyo, Japan) using 4× Plan Fluor objectives. Overview images were composed using the NIS-Elements AR 5.11.03 software by stitching together multiple image frames with the stitching method “optimal path” with an overlap of 15%. Alternatively, the Hamamatsu NanoZoomer 2.0-HT C9600 whole slide scanner (Hamamatsu Photonics, Tokyo, Japan) and NDP view v2.7.43 software was used to obtain overview pictures.
Pictures were edited using Adobe Photoshop CS6 (Adobe Inc., San José, USA) for adjustments of brightness, contrast, and white balance.
Quantification and scoring of immunostainings
For quantification of the area of remaining proliferative tumor tissue in relation to total cerebellar area (Ki67 positive cerebellum area/total cerebellum area in %), pictures of whole brain sections were taken as described above. Quantitative area analysis was performed with version 0.2.3 of the open-source software QuPath.21
For quantification of the immunostainings for PHH3, Cleaved Caspase 3, SOX2, OLIG2, IBA1, and iNOS high-power field images were acquired with the Olympus BX43 microscope and Olympus cellSens software as described above. Cell quantification was performed with ImageJ/Fiji (Wayne Rasband, National Institute of Health, USA). Per biological replicate, four non-overlapping high-power images adding up to an area of 1 mm² were evaluated. Representative images from the cerebellum depicting proliferative tumor tissue or, if absent, depicting the inner granule cell layer were chosen for analysis. For PHH3, Cleaved Caspase 3, SOX2, OLIG2, and iNOS stained sections, manual quantification of immunopositive cells per mm² was performed.
For IBA1 immunolabeled sections, digital image analysis was performed using ImageJ/Fiji. After applying the color deconvolution plugin as well as a consistent threshold to all images of the batch, the DAB-positive pixels (ie, IBA1 immunostaining) were quantified per square millimeter.
For all quantifications, at least three biological replicates were used, and the number of samples are indicated in the figure legends.
Column measurements
The column index and the cells in column analysis were performed using ImageJ/Fiji software as previously described.22 Briefly, H&E stained sections were imaged at 200× magnification and cropped to a region of 1800 × 600 pixels centered on the proliferative zone. A column was defined as at least three consecutive cells with an angle between each cell ranging from −155° to −179° or 155° to 180° and a maximal distance of 20 pixels (=2.2 µm) between the cells. The column index was calculated, as the percentage of cells in columns multiplied by the average column length. The percentage of cells in columns was calculated by dividing the total number of cells by the number of cells in columns.
DNA sequencing
DNA was isolated from tumor tissue and amplified by PCR using standard methods. The primers used for sequencing of Smo are listed in Table 1 (purchased at Metabion international AG/metabion GmbH 2018). Sequencing of the amplified DNA was performed at Microsynth Seqlab, Göttingen, Germany, and evaluated using the SnapGene software (www.snapgene.com) Version 4.3.8.1.
Table 1.
Primers used for sequencing of all 12 coding exons of Smo
| Exon (Smo) | Forward primer, 5´→ 3´ | Reverse primer, 5´→3´ |
|---|---|---|
| 1 | GGGCCCTCCCCCAGCCTCCGCGGAT | AGGCCTGAGCCTTTTCTTTCCGTCA |
| 2 | GGGTTACAACTGTATCAGTGGGTAA | GGCTCCCCAGCATCCAGTACAGTGC |
| 3 + 4 | GCCCAGTTTCCCAGACCCCCTTAAC | TCCTTCCTTCTAAGGAAAATGTGCA |
| 5 + 6 | ACCTGGGGACAAGGTGCAGTGACTG | AGCTACCATCCCACTTCCACAGAGT |
| 7 + 8 | GGCTCCCTCCATGCACCCCCCACCC | AGATGCAATGCTGCATTATAGCCGG |
| 9 + 10 | CTCCTATATGGATTCATGTTGGGTG | CCGTGTATCCAGACGGAAACCGGCC |
| 11 | AAGGACTGGCTAGGAAAGATGACCC | AGTTCTACAACCCCATACATTCATT |
| 12 | GAACAGGGATTCTGGGAGGGAAGGG | AAAGACCTCAGGAGTTCATGGAAGT |
Statistical analysis
For the comparison of multiple groups, one-way ANOVA (Graph Pad Prism; GraphPad Software, Inc., La Jolla, CA, USA) was performed and Dunnett’s multiple comparison test was applied when comparing the mean of each group with the mean of a single control group or Tukey’s test when comparing the mean of each group with the mean of every other group. In both cases, p values < .05 were considered statistically significant.
Symptom-free survival was evaluated by Kaplan–Meier curves. Animals sacrificed for reasons other than neurological symptoms (eg, weight loss) were censored. Significance between treatment groups was evaluated by Log rank (Mantel Cox) test, and a p value of <.05 was considered significant. Survival between CSF-placeboS1- and CSF-placeboS2-treated animals was not significantly different (n = 8 and n = 8, p = .1055). Therefore, we combined both CSF-placebo treatment groups in evaluations of survival, tumor response, and systemic side effects. We excluded one CSF-placeboS2-treated animal from analysis because of an atypically long symptom-free survival of 43 days. It was identified as a definite outlier with the ROUT method (robust regression followed by outlier elimination) using Q = 0.1%. OralS1-and OralS2-treated mice reached the end of observation in 4/7 and 6/9 cases, respectively, and the difference was not significant (n = 7 and n = 9, p > .99). Both oral treatment groups were, therefore, combined for further evaluations.
Results
Early antitumor effect of intraventricular Vismodegib treatment
Math1-cre::Ptch1 Fl/Fl mice develop SHH MB already in infancy with a penetrance of 100%. We treated Math1-cre::Ptch1Fl/Fl mice on P11, P12, and P13 by oral gavage with 200 mg/mg/d Vismodegib or by stereotactic intraventricular applications of either placebo (“CSF-placebo”), 0.2 mg/kg/d Vismodegib (“CSF-low”), or 1.6 mg/kg/d Vismodegib (“CSF-high”) (Workflow in Figure 1A). We investigated early treatment effects on the tumors at P15, that is 2 days after completed treatment. CSF-placebo as well as CSF-low treated mice showed prominent proliferative cerebellar tumors with strong NMYC expression and moderately elevated microglia activation estimated by IBA1 expression (Figure 1B–1M). In contrast, mice treated with CSF-high showed a lesser extent of remaining tumor, and in some animals, proliferation was drastically decreased (Figure 1N–1S). Oral treatment led to complete tumor regression (Figure 1T–1Y). Quantification of mitotic activity confirmed that CSF-high treatment resulted in drastically decreased mitotic activity in some animals, while others showed mitotic activity comparable to CSF-placebo-treated mice (Figure 1FF). Microglia activation was elevated in all tumor bearing mice compared to wild type cerebella, but not significantly different between treatment groups (Figure 1GG). Further markers for neuronal and glial differentiation as well as apoptosis were not differentially expressed between treatment groups at P15 (Supplementary Figure 1A–1U and 1QQ–1SS).
Figure 1.
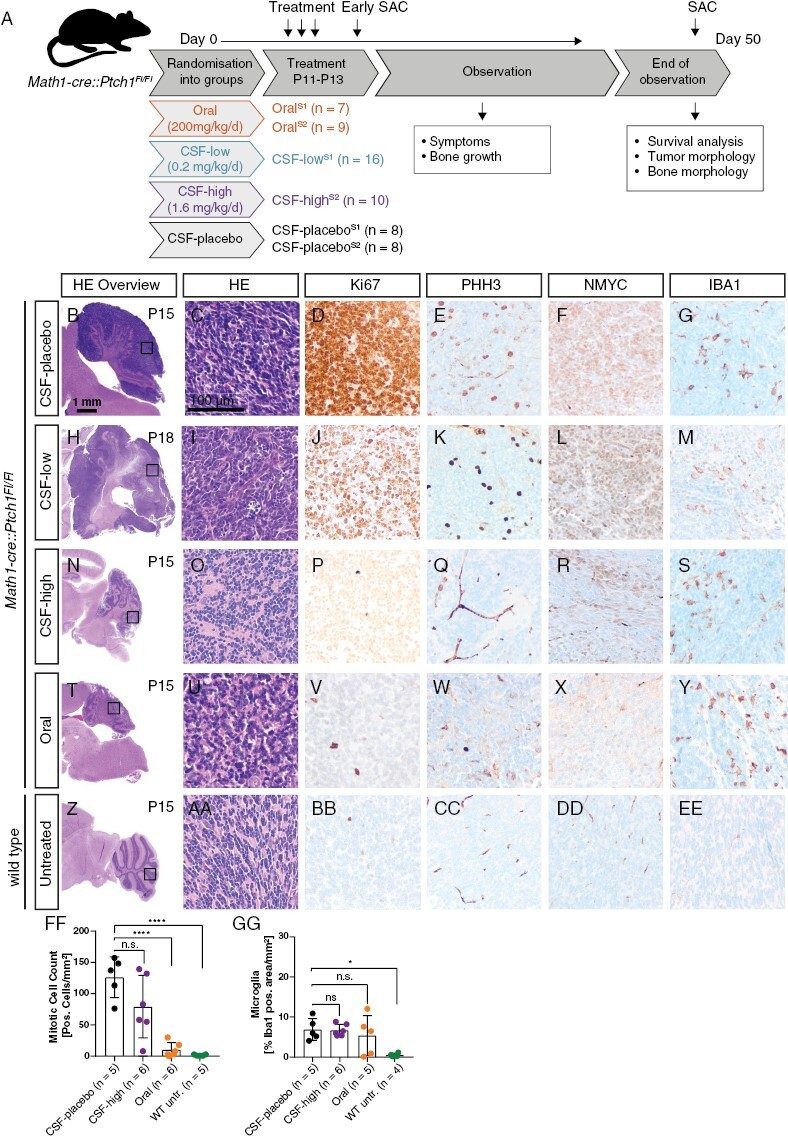
Treatment workflow and evaluation of early treatment effects. (A) Mice were randomized into three treatment groups as indicated. Treatment was performed on three consecutive days from P11 to P13 followed by an observation period until onset of neurological symptoms or latest until P50. Few animals were sacrificed at P15 to evaluate acute treatment response (“early SAC”). Oral treatment and CSF-placebo treatment were performed with both solvent 1 and solvent 2 (S1 and S2), CSF-low treatment with S1 and CSF-high treatment with S2. B–GG: Immunohistochemical evaluations after early sacrifice. Highly proliferative tumors with strong NMYC expression and moderate IBA1 expression after CSF-placebo treatment (B–G), comparable tumor histology after CSF-low treatment (H–M), reduced tumor mass and low proliferation activity after CSF-high treatment (N–S), complete tumor regression after oral treatment (T–Y), and unaffected cerebellum of a healthy wild type sibling without treatment (Z–EE). Quantification of PHH3-positive cells per area showed significantly reduced proliferation in untreated wild type and orally treated tumor mice but no significant reduction of proliferation after CSF-high treatment (FF) (****p < .0001). Microglia infiltration quantified by IBA1-positive area per total area was not significantly different between treatment groups (GG) but significantly lower in wild type untreated siblings (*p = .0147). Dunnett’s test was applied for comparison of each group to CSF-placebo in panel FF and GG.
We further assessed treatment response at P15 by quantification of the area of remaining proliferative tumor tissue in relation to total cerebellum area. While all CSF-placebo treated mice had remaining tumors covering more than 50% of the cerebellum area, orally treated mice showed complete remission with less than 5% proliferative cerebellar area (Supplementary Figure 2A–2J). CSF-high treatment resulted in a more heterogeneous response between individual cases with complete remission (n = 1), partial remission (n = 3), and fully remaining tumors (n = 2) (Supplementary Figure 2K–2Q).
We conclude from these findings that CSH-high treatment shows a promising acute effect on tumor growth and can lead to drastic reduction of tumor volume and proliferation within the remaining tumor tissue in a subset of treated animals.
Significantly increased survival after oral and intraventricular Vismodegib treatment
Next, we investigated the effect of different treatment strategies on symptom-free survival of Math1-cre::Ptch1Fl/Fl mice (Figure 2A). Both CSF-low and CSF-high treatment significantly improved symptom-free survival compared to CSF-placebo treatment (p < .0001). CSF-high treatment improved median symptom-free survival from P33 in CSF-low to P47 in CSF-high-treated mice; however, the difference between the two survival curves was not statistically significant (p = .19). Orally treated animals showed excellent symptom-free survival, which was significantly better than any other treatment group.
Figure 2.
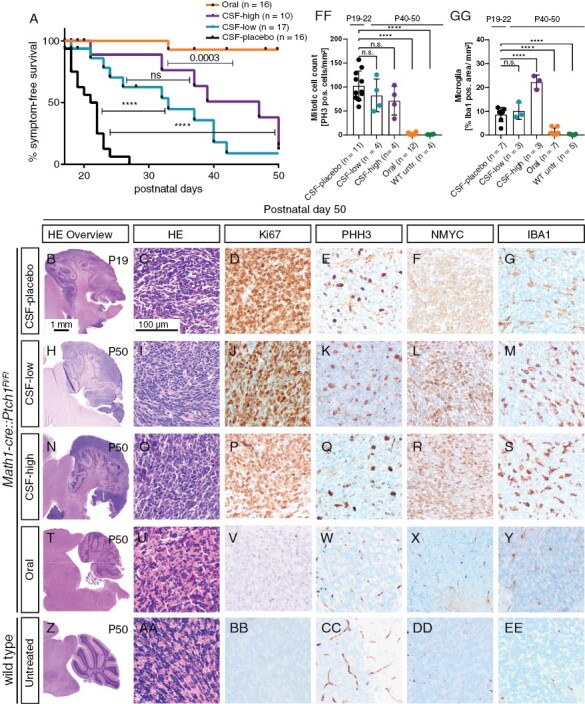
Intraventricular Vismodegib prolongs symptom-free survival. (A) Kaplan–Meier curves of all treatment groups showing symptom-free survival. Mice were sacrificed upon occurrence of neurological symptoms (ataxia, hydrocephalus). Mice were censored when they had to be sacrificed due to non-neurological reasons (weight loss/failure to thrive) or when they reached the end of observation without developing neurological symptoms. B–EE: Immunohistochemical evaluations at end of observation. CSF-placebo, CSF-low, and CSF-high-treated animals show tumors with comparable immunohistochemical characteristics regarding proliferation and NMYC expression (B–R). IBA1 expression was significantly higher after CSF-high treatment (S, GG). Orally treated and untreated wild type mice showed absence of proliferation markers and no NMYC or IBA1 expression (T–EE). (FF) Quantification of PHH3-positive cells per area showed significantly reduced proliferation in untreated wild-type siblings and orally treated mice (*p = .0001) but no significant reduction of proliferation after CSF-low and CSF-high treatment. GG: IBA1 expression was significantly elevated after CSF-high treatment compared to placebo, while orally treated and untreated mice had significantly lower IBA1 expression. Dunnett’s test was applied for comparison of each group to CSF-placebo in panel FF and GG. ****p < .0001.
All CSF-placebo-treated animals presented with highly proliferating tumors of the cerebellum at onset of symptoms (Figure 2B–2E). Tumors showed strong NMYC and moderate IBA1 expression (Figure 2F, 2G). While treatment with CSF-low and CSF-high led to prolonged symptom-free survival, all animals still showed prominent proliferative cerebellar tumors with immunohistochemical profiles comparable to CSF-placebo treatment at sacrifice (Figure 2H–2S, 2FF, Supplementary Figure 1V–1SS). However, microglial infiltration estimated by IBA1 positive staining was significantly elevated in CSF-high-treated mice compared to CSF-placebo (p = .0001, Figure 2GG). At the same time, the degree of M1 activated macrophages estimated by positive iNOS staining showed no significant differences between placebo and CSF-high treatment and almost no CD3 positive lymphocytes were detected in tumors of both treatment groups (Supplementary Figure S3). In summary, while CSF treatment with Vismodegib in two different dosages was able to prolong symptom-free survival, it did not completely eradicate the pre-existing tumor.
In contrast, oral treatment led to complete tumor regression in all animals (Figure 2T–2Y, 2FF). None of these animals showed significant traces of proliferative tumor in the cerebellum (Supplementary Figure S4). However, all animals showed pronounced abnormalities of bone growth that will be discussed in detail later.
Occurrence of mutations in Smo have been described as a mechanism of resistance to SHH Inhibition in mice and humans.23–27 We sequenced all 12 coding exons of Smo in tumors that developed between P16 and P50 after CSF-low (n = 11) or CFS-high (n = 7) treatment. None of the nucleotide sequence tracings of the 18 examined tumor samples showed any of the previously described or novel Smo mutations following SMO inhibition (data not shown).
Intact skeletal morphology after intraventricular Vismodegib treatment
SHH inhibition treatment for children is only feasible if systemic side effects on bone growth can be prevented. Therefore, we investigated the growth dynamics, anatomy, and histology of tibia and femur bones after treatment. As CSF-placebo treated Math1-cre::Ptch1Fl/Fl mice ("tumor mice") did not live until adulthood, untreated wild-type (WT) mice were analyzed as an additional control group. Tumor mice of all treatment groups showed decreased weight gain, but only orally treated tumor mice had a significantly lower body weight compared to untreated WT mice at P50 (Figure 3A and Supplementary Figure 5A, 5B). Tibia length was measured daily, and tumor mice of all treatment groups showed diminished tibia length compared to untreated WT siblings between P15 and P30 (Figure 3B, Supplementary Figure 5C, 5D). However, CSF-high and CSF-low-treated tumor mice then seemed to recover and finally showed a tibia length undistinguishable from untreated WT siblings at P50 (Figure 3C). In contrast, oral treatment led to distinctly shorter tibiae during the entire observation period, and bone growth stagnated from approximately P30 onward (Figure 3B, 3C).
Figure 3.
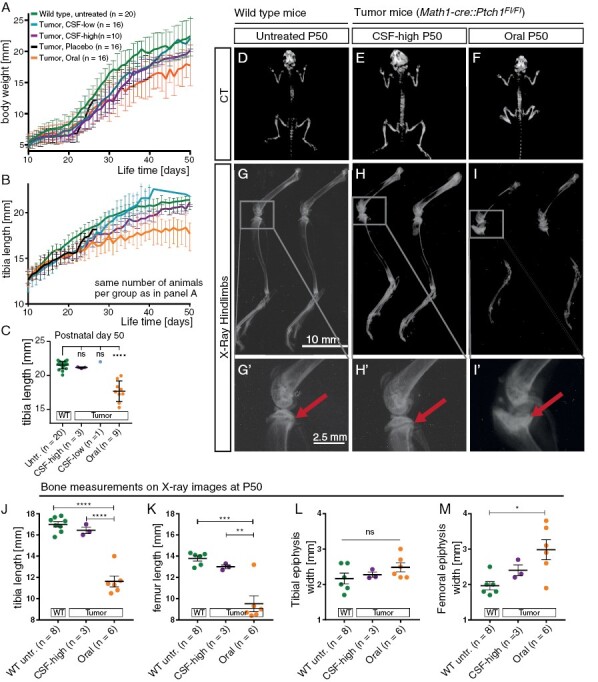
Intraventricular Vismodegib does not affect bone growth and macroscopic bone morphology. A: Body weight from P10 to P50 shows reduced weight gain in orally treated mice and to lesser extent in CSF-high-treated mice. B,C: Tibia length at P10 to P50 (B) and quantification of tibia length at P50 (C) show drastically reduced tibia length after oral Vismodegib treatment compared to untreated wild type siblings. D–F: CT images at end of observation show short stature and shortened tibiae in orally treated mice while CSF-high-treated mice show no anomalies of bone anatomy compared to untreated wild type siblings. G–I: X-rays of hindlimbs show shortened tibia and deformed fibula as well as dysmorphism of the proximal epiphysis of the tibia after oral treatment. No comparable changes were detected after CSF-high treatment. G’–I’: Close-ups of the knee joint confirm open growth plates of the tibia in healthy untreated siblings and CSF-high-treated siblings but premature closure after oral treatment (arrows). J–M: Quantification of tibia length, femur length, tibial epiphysis width and femoral epiphysis width measured on X-rays from mice at P50. ns = p > .05, *p < .05, **p < .01, ***p < .001, ****p < .0001. Dunnett’s test was applied for comparison of each group to untreated wild type in panel C. Tukey’s test was applied for comparing the mean of each group with the mean of every other group in panel J–M.
To closer characterize the effects of Vismodegib treatment strategies on bone growth and body stature, we performed whole body CT scans at P50. Because of the weaker treatment effect compared to CSF-high, we did not further investigate effects on bone growth in CSF-low-treated mice. On CT scans, orally treated tumor mice had shorter tibia and femur bones as well as thickened knee joints compared to untreated WT and CSF-high-treated tumor mice (Figure 3D–3F). X-rays of the hind-limbs confirmed intact anatomical structures with clearly discernible epiphyseal growth plates in untreated WT mice at P50 and CSF-high treated tumor mice between P39 and P50 (Figure 3G, 3G’, 3H, 3H’). In contrast, orally treated tumor mice showed severe dysmorphism of the tibia and femur, including calcification of the femoral and tibial growth plate, abnormally disfigured and broadened epiphyses, and metaphyses as well as severely shortened femur and tibia length, accompanied by a strongly curved fibula (Figure 3I–3M).
We next evaluated the histological morphology of femur and tibia after treatment using Von Kossa stainings that detect deposits of calcium or calcium salt. Untreated WT mice and CSF-high-treated tumor mice showed intact bone anatomy on overview sections at P50, whereas the shortening and severe dysmorphism of femora and tibiae of orally treated tumor mice was apparent in the histological sections (Figure 4A–4C). Untreated WT and CSF-high-treated tumor animals showed clearly discernible epiphyseal growth plates, whereas orally treated tumor mice showed disturbed distribution of mineralized tissue disrupting the epiphyseal growth plate (Figure 4D–4I, Supplementary Figure S6). Typical growth plate morphology was completely lost after oral treatment in most analyzed bones. In some cases, small islands of most likely cells from the residual zone of proliferation were surrounded by newly synthesized mineralized matrix (Figure 4F, 4I). H&E stainings confirmed intact chondrocyte column formation in untreated CSF-high-treated mice, whereas the columnar organization was completely abrogated after oral treatment (Figure 4J–4O).
Figure 4.
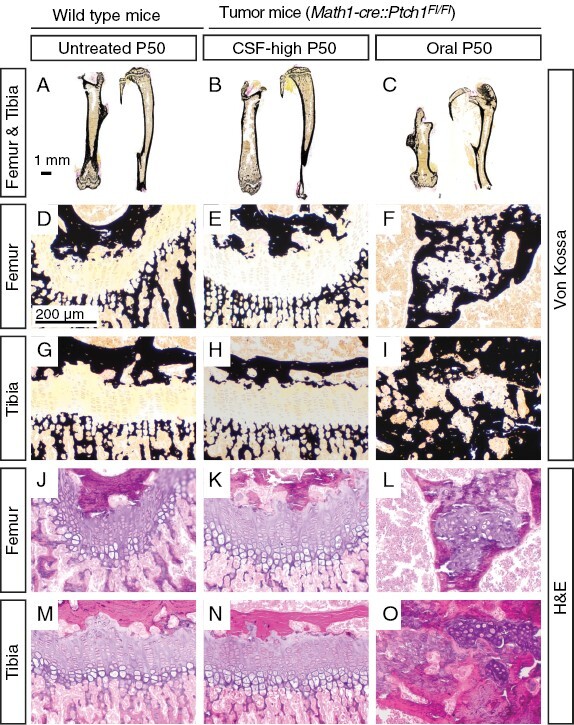
Intraventricular Vismodegib does not affect microscopic bone morphology. A–C: Overview sections of von Kossa stained femur and tibia bones show no aberrations und untreated wild type mice (A) and CSF-high treated tumor mice (B) but shortened bones in orally treated tumor mice (C). D-I: At 100× magnification, von Kossa stained sections of growth plates of orally treated mice show aberrant mineralized tissue, while growth plates of untreated wild-type siblings and CSF-high-treated mice show no irregular mineralization. J–O: Corresponding H&E stainings show highly disturbed columnar organization after oral treatment, while growth plates of untreated wild type siblings and CSF-high treated mice are intact. At least four animals per group were investigated. Representative images are shown.
Taken together, macroscopical and histological disruption of the growth plate was obvious in orally treated Math1-cre::Ptch1Fl/Fl mice but seemed absent after intraventricular Vismodegib treatment. However, genotype, tumor burden, and straining therapeutic interventions might have contributed to overall impaired thriving and complicated the evaluations. Also, CSF-placebo-treated Math1-cre::Ptch1Fl/Fl mice did not live long enough to allow evaluations at adult age and only three CSF-high-treated tumor mice lived until P50. Therefore, we additionally treated healthy WT siblings (Ptch1Fl/Wt or Ptch1Fl/Fl) with oral and CSF-high Vismodegib as well as CSF-placebo. CSF-high-treated WT mice displayed weight gain and tibia growth curves undistinguishable from CSF-placebo treated and untreated WT mice, whereas orally treated WT mice had significantly shorter bones and body weight (p < .0001, Figure 5A, 5B and Supplementary Figure 5E, 5F). X-rays of CSF-high treated, untreated, and CSF-placebo-treated WT mice showed normal growth plates at P15, whereas orally treated WT mice already showed discretely narrowed epiphyseal growth plates (Figure 5C–5F). X-rays at P50 recapitulated the results observed in Math1-cre::Ptch1Fl/Fl mice, showing pronounced dysmorphism of femora and tibiae only after oral treatment (Figure 5G–5J and Supplementary Figure 5G–5J). Histological analysis of growth plates at P15 showed intact columnar organization in untreated, CSF-high-treated and CSF-placebo-treated WT mice but disturbed columnar organization with rounded chondrocytes after oral treatment (Figure 5K–5Z, Supplementary Figure S7). We quantified the degree of column organization within the growth plate by measuring growth plate column index (CI) and the percentage of pre-hypertrophic cells in columns in tibia and femur at P15 and P50. Both parameters were significantly lower only in orally treated WT mice in tibiae and femora both at P15 and P50 (Figure 5AA–5HH).
Figure 5.
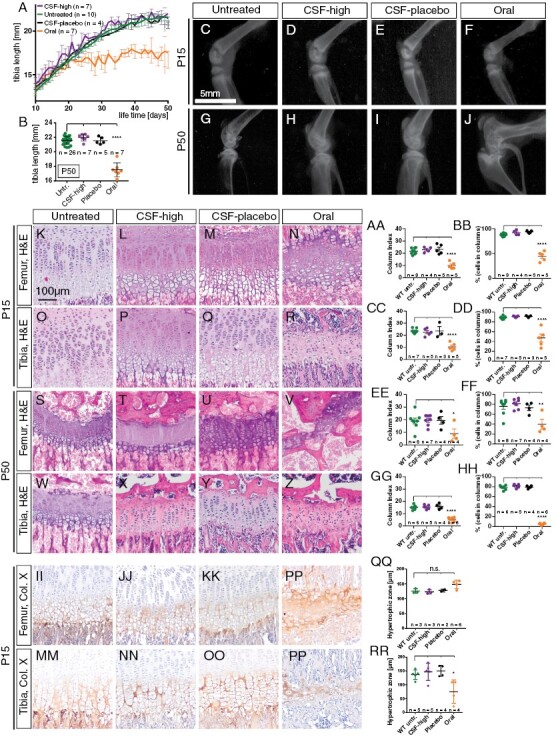
Intraventricular Vismodegib does not affect microscopic bone morphology of wild-type mice. A, B: Tibia length of CSF-placebo and CSF-high-treated mice is comparable to untreated mice but significantly shorter in orally treated mice at P50. C–J: Representative X-rays of the knee joint reveal normal anatomy with clearly discernible growth plate in untreated (n = 3), CSF-high (n = 4), and CSF-placebo (n = 3) treated mice but marked dysmorphism and a closed growth plate after oral treatment. K–Z: H&E stainings of the growth plate of femur and tibia at P15 and P50 reveal maintained columnar organization in untreated, CSF-high and CSF-placebo treated mice but disrupted column formation after oral treatment. AA–HH: Quantification of column index and percentage of cells in columns (*p = .0354 in OO, *p = .0019 in PP). II–PP: Immunohistological stainings for collagen X illustrate comparable width of the hypertrophic zone in untreated, CSF-high and CSF-placebo treated mice but premature closure of the epiphyseal plate after oral treatment with an absent proliferative zone. QQ–RR: Width of hypertrophic zone in femura does not differ between treatment groups but is significantly lower in tibiae of orally treated mice than in tibiae of untreated WT mice (*p = .0173). Dunnett’s test for was applied in all quantifications (AA–HH and QQ, RR), comparing each treatment group to untreated mice. ****p < .0001.
Kimura et al. found that an early effect of oral Vismodegib treatment is broadening of the femoral hypertrophic zone that is followed by premature closure of the growth plate.15 In order to quantify the width of the hypertrophic zone at P15, we used collagen X staining to detect hypertrophic differentiation in the growth plate as described previously.22 While the hypertrophic zone of tibia and femur showed similar width after no treatment, CSF-high, and CSF-placebo treatment, we observed complete absence of the pre-hypertrophic zone and strong and diffuse collagen X staining in the highly disorganized remnants of the epiphyseal growth plate in orally treated WT animals (Figure 5II–5PP). While the orally treated mice showed a slightly broader hypertrophic zone in femoral bones, the difference was not significant compared to untreated WT siblings (Figure 5QQ). Instead, the width of the hypertrophic zone in tibiae was significantly lower after oral treatment compared to untreated WT mice (Figure 5RR). This indicates an already advanced stage of growth plate disruption in orally treated mice at P15, while CSF-high treatment causes no measurable effects on growth plate development.
Discussion
So far, treatment with SHH pathway inhibitors has been limited in the field of pediatric oncology, due to severe systemic side effects on bone growth. Here, we investigated an alternative strategy for drug delivery in form of intraventricular application in order to circumvent systemic side effects. In our SHH MB mouse model, intraventricular Vismodegib application in infant mice did not induce any measurable bone growth defects on a macroscopic or microscopic level. We further found that intraventricular application of Vismodegib significantly improved survival compared to placebo treatment. In one out of six animals, we observed a strong early treatment effect with no detectable proliferative tumor residuum 2 days after completed treatment. Nonetheless, when observing the long term treatment effect, all mice eventually developed cerebellar MB after CSF-low and CSF-high treatment. This may be preventable in patients by using prolonged treatment regimens, for example, as children receive intraventricular methotrexate over 6–10 months as part of the HIT-SKK regimen.3 However, application of repeated cycles of intraventricular Vismodegib was not feasible in our mouse model. The tumors that developed after CSF-low and CSF-high treatment were morphologically and immunohistochemically undistinguishable from CSF-placebo-treated tumors. The only exception was macrophage marker IBA1, which was significantly higher expressed in tumors after CSF-high treatment than after placebo treatment. Tumor associated macrophages (TAMs) play an important role in the tumor microenvironment of MB, and the infiltration of TAMs is significantly higher in SHH MB than in other MB subgroups.28,29 In line with this, we observed elevated IBA1 expression in tumors of all treatment groups compared to untreated WT cerebella. The exact role of macrophages in tumor growth and progression is still controversial in MB.28,30–32 Therefore, it remains speculative whether the increase in macrophage activation after CSF-high treatment is a sign of good treatment response or of increasing treatment resistance. In both cases, a combination therapy aimed at targeting the activated macrophages in addition to the intraventricular SHH pathway inhibitions might further improve treatment response.28
CSF-high-treated tumors did not show any signs of resistance in form of changed tumor morphology, mutations of Smo or increase in SOX2- or OLIG2-positive tumor cells. This suggests that not a mechanisms of resistance against the SHH treatment but rather an insufficient treatment dosage led to (re)growth of the tumors in our mouse model. However, SHH pathway inhibitors have been described to induce drug resistance in clinical and preclinical studies.14,23,26 Therefore, SHH inhibition alone would not be a recommendable treatment for MB patients but would have to be considered as a part of a multimodal treatment regimen.
Our study has important limitations. Our results indicated that the applied dosages of intraventricular Vismodegib were not sufficient for an optimal treatment effect. Higher dosages of intraventricular Vismodegib could not be tested due to poor drug solubility. Therefore, an optimal drug concentration and respective systemic side effects at that concentration could not be determined. Another limitation is that we did not specifically investigate side effects on other organs besides bones. The clinical data from Robinson et al. suggested possible side effects on endocrine functions as two of three reported pediatric patients that received oral Vismodegib developed precocious puberty.16 More detailed investigations of other systemic side effects of intraventricular Vismodegib applications would, therefore, be advisable, including determinations of sex hormone blood concentrations and evaluation of maturation of genital organs. Furthermore, intrathecal drug administration could harbor the risk to impair normal neurocognitive development as discussed controversially regarding the administration of intrathecal MTX in patients with leukemia and with pediatric brain tumors.3,33,34 Therefore, testing of neurocognitive functions should be performed in a preclinical setting.
Moreover, we suggest that testing higher concentrations and more frequent applications of intraventricular SHH inhibition would ideally be performed in an animal model of larger size, as the repeated applications are very challenging to perform in infant mice. Robust methods for intraventricular catheterization are well established in rats, providing the opportunity to either apply repeated applications or a continuous infusion using minipumps.35–37 This model would also allow for investigations of Vismodegib plasma concentrations after drug administration in order to assess possible systemic leakage after intraventricular application and the according risk for systemic side effects at varying dosages. In addition, valuable information about pharmacokinetic characteristics and CNS penetrance of oral versus intraventricular applications for example could be complemented by measuring Vismodegib concentrations from brain and tumor tissue by liquid chromatography–mass spectrometry or by determining the spatial distribution of Vismodegib across sections of tumor and brain using IR-MALDESI.38,39 However, the advantage of a robust mouse model for SHH MB overweighed the benefits of any other animal model in this first study. The mouse model allowed us to test both tumor response and systemic side effects at the same time.
Recently, an elegant nanotherapeutic approach has been reported that targets endothelial tumor vasculature to enhance blood–brain barrier crossing of systemically applied Vismodegib.39 A slightly different mouse model was used, and treatment was performed starting at onset of severe symptoms which impedes exact comparison of survival effects to our study. Treatment was especially effective after a single dose X-ray irradiation with 0.25 Gy followed by eight doses of 10 mg/kg Fucoidan nanoparticles encapsulating Vismodegib (FiVis) given every second day. This treatment strategy prolonged survival to between 27 and 63 days post-treatment. Investigations of femur length and trabecular bone number did not show noticeable toxicities of treatment with FiVis at therapeutic concentrations in n = 3 investigated mice per treatment group. However, growth plate morphology or more detailed quantifications of columnar organization were not investigated. It would be interesting to see whether technologically advanced drug modifications like nanoparticle encapsulation would be of additional benefit upon intraventricular delivery.
In summary, we were able to report the following important observations: Application of intraventricular Vismodegib during a vulnerable phase of bone development can prevent side effects on bone growth in mice. At the same time, intraventricular Vismodegib treatment significantly improved survival in our mouse model of infant SHH MB. Therefore, we propose that intraventricular Vismodegib treatment combines the benefits of targeted delivery of the drug to the CSF and specific inhibition of the tumor-causing pathway in SHH MB. Addition of SHH pathway inhibitors to a multimodal treatment regimen might significantly improve treatment response and at the same time reduce the need of other, highly toxic treatment elements like CSI.
Supplementary Material
Acknowledgments
We would like to thank the animal facility of the Leibniz Institute of Virology Hamburg. We would like to thank Vanessa Thaden, Kim Kilian, Olga Winter, Andrea Thieke, and Annette Jung (all University Medical Center Hamburg-Eppendorf, Germany), and Kristin Hartmann (Mouse Pathology Facility, University Medical Center Hamburg-Eppendorf) for their excellent technical assistance.
Contributor Information
Catena Kresbach, Department of Pediatric Hematology and Oncology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany; Research Institute Children’s Cancer Center Hamburg, Hamburg, Germany; Center of Diagnostics, Institute of Neuropathology, Center of Diagnostics, University Medical Center Hamburg-Eppendorf, Hamburg, Germany; Mildred Scheel Cancer Career Center HaTriCS4, University Medical Center Hamburg- Eppendorf, Hamburg, Germany.
Lea Holst, Research Institute Children’s Cancer Center Hamburg, Hamburg, Germany.
Melanie Schoof, Department of Pediatric Hematology and Oncology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany; Research Institute Children’s Cancer Center Hamburg, Hamburg, Germany.
Tara Leven, Department of Pediatric Hematology and Oncology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany; Research Institute Children’s Cancer Center Hamburg, Hamburg, Germany.
Carolin Göbel, Department of Pediatric Hematology and Oncology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany; Research Institute Children’s Cancer Center Hamburg, Hamburg, Germany.
Sina Neyazi, Department of Pediatric Hematology and Oncology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany; Research Institute Children’s Cancer Center Hamburg, Hamburg, Germany.
Jacqueline Tischendorf, Department of Pediatric Hematology and Oncology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany; Research Institute Children’s Cancer Center Hamburg, Hamburg, Germany.
Carolin Loose, Research Institute Children’s Cancer Center Hamburg, Hamburg, Germany.
Antonina Wrzeszcz, Department of Pediatric Hematology and Oncology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany; Research Institute Children’s Cancer Center Hamburg, Hamburg, Germany.
Timur Yorgan, Department of Osteology and Biomechanics, University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
Stefan Rutkowski, Department of Pediatric Hematology and Oncology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
Ulrich Schüller, Department of Pediatric Hematology and Oncology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany; Research Institute Children’s Cancer Center Hamburg, Hamburg, Germany; Center of Diagnostics, Institute of Neuropathology, Center of Diagnostics, University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
Conflict of interest statement
S.R. received consulting fees from Bayer (advisory board), Novartis (advisory board), BMS (advisory board), Roche (advisory board), Celgene (DMSC), the German Children’s Cancer Foundation (Assessment) and MyChildsCancer (Consulting).
Funding
C.K. is sponsored by a fellowship of the Mildred Scheel Cancer Career Center Hamburg/Deutsche Krebshilfe. Further funding was provided by the Fördergemeinschaft Kinderkrebszentrum Hamburg e.V. (to U.S., C.K., and M.S.), the Gesellschaft für Kinderkrebsforschung e.V. (to U.S.), the Hamburger Krebsgesellschaft e.V. (to L.H.), the German Children’s Cancer Foundation (Deutsche Kinderkrebsstiftung) (Deutsche Kinderkrebsstiftung) (Deutsche Kinderkrebsstiftung) (Deutsche Kinderkrebsstiftung) (to S.R.), the Hubertus Wald Tumorzentrum, University Cancer Center Hamburg (to T.L. and C.K.) and the German Society for Pediatric Oncology and Hematology (to S.R.).
Authors contributions
C. Kresbach: Visualization, writing–original draft, investigation, methodology, L. Holst: investigation, M. Schoof: investigation, T. Leven: investigation, C. Göbel: investigation, S. Neyazi: investigation, J. Tischendorf: investigation, C. Loose: investigation, A. Wrzeszcz: investigation, T. Yorgan: investigation, S. Rutkowski: resources, advise, U. Schüller: Conceptualization, supervision, writing–review and editing.
References
- 1. Kiltie AE, Lashford LS, Gattamaneni HR.. Survival and late effects in medulloblastoma patients treated with craniospinal irradiation under three years old. Med Pediatr Oncol. 1997;28(5):348–354. [DOI] [PubMed] [Google Scholar]
- 2. Jenkin D, Danjoux C, Greenberg M.. Subsequent quality of life for children irradiated for a brain tumor before age four years. Med Pediatr Oncol. 1998;31(6):506–511. [DOI] [PubMed] [Google Scholar]
- 3. Mynarek M, von Hoff K, Pietsch T, et al. Nonmetastatic medulloblastoma of early childhood: results from the prospective clinical trial HIT-2000 and an extended validation cohort. J Clin Oncol. 2020;38(18):2028–2040. [DOI] [PubMed] [Google Scholar]
- 4. Rutkowski S, Bode U, Deinlein F, et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med. 2005;352(10):978–986. [DOI] [PubMed] [Google Scholar]
- 5. Rutkowski S, Gerber NU, von Hoff K, et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy and deferred radiotherapy. Neuro Oncol. 2009;11(2):201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moe PJ, Holen A.. High-dose methotrexate in childhood all. Pediatr Hematol Oncol. 2000;17(8):615–622. [DOI] [PubMed] [Google Scholar]
- 7. Mack F, Baumert BG, Schäfer N, et al. Therapy of leptomeningeal metastasis in solid tumors. Cancer Treat Rev. 2016;43:83–91. [DOI] [PubMed] [Google Scholar]
- 8. Pompe RS, von Bueren AO, Mynarek M, et al. Intraventricular methotrexate as part of primary therapy for children with infant and/or metastatic medulloblastoma: Feasibility, acute toxicity and evidence for efficacy. Eur J Cancer. 2015;51(17):2634–2642. [DOI] [PubMed] [Google Scholar]
- 9. Ottensmeier H, Schlegel PG, Eyrich M, et al. Treatment of children under 4 years of age with medulloblastoma and ependymoma in the HIT2000/HIT-REZ 2005 trials: neuropsychological outcome 5 years after treatment. PLoS One. 2020;15(1):e0227693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Northcott PA, Korshunov A, Witt H, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29(11):1408–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123(4):465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cirrone F, Harris CS.. Vismodegib and the hedgehog pathway: a new treatment for basal cell carcinoma. Clin Ther. 2012;34(10):2039–2050. [DOI] [PubMed] [Google Scholar]
- 13. Romer JT, Kimura H, Magdaleno S, et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/–)p53(–/–) mice. Cancer Cell. 2004;6(3):229–240. [DOI] [PubMed] [Google Scholar]
- 14. Robinson GW, Orr BA, Wu G, et al. Vismodegib exerts targeted efficacy against recurrent sonic hedgehog-subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC-025B and PBTC-032. J Clin Oncol. 2015;33(24):2646–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kimura H, Ng JM, Curran T.. Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell. 2008;13(3):249–260. [DOI] [PubMed] [Google Scholar]
- 16. Robinson GW, Kaste SC, Chemaitilly W, et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget. 2017;8(41):69295–69302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang ZJ, Ellis T, Markant SL, et al. Medulloblastoma can be initiated by deletion of patched in lineage-restricted progenitors or stem cells. Cancer Cell. 2008;14(2):135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pöschl J, Stark S, Neumann P, et al. Genomic and transcriptomic analyses match medulloblastoma mouse models to their human counterparts. Acta Neuropathol. 2014;128(1):123–136. [DOI] [PubMed] [Google Scholar]
- 19. Schinke T, Schilling AF, Baranowsky A, et al. Impaired gastric acidification negatively affects calcium homeostasis and bone mass. Nat Med. 2009;15(6):674–681. [DOI] [PubMed] [Google Scholar]
- 20. Vollersen N, Zhao W, Rolvien T, et al. The WNT1(G177C) mutation specifically affects skeletal integrity in a mouse model of osteogenesis imperfecta type XV. Bone Res. 2021;9(1):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bankhead P, Loughrey MB, Fernández JA, et al. QuPath: open source software for digital pathology image analysis. Sci Rep. 2017;7(1):16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Killion CH, Mitchell EH, Duke CG, Serra R.. Mechanical loading regulates organization of the actin cytoskeleton and column formation in postnatal growth plate. Mol Biol Cell. 2017;28(14):1862–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ocasio JK, Babcock B, Malawsky D, et al. scRNA-seq in medulloblastoma shows cellular heterogeneity and lineage expansion support resistance to SHH inhibitor therapy. Nat Commun. 2019;10(1):5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lospinoso Severini L, Ghirga F, Bufalieri F, et al. The SHH/GLI signaling pathway: a therapeutic target for medulloblastoma. Expert Opin Ther Targets. 2020;24(11):1159–1181. [DOI] [PubMed] [Google Scholar]
- 25. Dijkgraaf GJ, Alicke B, Weinmann L, et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011;71(2):435–444. [DOI] [PubMed] [Google Scholar]
- 26. Yauch RL, Dijkgraaf GJ, Alicke B, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326(5952):572–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu X, Zhang Y, Li Y, et al. Development of hedgehog pathway inhibitors by epigenetically targeting GLI through BET bromodomain for the treatment of medulloblastoma. Acta Pharmaceutica Sinica B. 2021;11(2):488–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Margol AS, Robison NJ, Gnanachandran J, et al. Tumor-associated macrophages in SHH subgroup of medulloblastomas. Clin Cancer Res. 2015;21(6):1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bockmayr M, Mohme M, Klauschen F, et al. Subgroup-specific immune and stromal microenvironment in medulloblastoma. Oncoimmunology. 2018;7(9):e1462430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang J, Wang T.. Immune cell landscape and immunotherapy of medulloblastoma. Pediatric Investig. 2021;5(4):299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee C, Lee J, Choi SA, et al. M1 macrophage recruitment correlates with worse outcome in SHH medulloblastomas. BMC Cancer. 2018;18(1):535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maximov V, Chen Z, Wei Y, et al. Tumour-associated macrophages exhibit anti-tumoural properties in Sonic Hedgehog medulloblastoma. Nat Commun. 2019;10(1):2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moleski M. Neuropsychological, neuroanatomical, and neurophysiological consequences of CNS chemotherapy for acute lymphoblastic leukemia. Arch Clin Neuropsychol. 2000;15(7):603–630. [PubMed] [Google Scholar]
- 34. Butler RW, Fairclough DL, Katz ER, et al. Intellectual functioning and multi-dimensional attentional processes in long-term survivors of a central nervous system related pediatric malignancy. Life Sci. 2013;93(17):611–616. [DOI] [PubMed] [Google Scholar]
- 35. Mazur C, Fitzsimmons B, Kamme F, et al. Development of a simple, rapid, and robust intrathecal catheterization method in the rat. J Neurosci Methods. 2017;280:36–46. [DOI] [PubMed] [Google Scholar]
- 36. Chao H, Lin C, Zuo Q, et al. Cardiolipin-dependent mitophagy guides outcome after traumatic brain injury. J Neurosci. 2019;39(10):1930–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tang J, Chen Q, Guo J, et al. Minocycline attenuates neonatal germinal-matrix-hemorrhage-induced neuroinflammation and brain edema by activating cannabinoid receptor 2. Mol Neurobiol. 2016;53(3):1935–1948. [DOI] [PubMed] [Google Scholar]
- 38. Hwang D, Dismuke T, Tikunov A, et al. Poly(2-oxazoline) nanoparticle delivery enhances the therapeutic potential of vismodegib for medulloblastoma by improving CNS pharmacokinetics and reducing systemic toxicity. Nanomed Nanotechnol Biol Med. 2021;32:102345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tylawsky DE, Kiguchi H, Vaynshteyn J, et al. P-selectin-targeted nanocarriers induce active crossing of the blood-brain barrier via caveolin-1-dependent transcytosis. Nat Mater. 2023;22(3):391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.