

Abstract
Complex traits often exhibit complex underlying genetic architectures resulting from a combination of evolution from standing variation, hard and soft sweeps, and alleles of varying effect size. Increasingly, studies implicate both large‐effect loci and polygenic patterns underpinning adaptation, but the extent that common genetic architectures are utilized during repeated adaptation is not well understood. Sea age or age at maturation represents a significant life history trait in Atlantic Salmon (Salmo salar), the genetic basis of which has been studied extensively in European Atlantic populations, with repeated identification of large‐effect loci. However, the genetic basis of sea age within North American Atlantic Salmon populations remains unclear, as does the potential for a parallel trans‐Atlantic genomic basis to sea age. Here, we used a large single‐nucleotide polymorphism (SNP) array and low‐coverage whole‐genome resequencing to explore the genomic basis of sea age variation in North American Atlantic Salmon. We found significant associations at the gene and SNP level with a large‐effect locus (vgll3) previously identified in European populations, indicating genetic parallelism, but found that this pattern varied based on both sex and geographic region. We also identified nonrepeated sets of highly predictive loci associated with sea age among populations and sexes within North America, indicating polygenicity and low rates of genomic parallelism. Despite low genome‐wide parallelism, we uncovered a set of conserved molecular pathways associated with sea age that were consistently enriched among comparisons, including calcium signaling, MapK signaling, focal adhesion, and phosphatidylinositol signaling. Together, our results indicate parallelism of the molecular basis of sea age in North American Atlantic Salmon across large‐effect genes and molecular pathways despite population‐specific patterns of polygenicity. These findings reveal roles for both contingency and repeated adaptation at the molecular level in the evolution of life history variation.
Keywords: Atlantic Salmon, genetic architecture, genomics, life history
Complex traits often exhibit complex underlying genetic architectures, and studies have begun to identify both large‐effect loci and polygenic patterns underpinning adaptation. Sea age represents a significant life history trait in Atlantic Salmon (Salmo salar), the genetic basis of which has been studied extensively in European populations, with repeated identification of large‐effect loci. In a survey of North American rivers, we found significant associations at the gene and SNP level with a large‐effect locus (vgll3) previously identified in European populations, but found that this pattern varied based on both sex and geographic region. Contrasting low genome‐wide parallelism, we uncovered a set of conserved molecular pathways associated with sea age that were consistently enriched among comparisons, indicating parallelism of the molecular basis of sea age in North American Atlantic Salmon across large‐effect genes and molecular pathways despite population‐specific patterns of polygenicity.
1. INTRODUCTION
A key component of understanding adaptive genetic variation is identifying the predictability of genomic patterns underlying repeated adaptation, thus providing insight into the possible molecular solutions for ecological challenges (Blount et al., 2018; Elmer & Meyer, 2011). Theoretical and empirical studies have indicated that variability among genetic architectures underlying adaptive traits may have consequences for their genomic parallelism (Bolnick et al., 2018; Yeaman et al., 2018). Parallelism at the genomic level is expected to occur with the greatest frequency in scenarios of shared ecological conditions and selection (Kaeuffer et al., 2012), phylogenetic similarity (Conte et al., 2012), shared standing variation (Ralph & Coop, 2015), and large‐effect loci controlling a large proportion of phenotypic variance (Yeaman, 2015). However, many traits exhibit polygenic architecture, and models of polygenic adaptation indicate small changes in allele frequency across many genetic pathways could reduce genomic parallelism (Barghi et al., 2019; Fagny & Austerlitz, 2021; Yeaman, 2015). Large‐scale follow‐up studies on the genetic basis of adaptive traits with previously identified large‐effect loci have also identified polygenic patterns explaining additional variation (Kreiner et al., 2021; Sinclair‐Waters et al., 2020), suggesting polygenicity may be common. Currently, the extent that simple and repeatable or polygenic and variable genetic architectures are more frequent during repeated adaptation remains unknown, requiring further study in the wild to characterize the genomic parallelism of adaptive traits.
Salmonids exhibit an extensive array of adaptive diversity (Klemetsen et al., 2003), with varying rates of underlying genomic parallelism (Jeffery et al., 2017; Salisbury et al., 2022). Atlantic Salmon (Salmo salar) is a culturally, ecologically, and economically significant species with an anadromous life cycle consisting of fresh water residency followed by a period at sea with enhanced growth rates prior to sexual maturation upon returning to fresh water to spawn (reviewed in Mobley et al., 2021). Sea age at first maturation varies both among individuals (Jonsson & Jonsson, 2007) and rivers (Hutchings & Jones, 1998) representing a variable life history strategy based on investment in reproduction timing and fecundity (Garant et al., 2003). This trait shows both genetic (Johnston et al., 2014) and environmental underpinnings (Friedland et al., 2000). Age at first maturation is demarcated by the number of winters spent at sea, also known as “sea age” or “sea winters”, with fish maturing after a single sea winter commonly known as 1SW or one‐sea‐winter salmon (1SW; Hutchings & Jones, 1998; Klemetsen et al., 2003) and fish first maturing after two or more winters at sea referred to as multi‐sea‐winter (MSW) salmon. Declines in Atlantic Salmon stocks across both Europe and North America (Lehnert, Kess, et al., 2019; Olmos et al., 2020), and reductions in the number of MSW salmon across years (Olmos et al., 2020) indicate recent selection against older age at first maturation (Czorlich et al., 2018) and highlight the need for greater understanding of this trait to inform conservation of life history variation within this species.
The genetic basis of sea age in Atlantic Salmon has been studied primarily in European populations (Barson et al., 2015; Sinclair‐Waters et al., 2022). Genomic investigation of this trait in rivers in Norway identified >30% of sea age variation was controlled by a large‐effect locus (vgll3, Barson et al., 2015) also associated with adiposity regulation and puberty onset in humans (Cousminer et al., 2013; Perry et al., 2014). However, significant associations with major effect loci in North American populations have varied among studies, indicating a complex genetic architecture (Boulding et al., 2019; Kusche et al., 2017; Mohamed et al., 2019).
Evolutionary history may play a role in explaining the discrepancy in identified genetic architectures underlying sea age between Europe and North America. Limited gene flow and trans‐Atlantic divergence may limit the genomic repeatability of this trait; differentiation between European and North American Atlantic Salmon populations is high (F ST = 0.26) and genome‐wide (Lehnert et al., 2020), reflecting extensive periods of isolation (~600,000 years) for the potential evolution of distinct genetic architectures of maturation. However, secondary contact between North American and European lineages during deglaciation has also been identified (Rougemont & Bernatchez, 2018). Adaptive introgression of structural variants has been found in Newfoundland and Labrador (Lehnert, Bentzen, et al., 2019), suggesting potential for a shared genetic basis of maturation in regions with retained European variation.
Variation in regional ecological factors has also been shown to play a role in the expression of different sea age phenotypes, and the interaction between local conditions and genetic variation may shape the architecture of this trait across different locations. At a broad spatial scale, sea age in North American populations exhibits a strong latitudinal gradient and shows associations with precipitation, and river discharge (Hutchings & Jones, 1998; Power, 1981). Both river size and access to lacustrine habitat have also been shown to impact sea age, which may intersect with predicted trade‐offs between growth, sexual maturation, and survival (Klemetsen et al., 2003). North American populations exhibiting greater sea ages that are comparable to Norwegian populations are found at lower latitudes, primarily in the Maritimes and Gulf of St. Lawrence, whereas Newfoundland and Labrador populations exhibit low rates of MSW fish (Hutchings & Jones, 1998). Analyses of 1SW proportions across Norwegian populations have also uncovered a role for marine temperature in shaping this trait across rivers, together indicating variation across both marine and freshwater environments contributes to maturation age (Vollset et al., 2022).
Genetic analyses of sea age across local environmental conditions also indicate variation in how this trait is controlled across environments. Studies in differing light and feed availability conditions experienced in aquaculture environments (Ayllon et al., 2019; Mohamed et al., 2019), temperature exposures (Åsheim et al., 2023), and from different source populations have led to different detected genetic architectures (Boulding et al., 2019). This variation in trait expression has also corresponded with different detected associations at large effect loci (vgll3, six6) in a Norwegian population over temporal comparisons, indicating that these loci may be significant contributors to sea age only under specific conditions (Besnier et al., 2023). Together, these findings reveal complex interactions at regional and individual scales with environmental conditions can contribute to both genetic and plastic determination of sea age, implicating different genes in different contexts. However, genome‐scale investigations into sea age variation in wild Atlantic Salmon populations across North America have not yet been carried out and have the potential to reveal the genetic architecture and parallelism of this important life history trait.
Here, we explore two remaining questions about the genetic architecture of sea age in Atlantic Salmon: (1) is there evidence of genomic parallelism associated with sea age variation at the population and individual level in the Northwest Atlantic, and (2) is there detectable genomic evidence of polygenic adaptation associated with sea age in Atlantic Salmon. We used low‐coverage whole‐genome resequencing (WGS, n = 582) and a single‐nucleotide polymorphism (SNP) array (n = 658) to conduct population structure inference, genome‐wide association (GWA), and genome scans for variation associated with sea age in Atlantic Salmon sampled from 29 (26 SNP array, 8 WGS) rivers across eastern North America. To detect polygenic patterns associated with sea age, we then used machine learning to predict individual sea age from a panel of genome‐wide markers. Finally, we used gene‐set enrichment to detect biological and molecular function exhibiting polygenic selection associated with sea age. Our findings here provide insight into factors driving parallelism of individual genes and higher‐level conserved pathways underlying traits with variable and polygenic backgrounds.
2. METHODS
2.1. Sampling and genotyping: SNP array
We first explored the relationship between genomic variation and sea age at the population‐level range‐wide acrossNorth American rivers. For range‐wide genomic analysis of population structure and sea age association, we used previously genotyped samples from published sources (combined from Lehnert et al., 2020; Wringe et al., 2018, full dataset described in Nugent et al., 2023), using a previously developed Atlantic Salmon 220K Axiom SNP array (Barson et al., 2015). We used genotype data at 97,566 SNPs that passed Axiom quality filters, with minor allele frequencies (maf) greater than 0.05. For all analyses, genotype data were retained for 658 individuals from 26 rivers of varying watershed size with population‐level mean proportion of maiden 1SW fish summarized from Canadian federal (Fisheries and Oceans Canada) and Quebec provincial (Ministère des Forêts, de la Faune et des Parcs) Atlantic Salmon counts from 1984 to 2020 (Figure 1a; Table 1). Individual genotypes were exported in plink format (Chang et al., 2015), and subsequent data filtration and conversions for analysis of SNP Array data were carried out in plink 1.90b6.16.
FIGURE 1.
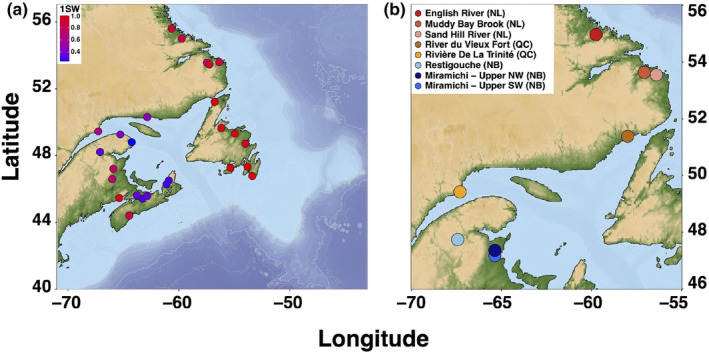
Map locations colored by 1SW proportion from 26 sampled rivers for individuals genotyped on the SNP array (a), and locations from eight sampled rivers for samples with individual sea age genotyped using whole‐genome resequencing (WGS) (b).
TABLE 1.
Sampled river names, locations, collection years, life stages, timeframe during which sea age data were collected, proportion 1SW, and number of sampled individuals for rivers genotyped on the SNP array.
| River | Latitude | Longitude | Region | Year collected | Life stage | Sea age data time period | Proportion 1SW | Sampled |
|---|---|---|---|---|---|---|---|---|
| Corneille | 50.28 | −62.88 | Gulf St. Lawrence and Quebec | 2018 | Adult | 1985–1988 | 0.462 | 28 |
| Madeleine | 49.23 | −65.32 | Gulf St. Lawrence and Quebec | 2018 | Adult | 1984–2020 | 0.409 | 28 |
| Matapedia | 48.18 | −67.142 | Gulf St. Lawrence and Quebec | 2018 | Adult | 1984–2020 | 0.334 | 15 |
| Miramichi—Northwest | 47.17 | −65.94 | Gulf St. Lawrence and Quebec | 2016 | Parr | 1993–2013 | 0.733 | 24 |
| Miramichi—Southwest | 46.55 | −66.04 | Gulf St. Lawrence and Quebec | 2016 | Parr | 1993–2013 | 0.719 | 23 |
| Northeast Margaree | 46.47 | −60.919 | Gulf St. Lawrence and Quebec | 2018 | Adult | 1987–1996 | 0.289 | 12 |
| Riviere de la Trinite | 49.42 | −67.305 | Gulf St. Lawrence and Quebec | 2012 | Adult | 1984–2019 | 0.634 | 49 |
| Saint‐Jean (Gaspesie) | 48.81 | −64.43 | Gulf St. Lawrence and Quebec | 2018 | Adult | 1984–2019 | 0.307 | 28 |
| Southwest Margaree | 46.24 | −61.122 | Gulf St. Lawrence and Quebec | 2018 | Adult | 1987–1996 | 0.289 | 14 |
| Eagle River | 53.53 | −57.467 | Labrador | n/a | n/a | 2000–2007, 2016 | 0.747 | 21 |
| English River | 54.97 | −59.75 | Labrador | 2010 | Parr | 2000–2019 | 0.859 | 27 |
| Hunt River | 55.57 | −60.67 | Labrador | n/a | n/a | 2000–2007 | 0.893 | 19 |
| Paradise River | 53.42 | −57.25 | Labrador | 2011 | Parr | 2001–2005, 2007–2020 | 0.924 | 19 |
| Big Salmon | 45.42 | −65.41 | Maritimes | 2014 | n/a | 2000–2019 | 0.974 | 22 |
| East River Pictou | 45.54 | −62.877 | Maritimes | 2018 | Parr | 1987–1996 | 0.34 | 23 |
| LaHave | 44.37 | −64.5 | Maritimes | n/a | n/a | 1979–2010 | 0.853 | 22 |
| North River NS | 45.38 | −63.31 | Maritimes | n/a | n/a | 1994–2019 | 0.299 | 22 |
| River Philip | 45.59 | −63.82 | Maritimes | 2018 | Parr | 1987–1996 | 0.352 | 17 |
| Sand Hill River | 53.57 | −56.35 | Labrador | n/a | n/a | 2002–2019 | 0.851 | 19 |
| Campbellton | 49.28 | −54.934 | Newfoundland | 2009 | Parr | 2011–2020 | 0.995 | 25 |
| Garnish | 47.23 | −55.35 | Newfoundland | 2009 | Parr | 2015–2020 | 0.992 | 22 |
| Great Rattling Brook—Exploits | 49.62 | −56.17 | Newfoundland | 2010 | Parr | 2000–2002, 2011–2020 | 0.971 | 26 |
| Northeast Brook Trepassey | 46.74 | −53.36 | Newfoundland | 2010 | Parr | 2000–2010, 2012 | 1 | 25 |
| Northeast Placentia River | 47.29 | −53.796 | Newfoundland | 2017–2019 | Parr | 2000, 2015–2019 | 1 | 81 |
| Terra Nova River | 48.67 | −54 | Newfoundland | 2009 | Parr | 2000–2001, 2005, 2011, 2014–2020 | 0.977 | 29 |
| Western Arm Brook | 51.19 | −56.765 | Newfoundland | 2016 | Adult | 2000–2020 | 0.996 | 18 |
2.2. Sampling and genotyping: WGS
To identify associations between individual sea age and genomic variation, we carried out low‐coverage whole‐genome resequencing as in Therkildsen and Palumbi (2017) of 582 male and female wild salmon from eight rivers in North America (Figure 1b), spanning southern rivers in Quebec and New Brunswick, and northern rivers in Quebec and Labrador. To minimize impacts of population structure or sex on individual GWA results, these individuals were grouped into separate sets of MSW and 1SW fish of each sex from each sampling region (Table 2). Atlantic Salmon were caught at fish‐counting fence traps or by angling and were sampled for fin clips for DNA extraction and scales to identify sea age. Sex was confirmed for each individual with PCR of the Salmo salar sex determining region sdY from Yano et al., 2013 (SS sdY S: “GGCCTATGCATTTCTGATGTTGA”, SS sdY AS: “AGAGGATTGAACGGTCAGAGGAG”). To prepare whole‐genome resequencing libraries, we extracted genomic DNA following Mayjonade et al. (2016), using Longmire's buffer (Longmire et al., 1997) for tissue lysis. We then used a modified version of the protocol described in Therkildsen and Palumbi (2017) and scaled down reaction volumes of Nextera DNA Flex Library Prep Kits (Illumina) to 0.13× the volume of the standard Illumina protocol. For library amplification, we used Kapa Hi‐Fidelity Library Amplification Kits (Roche) in 20‐μL reactions with 4‐μL Nextera Unique Dual Indexes Set A (Illumina). We then quantified libraries by Qubit (ThermoFisher) and checked average fragment size using an Agilent Bioanalyzer. We normalized libraries from 96‐well plates to equimolar concentrations prior to combining as a single pool per lane of sequencing. Libraries were then sequenced on six lanes of an Illumina NovaSeq6000 S4 at the Genome Quebec Centre d'Expertise et de Services.
TABLE 2.
Sample size of each sea age class and sex, with sampling locations and years, for WGS dataset.
| River | Region | Male—Single sea winter | Male—Multiple sea winters | Female—Single sea winter | Female—Multiple sea winters | Latitude | Longitude | Sampling years |
|---|---|---|---|---|---|---|---|---|
| Total | 155 | 116 | 140 | 171 | ||||
| Southern Rivers | 69 | 79 | 58 | 82 | ||||
| Miramichi Upper Northwest NB | 21 | 17 | 28 | 17 | 47.17 | −65.94 | 2018 | |
| Miramichi Upper Southwest NB | 12 | 16 | 9 | 20 | 46.55 | −66.04 | 2017–2018 | |
| Restigouche NB | 16 | 32 | 1 | 25 | 47.99 | −66.89 | 2019 | |
| Riviere de la Trinite QC | 20 | 14 | 20 | 20 | 49.42 | −67.3 | 2011–2017 | |
| Northern rivers | 86 | 37 | 82 | 89 | ||||
| English River NL | 23 | 9 | 21 | 26 | 54.97 | −59.75 | 2018–2019 | |
| Muddy Bay Brook NL | 20 | 0 | 19 | 20 | 53.638 | −57.0651 | 2016–2019 | |
| Sand Hill River NL | 23 | 20 | 22 | 23 | 53.548 | −56.393 | 2018–2019 | |
| River du Vieux Fort QC | 20 | 8 | 20 | 20 | 51.38 | −57.99 | 2011, 2015–2017 |
Quality of sequenced libraries was checked using FastQC, (Andrews, 2010). We used cutadapt 2.1 (Martin, 2011) to remove the leading 15 bases, adapter content, bases falling below of q score of 10, and any read with less than 40 remaining base pairs. Trimmed reads from each individual were aligned to the 29 chromosomal contigs from the ICSASG_V2 Salmo salar reference assembly (GCF_000233375.1) using bwa mem 0.7.17 (Li, 2013). Subsequent steps followed best practice recommendations for Genomic Analysis Toolkit (GATK 3.7, DePristo et al., 2011): We first removed duplicate reads using the PicardTools 2.20.6 MarkDuplicates function. Deduplicated reads were then sorted and realigned around potential insertions and deletions using RealignerTargetCreator and IndelRealigner functions in GATK. We estimated alignment depths on realigned .bam files using mosdepth 0.3.3 (Pedersen & Quinlan, 2018), and alignment rate and read numbers using samtools flagstat. This processing resulted in a set of deduplicated, realigned reads consisting of a mean of 54,538,983 paired reads per individual (median 56,011,607, SD = 13,145,588) with a mean 97.59% alignment rate (median = 97.59%, SD = 0.14%) on an average sequencing depth of 3.23×, (median = 3.31×, SD = 0.782).
Next, we estimated genotype likelihoods, as well as directly called genotypes, for each individual using Analysis of Next‐Generation Sequencing Data (ANGSD 0.935, Korneliussen et al., 2014). We output genotype likelihoods for each chromosome as beagle format likelihood files (Browning & Browning, 2007) for all SNPs passing quality filters (‐minMapQ 30 ‐minQ 20 ‐SNP_pval 2e‐6 ‐uniqueOnly 1 ‐remove_bads 1) and ensuring greater than 80% (‐minInd 470) of individuals had genotypes, and a minimum of 500 reads were present per locus (‐setMinDepth 500). Genotype calls from each chromosome were exported as bcf files using the ‐dobcf flag and converted to vcf files (Danecek et al., 2011) using bcftools 1.11 (Li et al., 2009). We then performed phasing and imputation of vcf files using Beagle 4.0 (Browning & Browning, 2007) resulting in 9,895,443 SNPs. To test the accuracy of imputed genotypes, we estimated allele frequency for the dataset using both the ‐freq function in vcftools 0.1.16 (Danecek et al., 2011) on imputed genotypes and the ‐domaf function on realigned, deduplicated bam files in ANGSD.
2.3. Population structure
To account for potential covariation in ancestry and sea age in GWA, we first estimated population structure of SNP array data using principal component analysis (PCA) in the R package pcadapt (Privé et al., 2020). For retaining relevant PC axes for GWA and outlier detection, we followed Cattell's methodology (1966) and retained PCs at break points in the amount of variation explained per axis, as suggested for pcadapt (Luu et al., 2017). Using this approach, we used scree plot visualization to identify K = 5 ancestral populations (Figure S1). We also estimated population structure using the sparse non‐negative matrix factorization algorithm (snmf, Frichot et al., 2014) implemented in the R package LEA (Frichot & Francois, 2015). This approach models ancestral populations from which contemporary genomes are derived based on observed allele frequencies. We selected K = 5, based on PCA results, and inspection of cross‐entropy criteria which showed highest rates of reduction in cross‐entropy until K = 5 (Figure S1). However, we acknowledge additional changes in percent variation explained and cross‐entropy values past K = 5, and alternative values of K are likely also valid using this approach (Lawson et al., 2018) especially given known hierarchical population structure in Atlantic Salmon (Bradbury et al., 2014; Lehnert et al., 2023). We focus on these first five primary ancestry sources, as our goal here is to characterize the main potential sources of broad‐scale population structure that may be associated with 1SW proportion.
We directly tested for covariation of 1SW proportion and population structure using variance partitioning (Capblancq & Forester, 2021; Legendre & Legendre, 2012) with the varpart function in the vegan R package (Oksanen et al., 2020). Using this function, we tested the extent that variation in 1SW proportion can be explained by the first five principal components of genetic variation from PCA. Significance of the variance partitioning model was explored by predicting 1SW proportion from genetic PCs in a redundancy analysis (RDA) model with the rda function, and the significance of this model was tested with the anova.cca function with 999 permutations.
Population structure in WGS samples was quantified using genotype likelihoods in PCAngsd 1.02 (Meisner & Albrechtsen, 2018). This method accommodates low‐coverage whole‐genome resequencing data through modeling genotype uncertainty in PCA for continuous estimation of population structure and in non‐negative matrix factorization for estimation of admixture components. Selection of the optimal number of principal components was handled here internally in PCANGSD using the minimum average partial test. Selection of the K number of ancestral populations was also carried out in PCANGSD based on likelihood convergence. Details of these methods can be found in Meisner and Albrechtsen (2018). As with the SNP array data, we also used variance partitioning to directly estimate the proportion and significance of variation in principal components of genetic structure that explained individual sea age, using the first two PCs from WGS data with all samples included.
2.4. PCA selection scan
We identified the genome‐wide landscape of differentiation associated with population structure by carrying out genome scans with per‐SNP significance estimates from PCA loadings. This approach leverages PCA to identify separate PC axes associated with different sources of population structure. Genomic regions that show strong associations with these axes are considered potential targets of divergent selection between populations based on allele frequency differences between projected clusters in ordination space (Duforet‐Frebourg et al., 2016). Loci exhibiting signatures of selection associated with population structure were detected in SNP array data by estimating p‐values for each SNP using the Mahalanobis method in pcadapt with K = 5. This method measures the strength of association of SNPs with K retained principal components; outliers will exhibit stronger associations with PC axes relative to the majority of loci genome‐wide (Luu et al., 2017). We adjusted p‐values estimated from pcadapt for false discovery rate (FDR, Storey & Tibshirani, 2003) using the qvalue R packages (Storey et al., 2015) and selected SNPs with q < 0.05 as exhibiting significant signatures of selection associated with population structure. This threshold has recently been identified to provide an acceptable trade‐off between false positive inclusion and failure to detect relevant loci due to test stringency (Chen et al., 2021).
To detect regions of elevated population structure associated with divergence in WGS data, we estimated p‐values for each SNP from its loading on each PC axis estimated in PCANGSD (Meisner et al., 2021), assuming a χ2 distribution of PC loadings, and then selected FDR‐adjusted q values <0.05.
2.5. Genome‐wide association with sea age
To explore the association between range‐wide genetic and sea age variation, we conducted genome‐wide association with SNP array data using population‐level 1SW proportions, which reflect the average number of 1SW fish per population. Per‐SNP associations with 1SW proportion were estimated using latent factor mixed models with LFMM 2 in lfmm (Caye et al., 2019), using the “gif” parameter to correct for genomic inflation driven by potential systematic biases in p‐values. We specified K = 1 to test for association without correction for population structure and K = 5 to account for variation associated with the retained PC axes which separated individuals by river and 1SW proportion (Figure 2b). To characterize multilocus associations with 1SW proportion, we also conducted RDA with the rda function in vegan, using 1SW proportion to predict genome‐wide variation, and selected the top 1% of SNPs based on absolute value of per‐SNP RDA scores.
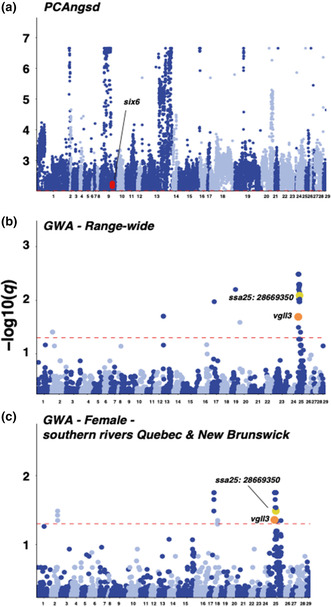
FIGURE 2.
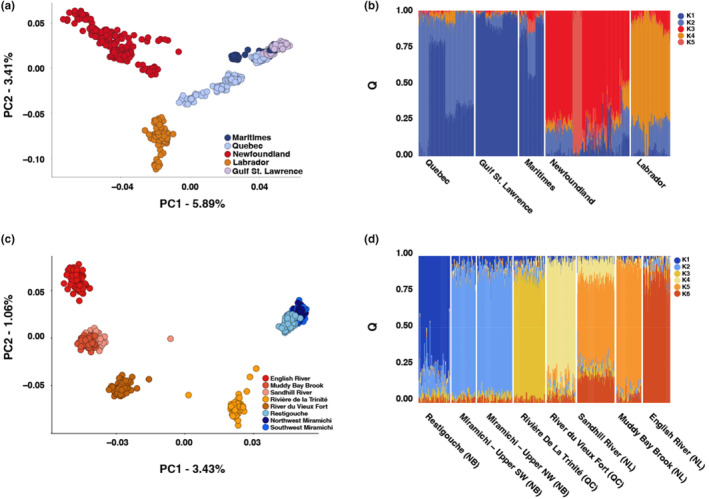
Population structure inferred from principal component (PC) analysis in pcadapt colored by geographic origin (a), individual admixture proportions identified from sparse non‐negative matrix factorization in LEA of individuals genotyped on the SNP array (b), population structure inferred from principal component analysis (c), and admixture proportions across regions identified from non‐negative matrix factorization (d) in PCANGSD using whole‐genome resequencing (WGS) samples.
For WGS data with individual phenotypes, we used the association functions in ANGSD to test for genomic association with individual sea ages from genotype likelihoods using a latent genotype model (‐doAsso 4, Jørsboe & Albrechtsen, 2022). Individual sea age was included as a binary phenotype, as all samples compared in this study exhibited a sea age at first maturity of 1 or 2 years, and only a small subset (n = 11) exhibited total sea age greater than two. These individuals were assigned an initial sea age of 1 years, indicating repeat spawning of 1SW fish rather than MSW maturation. Association analysis was carried out on genotype likelihoods from the phased and imputed vcf of all samples, without correction for population structure, as well as with the scores from the first two PCs as covariates. To identify the genetic architecture of this trait at finer spatial scales, we then repeated this analysis within each regional grouping (within southern rivers in Quebec and New Brunswick, and within northern rivers in Quebec and Labrador) and sex, with the first PC included in each analysis as a covariate to correct for population structure. For region‐level GWA, we re‐estimated PC scores per individual within each region separately (North and South). For all GWA with WGS data, we estimated the lambda inflation factor (Devlin & Roeder, 1999) as evidence of population structure‐driven confounding using the P_lamba function in the QCEWAS R package (Van der Most et al., 2017). As a second measure of genomic divergence associated with sea age, per‐locus Weir & Cockherham's F ST (Weir & Cockerham, 1984) was estimated with vcftools from phased and imputed vcf files between these same groups based on separation by sea age class.
2.6. Machine learning prediction of sea age from genotype dosage
As many genomic regions that fall below genome‐wide statistical significance may still carry substantial predictive information (McGaugh et al., 2021), we next tested the utility of a machine learning approach to predict individual sea age using WGS data. We first selected the top 100, 200, 300, 400, and 500 loci with the highest likelihood ratio test score from each population structure‐corrected GWA using WGS data to use as predictors in a random forest model (Breiman, 2001), as in Hess et al. (2016) and suggested in Brieuc et al. (2018) to minimize overfitting. We then used continuous genotype dosages, which allow for quantifying the uncertainty at individual loci from differences in read depth or SNP quality for each locus (Zheng et al., 2011), as predictors of trait variation. Prediction of sea age for each sex and regional grouping was carried out using a random forest classification model (Breiman, 2001) in the randomForest R package (Liaw & Wiener, 2002), setting 25,000 trees and 80% of SNP sample size for the mtry parameter for each analysis. To estimate the prediction accuracy of each model, we estimated out‐of‐bag error (OOB), which quantifies the number of misidentified samples from a testing dataset with known categories. For each tree, we specified a holdout of half of individuals from each sea age class, equalized by sample size, for testing and training, similar to the approach used by Brieuc et al. (2018). We then compared the accuracy of the selected panel of SNPs by generating panels of 100–500 randomly selected SNPs and calculated OOB for these panels to quantify classification error of random SNPs relative to those with elevated GWA significance. The trade‐off between sensitivity and specificity was estimated for the full model of all samples for 500 SNP prediction with a receiver operator characteristic (ROC) curve using the R package pROC.
2.7. Gene‐set enrichment
To identify signals of polygenic selection at the pathway level, we next used the gene‐set enrichment approach implemented in the polysel set of R tools (Daub et al., 2013), which tests for shared patterns of elevated values of a population‐genetic parameter among genes within pathways. This method differs from gene ontology enrichment methods by testing for elevation of parameters across all genes within a pathway as evidence of polygenic selection, whereas gene ontology enrichment relies on greater than expected similarity of function in a set of preidentified outlier genes. We downloaded gene information for Atlantic Salmon from http://www.ncbi.nlm.nih.gov/gene and KEGG pathway information (Ogata et al., 1998) for Atlantic Salmon from https://www.ncbi.nlm.nih.gov/biosystems/. We then selected the highest scoring SNP of each tested statistic for each gene and ran gene‐set enrichment on: Likelihood ratio test scores from genome‐wide association with sea age in the WGS datasets, per‐SNP PC1 scores from PCAngsd, −log10(p) LFMM scores from SNP array association with 1SW proportion at K = 1 and K = 5, and PC1 scores from pcadapt. We set a minimum of five genes per set, resulting in 237 sets and 11,437 genes analyzed for whole‐genome data, and 209 sets and 5507 genes analyzed for Axiom Array data. Significantly enriched KEGG pathways were identified using a q threshold of 0.05.
3. RESULTS
3.1. Population structure
Population structure analyses using the SNP array uncovered structuring associated with regional differences in North American Atlantic Salmon populations (or 1SW salmon; Figure 2a,b). We found that the first two PCs separated populations partially by regional differences in 1SW proportion among sampled rivers (primarily along the first PC axis, Figure S3), explaining most of the genetic variation, after which percent variation explained fell below 2% (Figure S1). Admixture analysis using snmf also supported multiple genetic clusters within broader geographic regions (Figure 1b). Variance partitioning of 1SW proportion by principal component scores of genetic variation identified a significant (p < .001) and large proportion of variation explained by PC1 (61%), with smaller but significant contributions from PC2 (1%) and PC5 (4%).
Consistent with our experimental design, we did not find a large proportion of genome‐wide population structure associated with individual age classes of WGS individuals, in contrast to population‐genetic clustering by river‐level 1SW proportion observed with the SNP array. Population structure inferred using genotype likelihoods in PCAngsd among the eight rivers in the whole‐genome resequencing dataset revealed regional separation between southern and northern rivers, and further clustering at the river level (Figure 2c). PCAngsd identified K = 6 as optimally describing population structure (Figure 2d) and revealed more similar ancestry among close rivers within the same region. Variance partitioning of sea age variation by PCs revealed a small but significant proportion of variance in individual sea age explained by PC1 (1.7%, p < .001), indicating reduced influence of population structure on individual‐level GWA statistics compared to SNP array data.
3.2. PCA‐based signatures of selection
Using PCA‐based scans for selection associated with population structure among populations genotyped on the SNP array, we uncovered many genomic regions exhibiting elevated divergence, including a previous sea age‐associated locus, six6 (Figure 3a). We identified 3593 SNPs with elevated significance in PCA using pcadapt (q < 0.05), distributed genome‐wide. These outliers overlapped 1139 genes (Table S1), as well as the introgressed European karyotypic variant regions (Lehnert, Bentzen, et al., 2019) on chromosomes ssa01and ssa23 (n = 559).
FIGURE 3.
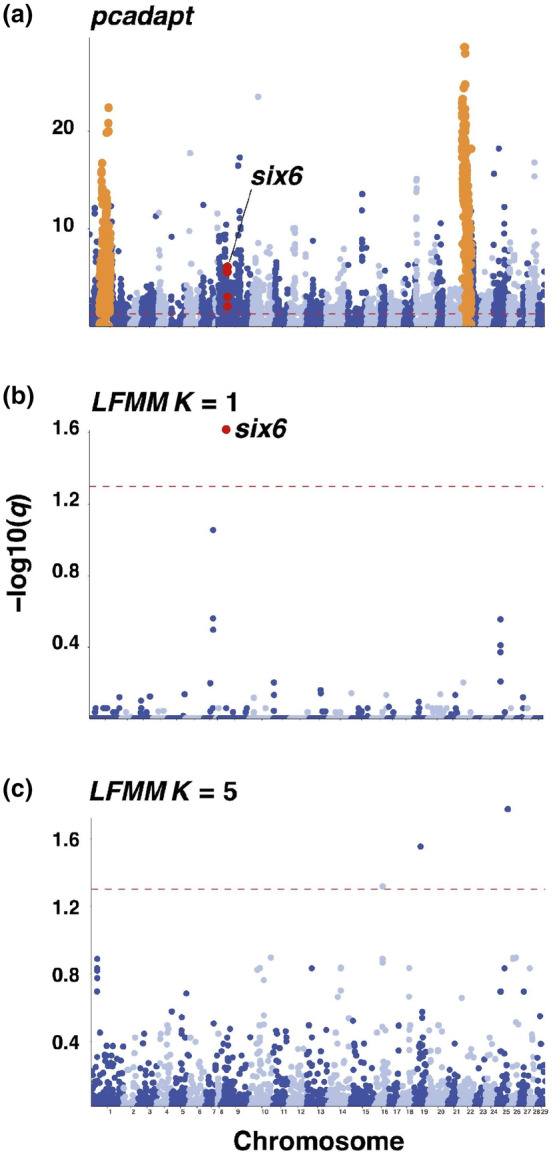
Genome‐wide distribution of Axiom array SNPs exhibiting significant association (red dotted line, q < 0.05) with population structure in pcadapt (a), 1SW proportion in river of origin in LFMM with K = 1 (b) with red circles indicating the location of SNPs within six6, and orange circles indicating location of karyotypic variants identified by Lehnert, Bentzen, et al. (2019).
Selection scans of WGS samples using PCAngsd identified 52,986 significant SNPs overlapping 3262 genes (Table S2), again including six6 (Figure 4a). Allele frequencies at the SNPs with the highest PCA loading differed substantially between North and South datasets but did not show divergence between sea age classes (Figure S4). Significant association of population structure with karyotype variants was negligible (n = 18, ssa01, n = 1 ssa23) among WGS individuals.
FIGURE 4.
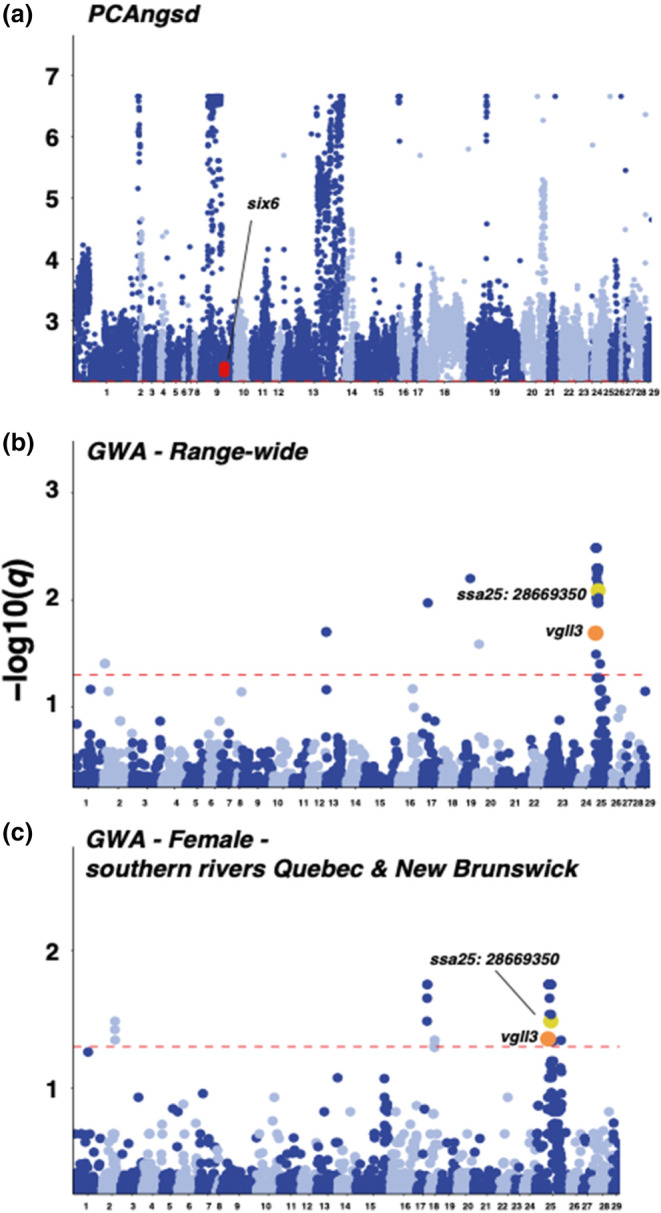
Genome‐wide distribution of outlier SNPs identified from low‐coverage whole‐genome resequencing (WGS) exhibiting significant association (red dotted line, q < 0.05) with population structure in PCAngsd (a), the top 5000 most significant SNPs associated with individual sea age across all samples (b) individual sea age within female fish sampled in southern rivers in Quebec and New Brunswick Rivers (c), with red circles indicating the location of SNPs within six6, orange indicating within vgll3, and yellow indicating the location of the SNP with highest explanatory power for sea age on ssa25 identified by Sinclair‐Waters et al. (2022).
3.3. Genome‐wide association with sea age
Using SNP array data and 1SW proportions from 26 rivers across North America, we found 11 associated SNPs at K = 1 and one overlapping gene: six6 (Figure 3b; Table S3). This pattern of association at six6 was also strongest among the 976 SNPs identified used RDA, overlapping 312 distinct genes (Table S4), and we found complete overlap of the 11 detected SNPs with K = 1 and RDA and pcadapt outliers. However, after controlling for population structure in LFMM using K = 5, we did not detect an association with six6 and instead identified six significantly associated SNPs overlapping two genes, with the strongest signal of association found at a tissue factor pathway inhibitor‐like gene (Table S3). We found both sets of SNPs detected with LFMM exhibited complete overlap with RDA outliers, and high overlap with pcadapt (K1 = 11, 100%, K5 = 3, 50%) outliers.
Genome‐wide association using individual phenotypes using the WGS data instead identified 32 significant loci, and an overlap with vgll3 as the only significantly associated gene in the total dataset analysis (Figure 4b; Table S5). The lambda inflation factor was 1.0635, consistent with that identified in Barson et al. (2015). In a finer‐scale analysis of female fish from southern rivers, we identified 35 significant SNPs, overlapping with three genes: a predicted glutamate receptor, NMDA 2B‐like on ssa02, a butyrophilin subfamily 2 member A1‐like gene on ssa18, and vgll3 (Figure 4c; Table S5). In both comparisons, we uncovered a significant association at a SNP on ssa25 (Figure 4c; Figures S4 and S5) showing the strongest sea age association in European Atlantic salmon, recently identified by Sinclair‐Waters et al. (2022). No significant associations were identified within the other subsets at q < 0.05. In comparison with the genome‐wide average, we identified elevated F ST values among the top 100 most significant SNPs, but no increased PC1 association (Table 3), indicating that SNPs identified in GWA show elevated differentiation between sea age classes, but do not show elevated differentiation associated with population structure. Comparing F ST to uncorrected GWA LRT revealed high and significant correlation (r 2 = .914, p < 1 × 10−15), indicating imputation and genotype likelihood estimates captured similar allele frequency variation across the genome. Similarly, direct comparison of allele frequencies estimated in ANGSD from genotype likelihoods or vcftools from imputed genotypes showed strong correlation (r 2 = .986, p < 1 × 10−15).
TABLE 3.
F ST between 1SW and MSW, PC1 loading in PCANGSD for the top 100 associated SNPs and genome‐wide loci across separate GWA of sea age, with * denoting a significant difference in parameters between top SNP and genome‐wide SNP comparisons.
| GWA group | F ST* | PC1 loading | ||
|---|---|---|---|---|
| Top 100 SNPs | Genome‐wide SNPs | Top 100 SNPs | Genome‐wide SNPs | |
| Female—southern rivers Quebec and New Brunswick | 0.161 | 0.0007 | 0.759 | 1.02 |
| Male—southern rivers Quebec and New Brunswick | 0.0928 | 0.0002 | 0.945 | 1.02 |
| Female—northern rivers Quebec and Labrador | 0.0854 | 0.00009 | 0.923 | 1.02 |
| Male—northern rivers Quebec and Labrador | 0.1533 | 0.0003 | 0.677 | 1.04 |
3.4. Machine learning prediction of sea age
Across datasets, the 100 most significant SNPs based on LRT score could predict sea age with greater than 75% accuracy (Figure 5). Increasing the number of SNPs within each dataset increased accuracy to greater than 80% overall, and 90% in all but the female southern rivers dataset and total dataset models. Investigation of the ROC curve for the total dataset identified both high sensitivity and specificity of random forest models using 500 sea age‐associated SNPs, inferred from rapid rise above the diagonal slope of a random classifier (Figure S6). Across models with randomly selected SNPs, we found consistently low prediction accuracy (~45%), even with the 500 SNP panel, indicating reduced predictive capacity of comparably sized panels of randomly selected genome‐wide SNPs.
FIGURE 5.
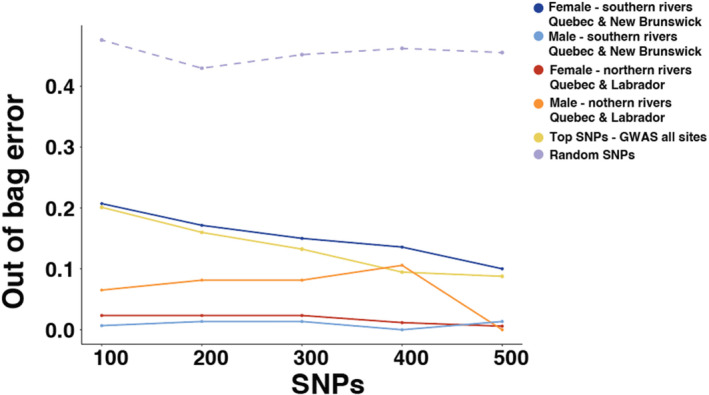
Random forest out‐of‐bag classification error rates across SNP panel sizes (100–500) for the highest ranking SNPs in GWAS for all samples stratified by sex and region from whole‐genome resequencing (WGS). Randomly selected SNPs are represented with a dashed line.
3.5. Gene‐set and gene ontology enrichment
Using gene‐set enrichment, we identified 37 significantly enriched KEGG pathways among GWA comparisons of individual sea age in the WGS data, many of which were significantly enriched among multiple comparisons (Figure 6; Table S6). In contrast, we did not identify significant enrichment among PC scores from WGS or SNP array analyses, or −log10(p) values from range‐wide SNP array LFMM analyses. Hierarchical clustering of −log10(q) values from gene‐set enrichment revealed a core set of nine processes corresponding to developmental, osmoregulatory, neurological, and cardiovascular functions that were most significantly enriched among all groups, such as the phosphatidylinositol signaling system, adrenergic signaling in cardiomyocytes, the GnRH signaling pathway, and focal adhesion. We found the most significant enrichment among all groups in the calcium signaling pathway (Figure 6).
FIGURE 6.
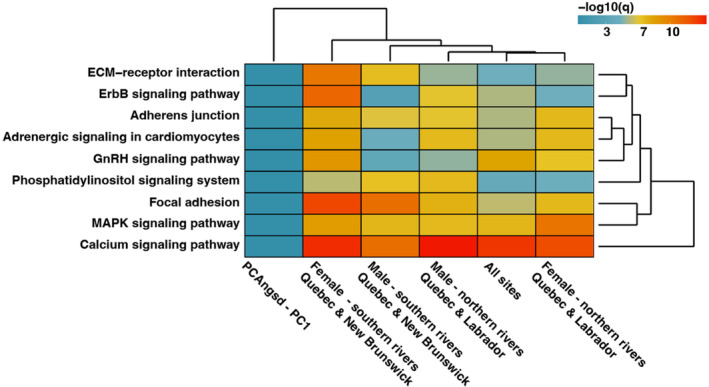
Hierarchical clustering of the nine parallel and highly significant (q < 0.001) KEGG pathways identified using gene‐set enrichment across groups using GWAS scores from whole‐genome resequencing (WGS) data, with gene‐set enrichment PCANGSD results for WGS data included for comparison.
4. DISCUSSION
Identifying the genomic basis of adaptive traits in wild populations is key to understanding how evolutionary processes give rise to diverse and ecologically important phenotypes (Bolnick et al., 2018). In Atlantic Salmon, evolutionary history, marine and freshwater ecological and habitat variability, and plasticity likely shape the genomic repeatability of sea age, but its genomic basis in North America has yet to be identified. Here, we characterized the genetic architecture of sea age variation in North American Atlantic Salmon using SNP array and WGS genomic datasets to test whether this genetic architecture is repeatable at two spatial scales: between European and North American populations, and within North American populations. Consistent with the expectation that large‐effect loci will exhibit higher rates of parallelism (Barghi et al., 2019; Yeaman, 2015), significant associations with sea age in North America overlapped with a large‐effect European sea age locus at the gene and SNP level (vgll3). Within North America, we also found polygenic adaptation within regions and sexes with no overlap of associated loci, whereas gene‐set enrichment revealed repeatable patterns of selection within conserved pathways associated with maturation. Our findings indicate contingency in the genetic architecture of sea age in Atlantic Salmon, but high parallelism of core molecular pathways and large‐effect loci in generating a diversity of at‐sea maturation strategies.
4.1. Detection of known maturation loci
Our results across association analyses in the WGS dataset revealed parallel genetic architecture of sea age variation between North American and European Atlantic Salmon at the previously identified large‐effect locus vgll3. Significant GWA results using WGS data paired with individual phenotypes and elevated differentiation (F ST) indicated strong selection at vgll3 in female fish in southern rivers in Quebec and New Brunswick. This finding is concordant with past research in European populations, both in identifying strong selection and sea age association at vgll3, and in finding sex‐specific patterns of association and dominance at vgll3 (Barson et al., 2015). Similar patterns of association with large‐effect loci underlying sex‐specific maturation and migration have also been revealed across the broader salmonid lineage, indicating large‐effect loci may be common in genetic architectures for distinct life history strategies (Pearse et al., 2019; Thompson et al., 2020).
Additionally, we uncovered a SNP‐level pattern of parallelism at a vgll3‐adjacent region between European and North American Atlantic Salmon. This region has previously been identified as highly significant in explaining sea age variation in European Atlantic Salmon in a recent candidate gene study (Sinclair‐Waters et al., 2022). Our observation is consistent with the expectation that large‐effect loci are more likely to exhibit parallelism (Yeaman, 2015). The source of this parallelism, either through ancestral variation, repeated evolution, or migration, remains unknown and is a clear goal for follow‐up studies (Lee & Coop, 2017). However, our results suggest that associations at vgll3 and the vgll3‐adjacent SNP varied across rivers and sexes, indicating heterogeneity in the genetic architecture of maturation across the North American range despite gene and SNP‐level genetic parallelism. This finding is consistent with a past amplicon‐based study (Kusche et al., 2017), which similarly identified significant variation in associations of vgll3 and sea age dependent on sampled river. Variation in associations with vgll3 and sea age across North American rivers observed by Kusche et al., 2017 was attributed to a potential low proportion of MSW fish with advanced sea age (>3SW) in amplicon‐genotyped samples. Similarly, our sampling of fish across eight North American locations identified predominantly 2SW fish and the small subset of fish with age greater than 2SW were repeat spawners with a 1‐year virgin sea age (n = 11, 1.9%). This finding is concordant with the low proportion of older MSW fish among several rivers studied here (Kusche et al., 2017) and does not preclude vgll3 exhibiting consistent associations with advanced sea age among rivers in North America as observed in Norwegian populations. Surprisingly, we do not find evidence of allelic associations at vgll3 in population‐level tests of association with 1SW proportion, despite it being identified with individual resequencing data. The lack of this association at the population level may be a result of the sex and region‐specific nature of this association, as well as the lack of known sea age classes in samples genotyped on the SNP array.
Using SNP array data, we found an association with river‐level 1SW proportion and six6, which covaried with population structure. The significant signal associated with population structure revealed in PCA, LFMM, and RDA analyses is also consistent with past associations of this locus with fine‐scale population structure and river‐specific features such as catchment area. This finding supports the possibility that this association may also correspond to a relationship with a correlated trait that is influenced by local environment rather than maturation itself (Pritchard et al., 2018; Zueva et al., 2021). This pattern reflects covariation between genetic architecture and population structure across regions exhibiting genome‐wide differentiation as revealed in PCA, as well as differences in 1SW proportion. GWA of individual‐level phenotypes and WGS data also failed to replicate this association in the present study, but we did observe differentiation at six6 among northern and southern rivers which exhibit differences in 1SW proportions, as seen in allele frequency comparisons and PCA of WGS data. Together, these results indicate that six6 itself may not be a major determinant of maturation across North American populations sampled here but is associated with broad‐scale variation in grilse proportion, also consistent with association with a correlated trait. In contrast, GWA of a European multigeneration aquaculture line uncovered significant associations in genomic regions near six6 (Sinclair‐Waters et al., 2020), and this locus was also identified using haplotype‐based and single SNP association models in follow‐up analyses of wild individuals in Europe (Sinclair‐Waters et al., 2022), indicating a more direct role for six6 in maturation among European populations compared to North America.
Our combined results indicate high context dependence of observed parallelism at vgll3 and six6. These genes have shown associations with maturation timing and growth across the broader vertebrate lineage, indicating a conserved genetic role in control of maturation (Cousminer et al., 2013; Perry et al., 2014). Within salmonids, variable patterns of association have also been identified, with recurring associations with maturation across the lineage identified only at six6 (Waters et al., 2021). However, we demonstrate that this relationship is variable even within Atlantic Salmon based on trans‐Atlantic divergence, sex, and regional population structure. Similar patterns of variable reuse of key genes implicated in intraspecific ecological divergence have also been observed in studies of Pungitius and Gasterosteus sticklebacks (Fang et al., 2021), as well as between Timema stick insect species (Villoutreix et al., 2020). Given the diversity of habitats colonized by Atlantic Salmon (Klemetsen et al., 2003), local environmental variation could play a role in biasing genetic parallelism by impacting the fitness landscape associated with trait/gene combinations. Environmental variation has been shown to significantly drive genome‐wide population structuring in Atlantic Salmon (Bradbury et al., 2014; Moore et al., 2014). Sea age has been shown to exhibit a strong latitudinal gradient in North America, with the lowest sea ages observed in Northern populations and associated with precipitation, river size, and lacustrine habitat access, as well as individual survival rates (Hutchings & Jones, 1998; Klemetsen et al., 2003). North American populations across Quebec and the Maritimes exhibit maturation ages more comparable to those in Norway, and we find that locations with lower 1SW proportions also exhibit SNP‐level parallelism with Northern European populations. Together, these findings indicate regional ecological conditions may impact selection for large‐effect loci. Future genomic studies may explore the relationship between life history, genomic, and environmental variation across the range as has recently been conducted in other systems, to quantify the level of overall genomic parallelism across traits and ecological contexts (Rennison et al., 2019).
4.2. Signals of polygenic adaptation
In addition to previously identified large‐effect loci, we uncovered nonparallel polygenic architecture underlying sea age variation across North American rivers. We build on recent detections of polygenic trait architecture underlying maturation detected within aquaculture (Mohamed et al., 2019; Sinclair‐Waters et al., 2020) and breeding experiments (Debes et al., 2021) by identifying sets of predictive loci for maturation. We achieved high prediction accuracy when accounting for population and sex‐level variation in per‐locus association with maturation and found nonparallel sets of loci enable accurate sea age prediction within regional groups. Consistent with other recent polygenic prediction studies in wild populations (Fuller et al., 2020; Hess et al., 2016; Lehnert, Kess, et al., 2019), our results highlight the utility of prediction approaches when accounting for genotype uncertainty and reveal a high capacity to predict sea age in wild Atlantic Salmon populations from genomic data.
The lack of overlap in the set of highly predictive SNPs used in each location and sex indicates high variability in the overall genetic architecture of sea age, consistent with polygenicity. Both variability in environmental conditions and drift likely impact this lack of parallelism, as maturation has been shown to be impacted by population‐specific genetic and environmental factors (Åsheim et al., 2023; Good & Davidson, 2016; Mobley et al., 2021). Atlantic Salmon exhibit high levels of environment‐driven genetic structuring, potentially altering the available set of standing adaptive variation across locations (Bradbury et al., 2014). Population structure has been shown to constrain rates of parallelism and promote heterogeneity in genetic architecture through stochastic loss of standing variation in other systems (Fang et al., 2021), and identification of high postcolonization drift in this and past studies is consistent with this process (Bradbury et al., 2014). Lastly, genome duplications may facilitate nonparallelism through diversification from duplicated gene copies (Ohno, 1970), and whole‐genome duplication within salmonids (Allendorf & Thorgaard, 1994; Lien et al., 2016) may have provided many genomic substrates for adaptation (Campbell et al., 2021). This variation in colonized environments, available adaptive variation, and the environmental variability of maturation together could drive variation in the individual genomic regions associated with maturation.
Our findings from gene‐set enrichment tests (Daub et al., 2013) reveal parallelism in pathways and molecular processes associated with maturation across the broader vertebrate lineage. Parallel enrichment at calcium signaling pathways is consistent with a significant role for calcium metabolism during oocyte maturation (Tosti, 2006) and osmoregulation changes during river spawning (Persson et al., 1998). Additional pathways also revealed significant associations with sexual maturation, including MAPK signaling, which plays a role in meiotic maturation (Kishimoto, 2003), phosphatidylinositol signaling associated with oocyte maturation, and previously in association with polygenic sea age variation in aquaculture fish (Hoshino et al., 2004; Mohamed et al., 2019), and gonadotropin releasing hormone (GnRH), implicated in male maturation (Wen et al., 2010). Neurological processes which may underlie necessary behavioral changes for both sexual maturation and migration (Mobley et al., 2021) were also enriched across comparisons, and we find enrichment in ErbB signaling, which has been linked to neural development and aggression (Barros et al., 2009), and phosphatidylinositol signaling, which has also been shown to mediate nervous system function (Raghu et al., 2019). Detection of significant enrichment among pathways implicated in cardiovascular function is also consistent with differences in energetic costs of large body size and migration in older sea winter fish (Jonsson et al., 1997) and elevated vgll3 expression in heart tissue during maturation (Verta et al., 2020).
We find a high degree of parallelism at the pathway level despite no gene reuse across North American populations, consistent with increasing parallelism at higher levels of organization. Studies of parallelism have frequently identified high parallelism at the level of integrated traits that declines at finer scales of comparison (Bolnick et al., 2018). At the molecular level, recent investigations into parallelism in other salmonids (Jacobs et al., 2020) and humans (Bergey et al., 2018) have similarly uncovered a high degree of parallelism of enriched pathways despite low genetic overlap, suggesting that polygenic architectures underlying complex traits may map on to relatively limited molecular processes. Our results indicate largely idiosyncratic genomic changes at the gene and SNP level across regions and sexes contribute to a few shared higher‐level molecular pathways, corresponding to discrete differences in sea age. The observed molecular parallelism at the pathway level indicates that conserved processes with associations in other vertebrate lineages also underlie maturation in Atlantic Salmon. Interestingly, we find that this relationship holds despite large‐scale differences in 1SW proportions between sampled populations, as well as large variation in environmental conditions, and stock characteristics (e.g. growth rate, survival rate) associated with maturation age (Hutchings & Jones, 1998; Power, 1981). Our results indicate that despite significant plasticity in sea age and corresponding heterogeneity in genetic architecture (Ayllon et al., 2019; Besnier et al., 2023), the pathways implicated in maturation are repeatable between locations. These findings also indicate that ecological variation may play only a limited role in altering the set of conserved molecular functions associated with complex traits and suggest these functions may be predictable across both species and environmental contexts. Future research in this system may benefit from testing the generality of pathway level parallelism of maturation across European populations, as well as other traits, and salmonid and fish species.
4.3. Limitations and future directions
Our study provides the first genomic investigation into polygenic and molecular components of sea age variation in North American Atlantic Salmon, but follow‐up studies are necessary to build on uncertainty from limitations to the approaches used here. Our identification of different genes associated with sea age variation across individual‐level (WGS) and river‐level (SNP array) datasets indicates remaining sensitivity to population structure in analyses of population‐level phenotypic information. Replicability and informativeness of GWA studies can be significantly reduced when population structure is not appropriately controlled (Korte & Farlow, 2013); this challenge is exacerbated in instances of overlap between population structure and adaptation making population structure correction overly conservative in some instances (François et al., 2016). However, our individual‐level resequencing GWA showed only a weak association with major axes of population structure, and across methods, we uncovered previously identified large‐effect sea age loci, highlighting the utility of range‐wide sampling and individual phenotypes for GWA in wild populations. Our sample sizes here are comparable to those used in detection of sex‐specific large effect loci in Atlantic Salmon (Ayllon et al., 2015; Barson et al., 2015) and other salmonids (McKinney et al., 2021; Thompson et al., 2020), and in identification of polygenic patterns identified in gene‐set enrichment studies (Bergey et al., 2018; Foll et al., 2014). However, our study remains much smaller than large‐scale human and agricultural GWA studies, and follow‐up validation with larger, independent datasets and meta‐analysis of GWA scores will provide greater capacity to detect polygenicity in Atlantic Salmon using both GWA and gene‐set enrichment methods.
An additional potential source of uncertainty is that our WGS analyses were restricted to low‐depth (~3x) resequenced SNPs. Recent studies indicate significant roles for structural and epigenomic variation in mediating adaptation across many systems (Layton & Bradbury, 2022), and future assay of these types of variation may identify additional molecular mechanisms important in sea age variation. Low‐depth resequencing also introduces some uncertainty about variants detected. However, we employed large sample size (n > 500), and statistical control for genotype uncertainty through genotype likelihood approaches and imputation. Recent simulations have shown that with sample sizes, sequencing depths, and levels of population structure used here, both likelihood and imputation approaches should accurately capture allele frequencies (Lou et al., 2021), and we find very high and significant correlation among allele frequencies, F ST from imputed data, and genotype likelihood GWA scores. However, as sequencing quality and depth improves, sequencing larger panels of individuals with technologies that capture more classes of variant (e.g., epigenomic modifications and structural variants) will aid in characterizing the genomic basis of sea age variation.
5. CONCLUSIONS
Adaptation of complex phenotypes such as life history variation often presents comparably complex underlying architectures. These traits may evolve through a combination of evolution from standing variation, hard and soft sweeps, and alleles of varying effect size. Here, we used GWA, genome scans, and random forest‐based polygenic prediction to explore the genomic basis of sea age variation in North American Atlantic Salmon. We found evidence of significant association with previously identified large‐effect genes and individual variants in vgll3 as well as varying patterns of association based on both sex and region, suggesting both parallelism at large‐effect loci, and a high degree of genetic redundancy and polygenicity in this trait. Despite low overlap of the most strongly associated genes, we found a set of molecular pathways with conserved roles in maturation were consistently enriched among comparisons, revealing a core set of molecular mechanisms that underlie sea age variation in North American Atlantic Salmon. Our findings demonstrate clear pathways and genes for future investigations of maturation traits, and show how methods aimed at resolving polygenic patterns can uncover the molecular basis of a complex phenotype with vary genetic architectures across wild populations.
AUTHOR CONTRIBUTIONS
Tony Kess: Conceptualization (lead); data curation (lead); formal analysis (lead); investigation (lead); methodology (lead); project administration (supporting); software (lead); validation (lead); visualization (lead); writing – original draft (lead); writing – review and editing (lead). Sarah J. Lehnert: Data curation (lead); resources (equal); validation (equal); writing – original draft (equal); writing – review and editing (equal). Paul Bentzen: Data curation (supporting); funding acquisition (supporting); investigation (equal); methodology (supporting); project administration (supporting); resources (equal); supervision (equal); writing – original draft (equal); writing – review and editing (equal). Steven Duffy: Conceptualization (lead); data curation (lead); methodology (lead); project administration (lead); resources (lead); supervision (lead); writing – original draft (equal); writing – review and editing (equal). Amber Messmer: Conceptualization (equal); data curation (equal); methodology (lead); project administration (equal); resources (equal); writing – original draft (equal); writing – review and editing (equal). J. Brian Dempson: Conceptualization (equal); data curation (equal); investigation (equal); resources (equal); writing – original draft (equal); writing – review and editing (equal). Jason Newport: Formal analysis (equal); methodology (equal); software (equal). Christopher Whidden: Formal analysis (equal); methodology (equal); software (equal); validation (equal); writing – original draft (equal); writing – review and editing (equal). Martha J. Robertson: Data curation (equal); resources (equal); writing – original draft (equal); writing – review and editing (equal). Gerald Chaput: Data curation (equal); methodology (equal); resources (equal); validation (equal); writing – original draft (equal); writing – review and editing (equal). Cindy Breau: Data curation (equal); methodology (equal); resources (equal); writing – original draft (equal); writing – review and editing (equal). Julien April: Data curation (equal); methodology (equal); resources (equal); writing – original draft (equal); writing – review and editing (equal). Carole‐Anne Gillis: Data curation (equal); methodology (equal); resources (equal); writing – original draft (equal); writing – review and editing (equal). Matthew Kent: Data curation (equal); methodology (equal); resources (equal); writing – original draft (equal); writing – review and editing (equal). Cameron M. Nugent: Formal analysis (supporting); methodology (supporting); software (supporting); validation (supporting); writing – original draft (equal); writing – review and editing (equal). Ian R. Bradbury: Conceptualization (lead); data curation (equal); formal analysis (supporting); funding acquisition (lead); investigation (lead); methodology (equal); project administration (lead); resources (lead); supervision (lead); validation (supporting); visualization (supporting); writing – original draft (lead); writing – review and editing (lead).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
Supporting information
Figures S1–S7
Tables S1–S7
ACKNOWLEDGMENTS
We thank DFO staff and private partners for sampling and scale reading for sea age inference, CIGENE for SNP genotyping and data processing, and Genome Quebec for whole‐genome sequencing. Thanks to Brendan Wringe for assistance with regional assignments of SNP array populations. This study was supported by the Genomics Research and Development Initiative (GRDI) and the Program for Aquaculture Regulatory Research (PARR) of the Department of Fisheries and Oceans Canada (DFO) and the Natural Sciences Engineering and Research Council Canada (NSERC). Samples for the Restigouche River were generously donated by Listuguj Mi'gmaq fishers to GINU to contribute to the advancement of Atlantic salmon research.
Kess, T. , Lehnert, S. J. , Bentzen, P. , Duffy, S. , Messmer, A. , Dempson, J. B. , Newport, J. , Whidden, C. , Robertson, M. J. , Chaput, G. , Breau, C. , April, J. , Gillis, C.‐A. , Kent, M. , Nugent, C. M. , & Bradbury, I. R. (2024). Variable parallelism in the genomic basis of age at maturity across spatial scales in Atlantic Salmon. Ecology and Evolution, 14, e11068. 10.1002/ece3.11068
DATA AVAILABILITY STATEMENT
SNP array genotypes in plink format and phenotype data for association analyses are available on dryad at https://doi.org/10.5061/dryad.g1jwstqz3. Raw reads from WGS data have been uploaded to the NCBI SRA with accession number PRJNA1083490. All scripts used for analysis in this study are available at: https://github.com/TonyKess/seaage_GWAS.
REFERENCES
- Allendorf, F. W. , & Thorgaard, G. H. (1994). Tetraploidy and the evolution of salmonid fishes. In Evolutionary genetics of fishes (pp. 1–53). Springer. [Google Scholar]
- Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- Åsheim, E. R. , Debes, P. V. , House, A. , Liljeström, P. , Niemelä, P. T. , Siren, J. P. , Erkinaro, J. , & Primmer, C. R. (2023). Atlantic salmon (Salmo salar) age at maturity is strongly affected by temperature, population and age‐at‐maturity genotype. Conservation Physiology, 11(1), coac086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayllon, F. , Kjærner‐Semb, E. , Furmanek, T. , Wennevik, V. , Solberg, M. F. , Dahle, G. , Taranger, G. L. , Glover, K. A. , Almén, M. S. , Rubin, C. J. , & Edvardsen, R. B. (2015). The vgll3 locus controls age at maturity in wild and domesticated Atlantic salmon (Salmo salar L.) males. PLoS Genetics, 11(11), e1005628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayllon, F. , Solberg, M. F. , Glover, K. A. , Mohammadi, F. , Kjærner‐Semb, E. , Fjelldal, P. G. , Andersson, E. , Hansen, T. , Edvardsen, R. B. , & Wargelius, A. (2019). The influence of vgll3 genotypes on sea age at maturity is altered in farmed mowi strain Atlantic salmon. BMC Genetics, 20, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barghi, N. , Tobler, R. , Nolte, V. , Jakšić, A. M. , Mallard, F. , Otte, K. A. , Dolezal, M. , Taus, T. , Kofler, R. , & Schlötterer, C. (2019). Genetic redundancy fuels polygenic adaptation in drosophila. PLoS Biology, 17(2), e3000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros, C. S. , Calabrese, B. , Chamero, P. , Roberts, A. J. , Korzus, E. , Lloyd, K. , Stowers, L. , Mayford, M. , Halpain, S. , & Müller, U. (2009). Impaired maturation of dendritic spines without disorganization of cortical cell layers in mice lacking NRG1/ErbB signaling in the central nervous system. Proceedings of the National Academy of Sciences, 106(11), 4507–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barson, N. J. , Aykanat, T. , Hindar, K. , Baranski, M. , Bolstad, G. H. , Fiske, P. , Jacq, C. , Jensen, A. J. , Johnston, S. E. , Karlsson, S. , Kent, M. , Moen, T. , Niemelä, E. , Nome, T. , Næsje, T. F. , Orell, P. , Romakkaniemi, A. , Sægrov, H. , Urdal, K. , … Primmer, C. R. (2015). Sex‐dependent dominance at a single locus maintains variation in age at maturity in salmon. Nature, 528(7582), 405–408. [DOI] [PubMed] [Google Scholar]
- Bergey, C. M. , Lopez, M. , Harrison, G. F. , Patin, E. , Cohen, J. A. , Quintana‐Murci, L. , Barreiro, L. B. , & Perry, G. H. (2018). Polygenic adaptation and convergent evolution on growth and cardiac genetic pathways in African and Asian rainforest hunter‐gatherers. Proceedings of the National Academy of Sciences, 115(48), 11256–11263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnier, F. , Skaala, Ø. , Wennevik, V. , Ayllon, F. , Utne, K. R. , Fjeldheim, P. T. , Andersen‐Fjeldheim, K. , Knutar, S. , & Glover, K. A. (2023). Overruled by nature: A plastic response to environmental change disconnects a gene and its trait. Molecular Ecology, 33, e16933. [DOI] [PubMed] [Google Scholar]
- Blount, Z. D. , Lenski, R. E. , & Losos, J. B. (2018). Contingency and determinism in evolution: Replaying life's tape. Science, 362, eaam5979. [DOI] [PubMed] [Google Scholar]
- Bolnick, D. I. , Barrett, R. D. , Oke, K. B. , Rennison, D. J. , & Stuart, Y. E. (2018). (Non) parallel evolution. Annual Review of Ecology, Evolution, and Systematics, 49, 303–330. [Google Scholar]
- Boulding, E. G. , Ang, K. P. , Elliott, J. A. , Powell, F. , & Schaeffer, L. R. (2019). Differences in genetic architecture between continents at a major locus previously associated with sea age at sexual maturity in European Atlantic salmon. Aquaculture, 500, 670–678. [Google Scholar]
- Bradbury, I. R. , Hamilton, L. C. , Robertson, M. J. , Bourgeois, C. E. , Mansour, A. , & Dempson, J. B. (2014). Landscape structure and climatic variation determine Atlantic salmon genetic connectivity in the Northwest Atlantic. Canadian Journal of Fisheries and Aquatic Sciences, 71(2), 246–258. [Google Scholar]
- Breiman, L. (2001). Random forests. Machine Learning, 45(1), 5–32. [Google Scholar]
- Brieuc, M. S. , Waters, C. D. , Drinan, D. P. , & Naish, K. A. (2018). A practical introduction to random forest for genetic association studies in ecology and evolution. Molecular Ecology Resources, 18(4), 755–766. [DOI] [PubMed] [Google Scholar]
- Browning, S. R. , & Browning, B. L. (2007). Rapid and accurate haplotype phasing and missing data inference for whole genome association studies by use of localized haplotype clustering. American Journal of Human Genetics, 81, 1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, M. A. , Anderson, E. C. , Garza, J. C. , & Pearse, D. E. (2021). Polygenic basis and the role of genome duplication in adaptation to similar selective environments. Journal of Heredity, 112(7), 614–625. [DOI] [PubMed] [Google Scholar]
- Capblancq, T. , & Forester, B. R. (2021). Redundancy analysis: A Swiss Army knife for landscape genomics. Methods in Ecology and Evolution, 12, 2298–2309. 10.1111/2041-210X.13722 [DOI] [Google Scholar]
- Cattell, R. B. (1966). The scree test for the number of factors. Multivariate Behavioral Research, 1, 245–276. [DOI] [PubMed] [Google Scholar]
- Caye, K. , Jumentier, B. , Lepeule, J. , & François, O. (2019). LFMM 2: Fast and accurate inference of gene‐environment associations in genome‐wide studies. Molecular Biology and Evolution, 36(4), 852–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, C. C. , Chow, C. C. , Tellier, L. C. , Vattikuti, S. , Purcell, S. M. , & Lee, J. J. (2015). Second‐generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience, 4, 7. 10.1186/s13742-015-0047-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z. , Boehnke, M. , Wen, X. , & Mukherjee, B. (2021). Revisiting the genome‐wide significance threshold for common variant GWAS. G3, 11(2), jkaa056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte, G. L. , Arnegard, M. E. , Peichel, C. L. , & Schluter, D. (2012). The probability of genetic parallelism and convergence in natural populations. Proceedings of the Royal Society B: Biological Sciences, 279(1749), 5039–5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousminer, D. L. , Berry, D. J. , Timpson, N. J. , Ang, W. , Thiering, E. , Byrne, E. M. , Taal, H. R. , Huikari, V. , Bradfield, J. P. , Kerkhof, M. , Groen‐Blokhuis, M. M. , Kreiner‐Møller, E. , Marinelli, M. , Holst, C. , Leinonen, J. T. , Perry, J. R. B. , Surakka, I. , Pietiläinen, O. , Kettunen, J. , … for the Early Growth Genetics (EGG) Consortium . (2013). Genome‐wide association and longitudinal analyses reveal genetic loci linking pubertal height growth, pubertal timing and childhood adiposity. Human Molecular Genetics, 22, 2735–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czorlich, Y. , Aykanat, T. , Erkinaro, J. , Orell, P. , & Primmer, C. R. (2018). Rapid sex‐specific evolution of age at maturity is shaped by genetic architecture in Atlantic salmon. Nature Ecology & Evolution, 2(11), 1800–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C. A. , Banks, E. , DePristo, M. A. , Handsaker, R. E. , Lunter, G. , Marth, G. T. , Sherry, S. T. , & McVean, G. (2011). The variant call format and VCFtools. Bioinformatics, 27(15), 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub, J. T. , Hofer, T. , Cutivet, E. , Dupanloup, I. , Quintana‐Murci, L. , Robinson‐Rechavi, M. , & Excoffier, L. (2013). Evidence for polygenic adaptation to pathogens in the human genome. Molecular Biology and Evolution, 30(7), 1544–1558. [DOI] [PubMed] [Google Scholar]
- Debes, P. V. , Piavchenko, N. , Ruokolainen, A. , Ovaskainen, O. , Moustakas‐Verho, J. E. , Parre, N. , Aykanat, T. , Erkinaro, J. , & Primmer, C. R. (2021). Polygenic and major‐locus contributions to sexual maturation timing in Atlantic salmon. Molecular Ecology, 30(18), 4505–4519. [DOI] [PubMed] [Google Scholar]
- DePristo, M. A. , Banks, E. , Poplin, R. , Garimella, K. V. , Maguire, J. R. , Hartl, C. , Philippakis, A. A. , Del Angel, G. , Rivas, M. A. , Hanna, M. , & McKenna, A. (2011). A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nature Genetics, 43(5), 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin, B. , & Roeder, K. (1999). Genomic control for association studies. Biometrics, 55(4), 997–1004. [DOI] [PubMed] [Google Scholar]
- Duforet‐Frebourg, N. , Luu, K. , Laval, G. , Bazin, E. , & Blum, M. G. (2016). Detecting genomic signatures of natural selection with principal component analysis: Application to the 1000 genomes data. Molecular Biology and Evolution, 33(4), 1082–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmer, K. R. , & Meyer, A. (2011). Adaptation in the age of ecological genomics: Insights from parallelism and convergence. Trends in Ecology & Evolution, 26, 298–306. [DOI] [PubMed] [Google Scholar]
- Fagny, M. , & Austerlitz, F. (2021). Polygenic adaptation: Integrating population genetics and gene regulatory networks. Trends in Genetics, 7(7), 631–638. [DOI] [PubMed] [Google Scholar]
- Fang, B. , Kemppainen, P. , Momigliano, P. , & Merilä, J. (2021). Population structure limits parallel evolution in sticklebacks. Molecular Biology and Evolution, 38(10), 4205–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foll, M. , Gaggiotti, O. E. , Daub, J. T. , Vatsiou, A. , & Excoffier, L. (2014). Widespread signals of convergent adaptation to high altitude in Asia and America. The American Journal of Human Genetics, 95(4), 394–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- François, O. , Martins, H. , Caye, K. , & Schoville, S. D. (2016). Controlling false discoveries in genome scans for selection. Molecular Ecology, 25(2), 454–469. [DOI] [PubMed] [Google Scholar]
- Frichot, E. , & Francois, O. (2015). LEA: An R package for landscape and ecological association studies. Methods in Ecology and Evolution, 6, 925–929. [Google Scholar]
- Frichot, E. , Mathieu, F. , Trouillon, T. , Bouchard, G. , & François, O. (2014). Fast and efficient estimation of individual ancestry coefficients. Genetics, 196, 973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland, K. D. , Hansenm, L. P. , Dunkley, D. A. , & MacLean, J. C. (2000). Linkage between ocean climate, post‐smolt growth, and survival of Atlantic salmon (Salmo salar L.) in the North Sea area. ICES Journal of Marine Science, 57(2), 419–429. [Google Scholar]
- Fuller, Z. L. , Mocellin, V. J. , Morris, L. A. , Cantin, N. , Shepherd, J. , Sarre, L. , Peng, J. , Liao, Y. , Pickrell, J. , Andolfatto, P. , Matz, M. , Bay, L. K. , & Przeworski, M. (2020). Population genetics of the coral Acropora millepora: Toward genomic prediction of bleaching. Science, 369(6501), eaba4674. [DOI] [PubMed] [Google Scholar]
- Garant, D. , Dodson, J. J. , & Bernatchez, L. (2003). Differential reproductive success and heritability of alternative reproductive tactics in wild Atlantic salmon (Salmo salar L.). Evolution, 57(5), 1133–1141. [DOI] [PubMed] [Google Scholar]
- Good, C. , & Davidson, J. (2016). A review of factors influencing maturation of Atlantic salmon, Salmo salar, with focus on water recirculation aquaculture system environments. Journal of the World Aquaculture Society, 47(5), 605–632. [Google Scholar]
- Hess, J. E. , Zendt, J. S. , Matala, A. R. , & Narum, S. R. (2016). Genetic basis of adult migration timing in anadromous steelhead discovered through multivariate association testing. Proceedings of the Royal Society B: Biological Sciences, 283(1830), 20153064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino, Y. , Yokoo, M. , Yoshida, N. , Sasada, H. , Matsumoto, H. , & Sato, E. (2004). Phosphatidylinositol 3‐kinase and Akt participate in the FSH‐induced meiotic maturation of mouse oocytes. Molecular Reproduction and Development: Incorporating Gamete Research, 69(1), 77–86. [DOI] [PubMed] [Google Scholar]
- Hutchings, J. A. , & Jones, M. E. (1998). Life history variation and growth rate thresholds for maturity in Atlantic salmon, Salmo salar . Canadian Journal of Fisheries and Aquatic Sciences, 55(S1), 22–47. [Google Scholar]
- Jacobs, A. , Carruthers, M. , Yurchenko, A. , Gordeeva, N. V. , Alekseyev, S. , Hooker, O. , Leong, J. , Minkley, D. R. , Rondeau, E. , Koop, B. , Adams, C. , & Elmer, K. R. (2020). Parallelism in eco‐morphology and gene expression despite variable evolutionary and genomic backgrounds in a Holarctic fish. PLoS Genetics, 16(4), e1008658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery, N. W. , Stanley, R. R. , Wringe, B. F. , Guijarro‐Sabaniel, J. , Bourret, V. , Bernatchez, L. , Bentzen, P. , Beiko, R. G. , Gilbey, J. , Clément, M. , & Bradbury, I. R. (2017). Range‐wide parallel climate‐associated genomic clines in Atlantic salmon. Royal Society Open Science, 4(11), 171394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, S. E. , Orell, P. , Pritchard, V. L. , Kent, M. P. , Lien, S. , Niemelä, E. , Erkinaro, J. , & Primmer, C. R. (2014). Genome wide SNP analysis reveals a genetic basis for sea age variation in a wild population of Atlantic salmon (Salmo salar). Molecular Ecology, 23(14), 3452–3468. [DOI] [PubMed] [Google Scholar]
- Jonsson, N. , & Jonsson, B. (2007). Sea growth, smolt age and age at sexual maturation in Atlantic salmon. Journal of Fish Biology, 71(1), 245–252. [Google Scholar]
- Jonsson, N. , Jonsson, B. , & Hansen, L. P. (1997). Changes in proximate composition and estimates of energetic costs during upstream migration and spawning in Atlantic salmon Salmo salar . Journal of Animal Ecology, 66, 425–436. [Google Scholar]
- Jørsboe, E. , & Albrechtsen, A. (2022). Efficient approaches for large‐scale GWAS with genotype uncertainty. G3: Genes, Genomes, Genetics, 12(1), jkab385. 10.1093/g3journal/jkab385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeuffer, R. , Peichel, C. L. , Bolnick, D. I. , & Hendry, A. P. (2012). Parallel and nonparallel aspects of ecological, phenotypic, and genetic divergence across replicate population pairs of lake and stream stickleback. Evolution, 66(2), 402–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto, T. (2003). Cell‐cycle control during meiotic maturation. Current Opinion in Cell Biology, 15(6), 654–663. [DOI] [PubMed] [Google Scholar]
- Klemetsen, A. , Amundsen, P. A. , Dempson, J. B. , Jonsson, B. , Jonsson, N. , O'connell, M. F. , & Mortensen, E. (2003). Atlantic salmon Salmo salar (L)., brown trout Salmo trutta (L). and Arctic charr Salvelinus alpinus (L.): A review of aspects of their life histories. Ecology of Freshwater Fish, 12(1), 1–59. [Google Scholar]
- Korneliussen, T. S. , Albrechtsen, A. , & Nielsen, R. (2014). ANGSD: Analysis of next generation sequencing data. BMC Bioinformatics, 15(1), 356. 10.1186/s12859-014-0356-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte, A. , & Farlow, A. (2013). The advantages and limitations of trait analysis with GWAS: A review. Plant Methods, 9(1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiner, J. M. , Tranel, P. J. , Weigel, D. , Stinchcombe, J. R. , & Wright, S. I. (2021). The genetic architecture and population genomic signatures of glyphosate resistance in Amaranthus tuberculatus . Molecular Ecology, 30(21), 5373–5389. [DOI] [PubMed] [Google Scholar]
- Kusche, H. , Côté, G. , Hernandez, C. , Normandeau, E. , Boivin Delisle, D. , & Bernatchez, L. (2017). Characterization of natural variation in north American Atlantic Salmon populations (Salmonidae: Salmo salar) at a locus with a major effect on sea age. Ecology and Evolution, 7(15), 5797–5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson, D. J. , van Dorp, L. , & Falush, D. (2018). A tutorial on how not to over‐interpret STRUCTURE and ADMIXTURE bar plots. Nature Communications, 9, 3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layton, K. K. , & Bradbury, I. R. (2022). Harnessing the power of multi‐omics data for predicting climate change response. Journal of Animal Ecology, 91, 1064–1072. 10.1111/1365-2656.13619 [DOI] [PubMed] [Google Scholar]
- Lee, K. M. , & Coop, G. (2017). Distinguishing among modes of convergent adaptation using population genomic data. Genetics, 207(4), 1591–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre, P. , & Legendre, L. (2012). Numerical ecology. Elsevier. [Google Scholar]
- Lehnert, S. J. , Bentzen, P. , Kess, T. , Lien, S. , Horne, J. B. , Clément, M. , & Bradbury, I. R. (2019). Chromosome polymorphisms track trans Atlantic divergence and secondary contact in Atlantic salmon. Molecular Ecology, 28(8), 2074–2087. [DOI] [PubMed] [Google Scholar]
- Lehnert, S. J. , Bradbury, I. R. , Wringe, B. F. , Van Wyngaarden, M. , & Bentzen, P. (2023). Multifaceted framework for defining conservation units: An example from Atlantic salmon (Salmo salar) in Canada. Evolutionary Applications, 16, 1568–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnert, S. J. , Kess, T. , Bentzen, P. , Clément, M. , & Bradbury, I. R. (2020). Divergent and linked selection shape patterns of genomic differentiation between European and north American Atlantic salmon (Salmo salar). Molecular Ecology, 29(12), 2160–2175. [DOI] [PubMed] [Google Scholar]
- Lehnert, S. J. , Kess, T. , Bentzen, P. , Kent, M. P. , Lien, S. , Gilbey, J. , Clément, M. , Jeffery, N. W. , Waples, R. S. , & Bradbury, I. R. (2019). Genomic signatures and correlates of widespread population declines in salmon. Nature Communications, 10(1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv, 1303, 3997.
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. , & Durbin, R. (2009). The sequence alignment/map format and SAMtools. Bioinformatics, 25(16), 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw, A. , & Wiener, M. (2002). Classification and regression by randomForest. R News, 2(3), 18–22. [Google Scholar]
- Lien, S. , Koop, B. F. , Sandve, S. R. , Miller, J. R. , Kent, M. P. , Nome, T. , Hvidsten, T. R. , Leong, J. S. , Minkley, D. R. , Zimin, A. , Grammes, F. , Grove, H. , Gjuvsland, A. , Walenz, B. , Hermansen, R. A. , von Schalburg, K. , Rondeau, E. B. , di Genova, A. , Samy, J. K. A. , … Davidson, W. S. (2016). The Atlantic salmon genome provides insights into rediploidization. Nature, 533, 200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longmire, J. , Maltbie, M. , & Baker, R.J. (1997). Use of "Lysis Buffer" in DNA isolation and its implication for museum collections . Museum of Texas Tech University. Occasional papers.
- Lou, R. N. , Jacobs, A. , Wilder, A. P. , & Therkildsen, N. O. (2021). A beginner's guide to low‐coverage whole genome sequencing for population genomics. Molecular Ecology, 30(23), 5966–5993. [DOI] [PubMed] [Google Scholar]
- Luu, K. , Bazin, E. , & Blum, M. G. (2017). pcadapt: An R package to perform genome scans for selection based on principal component analysis. Molecular Ecology Resources, 17(1), 67–77. 10.1111/1755-0998.12592 [DOI] [PubMed] [Google Scholar]
- Martin, M. (2011). Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet. Journal, 17(1), 10–12. [Google Scholar]
- Mayjonade, B. , Gouzy, J. , Donnadieu, C. , Pouilly, N. , Marande, W. , Callot, C. , Langlade, N. , & Muños, S. (2016). Extraction of high‐molecular‐weight genomic DNA for long‐read sequencing of single molecules. BioTechniques, 61(4), 203–205. [DOI] [PubMed] [Google Scholar]
- McGaugh, S. E. , Lorenz, A. J. , & Flagel, L. E. (2021). The utility of genomic prediction models in evolutionary genetics. Proceedings of the Royal Society B, 288(1956), 20210693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney, G. J. , Nichols, K. M. , & Ford, M. J. (2021). A mobile sex‐determining region, male‐specific haplotypes and rearing environment influence age at maturity in Chinook salmon. Molecular Ecology, 30(1), 131–147. [DOI] [PubMed] [Google Scholar]
- Meisner, J. , & Albrechtsen, A. (2018). Inferring population structure and admixture proportions in low‐depth NGS data. Genetics, 210(2), 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisner, J. , Albrechtsen, A. , & Hanghøj, K. (2021). Detecting selection in low‐coverage high‐throughput sequencing data using principal component analysis. BMC Bioinformatics, 22, 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobley, K. B. , Aykanat, T. , Czorlich, Y. , House, A. , Kurko, J. , Miettinen, A. , Moustakas‐Verho, J. , Salgado, A. , Sinclair‐Waters, M. , Verta, J. P. , & Primmer, C. R. (2021). Maturation in Atlantic salmon (Salmo salar, Salmonidae): A synthesis of ecological, genetic, and molecular processes. Reviews in Fish Biology and Fisheries, 31, 523–571. [Google Scholar]
- Mohamed, A. R. , Verbyla, K. L. , Al‐Mamun, H. A. , McWilliam, S. , Evans, B. , King, H. , Kube, P. , & Kijas, J. W. (2019). Polygenic and sex specific architecture for two maturation traits in farmed Atlantic salmon. BMC Genomics, 20, 139. 10.1186/s12864-019-5525-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, J. S. , Bourret, V. , Dionne, M. , Bradbury, I. R. , O'Reilly, P. , Kent, M. , Chaput, G. , & Bernatchez, L. (2014). Conservation genomics of anadromous Atlantic salmon across its north American range: Outlier loci identify the same patterns of population structure as neutral loci. Molecular Ecology, 23(23), 5680–5697. [DOI] [PubMed] [Google Scholar]
- Nugent, C. M. , Kess, T. , Brachmann, M. K. , Langille, B. L. , Holborn, M. K. , Beck, S. V. , Smith, N. , Duffy, S. J. , Lehnert, S. J. , Wringe, B. F. , Bentzen, P. , & Bradbury, I. R. (2023). Genomic and machine learning‐based screening of aquaculture‐associated introgression into at‐risk wild north American Atlantic salmon (Salmo salar) populations. Molecular Ecology Resources, 00, 1–17. 10.1111/1755-0998.13811 [DOI] [PubMed] [Google Scholar]
- Ogata, H. , Goto, S. , Fujibuchi, W. , & Kanehisa, M. (1998). Computation with the KEGG pathway database. Bio Systems, 47(1–2), 119–128. [DOI] [PubMed] [Google Scholar]
- Ohno, S. (1970). The enormous diversity in genome sizes of fish as a reflection of nature's extensive experiments with gene duplication. Transactions of the American Fisheries Society, 99(1), 120–130. [Google Scholar]
- Oksanen, J. , Guillaume, F. G. , Friendly, M. , Kindt, R. , Legendre, P. , McGlinn, D. , Minchin, P. R. , O'Hara, R. B. , Simpson, G. L. , Solymos, P. , Stevens, M. H. H. , Szoecs, E. , & Wagner, H. (2020). Vegan: Community ecology package. R package version 2.5‐7. https://CRAN.R‐project.org/package=vegan
- Olmos, M. , Payne, M. R. , Nevoux, M. , Prévost, E. , Chaput, G. , Du Pontavice, H. , Guitton, J. , Sheehan, T. , Mills, K. , & Rivot, E. (2020). Spatial synchrony in the response of a long range migratory species (Salmo salar) to climate change in the North Atlantic Ocean. Global Change Biology, 26, 1319–1337. [DOI] [PubMed] [Google Scholar]
- Pearse, D. E. , Barson, N. J. , Nome, T. , Gao, G. , Campbell, M. A. , Abadía‐Cardoso, A. , Anderson, E. C. , Rundio, D. E. , Williams, T. H. , Naish, K. A. , Moen, T. , Liu, S. , Kent, M. , Moser, M. , Minkley, D. R. , Rondeau, E. B. , Brieuc, M. S. O. , Sandve, S. R. , Miller, M. R. , … Lien, S. (2019). Sex‐dependent dominance maintains migration supergene in rainbow trout. Nature Ecology & Evolution, 3(12), 1731–1742. [DOI] [PubMed] [Google Scholar]
- Pedersen, B. S. , & Quinlan, A. R. (2018). Mosdepth: Quick coverage calculation for genomes and exomes. Bioinformatics, 34(5), 867–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry, J. R. B. , Day, F. , Elks, C. E. , Sulem, P. , Thompson, D. J. , Ferreira, T. , He, C. , Chasman, D. I. , Esko, T. , Thorleifsson, G. , Albrecht, E. , Ang, W. Q. , Corre, T. , Cousminer, D. L. , Feenstra, B. , Franceschini, N. , Ganna, A. , Johnson, A. D. , Kjellqvist, S. , … Ong, K. (2014). Parent‐of‐origin‐specific allelic associations among 106 genomic loci for age at menarche. Nature, 514, 92–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson, P. , Sundell, K. , Björnsson, B. T. , & Lundqvist, H. (1998). Calcium metabolism and osmoregulation during sexual maturation of river running Atlantic salmon. Journal of Fish Biology, 52(2), 334–349. [Google Scholar]
- Power, G. (1981). Stock characteristics and catches of Atlantic salmon (Salmo salar) in Quebec, and Newfoundland and Labrador in relation to environmental variables. Canadian Journal of Fisheries and Aquatic Sciences, 38(12), 1601–1611. [Google Scholar]
- Pritchard, V. L. , Mäkinen, H. , Vähä, J. P. , Erkinaro, J. , Orell, P. , & Primmer, C. R. (2018). Genomic signatures of fine‐scale local selection in Atlantic salmon suggest involvement of sexual maturation, energy homeostasis and immune defence‐related genes. Molecular Ecology, 27(11), 2560–2575. [DOI] [PubMed] [Google Scholar]
- Privé, F. , Luu, F. , Vilhjálmsson, B. J. , & Blum, M. G. B. (2020). Performing highly efficient genome scans for local adaptation with R package pcadapt version 4. Molecular Biology and Evolution, 37(7), 2153–2154. [DOI] [PubMed] [Google Scholar]
- Raghu, P. , Joseph, A. , Krishnan, H. , Singh, P. , & Saha, S. (2019). Phosphoinositides: Regulators of nervous system function in health and disease. Frontiers in Molecular Neuroscience, 12, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph, P. L. , & Coop, G. (2015). The role of standing variation in geographic convergent adaptation. The American Naturalist, 186(S1), S5–S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennison, D. J. , Stuart, Y. E. , Bolnick, D. I. , & Peichel, C. L. (2019). Ecological factors and morphological traits are associated with repeated genomic differentiation between lake and stream stickleback. Philosophical Transactions of the Royal Society B, 374(1777), 20180241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougemont, Q. , & Bernatchez, L. (2018). The demographic history of Atlantic salmon (Salmo salar) across its distribution range reconstructed from approximate Bayesian computations. Evolution, 72(6), 1261–1277. [DOI] [PubMed] [Google Scholar]
- Salisbury, S. , McCracken, G. R. , Perry, R. , Keefe, D. , Layton, K. K. , Kess, T. , Nugent, C. M. , Leong, J. S. , Bradbury, I. R. , Koop, B. F. , Ferguson, M. M. , & Ruzzante, D. E. (2022). The genomic consistency of the loss of anadromy in an Arctic fish (Salvelinus alpinus). The American Naturalist, 199(5), 617–635. [DOI] [PubMed] [Google Scholar]
- Sinclair‐Waters, M. , Nome, T. , Wang, J. , Lien, S. , Kent, M. P. , Sægrov, H. , Florø‐Larsen, B. , Bolstad, G. H. , Primmer, C. R. , & Barson, N. J. (2022). Dissecting the loci underlying maturation timing in Atlantic salmon using haplotype and multi‐SNP based association methods. Heredity, 129, 356–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair‐Waters, M. , Ødegård, J. , Korsvoll, S. A. , Moen, T. , Lien, S. , Primmer, C. R. , & Barson, N. J. (2020). Beyond large‐effect loci: Large‐scale GWAS reveals a mixed large‐effect and polygenic architecture for age at maturity of Atlantic salmon. Genetics Selection Evolution, 52, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey, J. D. , Bass, A. J. , Dabney, A. , & Robinson, D. (2015). Qvalue: Q‐value estimation for false discovery rate control. R Package Version . http://github.com/jdstorey/qvalue
- Storey, J. D. , & Tibshirani, R. (2003). Statistical significance for genomewide studies. Proceedings of the National Academy of Sciences of the United States of America, 100, 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therkildsen, N. O. , & Palumbi, S. R. (2017). Practical low‐coverage genome‐wide sequencing of hundreds of individually barcoded samples for population and evolutionary genomics in non‐model species. Molecular Ecology Resources, 17(2), 194–208. [DOI] [PubMed] [Google Scholar]
- Thompson, N. F. , Anderson, E. C. , Clemento, A. J. , Campbell, M. A. , Pearse, D. E. , Hearsey, J. W. , Kinziger, A. P. , & Garza, J. C. (2020). A complex phenotype in salmon controlled by a simple change in migratory timing. Science, 370(6516), 609–613. [DOI] [PubMed] [Google Scholar]
- Tosti, E. (2006). Calcium ion currents mediating oocyte maturation events. Reproductive Biology and Endocrinology, 4(1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Most, P. J. , Küpers, L. K. , Snieder, H. , & Nolte, I. (2017). QCEWAS: Automated quality control of results of epigenome‐wide association studies. Bioinformatics, 33(8), 1243–1245. [DOI] [PubMed] [Google Scholar]
- Verta, J. P. , Debes, P. V. , Piavchenko, N. , Ruokolainen, A. , Ovaskainen, O. , Moustakas‐Verho, J. E. , Tillanen, S. , Parre, N. , Aykanat, T. , Erkinaro, J. , & Primmer, C. R. (2020). Cis‐regulatory differences in isoform expression associate with life history strategy variation in Atlantic salmon. PLoS Genetics, 16(9), e1009055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villoutreix, R. , de Carvalho, C. F. , Soria‐Carrasco, V. , Lindtke, D. , De‐la‐Mora, M. , Muschick, M. , Feder, J. L. , Parchman, T. L. , Gompert, Z. , & Nosil, P. (2020). Large‐scale mutation in the evolution of a gene complex for cryptic coloration. Science, 369(6502), 460–466. [DOI] [PubMed] [Google Scholar]
- Vollset, K. W. , Urdal, K. , Utne, K. , Thorstad, E. B. , Sægrov, H. , Raunsgard, A. , Skagseth, Ø. , Lennox, R. J. , Østborg, G. M. , Ugedal, O. , Jensen, A. J. , Bolstad, G. , & Fiske, P. (2022). Ecological regime shift in the Northeast Atlantic Ocean revealed from the unprecedented reduction in marine growth of Atlantic salmon. Science Advances, 8(9), eabk2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters, C. D. , Clemento, A. , Aykanat, T. , Garza, J. C. , Naish, K. A. , Narum, S. , & Primmer, C. R. (2021). Heterogeneous genetic basis of age at maturity in salmonid fishes. Molecular Ecology, 30(6), 1435–1456. [DOI] [PubMed] [Google Scholar]
- Weir, B. S. , & Cockerham, C. C. (1984). Estimating F‐statistics for the analysis of population structure. Evolution, 38, 1358–1370. [DOI] [PubMed] [Google Scholar]
- Wen, S. , Ai, W. , Alim, Z. , & Boehm, U. (2010). Embryonic gonadotropin‐releasing hormone signaling is necessary for maturation of the male reproductive axis. National Academy of Sciences of the United States of America, 107(37), 16372–16377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wringe, B. F. , Jeffery, N. W. , Stanley, R. R. , Hamilton, L. C. , Anderson, E. C. , Fleming, I. A. , Grant, C. , Dempson, J. B. , Veinott, G. , Duffy, S. J. , & Bradbury, I. R. (2018). Extensive hybridization following a large escape of domesticated Atlantic salmon in the Northwest Atlantic. Communications Biology, 1, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano, A. , Nicol, B. , Jouanno, E. , Quillet, E. , Fostier, A. , Guyomard, R. , & Guiguen, Y. (2013). The sexually dimorphic on the Y‐chromosome gene (sdY) is a conserved male‐specific Y‐chromosome sequence in many salmonids. Evolutionary Applications, 6, 486–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeaman, S. (2015). Local adaptation by alleles of small effect. The American Naturalist, 186(S1), S74–S89. [DOI] [PubMed] [Google Scholar]
- Yeaman, S. , Gerstein, A. C. , Hodgins, K. A. , & Whitlock, M. C. (2018). Quantifying how constraints limit the diversity of viable routes to adaptation. PLoS Genetics, 14(10), e1007717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, J. , Li, Y. , Abecasis, G. R. , & Scheet, P. A. (2011). Comparison of approaches to account for uncertainty in analysis of imputed genotypes. Genetic Epidemiology, 35(2), 102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zueva, K. J. , Lumme, J. , Veselov, A. E. , Primmer, C. R. , & Pritchard, V. L. (2021). Population genomics reveals repeated signals of adaptive divergence in the Atlantic salmon of north‐eastern Europe. Journal of Evolutionary Biology, 4(6), 866–878. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S7
Tables S1–S7
Data Availability Statement
SNP array genotypes in plink format and phenotype data for association analyses are available on dryad at https://doi.org/10.5061/dryad.g1jwstqz3. Raw reads from WGS data have been uploaded to the NCBI SRA with accession number PRJNA1083490. All scripts used for analysis in this study are available at: https://github.com/TonyKess/seaage_GWAS.