

Abstract
BACKGROUND:
Intracerebral hemorrhage (ICH) is characterized by bleeding into the brain parenchyma. During an ICH, iron released from the breakdown of hemoglobin creates a cytotoxic environment in the brain through increased oxidative stress. Interestingly, the loss of iron homeostasis is associated with the pathological process of other neurological diseases. However, we have previously shown that the H63D mutation in the homeostatic iron regulatory (HFE) gene, prevalent in 28% of the White population in the United States, acts as a disease modifier by limiting oxidative stress. The following study aims to examine the effects of the murine homolog, H67D HFE, on ICH.
METHODS:
An autologous blood infusion model was utilized to create an ICH in the right striatum of H67D and wild-type mice. The motor recovery of each animal was assessed by rotarod. Neurodegeneration was measured using fluorojade-B and mitochondrial damage was assessed by immunofluorescent numbers of CytC+ (cytochrome C) neurons and CytC+ astrocytes. Finally, the molecular antioxidant response to ICH was quantified by measuring Nrf2 (nuclear factor-erythroid 2 related factor), GPX4 (glutathione peroxidase 4), and FTH1 (H-ferritin) levels in the ICH-affected and nonaffected hemispheres via immunoblotting.
RESULTS:
At 3 days post-ICH, H67D mice demonstrated enhanced performance on rotarod compared with wild-type animals despite no differences in lesion size. Additionally, H67D mice displayed higher levels of Nrf2, GPX4, and FTH1 in the ICH-affected hemisphere; however, these levels were not different in the contralateral, non-ICH–affected hemisphere. Furthermore, H67D mice showed decreased degenerated neurons, CytC+ Neurons, and CytC+ astrocytes in the perihematomal area.
CONCLUSIONS:
Our data suggest that the H67D mutation induces a robust antioxidant response 3 days following ICH through Nrf2, GPX4, and FTH1 activation. This activation could explain the decrease in degenerated neurons, CytC+ neurons, and CytC+ astrocytes in the perihematomal region, leading to the improved motor recovery. Based on this study, further investigation into the mechanisms of this neuroprotective response and the effects of the H63D HFE mutation in a population of patients with ICH is warranted.
GRAPHIC ABSTRACT:
A graphic abstract is available for this article.
Keywords: astrocytes, brain, cerebral hemorrhage, mutation, neurons
Intracerebral hemorrhage (ICH) is characterized by bleeding into the brain parenchyma. This condition ranks as the second most common cause of stroke behind ischemic stroke; however, ICH results in a disproportionate cause of morbidity and mortality.1 Overall, ICH is responsible for 2 million out of the 17 million stroke admissions worldwide and has a 1-year mortality rate of 45.4%.2 Unfortunately, only 20% of survivors gain functional independence by 6 months.1 Despite advances in research and management protocols, there has been no decrease in ICH mortality over the past decade.3
In the setting of an ICH, abundant amounts of heme are released from the hemoglobin of lysed erythrocytes which is then degraded by heme oxygenase into carbon monoxide, iron, and biliverdin.4 Notably, excess iron has been identified as a key component of stroke-induced neurodegeneration. This occurs as ferrous iron catalyzes the formation of hydroxide free radicals and reactive oxygen species (ROS) from hydrogen peroxide via the Fenton reaction.5–8 The buildup of ROS leads to increased oxidative stress, mitochondrial damage, excitotoxicity, and an iron-dependent form of cell death known as ferroptosis. However, there exists an innate antioxidant response system to limit oxidative stress and ROS production. This system is regulated by Nrf2 (nuclear factor-erythroid 2 related factor), the major transcription factor for the antioxidant response, and GPX4 (glutathione peroxidase 4), an enzyme that directly reduces lipid peroxides.9 Furthermore, Nrf2 also acts as a major transcription factor for FTH1 (H-ferritin), an important protein for iron storage and delivery.10
The loss of iron homeostasis has been demonstrated as a crucial part of the pathological process in several other neurological diseases. For example, loss in iron homeostasis leading to increased brain iron exacerbates the pathological processes such as neuroinflammation and oxidative stress. This has led to the investigation of iron regulatory proteins and their associated genes on iron homeostasis in the context of neurological diseases. HFE (homeostatic iron regulator) is an iron regulatory protein that modulates iron bioavailability in all cells. It functions as an MHC (major histocompatibilty complex) class I–like protein and binds with the transferrin receptor at the cell membrane to negatively regulate iron uptake.11 Additionally, the HFE gene has the highest prevalence of polymorphisms among iron regulatory genes known to alter brain iron and structure.12–14 One such variant, H63D, is found in 28% of the non-Hispanic white population in the United States.15 This mutation decreases the affinity of HFE to bind with the transferrin receptor allowing greater uptake of iron, therefore, leading to a subclinical iron overload in the brain and increased oxidative stress.16
Studies in our laboratory and others have linked the H63D variant to several neurological diseases such as Parkinson disease (PD), Alzheimer disease, and amyotrophic lateral sclerosis as a disease progression modifier.17,18 Initially, it was thought that the H63D variant posed a risk factor for neurological diseases owing to its prevalence in several neurological disease patient populations. However, recent discoveries suggest that patients with the H63D variant have a slower disease progression and are over-represented within their respective patient population. Moreover, studies with our animal model of this HFE mutation have shown that the mutation is neuroprotective.18 For example, PD is caused pathologically by the loss of dopaminergic neurons in the substantia nigra, it can be further characterized by brain iron accumulation as well as intracellular aggregates containing α-synuclein called Lewy bodies in the substantia nigra.19–21 In both animal and cell culture studies, the expression of the H63D variant protected against the toxicity associated with α-synuclein.14 Studies in patients with PD indicate that the H63D variant is also associated with slower disease progression.14 Additionally, toxin-induced PD models have shown that astrocytes from the murine homolog equivalent, H67D HFE, are less vulnerable to the toxic and parkinsonian-inducing effects of paraquat.22 This may be due to alterations in the antioxidant defense system as seen by increased levels of Nrf2 expression in paraquat-treated H67D astrocytes.23 Furthermore, H63D heterozygosity was associated with decreased neurotoxicity in patients with PD and known paraquat exposures.24 Other researchers have found increases in NQO1 (NAD(P)H quinone oxidoreductase 1), an enzyme that prevents the formation of ROS, in cerebrospinal fluid samples of H63D carries with PD. Studies on the H63D variant and Alzheimer disease have mixed conclusions. Some reports suggest that H63D carriers have an increased chance to develop early-onset Alzheimer disease, whereas others refute this claim completely. Others suggest that the negative impact of the H63D variant on Alzheimer disease is only seen in males or those with other genetic risk factors for the disease, such as ApoE ε4 carriers. Finally, amyotrophic lateral sclerosis patients carrying the H63D variant were found to have a slower disease progression.25
Overall, these studies illustrate that the H67D/H63D variants have fundamental phenotypic differences that could limit the amount of oxidative stress-induced injury to possibly modify the degenerative and reparative processes in neurodegenerative disorders. Given this, the objective of our current study was to observe the effect of the H67D mutation on neuronal degeneration, motor recovery, and key iron storage and antioxidant protein levels following ICH induction in a mouse model.
METHODS
The data for this study are made available on request to the corresponding author and were obtained under ARRIVE guidelines (Animal Research: Reporting of In Vivo Experiments).26 Detailed methods can be found in the Supplemental Material.
Animal Model
An autologous blood infusion model was utilized to create a moderate-sized hemorrhage in the right caudate of 6-month-old C57BL/6J×129 mice carrying the H67D HFE (n=25, male=11, female=14) and wild-type (WT) HFE (n=25, male=13, female=12) by previously published methods.27 Additional sham surgeries were performed on both WT and mutant mice.27 The experiments performed followed the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and the protocol was approved by the Penn State Institutional Animal Care and Use Committee (Protocol: 20190115). Tissue was harvested, as previously described.12
Motor Coordination and Impairment
Each animal had their motor function assessed by rotarod testing according to a previously published ramp-up paradigm.28 Mice were trained 3 days before ICH surgery and tested each day following until euthanization.
Hematoxylin and Eosin Staining and Lesion Volume Analysis
Thirty-micrometer serial, coronal sections were taken through the entirety of the hematoma of H67D (n=3, male=2, female=1) and WT (n=3 male=2, female=1) mice. Each section was washed and stained with Hematoxylin & Eosin according to manufacturer’s instructions (ab245880; Abcam). A blinded researcher then calculated the cross-sectional area of each section using ImageJ.
Blood Iron and Hematocrit Analysis
Whole blood iron levels and hematocrit were measured in H67D (n=4, male=2, female=2) and WT (n=4, male=2, female=2) mice using inductively coupled plasma mass spectrometry and a Heska HT5 Veterinary Hematology Analyzer, respectively, as previously published.29
Fluorojade-B Staining
Fluorojade-B staining was conducted according to the manufacturer’s instructions (VWR 76459–462) in 3 consecutive 15 μm coronal sections within 100 μm of the needle track of H67D (n=7, male=3, female=4) and WT (n=7, male=4, female=3) mice. Total perihematomal fluorojade-B–positive cells within the striatum were quantified blindly using ImageJ software.
Immunofluorescence and Cytochrome C Analysis
Three consecutive 15 μm coronal sections within 100 μm of the needle track of H67D (n=7 male=3, female=4) and WT (n=7 male=3, female=4) mice were immunostained, as previously described.30 Blocking of each section used 0.3% triton/10% normal goat serum; primary antibodies included of NeuN (rabbit polyclonal, ABN78; 1:500; Millipore) or GFAP ([glial fibrillary acidic protein]; rabbit polyclonal, ab68428; 1:500) and CytC (cytochrome C; mouse monoclonal, ab13575; 1:100); secondary antibodies included goat anti-rabbit 1:500, conjugated with Alexa Flour-546 (red), (Life Technologies A11010) and goat anti-mouse 1:500, conjugated for Alexa Flour-488 (green; Life Technologies A11029).
Immunoblotting
Immunoblotting followed previously published methods using a standard polyvinylidene difluoride membrane that was probed for FTH1 (1:1000, 4393S; Cell Signaling Technology), Nrf2 (1:1000, ab92946; Abcam), GPX4 (1:1000, 125066; Abcam), or beta-actin (1:1000, ab115777; Abcam) in tissue collected from H67D (n=8 male=3, female=5) and WT (n=8 male=4, female=4) mice.31
Statistics
Statistical analysis was conducted using GraphPad Prism 9. Unpaired t tests were used to detect statistical significance between the 2 groups while 1-way ANOVA and Tukey post hoc test were used to compare multiple groups. All data are expressed as means and SDs. A P<0.05 was considered significant.
RESULTS
Striatal Lesion Volume and Blood Iron Levels Are Not Different Between H67D and WT Animals
Serial coronal sections through the striatum of both WT and H67D animals were obtained to determine fundamental genotypic differences in the lesion volume. Overall, the ICH in each animal was created in the right striatum as intended (Figure 1A). These volumes were not found to be significantly different between WT and H67D animals (P=0.602; Figure 1B). Additionally, because serum iron could contribute to the stroke damage, whole blood iron and hematocrit levels were measured. Neither whole blood iron (H67D: mean, 517.3, SD=97.82; WT: mean, 488.4, SD=22.51; P=0.585; Figure 1C) nor hematocrit levels (H67D: mean, 45.38, SD=1.60; WT: mean, 43.40, SD=1.94; P=0.167; Figure 1D) were significantly different between groups.
Figure 1.
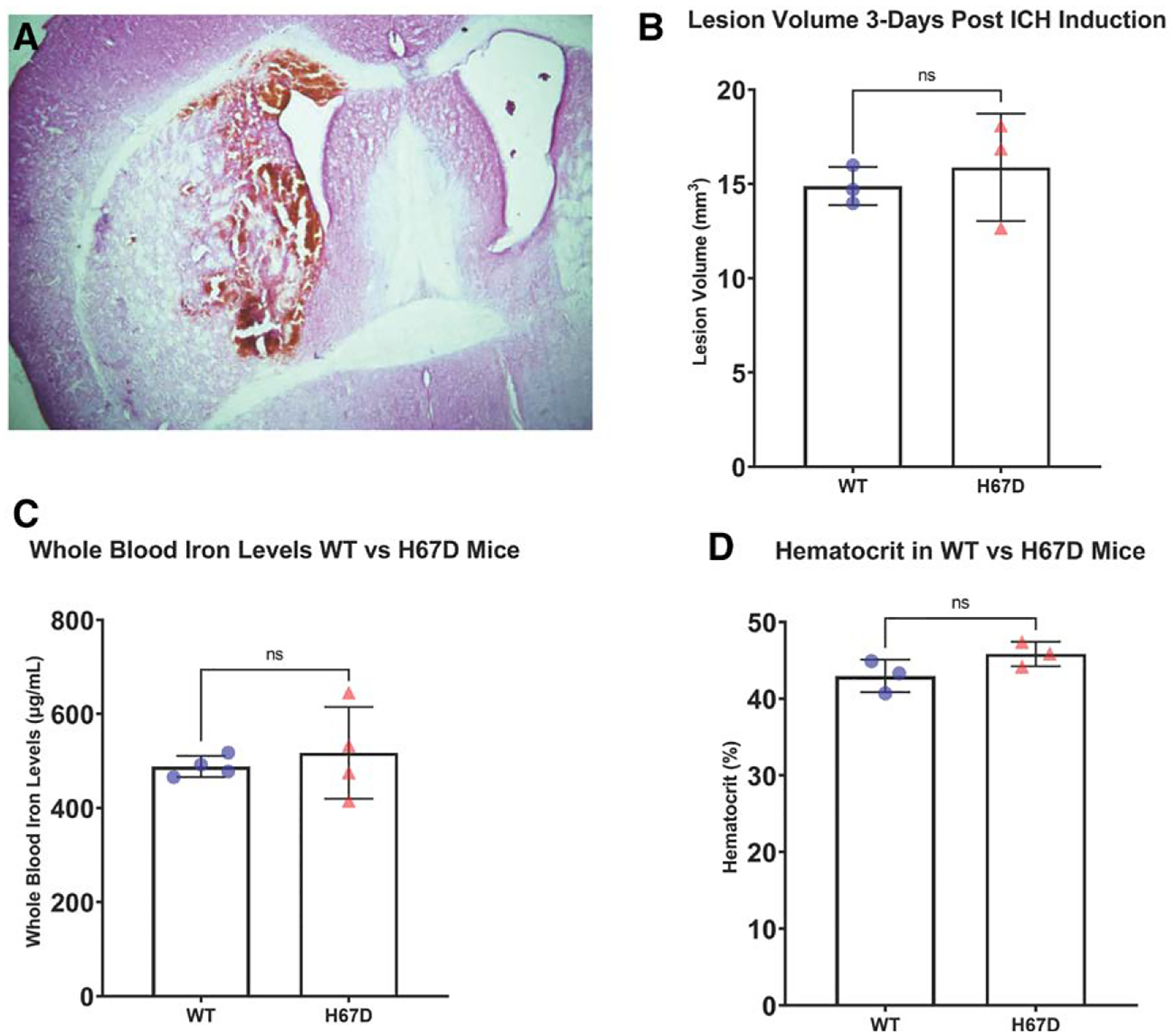
Representation of intracerebral hemorrhage (ICH) volume and blood iron levels.
A, Representation of ICH developed at 3 d postinfusion of 30 μL of autologous blood into the right striatum of wild-type (WT) C57BL/6J mouse. Animals were euthanized 3 d poststroke. The brains were harvested and cut into 30 μM sections through the entirety of the hematoma. Each cross-sectional area was calculated using ImageJ and multiplied by the thickness of the section. Each section was summed to determine to total hematoma size. B, No differences in hematoma were appreciated at 3 d poststroke in WT and H67D mice. C, Whole blood iron levels measured using inductively coupled plasma mass spectrometry of WT vs H67D mice. D, Hematocrit levels in WT vs H67D mice. Error bars reported as SD.
H67D Mice Show Enhanced Functional Motor Recovery 3 Days After ICH Induction Compared With WT
To investigate genotypic differences in functional recovery following ICH, motor coordination was assessed by latency to fall from the rotarod. Initially, H67D and WT mice displayed no differences in functional recovery on day-1 post-ICH induction. However, at day-2 and day-3 post-ICH induction, H67D mice are significantly less likely to fall from the rotarod when compared with WT animals (Figure 2). WT mice generally showed no changes in performance over the course of this study. No differences were observed between sham H67D and sham WT mice (Figure S1).
Figure 2.
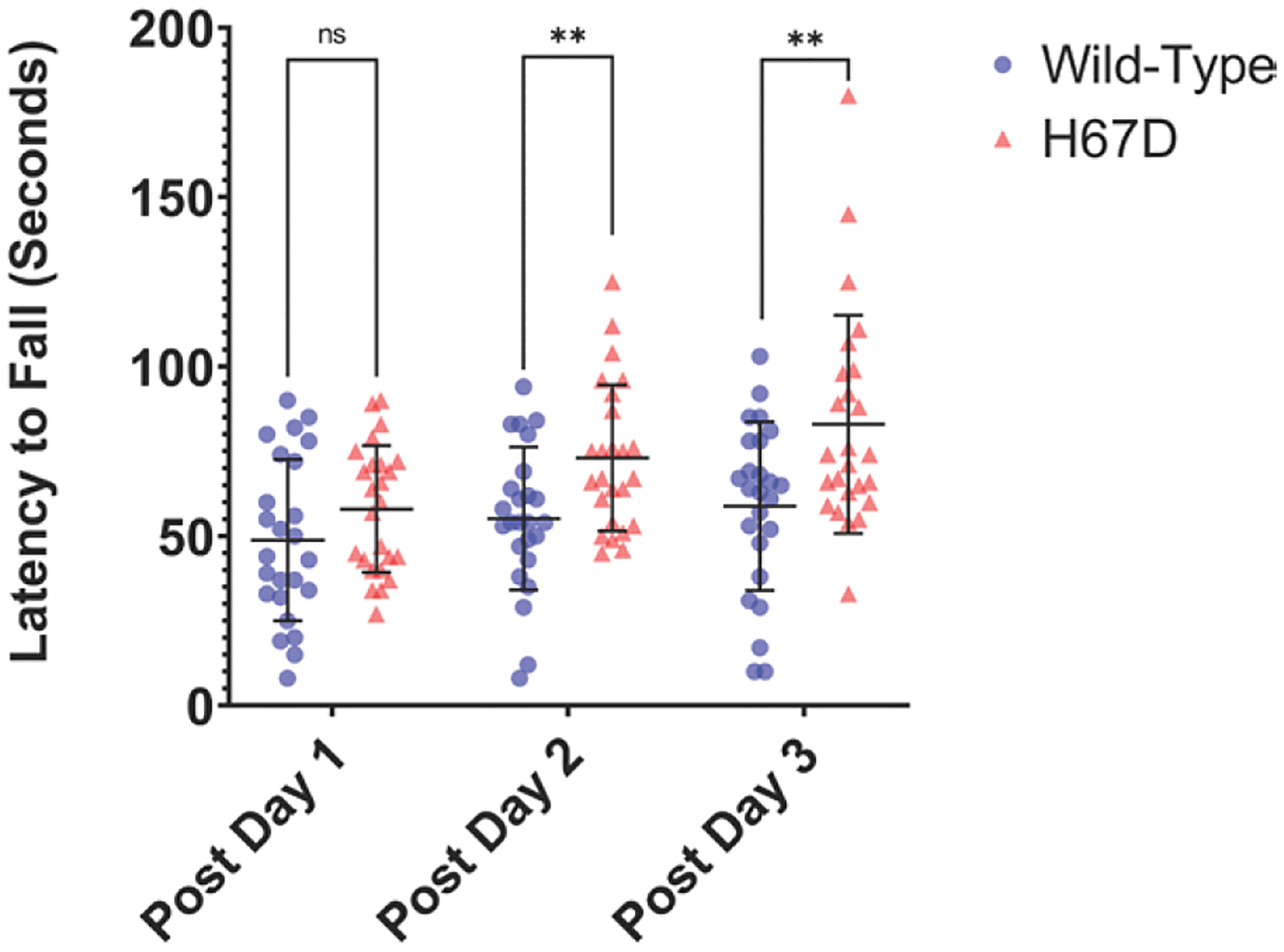
H67D mice perform better on rotarod 3 d after intracerebral hemorrhage (ICH) compared with wild-type (WT).
Functional motor recovery was measured by latency to fall from rotarod. H67D mice (n=24, mean, 48.8, SD=23.81) initially display no differences in functional recovery at day 1 poststroke when compared with WT (n=24, mean, 58.00, SD=18.63; P=0.134). However, at day 2 (H67D: mean, 73.04, SD=21.53; WT: mean, 55.16, SD=21.08; P=0.0047) and 3 poststroke (H67D: mean, 82.96, SD=32.16; WT: mean, 58.8, SD=24.88; P=0.0046), H67D mice are significantly less vulnerable to falling when compared with WT. Error bars reported as SD, *P<0.05, **P<0.001.
H67D Mice Display Fewer Degenerated Neurons in the Perihematomal Area Compared With WT
The effects of ICH on the neuronal population were assessed through fluorojade-B staining to determine differences in the number of degenerated neurons in the perihematomal region in WT mice and H67D mice at 3 days following ICH (Figure 3A). This staining revealed that H67D mice had significantly fewer degenerated neurons (mean, 166.1, SD=44.02) in the striatal perihematomal region when compared with WT animals (mean, 277, SD=109.9; P=0.0291; Figure 3B). No differences were observed between sham H67D and sham WT mice (Figure S2).
Figure 3.
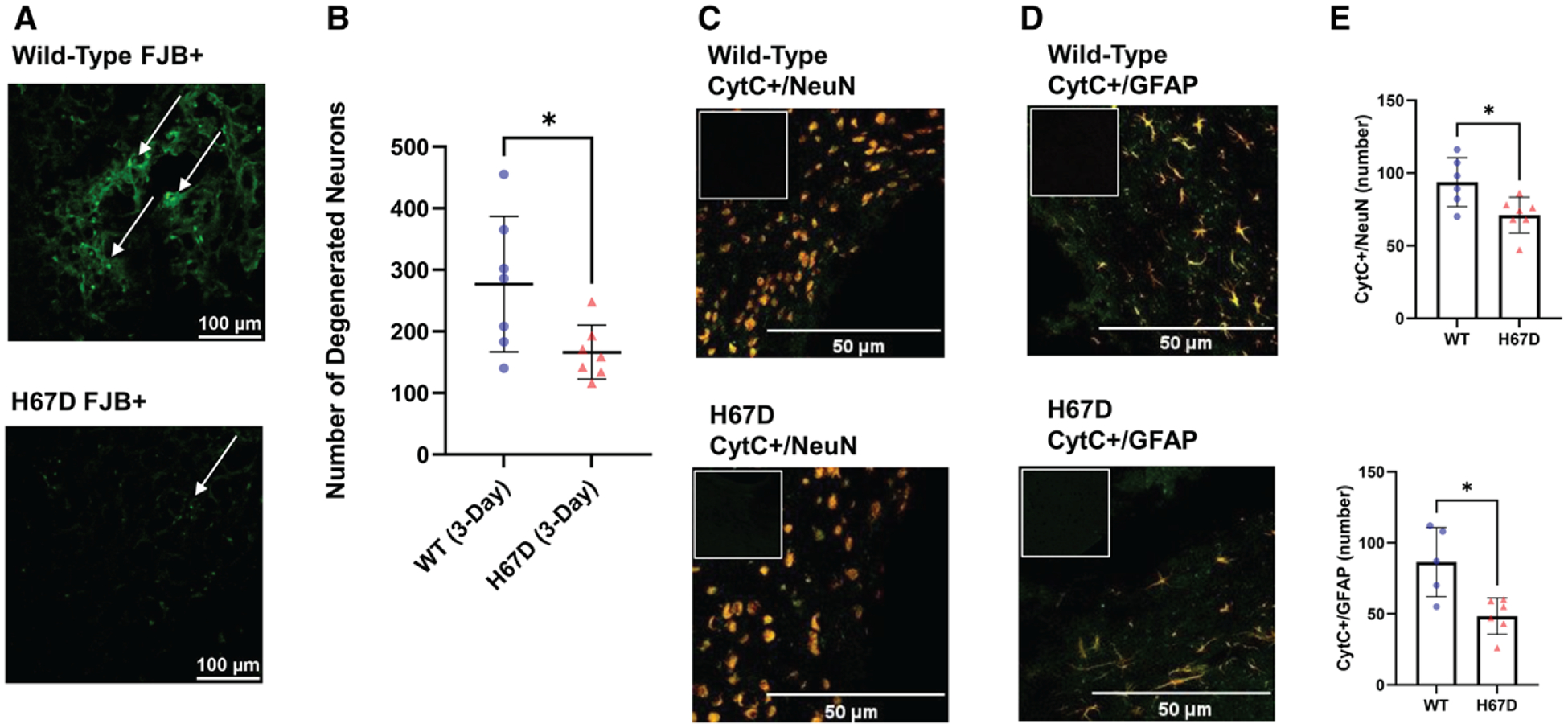
The neuroprotective effects of the H67D mice following intracerebral hemorrhage (ICH).
Fluorojade-B (FJB) staining determined differences in the number of degenerated neurons in the perihematomal region in (A) wild-type (WT) mice (top) and H67D mice (bottom) at 3 d following ICH; arrows point to FJB-positive cells. B, Quantification of FJB-positive cells done using ImageJ. The H67D showed significantly decreased FJB-positive cells (n=7, mean, 166.1, SD=44.02) in the perihematomal region at 3 d post-ICH compared with WT mice (n=7, mean, 277, SD=109.9; P=0.0291). CytC (cytochrome C; green) and NeuN ([neuronal nuclear protein]; red) colocalization in the perihematomal area of (C) WT mice (top) and H67D mice (bottom) at 3 d following ICH; CytC (green) and GFAP ([glial fibrillary acidic protein]; red) colocalization in the perihematomal area in (D) WT mice (top) and H67D mice (bottom); (E) Quantification of CytC+ NeuN cells and CytC+ GFAP cells. H67D showed significantly fewer CytC+ NeuN cells (n=7, mean, 71, SD=12.26) in the perihematomal region at 3 d post-ICH compared with WT mice (n=6, mean, 93.67, SD=16.81; P=0.0210). H67D also showed significantly fewer CytC+ GFAP cells (n=6, mean, 48.33, SD=12.83) in the perihematomal region at 3 d post-ICH compared with WT mice (n=5, mean, 85.40, SD=24.38; P=0.0216). Respective negative immunofluorescent staining is displayed in upper left corner. Error bars reported as SD, *P<0.05.
H67D Mice Show Reduced Numbers of CytC+ Neurons and Astrocytes in the Perihematomal Region
The leakage of CytC from mitochondrial membranes into the cytosol has been linked to mitochondrial dysfunction as well as increases in oxidative stress that are seen in ICH (Figure 3C through 3E).32 Therefore, to understand the extent of oxidative stress, we measured the number of CytC+ Neurons and astrocytes in the perihematomal region. We found that H67D mice demonstrate significantly fewer CytC+ neurons (n=7, mean, 69.86, SD=11.87) than WT animals (n=6, mean, 93.67, SD=16.81; P=0.0124). Similarly, H67D mice demonstrated significantly fewer CytC+ astrocytes (n=6, mean, 48.33, SD=12.83) than WT animals (n=5, mean, 85.40, SD=24.38).
H67D Mice Display Higher Levels of Nrf2, GPX4, and FTH1 in the Stroke-Affected Hemisphere
Given that blood breakdown following ICH leads to increased oxidative stress and excess iron release in the surrounding tissue, we next evaluated relative levels of iron storage and antioxidant response proteins in FTH1, Nrf2, and GPX4. We found that FTH1, Nrf2, and GPX4 levels in the ipsilateral, ICH-affected hemisphere were significantly higher than those of WT animals (Figures 4 through 6). Furthermore, we found that the levels of FTH1, Nrf2, and GPX4 were not significantly different in the contralateral, non-ICH–affected hemisphere. Thus, indicating a more robust antioxidant response to ICH in H67D animals at 3 days post-ICH. No differences were observed between sham H67D and sham WT mice (Figure S3).
Figure 4.
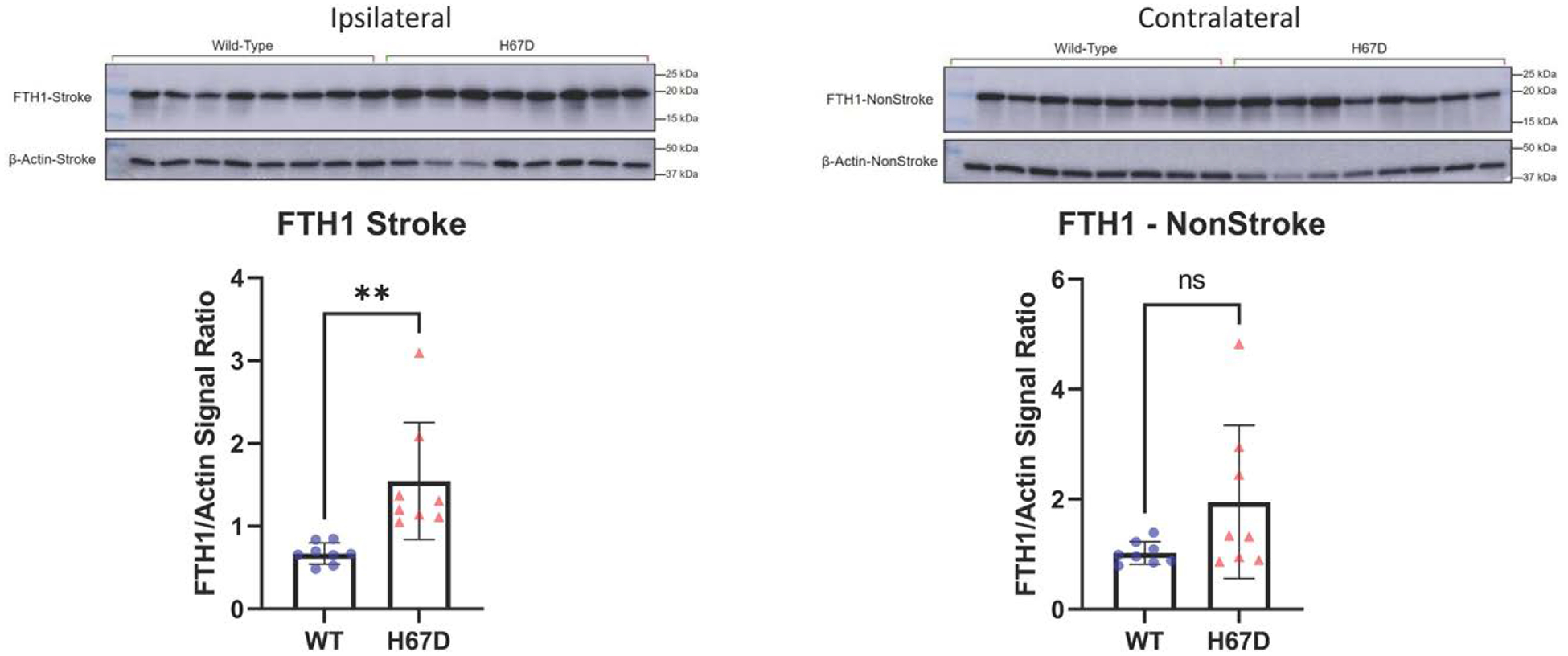
H67D mice show increased levels of FTH1 (H-ferritin) in response to intracerebral hemorrhage (ICH).
FTH1 levels in ipsilateral (ICH-induced hemisphere) of H67D mice (n=8, mean, 1.546, SD=0.706) are significantly higher when compared with wild-type (WT) mice (n=8, mean, 0.669, SD=0.128; P=0.0039). Error reported as SD, **P<0.01. FTH1 levels in contralateral (non-ICH–induced hemisphere) of H67D mice (n=8, mean, 1.950, SD=1.390) are not significantly difference when compared with WT mice (n=8, mean, 1.022, SD=0.204; P=0.082). Error reported as SD.
Figure 6.
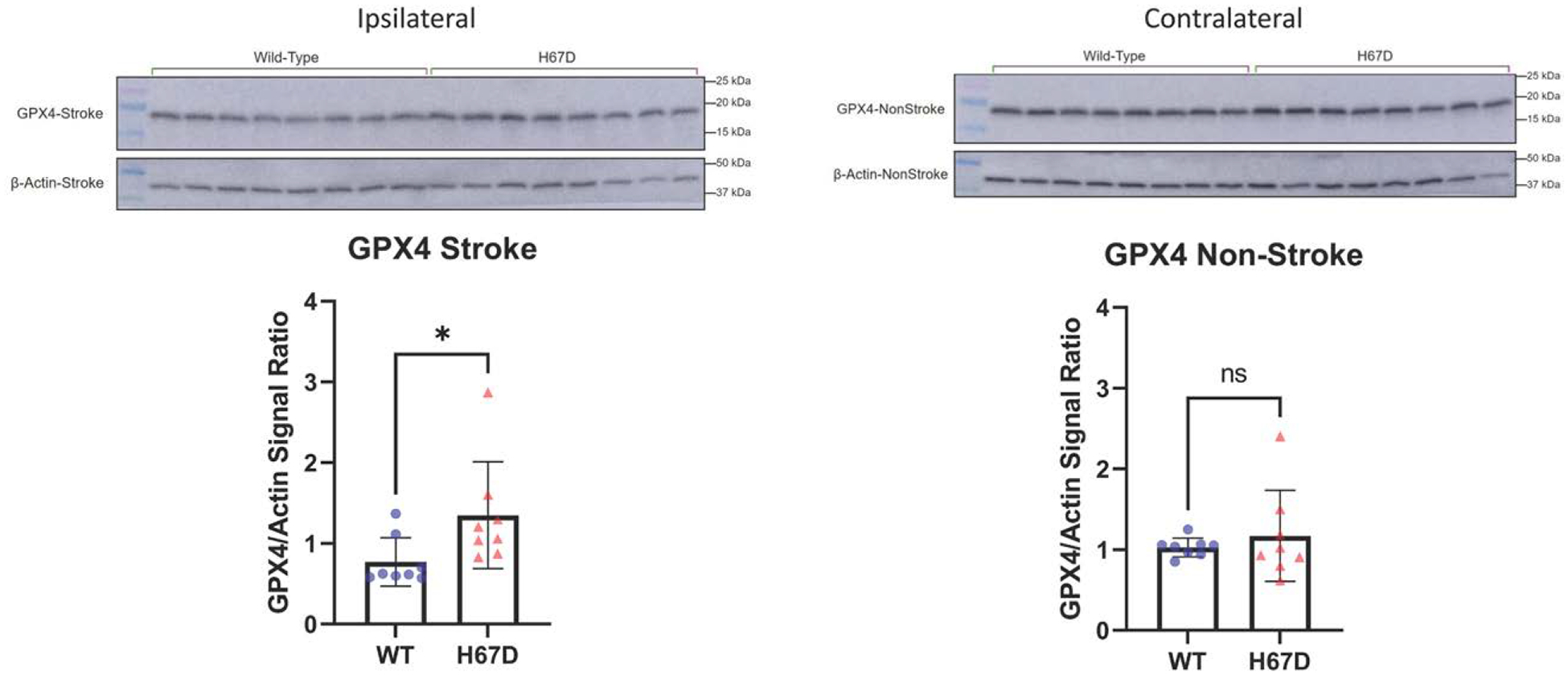
H67D mice show increased levels of antioxidant protein, GPX4 (glutathione peroxidase 4), in response to intracerebral hemorrhage (ICH).
GPX4 levels in ipsilateral (ICH-induced hemisphere) of H67D mice (n=8, mean, 1.349, SD=0.662) are significantly higher when compared with wild-type (WT) mice (n=8, mean, 0.772, SD=0.300; P=0.0413). Error reported as SD, *P<0.05. GPX4 levels in contralateral (non-ICH–induced hemisphere) of H67D mice (n=8, mean, 1.172, SD=0.564) are not significantly different when compared with WT mice (n=8, mean, 1.030, SD=0.116; P=0.495). Error reported as SD.
DISCUSSION
Our current study sought to determine the effects of the H67D HFE mutation on ICH. We discovered that H67D mice have elevated levels of Nrf2 and GPX4 in the ICH-affected hemisphere when compared with WT mice. Physiologically, Nrf2 is constitutively expressed, but once translated, is quickly degraded by Keap1 (Kelch-like ECH-associated protein 1)-mediated ubiquitination.33 In the event of an ICH, excess ferrous iron from hemoglobin breakdown catalyzes the formation of ROS through the Fenton Reaction. These ROS react with the polyunsaturated fatty acids in cell membranes leading to lipid peroxidation, mitochondria dysfunction, and subsequent oxidative damage. Under these conditions seen in ICH, the interaction between Nrf2 and Keap1 is disrupted allowing Nrf2 to function as a transcription factor by binding to DNA sequences called antioxidant response elements. In addition, GPX4 is a downstream target of Nrf2 and plays a pivotal role in preventing the buildup of lipid peroxides by directly reducing them to lipid alcohols.34 Depletions in GPX4 are associated with higher levels of ferroptosis, an iron-dependent form of cell death characterized by membrane dysfunction and lipid peroxidation.35 Importantly, we did not observe significant differences in lesion volume, whole blood iron levels, or hematocrit levels between H67D and WT mice, suggesting that both groups received similar injuries and iron loads. Another downstream target of Nrf2, FTH1 is also elevated in the H67D mice. FTH1 is 1 of 2 subunits that compose ferritin, a multimeric nanocage that stores iron intracellularly. Importantly, FTH1 contains a ferroxidase residue that oxidizes ferrous iron into its catalytically inactive ferric state where it is then sequestered within the ferritin core. In addition to iron storage, FTH1 has also been implicated in iron delivery and transport to other tissues and cells.36,37 Overall, the increase in Nrf2, GPX4, and FTH1, combine to provide a compelling argument that our study demonstrates that H67D mice have a more robust antioxidant response than the WT mice. This response could contribute to decreased neuronal degeneration and glial mitochondrial damage which could in turn contribute to improved functional recovery in H67D animals.
The seemingly neuroprotective milieu developed within H67D animals can perhaps be explained as an adaptive response to stress. This concept in toxicology, known as hormesis, refers to a biphasic dose response in which a stressor at a low dose confers long-term protection against similar stresses at a moderate to high dose.38 In the setting of the H67D HFE mutation, reduced negative inhibition of the HFE protein to the transferrin receptor results in higher subclinical levels of intracellular iron and increased oxidative stress in the brain.12 It is possible that this low-dose stressor primes the neurons and glial cells of H67D animals through the Nrf2 and antioxidant system to better handle the high doses of iron and oxidative stress seen following ICH. However, it is difficult to determine from our current study if this potential hormetic phenomenon is innate to neurons or if it enables astrocytes, in which most of the Nrf2 in brain is found, to more broadly provide support to susceptible neighboring cells.39 Immunofluorescent staining within the ICH-affected hemisphere of these animals demonstrates colocalization of Nrf2 with neurons and astrocytes, GPX4 with astrocytes, and FTH1 with astrocytes and oligodendrocytes (Figure S4). This suggests that the HFE mutation activates specific pathways in astrocytes for them to offer a central and broad supportive role to the other cell types.
Our findings are consistent with numerous animal studies that have demonstrated that Nrf2 plays a critical role in limiting oxidative damage in the perihematomal region. For example, Nrf2 knockout mice display greater neurological deficits and larger hematoma volumes when compared with WT mice. The Nrf2 knock-outs also demonstrate extensive cellular damage related to ROS production such as DNA damage, CytC release, and increased neuronal death.32 Molecular studies indicate that Nrf2 expression significantly increases in the perihematomal region within 2-hours of an ICH and remains elevated for up to 10 days.40 Furthermore, Nrf2 activation has been shown to decrease mitochondrial dysfunction by downregulating mitochondrial ROS formation.41,42 These studies suggest that Nrf2 is rapidly activated in response to excess iron and thus demonstrate a compensatory mechanism to battle the perturbed iron homeostasis and oxidative stress and limit ferroptosis seen in the setting of ICH.32,43,44 Additionally, an extensive body of literature exists showing favorable outcomes in ICH mouse models treated with Nrf2 analogs and activators.45–48 These improvements have been attributed to the reduction in iron-dependent oxidative stress and are considered well-tolerated. In parallel, Deferoxamine, a hydrophobic iron chelator, has been found to improve functional recovery and attenuate neuronal death in additional ICH animal models.49–51 Although promising, neither the effects of iron chelators nor Nrf2 analogs have been translated with success into clinical studies.52,53 It is important to note that the majority of these pharmacological treatments, such as sulforaphane, dimethyl fumarate, tert-butyl hydroquinone, and others, boost Nrf2 by targeting Nrf2-Keap1 dissociation.54,55 The animal model data, including ours in this study, continue to support that activation of Nrf2 is an appropriate target; however, clinical studies should identify alternative pathways for Nrf2 activation. For example, GSK3β (Glycogen Synthase Kinase 3 Beta) directly phosphorylates Nrf2 to initiate its export out of the nucleus for subsequent degradation.56 Previous work in our laboratory also indicates that inactivating GSK3β using CHIR9902, an aminopyrimidine derivative, increases Nrf2 nuclear accumulation and successfully rescues astrocytes from cell death after exposure to compounds that increase ROS and oxidative stress.57 Inhibition of the GSK3β pathway is emerging as a potential therapeutic for neurodegenerative diseases such as PD and ischemic stroke but has yet to be studied in ICH.58,59
Despite the difficulties in the translation of bench-to-bedside therapies, our model shows evidence of an innate, genotype-specific neuroprotective milieu associated with the HFE mutation. In practice, this model can be lever-aged to further study the disease course of ICH and identify alternative Nrf2 activating pathways and events that lead to improved outcomes. Furthermore, our model is translational in nature in that the H67D HFE mutation is the mouse homolog to the commonly occurring H63D mutation in the human population. To our knowledge, ICH outcomes in patients carrying the H63D mutation and other HFE mutations have not been studied but our study argues that outcomes studies in the population of patients with ICH should be stratified according to HFE genotype.
CONCLUSIONS
In summary, we observed the effect of the H67D mutation on neurodegeneration and functional recovery in a mouse model. We identified key changes in levels of iron storage and antioxidant proteins that potentially contribute to the neuroprotective milieu that limits neurodegeneration, oxidative stress, and improves functional recovery in mice. These results continue to support previous studies that recognize the antioxidant and neurodegenerative disease-modifying capabilities of the H67D gene mutation and extend the findings to ICH. In general, these findings continue to support the proposition that HFE genotype will impact disease progression and clinical outcome studies should take into consideration the common H63D mutation.
Supplementary Material
Figure 5.
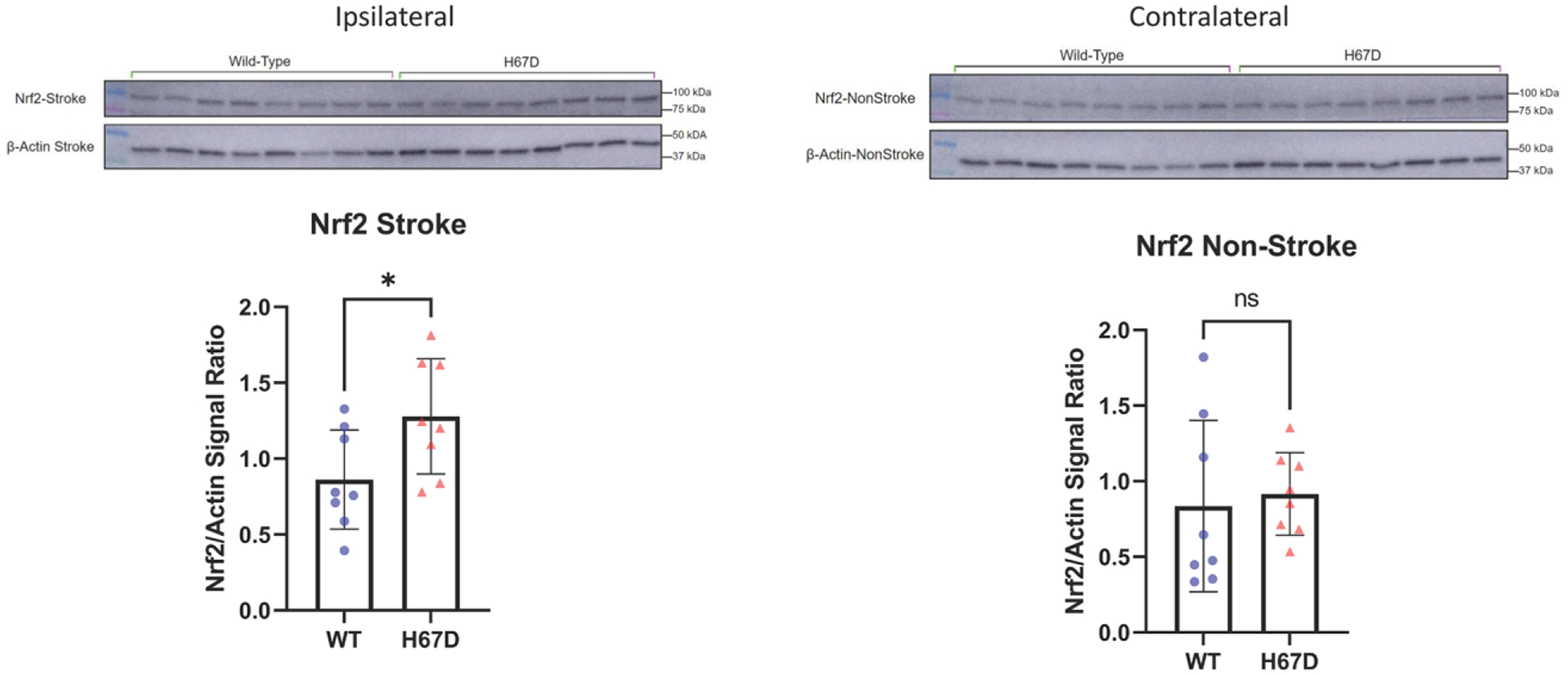
H67D mice show increased levels of antioxidant response regulation protein, Nrf2 (nuclear factor-erythroid 2 related factor), in response to intracerebral hemorrhage (ICH).
Nrf2 Levels in ipsilateral (ICH-induced hemisphere) of H67D mice (n=8, mean, 1.28, SD=0.38) are significantly higher when compared with wild-type (WT) mice (n=8, mean, 0.862, SD=0.327; P=0.0339). Error reported as SD, *P<0.05. Nrf2 Levels in contralateral (non-ICH–induced hemisphere) of H67D mice (n=8, mean, 0.916, SD=0.273) are not significantly different when compared with WT mice (n=8, mean, 0.836, SD=0.567; P=0.724). Error reported as SD, *P<0.05.
Acknowledgments
T.B. Helmuth, Drs Kumari, Palsa, E.B. Neely, B. Slagle-Webb, Drs Simon, and Connor contributed to methodology; T.B. Helmuth, Drs Simon, and Kumari contributed to investigation; T.B. Helmuth drafted the original article, Dr Kumari, Simon, Connor, E.B. Neely, B. Slagle-Webb contributed to article review and editing; Drs Simon and Connor participated in supervision.
Sources of Funding
Funding was provided by the Penn State Clinical and Translational Science Institute (2UL1TR002014-05A1).
Nonstandard Abbreviations and Acronyms
- CytC
cytochrome C
- FTH1
H-ferritin
- GPX4
glutathione peroxidase 4
- HFE
homeostatic iron regulator
- ICH
intracerebral hemorrhage
- Nrf2
nuclear factor-erythroid 2 related factor
- PD
Parkinson disease
- ROS
reactive oxygen species
Footnotes
Disclosures
None.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/STROKEAHA.123.043799.
REFERENCES
- 1.An SJ, Kim TJ, Yoon BW. Epidemiology, risk factors, and clinical features of intracerebral hemorrhage: an update. J Stroke. 2017;19:3. doi: 10.5853/jos.2016.00864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fernando SM, Qureshi D, Talarico R, Tanuseputro P, Dowlatshahi D, Sood MM, Smith EE, Hill MD, McCredie VA, Scales DC, et al. Intracerebral hemorrhage incidence, mortality, and association with oral anticoagulation use: a population study. Stroke. 2021;52:1673–1681. doi: 10.1161/strokeaha.120.032550 [DOI] [PubMed] [Google Scholar]
- 3.Zahuranec DB, Lisabeth LD, Sánchez BN, Smith MA, Brown DL, Garcia NM, Skolarus LE, Meurer WJ, Burke JF, Adelman EE, et al. Intracerebral hemorrhage mortality is not changing despite declining incidence. Neurology. 2014;82:2180–2186. doi: 10.1212/WNL.0000000000000519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fraser ST, Midwinter RG, Berger BS, Stocker R. Heme oxygenase-1: a critical link between iron metabolism, erythropoiesis, and development. Adv Hematol. 2011;2011:473709. doi: 10.1155/2011/473709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeGregorio-Rocasolano N, Martí-Sistac O, Gasull T. Deciphering the iron side of stroke:Neurodegeneration at the crossroads between iron dyshomeostasis, excitotoxicity, and ferroptosis. Front Neurosci. 2019;13:85. doi: 10.3389/fnins.2019.00085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu X, Tao C, Gan Q, Zheng J, Li H, You C. Oxidative stress in intracerebral hemorrhage: sources, mechanisms, and therapeutic targets. Oxid Med Cell Longev. 2016;2016:3215391. doi: 10.1155/2016/3215391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wan J, Ren H, Wang J. Iron toxicity, lipid peroxidation and ferroptosis after intracerebral haemorrhage. Stroke Vasc Neurol. 2019;4:93–95. doi: 10.1136/svn-2018-000205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, Sun B, Wang GF. Past, present and future. Cell Death Dis. 2020;11:88. doi: 10.1038/s41419-020-2298-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao X, Sun G, Zhang J, Strong R, Dash PK, Kan YW, Grotta JC, Aronowski J. Transcription factor Nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke. 2007;38:3280–3286. doi: 10.1161/STROKEAHA.107.486506 [DOI] [PubMed] [Google Scholar]
- 10.Cairo G, Tacchini L, Pogliaghi G, Anzon E, Tomasi A, Bernelli-Zazzera A. Induction of ferritin synthesis by oxidative stress. J Biol Chem. 1995;270:700–703. doi: 10.1074/jbc.270.2.700 [DOI] [PubMed] [Google Scholar]
- 11.Giannetti AM, Björkman PJ. HFE and transferrin directly compete for transferrin receptor in solution and at the cell surface. J Biol Chem. 2004;279:25866–25875. doi: 10.1074/jbc.M401467200 [DOI] [PubMed] [Google Scholar]
- 12.Nandar W, Neely EB, Unger E, Connor JR. A mutation in the HFE gene is associated with altered brain iron profiles and increased oxidative stress in mice. Biochim Biophys Acta. 2013;1832:729–741. doi: 10.1016/j.bbadis.2013.02.009 [DOI] [PubMed] [Google Scholar]
- 13.Jahanshad N, Kohannim O, Hibar DP, Stein JL, McMahon KL, De Zubicaray GI, Medland SE, Montgomery GW, Whitfield JB, Martin NG, et al. Brain structure in healthy adults is related to serum transferrin and the H63D polymorphism in the HFE gene. Proc Natl Acad Sci U S A. 2012;109:E851–E859. doi: 10.1073/pnas.1105543109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim Y, Stahl MC, Huang X, Connor JR. H63D variant of the homeostatic iron regulator (HFE) gene alters α-synuclein expression, aggregation, and toxicity. J Neurochem. 2020;155:177–190. doi: 10.1111/jnc.15107 [DOI] [PubMed] [Google Scholar]
- 15.Acton RT, Barton JC, Snively BM, McLaren CE, Adams PC, Harris EL, Speechley MR, McLaren GD, Dawkins FW, Leiendecker-Foster C, et al. ; Hemochromatosis and Iron Overload Screening Study Research Investigators. Geographic and racial/ethnic differences in HFE mutation frequencies in the Hemochromatosis and Iron Overload Screening (HEIRS) Study. Ethn Dis. 2006;16:815–821. doi: 10.1097/00000441-200302000-00001 [DOI] [PubMed] [Google Scholar]
- 16.Reuben A, Chung JW, Lapointe R, Santos MM. The hemochromatosis protein HFE 20 years later: an emerging role in antigen presentation and in the immune system. Immunity, Inflamm Dis. 2017;5:218. doi: 10.1002/iid3.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hollerer I, Bachmann A, Muckenthaler MU. Pathophysiological consequences and benefits of HFE mutations: 20 years of research. Haematologica. 2017;102:809–817. doi: 10.3324/haematol.2016.160432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim Y, Connor JR. The roles of iron and HFE genotype in neurological diseases. Mol Aspects Med. 2020;75:100867. doi: 10.1016/j.mam.2020.100867 [DOI] [PubMed] [Google Scholar]
- 19.Lee JH, Lee MS. Brain iron accumulation in atypical parkinsonian syndromes: In vivo MRI evidences for distinctive patterns. Front Neurol. 2019;10:74. doi: 10.3389/fneur.2019.00074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mochizuki H, Yasuda T. Iron accumulation in Parkinson’s disease. J Neural Transm. 2012;119:1511–1514. doi: 10.1007/s00702-012-0905-9 [DOI] [PubMed] [Google Scholar]
- 21.Tong J, Wong H, Guttman M, Ang LC, Forno LS, Shimadzu M, Rajput AH, Muenter MD, Kish SJ, Hornykiewicz O, et al. Brain α-synuclein accumulation in multiple system atrophy, Parkinson’s disease and progressive supranuclear palsy: a comparative investigation. Brain. 2010;133:172–188. doi: 10.1093/brain/awp282 [DOI] [PubMed] [Google Scholar]
- 22.Nixon AM, Meadowcroft MD, Neely EB, Snyder AM, Purnell CJ, Wright J, Lamendella R, Nandar W, Huang X, Connor JR. HFE genotype restricts the response to Paraquat in a mouse model of neurotoxicity. J Neurochem. 2018;145:299–311. doi: 10.1111/jnc.14299 [DOI] [PubMed] [Google Scholar]
- 23.Song IY, Snyder AM, Kim Y, Neely EB, Wade QW, Connor JR. The Nrf2-mediated defense mechanism associated with HFE genotype limits vulnerability to oxidative stress-induced toxicity. Toxicology. 2020;441:152525. doi: 10.1016/j.tox.2020.152525 [DOI] [PubMed] [Google Scholar]
- 24.Wang EW, Trojano ML, Lewis MM, Du G, Chen H, Brown GL, Jellen LC, Song I, Neely E, Kong L, et al. HFE H63D limits nigral vulnerability to paraquat in agricultural workers. Toxicol Sci. 2021;181:47–57. doi: 10.1093/toxsci/kfab020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Su XW, Lee SY, Mitchell RM, Stephens HE, Simmons Z, Connor JR. H63D HFE polymorphisms are associated with increased disease duration and decreased muscle superoxide dismutase-1 expression in amyotrophic lateral sclerosis patients. Muscle Nerve. 2013;48:242–246. doi: 10.1002/mus.23740 [DOI] [PubMed] [Google Scholar]
- 26.du Sert NP, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U, et al. The arrive guidelines 20: updated guidelines for reporting animal research. PLoS Biol. 2020;18:1–12. doi: 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rynkowski MA, Kim GH, Komotar RJ, Otten ML, Ducruet AF, Zacharia BE, Kellner CP, Hahn DK, Merkow MB, Garrett MC, et al. A mouse model of intracerebral hemorrhage using autologous blood infusion. Nat Protoc. 2008;3:122–128. doi: 10.1038/nprot.2007.513 [DOI] [PubMed] [Google Scholar]
- 28.Ruan J, Yao Y. Behavioral tests in rodent models of stroke. Brain Hemorrhages. 2020;1:171–184. doi: 10.1016/j.hest.2020.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kondaiah P, Palika R, Mashurabad P, Singh Yaduvanshi P, Sharp P, Pullakhandam R. Effect of zinc depletion/repletion on intestinal iron absorption and iron status in rats. J Nutr Biochem. 2021;97:108800. doi: 10.1016/j.jnutbio.2021.108800 [DOI] [PubMed] [Google Scholar]
- 30.Kumari R, Willing LB, Patel SD, Baskerville KA, Simpson IA. Increased cerebral matrix metalloprotease −9 activity is associated with compromised recovery in the diabetic db/db mouse following a stroke. J Neurochem. 2011;119:1029–1040. doi: 10.1111/j.1471-4159.2011.07487.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palsa K, Baringer SL, Shenoy G, Simpson IA, Connor JR. Exosomes are involved in iron transport from human blood-brain barrier endothelial cells and are modified by endothelial cell iron status. J Biol Chem. 2023;299:102868. doi: 10.1016/j.jbc.2022.102868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J, Fields J, Zhao C, Langer J, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Doré S. Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic Biol Med. 2007;43:408–414. doi: 10.1016/j.freeradbiomed.2007.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vomund S, Schäfer A, Parnham MJ, Brüne B, Von Knethen A. Nrf2, the master regulator of anti-oxidative responses. Int J Mol Sci. 2017;18:2772–2719. doi: 10.3390/ijms18122772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107. doi: 10.1016/j.redox.2019.101107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang D, Kroemer G. Ferroptosis. Curr Biol. 2020;30:R1292–R1297. doi: 10.1016/j.cub.2020.09.068 [DOI] [PubMed] [Google Scholar]
- 36.Fisher J, Devraj K, Ingram J, Slagle-Webb B, Madhankumar AB, Liu X, Klinger M, Simpson IA, Connor JR. Ferritin: a novel mechanism for delivery of iron to the brain and other organs. Am J Physiol - Cell Physiol. 2007;293:C641. doi: 10.1152/ajpcell.00599.2006 [DOI] [PubMed] [Google Scholar]
- 37.Chiou B, Neely EB, Mcdevitt DS, Simpson IA, Connor JR. Transferrin and H-ferritin involvement in brain iron acquisition during postnatal development: impact of sex and genotype. J Neurochem. 2020;152:381–396. doi: 10.1111/jnc.14834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mattson MP. Hormesis defined. Ageing Res Rev. 2008;7:1–7. doi: 10.1016/j.arr.2007.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Habas A, Hahn J, Wang X, Margeta M. Neuronal activity regulates astrocytic Nrf2 signaling. Proc Natl Acad Sci U S A. 2013;110:18291–18296. doi: 10.1073/pnas.1208764110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shang H, Yang D, Zhang W, Li T, Ren X, Wang X, Zhao W. Time course of Keap1-Nrf2 pathway expression after experimental intracerebral haemorrhage: correlation with brain oedema and neurological deficit. Free Radic Res. 2013;47:368–375. doi: 10.3109/10715762.2013.778403 [DOI] [PubMed] [Google Scholar]
- 41.Bento-Pereira C, Dinkova-Kostova AT. Activation of transcription factor Nrf2 to counteract mitochondrial dysfunction in Parkinson’s disease. Med Res Rev. 2021;41:785–802. doi: 10.1002/med.21714 [DOI] [PubMed] [Google Scholar]
- 42.Tan X, Yang Y, Xu J, Zhang P, Deng R, Mao Y, He J, Chen Y, Zhang Y, Ding J, et al. Luteolin exerts neuroprotection via modulation of the p62/Keap1/Nrf2 pathway in intracerebral hemorrhage. Front Pharmacol. 2020;10:1–15. doi: 10.3389/fphar.2019.01551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song X, Long D. Nrf2 and ferroptosis: a new research direction for neurodegenerative diseases. Front Neurosci. 2020;14:267. doi: 10.3389/fnins.2020.00267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen-Roetling J, Regan RF. Targeting the Nrf2-heme oxygenase-1 axis after intracerebral hemorrhage. Curr Pharm Des. 2016;23:2226. doi: 10.2174/1381612822666161027150616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Imai T, Matsubara H, Hara H. Potential therapeutic effects of Nrf2 activators on intracranial hemorrhage. J Cereb Blood Flow Metab. 2021;41:1483–1500. doi: 10.1177/0271678X20984565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao X, Sun G, Zhang J, Ting SM, Gonzales N, Aronowski J. Dimethyl fumarate protects brain from damage produced by intracerebral hemorrhage by mechanism involving Nrf2. Stroke. 2015;46:1923–1928. doi: 10.1161/STROKEAHA.115.009398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang CF, Cho S, Wang J. (−)-Epicatechin protects hemorrhagic brain via synergistic Nrf2 pathways. Ann Clin Transl Neurol. 2014;1:258–271. doi: 10.1002/acn3.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sukumari-Ramesh S, Alleyne CH. Post-injury administration of tert-butylhydroquinone attenuates acute neurological injury after intracerebral hemorrhage in mice. J Mol Neurosci. 2016;58:525–531. doi: 10.1007/s12031-016-0722-y [DOI] [PubMed] [Google Scholar]
- 49.Nakamura T, Keep RF, Hua Y, Schallert T, Hoff JT, Xi G. Deferoxamine-induced attenuation of brain edema and neurological deficits in a rat model of intracerebral hemorrhage. J Neurosurg. 2004;100:672–678. doi: 10.3171/jns.2004.100.4.0672 [DOI] [PubMed] [Google Scholar]
- 50.Hatakeyama T, Okauchi M, Hua Y, Keep RF, Xi G. Deferoxamine reduces neuronal death and hematoma lysis after intracerebral hemorrhage in aged rats. Transl Stroke Res. 2013;4:546–553. doi: 10.1007/s12975-013-0270-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zeng L, Tan L, Li H, Zhang Q, Li Y, Guo J. Deferoxamine therapy for intracerebral hemorrhage: a systematic review. PLoS One. 2018;13:e0193615. doi: 10.1371/journal.pone.0193615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Selim M, Foster LD, Moy CS, Xi G, Hill MD, Morgenstern LB, Greenberg SM, James ML, Singh V, Clark WM, et al. ; i-DEF Investigators. Deferoxamine mesylate in patients with intracerebral haemorrhage (i-DEF): a multicentre, randomised, placebo-controlled, double-blind phase 2 trial. Lancet Neurol. 2019;18:428–438. doi: 10.1016/S1474-4422(19)30069-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fouda AY, Newsome AS, Spellicy S, Waller JL, Zhi W, Hess DC, Ergul A, Edwards DJ, Fagan SC, Switzer JA. Minocycline in acute cerebral hemorrhage: an early phase randomized trial. Stroke. 2017;48:2885–2887. doi: 10.1161/STROKEAHA.117.018658 [DOI] [PubMed] [Google Scholar]
- 54.Scannevin RH, Chollate S, Jung MY, Shackett M, Patel H, Bista P, Zeng W, Ryan S, Yamamoto M, Lukashev M, et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J Pharmacol Exp Ther. 2012;341:274–284. doi: 10.1124/jpet.111.190132 [DOI] [PubMed] [Google Scholar]
- 55.Satoh T, Saitoh S, Hosaka M, Kosaka K. Simple ortho- and para-hydroquinones as compounds neuroprotective against oxidative stress in a manner associated with specific transcriptional activation. Biochem Biophys Res Commun. 2009;379:537–541. doi: 10.1016/j.bbrc.2008.12.106 [DOI] [PubMed] [Google Scholar]
- 56.Shaw P, Chattopadhyay A. Nrf2–ARE signaling in cellular protection: mechanism of action and the regulatory mechanisms. J Cell Physiol. 2020;235:3119–3130. doi: 10.1002/jcp.29219 [DOI] [PubMed] [Google Scholar]
- 57.Song I Adaptive defense mechanism associated with HFE gene variant. 2020. Accessed April 13, 2023. https://all3dp.com/2/fused-deposition-modeling-fdm-3d-printing-simply-explained/
- 58.Pang T, Wang YJ, Gao YX, Xu Y, Li Q, Zhou YB, Xu L, Huang ZJ, Liao H, Zhang LY, et al. A novel GSK-3β inhibitor YQ138 prevents neuronal injury induced by glutamate and brain ischemia through activation of the Nrf2 signaling pathway. Acta Pharmacol Sin. 2016;37:741–752. doi: 10.1038/aps.2016.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gu C, Zhang Y, Hu Q, Wu J, Ren H, Liu CF, Wang G. P7c3 inhibits gsk3β activation to protect dopaminergic neurons against neurotoxin-induced cell death in vitro and in vivo. Cell Death Dis. 2017;8:e2858–e2810. doi: 10.1038/cddis.2017.250 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.