

Abstract
Murine FAM72A, mFAM72A, binds the nuclear form of uracil-DNA glycosylase, mUNG2, inhibits its activity and causes its degradation. In immunoprecipitation assays the human paralog, hFAM72A, binds hUNG2 and is a potential anti-cancer drug target because of its high expression in many cancers. Using purified mFAM72A, and mUNG2 proteins we show that mFAM72A binds mUNG2, and the N-terminal 25 amino acids of mUNG2 bind mFAM72A at a nanomolar dissociation constant. We also show that mFAM72A is present throughout the cells, and mUNG2 helps localize it to nuclei. Based on in silico models of mFAM72A-mUNG2 interactions, we constructed several mutants of mFAM72A and found that while they have reduced ability to deplete mUNG2, the mutations also destabilized the former protein. We confirmed that Withaferin A, a predicted lead molecule for the design of FAM72A inhibitors, binds mFAM72A with micromolar affinity but has little affinity to mUNG2. We identified two potential metal-binding sites in mFAM72A and show that one of the sites contains an Fe-S cluster. This redox-sensitive cluster is involved in the mFAM72A-mUNG2 interaction and modulates mFAM72A activity. Hydrogen peroxide treatment of cells increases mUNG2 depletion in a FAM72A-dependent fashion suggesting that mFAM72A activity is redox-sensitive.
1. Introduction
FAM72 is a family of four paralogous genes (hFAM72A through hFAM72D) in the human genome that is associated with cell division, proliferation, and differentiation in cancer cells [1–3]. Among these paralogs, hFAM72A is also important for normal brain development and is upregulated in many types of malignancies including cancers of the colon, head and neck, brain, and B cells [2, 4–6]. Using immunoprecipitation assays, hFAM72A was shown to bind the uracil-DNA glycosylase isoform 2 (hUNG2) protein in cell-free extracts from a colon cancer-derived cell line [4]. Further investigations of this interaction through competition and immunoprecipitation experiments found that hFAM72A binds a polypeptide containing N-terminal 25 amino acids of hUNG2 [4]. These investigators reported that in cell lysates this interaction had no effect on the uracil excision activity of hUNG2, but we recently showed that the activity of the murine ortholog of hUNG2, mUNG2, is moderately inhibited by mFAM72A [7]. Moreover, we and others showed that mFAM72A decreases the cellular levels of mUNG2 in B lymphocytes, depleting the latter during the antibody maturation processes of somatic hypermutation (SHM) and class-switch recombination (CSR) [7, 8]. The depletion of mUNG2 by mFAM72A is likely to be due to the degradation of the former protein, because proteosome inhibitor MG-132 alleviates this depletion [[7] and Supplementary Fig. S1].
During the antibody maturation process an enzyme that is essential for both SHM and CSR, activation-induced deaminase, AID, converts cytosines in DNA to uracil [9–12] creating U•G mispairs. These mismatched uracils may be recognized by two DNA repair pathways, the base excision repair (BER) and a non-canonical mismatch repair (ncMMR), with the former requiring the activity of UNG2. The processing of these mispairs by DNA repair leads to mutations that shape SHM and creates strand breaks that are essential for CSR [13, 14]. Recent work regarding inhibition and degradation of mUNG2 by mFAM72A suggests that the latter protein helps maintain U•G pairs in the immunoglobulin genes during SHM and CSR to engage ncMMR in antibody maturation [7, 8]. The FAM72A-UNG2 interaction and the subsequent depletion of UNG2 is particularly intriguing as it applies to cancers- especially tumors that overexpress AID or the other human DNA-cytosine deaminases, the APOBEC3 proteins [15]. In these cells, suppression of BER through the expression of FAM72 paralogs may promote AID and APOBEC signature mutations [16, 17] and genomic instability. These findings make the FAM72A-UNG2 interaction an attractive therapeutic target.
However, the biochemical properties of the FAM72 proteins have not been studied and their structures have not been determined. These 149 aa proteins share poor sequence homology with other proteins in protein databases, but investigators have generated models of the human and murine FAM72A using computational tools [18, 19]. It was proposed that hFAM72A has two distinct cofactor-binding sites; one site consisting of C18, C21, C74, and C77 residues and was predicted to coordinate a metal cofactor (Zn2+ or Fe3+ ion), while the other site consisting of Y83, V85, C96, and N97 residues was predicted to bind an organic molecule [19]. Additionally, in the mFAM72A model, a handful of residues were proposed to interact directly with mUNG2’s N-terminal residues [18]. The computational models suggest a role for mFAM72A W125 residue in the between hUNG2 and hFAM72A [18] and there is experimental evidence that supports this prediction [4, 7, 8]. We initiated a study to characterize the mFAM72A protein and its interaction with mUNG2, instead of their human orthologs because mice have only one ortholog of the hFAM72A gene and the role of mFAM72A in SHM and CSR is already established [7, 8]. We wished to identify residues of mFAM72A necessary for the selective depletion of mUNG2 and better understand subcellular localization of this protein. To accomplish this, we created a cellular transfection assay, visualized the proteins in cells and monitored the ability of the wild-type and mutant mFAM72A proteins to deplete cellular mUNG2 levels. We also purified mFAM72A to near homogeneity and found that it has an Fe-S cluster that is redox sensitive. We describe these and other structural studies of mFAM72A below.
2. Methods and Materials
2.1. Cell lines and growth
Human cell lines HeLa and HEK293T cells were obtained from American Type Culture Collection. They were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) of penicillin/streptomycin. Cell counts and viability was determined using trypan blue staining (0.4% stain solution, Sigma) and a TC20 cell automated counter (Bio-rad).
2.2. Construction of plasmids for protein expression
The primers used for cloning are listed in Supplementary Table 1. The pcDNA3.1-mUNG2-FLAG (pmUNG2-FLAG) expression plasmid was described previously [7]. The pΔ83mUNG2-FLAG expression plasmid used the same vector backbone and was synthesized and obtained from ATUM. The mUNG1 gene was amplified from the pcDNA-mUNG1-GFP expression construct, obtained from Dr. Bodil Kavli (Norwegian University of Science and Technology) and cloned into FLAG-HA-pcDNA3.1(Addgene, plasmid #52535) using a Gibson Assembly kit (New England Biolabs) to create pmUNG1-FLAG. The pET28a-polyHis-SUMO vector was obtained from Dr. Xiaodong Cheng (The University of Texas Health Science Center at Houston) and the mUNG2 gene cloned into this vector as a SacI/NdeI fragment to create ppolyHis-SUMO-mUNG2. The FLAG tag was deleted from pcDNA3.1-HA-FLAG plasmid and mFAM72A gene was cloned into this plasmid creating pHA-mFAM72A plasmid. The pRSET-mFAM72A expression construct was received from Dr. Alberto Martin (University of Toronto) and has been described previously [7]. The mFAM72A gene was amplified and inserted into pET28a to create pHis-FAM72A. FAM72A mutants C18A, C21A, C18/21A, C65A, C89A, C65/89A, F61A, F104Y, W125A, W125R, and T131A were constructed in this plasmid using site-directed mutagenesis. The primers used for mutagenesis are listed in Supplementary Table 2. Sequences of all the recombinant plasmids were verified using Sanger sequencing (Michigan State University RTSF Genomics Core).
2.3. Expression and purification of proteins
All protein expression constructs were introduced into the E. coli strain BL21(DE3) and grown to an OD600 of 0.6–0.8 prior to induction of protein expression using 0.1 mM isopropyl ß-D-1-thiogalactopyranoside (IPTG) for 18–24 hours at 18 °C. The cells containing pHis-FAM72A were grown in Luria-Bertani (LB) media containing appropriate antibiotic and FeCl3 was added to the media to 0.01% just prior to protein induction. The cells were harvested and resuspended in lysis buffer I (50 mM sodium phosphate pH 8.0, 150 mM NaCl, 10 mM imidazole) which was supplemented with cOmplete protease inhibitor cocktail (Thermofisher). The cells were broken using a French Press at 1500 pounds per square inch pressure and the lysate clarified using centrifugation at 16,000 g for 30 minutes. The Ni-NTA agarose (Thermofisher) was washed in lysis buffer I and added to the clarified lysate. The suspension was rotated at 4°C for 2 hours. The Ni-NTA agarose was successively washed with Buffer A (50 mM sodium phosphate pH 8.0, 1 mM DTT, 25 mM imidazole) containing 150, 300, or 600 mM NaCl. The protein was eluted using 500 μL aliquots of Buffer A containing 150 mM NaCl and 250 mM imidazole. The eluted fractions were subjected to SDS-PAGE gel electrophoresis and the fractions containing the protein were combined and concentrated using 10 kDa MWCO centrifugal filters (ThermoFisher) while exchanging the buffer to Buffer B (25 mM HEPES pH 8, 10 mM NaCl, 1 mM DTT). The protein was then loaded onto an ion-exchange column containing Q-Sepharose equilibrated in Buffer B and the column was washed with 10 column volumes of the same buffer. The protein was eluted in five column volumes of Buffer C (25 mM HEPES pH 8, 300 mM NaCl, 1 mM DTT). The eluted protein was purified on an AKTA-pure FPLC equipped with a Superdex 75 Increase 10/300 GL column (Cytiva) in a buffer containing 50 mM sodium phosphate pH 7.5, 300 mM NaCl, 1 mM DTT, 0.01% TritonX-100, 2.5% glycerol). The fractions containing mFAM72A protein were concentrated, and buffer exchanged to mFAM72A storage buffer (25 mM HEPES pH 7.5, 500 mM NaCl, 2 mM DTT, 10% glycerol) and stored at −80°C.
The cells containing polyHis-SUMO-mUNG2 were induced for expression using 0.25 mM IPTG and the cell pellets were resuspended in lysis buffer II (25 mM HEPES 7.5, 200 mM NaCl, 1 mM TCEP) containing 10 mM imidazole. The cells were broken using a French Press at 1500 pounds per square inch pressure and the lysate clarified using centrifugation at 16,000 g for 30 minutes The Ni-NTA agarose was washed in lysis buffer and then added to the clarified lysate and the mixture was rotated at 4°C for 2 hours. The agarose was then successively washed with Buffer D (25 mM HEPES 7.5, 1 mM TCEP) containing 200, 400, or 600 mM NaCl supplemented with 25 mM imidazole. The His-SUMO-mUNG2 protein was eluted using 500 μL aliquots of Buffer D containing 200 mM NaCl and 250 mM imidazole. The His-SUMO tag was cleaved by the addition of a His-tagged SUMO protease (Millipore Sigma; 4 units/μg) in 1X SUMO buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM DTT, 0.2% Tween-20) followed by an incubation at 4°C for 2 hours. Both the SUMO protein and the protease have a poly-His tag and were removed using Ni-NTA agarose beads. The unbound protein, untagged mUNG2, was washed from the Ni-NTA beads using lysis buffer II containing 25 mM Imidazole. The eluted untagged mUNG2 was collected and purified by the same size exclusion chromatography system described above (Cytivia; Buffer: 25 mM HEPES 7.5, 200 mM NaCl, 1 mM TCEP, 0.01% TritonX-100, 2.5% glycerol). The fractions containing mUNG2 were concentrated to 10 mg/mL using a 10 kDa MWCO filter and the buffer was exchanged for UNG storage buffer (25 mM HEPES 7.5, 200 mM NaCl, 1 mM TCEP, 10% glycerol) and stored at −80°C.
The cells containing Δ83mUNG2-FLAG were induced for expression using 0.2 mM IPTG and the cell pellets were resuspended in lysis buffer II. Cells were broken using a French Press and clarified under the same conditions as described above. Anti-FLAG® M2 Magnetic Beads (Sigma) were washed in 1X TBS-T (50 mM Tris-Cl pH 7.6, 150 mM NaCl with 1% Tween-20), added to the clarified lysate and the mixture was rotated at 4°C overnight. The magnetic beads were washed three times with 1XTBS-T then incubated with 3XFLAG peptide (APExBIO) in 1XTBS at 4 °C for 2 hours in order to release the FLAG-tagged protein. The elution step was repeated one more time and the two eluates were pooled and concentrated using a 10 kDa MWCO filter. The concentrated protein was purified by the same size exclusion chromatography system as described above (Cytivia; Buffer: 50 mM Tris-Cl pH 7.5, 200 mM NaCl, 1 mM DTT, 2.5% glycerol) and the fractions containing Δ83mUNG2-FLAG protein were concentrated and the buffer was exchanged for UNG storage buffer.
2.4. Binding studies using Biolayer Interferometry
BLI measurements were done using a BLItz system (ForteBio) to evaluate the binding between mFAM72A and mUNG2 or 25 aa N-terminal polypeptide from mUNG2, the 25 aa N-terminal polypeptide from mUNG2, or the catalytic domain of mUNG2 with the N-terminus deleted (83ΔmUNG2). It was also used to study binding between Withaferin A and mFAM72A or mUNG2. The His-tagged mFAM72A was immobilized on a hydrated Ni-NTA bioprobe (Sartorius) using a solution containing 10 μM of protein in BLI buffer (10 mM Tris–HCl pH 8.0, 100 mM NaCl, 1 mM DTT, 1 mM EDTA, 0.1% Trition-X100) supplemented with 0.5 mg/mL BSA. Different concentrations of mUNG2 were prepared in BLI buffer in black microcentrifuge tubes (Argos Technologies). The association and dissociation rates were each measured at 120 second intervals for every concentration of the mUNG2 protein. The experiments were repeated three times and the KD values were determined using association and dissociation rate constants obtained using the BLItz Pro software.
The first 25 aa of mUNG2 were synthesized with a C-terminal biotin tag (Genescript). The His-tagged mFAM72A was immobilized on a hydrated Ni-NTA bioprobe and then dipped into solutions of BLI buffer containing the 25 aa peptide. Three replicates were performed for each peptide concentration and the KD values were determined as described above.
To evaluate the binding of Withaferin A (Selleckchem) to mFAM72A or mUNG2, BLI measurements were done using a Ni-NTA hydrated bioprobe saturated with either His-tagged mFAM72A or polyHis-SUMO-mUNG2. The Withaferin A concentrations which had a clear association and dissociation signal and an R2 value >0.9 were used to estimate the KD values for the binding of Withaferin A to mFAM72A. The binding of mUNG2 to mFAM72A was also evaluated in the presence of Withaferin A. The baseline, association and dissociation solutions contain BLI buffer and a 10X concentration of Withaferin A compared to the concentration of 25 aa peptide to be used (ie. 50 μM Withaferin A with 5 μM 25 aa peptide). Three concentrations of mUNG2 25 aa peptide were used to estimate its new KD value under these conditions.
2.5. mUNG2 Depletion Assay
HEK293T cells were grown in 12-well plates to 30 to 40% confluency and transfected with pmUNG1-FLAG or pmUNG2-FLAG along with pHA-mFAM72A or pHA-EV plasmids using Lipofectamine 3000. The cells were grown for 48 hours and harvested using trypsin digestion. For ROS experiments, co-transfected cells were treated with either 50, 100, or 250 μM H2O2 for 6 hours prior to cell harvesting. For proteasome inhibition experiments, co-transfected cells were treated with 10 μM MG-132 (Cell Signaling) for 4 hours prior to cell harvesting. The cells were pelleted at 500 g for 5 minutes, washed with 1XPBS and whole cell extracts were prepared by incubating cells with lysis buffer (20 mM Tris-HCl pH 7. 4, 150 mM NaCl, 1 mM EDTA, 1% Triton-X100, 1% sodium deoxycholate, 0.1% SDS) containing 1x Halt protease inhibitor cocktail for 30 minutes at 4°C followed by sonication and centrifugation at 16,000 g for 3 minutes to clear the lysate. The total protein lysate concentration was measured using Bio-Rad protein assay dye reagent concentrate (Bio-Rad). Protein lysates (5 to 10 μg) were separated on 18% SDS-PAGE gel and transferred to a PVDF membrane. The membrane was blocked with 5% (w/v) non-fat milk and probed with mouse anti-FLAG M2 monoclonal antibody (Millipore Sigma; 1:2000 dilution), mouse anti-α-tubulin antibody (Cell Signaling; 1:2000 dilution), mouse anti-HA tag antibody (Cell signaling; 1:2000), and rabbit anti-Histone3 antibody (Cell Signaling; 1:2000 dilution). This was followed by incubation with goat anti-mouse IgG HRP-conjugated antibody (Cell signaling; 1:1000) or goat anti-rabbit IgG HRP-conjugated antibody (Cell Signaling; 1:1000). The protein bands were visualized by the addition of SuperSignal West Pico Plus chemiluminescence substrate (ThermoFisher) and detected using an Invitrogen iBright 1500 imager (Thermo Fisher Scientific). Biological replicates of mUNG2 and mFAM72A pair were analyzed to determine the relative reduction in mUNG1 or mUNG2 protein level. Briefly, ImageJ software was used to determine the mUNG2 signal intensity in each sample which was then normalized to the corresponding α-tubulin signal. The fraction of mUNG2 normalized signal in EV to the mFAM72A samples was used to determine the amount of mUNG2 protein remaining. The data were plotted using GraphPad Prism and the statistical significance of difference between samples was evaluated using an unpaired t-test with Welch’s correction.
2.6. Visualization of the of FAM72A and mUNG2 proteins in cells
HeLa cells were grown to 70–80% confluency in an 8-well chamber slide (Ibidi) and transfected with expression plasmid for mFAM72A alone or along with mUNG2 expression plasmid using Lipofectamine3000 (ThermoFisher). The cells were grown for 24 hr and MitoTracker® Red CMXRos dye (Cell Signaling) was added to the cells at a 200 nM concentration. The cells were incubated in a 37°C incubator for 15 minutes, fixed with 4% paraformaldehyde, permeabilized with 0.3% Triton-X100, then blocked for 1 hour at 25°C with 10% goat serum (Gibco) in 1X PBS. The cells were incubated with mouse anti-HA tag antibody (Cell Signaling; 1:500), rabbit anti-FLAG antibody (Cell Signaling; 1:500), and mouse anti-FLAG tag M2 monoclonal antibody (Millipore Sigma; 1:500) for 2 h at 25°C. All antibodies were diluted in 1.5% goat serum. The secondary antibodies were goat anti-mouse IgG (H+L) Cy3-conjugated antibody (ThermoFisher; 1:1000), goat anti-mouse IgG (H+L) AlexaFluor488-conjugated antibody (ThermoFisher; 1:1000), and goat anti-rabbit IgG (H+L) AlexaFluor647-conjugated antibody (ThermoFisher; 1:1000). Following a one-hour incubation at 25°C the slides were mounted with ProLong™ Gold Antifade Mountant with DAPI (ThermoFisher). Images were acquired using an Axiovert.A1 inverted microscope (Carl Zeiss) with an oil immersion 63X objective lens. Image processing was done using Zen lite software (Carl Zeiss).
2.7. Homology modeling of mFAM72A and docking with mUNG2
The 3D structure of mFAM72A was predicted by submitting its sequence to the RaptorX web server (http://raptorx.uchicago.edu/) [20] to generate homology models. The top-ranked models (RMSD ≤ 4 Å) generated by RaptorX were compared to the model deposited in AlphaFold (Q8BFZ8; accessed on 4 April 2022 from https://alphafold.ebi.ac.uk/entry/Q8BFZ8) by superimposing the structures in PyMOL Molecular Graphics System (Version 2.0, Schrödinger, LLC). The disordered regions modelled with very low confidence (<50 per-residue confidence score) in AlphaFold [21, 22] and atoms with poor RMSD values (≥ 2.0 Å) in RaptorX were removed from the structures using Pymol “remove atoms” function.
The pyDock web server (https://life.bsc.es/pid/pydock/) [23] was used to dock mUNG2 with mFAM72A. As the X-ray crystal structure of hUNG2 protein is missing amino terminal 84 residues and the N-terminal UNG2 residues are important for its intearction with mFAM72A, we used the AlphaFold deposited structure of mUNG2 (https://alphafold.ebi.ac.uk/entry/P97931, accessed on 10 July 2021). The mFAM72A and mUNG2 AlphaFold models were docked using zero restraints and the top ranked model (Electrostatic energy: −31.61, desolvation energy: −16.68, Van der Waals energy: 17.17, total binding energy: −46.58) was analyzed further in PyMOL. Any contacts within 3Å of the mFAM72A-mUNG2 interface were used to find potential interacting residues. The vacuum electrostatics function in PyMOL was used to generate an electrostatic surface potential of mFAM72A and images were captured using the “ray” function.
2.8. UV-VIS spectral analysis of mFAM72A iron-sulfur cluster
UV-VIS absorbance measurements were recorded at room temperature using a DU 730 Life Science UV/Vis spectrophotometer (Beckman Coulter). Protein samples were prepared in mFAM72A storage buffer, and the concentrations were adjusted to 80 μM. The samples were scanned from 300 nm to 500 nm and absorbance was recorded at 1 nm increments. The reduced and oxidized mFAM72A samples were respectively prepared by the addition of 5 mM sodium dithionite to protein in a sealed cuvette or keeping a cuvette containing the protein open to air respectively. The samples were kept at room temperature and at the indicated times scanned as described above. The iron-sulfur cluster in mFAM72A was reconstituted as described by Guo et al [24]. Specifically, DTT (5 mM), ferrous ammonium sulfate (1 mM, Sigma Aldrich), and sodium sulfide (1 mM, Sigma Aldrich) were sequentially added to the protein solution to the indicated final concentrations and the mixture was incubated at room temperature for 3 hr. The buffer was exchanged three times with the mFAM72A storage buffer, the protein was concentrated to 80 μM and scanning was performed as described above. The absorbance values for each sample were exported into Microsoft Excel and converted into molar absorptivity using the Beer-Lambert Law. The resulting molar absorptivity values were plotted using GraphPad Prism software.
3. Results
3.1. mUNG2 helps localize mFam72A to the nucleus
hFAM72A was reported to be nuclear in the human colon cancer cell line SW480 that expresses hFAM72A at high levels [4], but other studies reported that it was largely associated with organelles, vesicles and membranes in the cytoplasm of other human cells [[2] and Human protein Atlas]. To determine the subcellular distribution of mFAM72A, HEK293T cells were transfected with plasmids encoding mFAM72A or mUNG2 individually or together. Representative cells were stained with appropriate antibodies and visualized.
When only the mFAM72A plasmid was present in HEK293T cells, the protein was found throughout the cells and appeared to be granular (Fig. 1, left panel and enlarged figure in Supplementary Fig. S2). The apparent granularity of the distribution is consistent with the suggestion that FAM72A is localized in subcellular vesicles [2]. However, some mFAM72A was also seen in nuclei which is consistent with the earlier observation of Guo et al [4].
Figure 1.
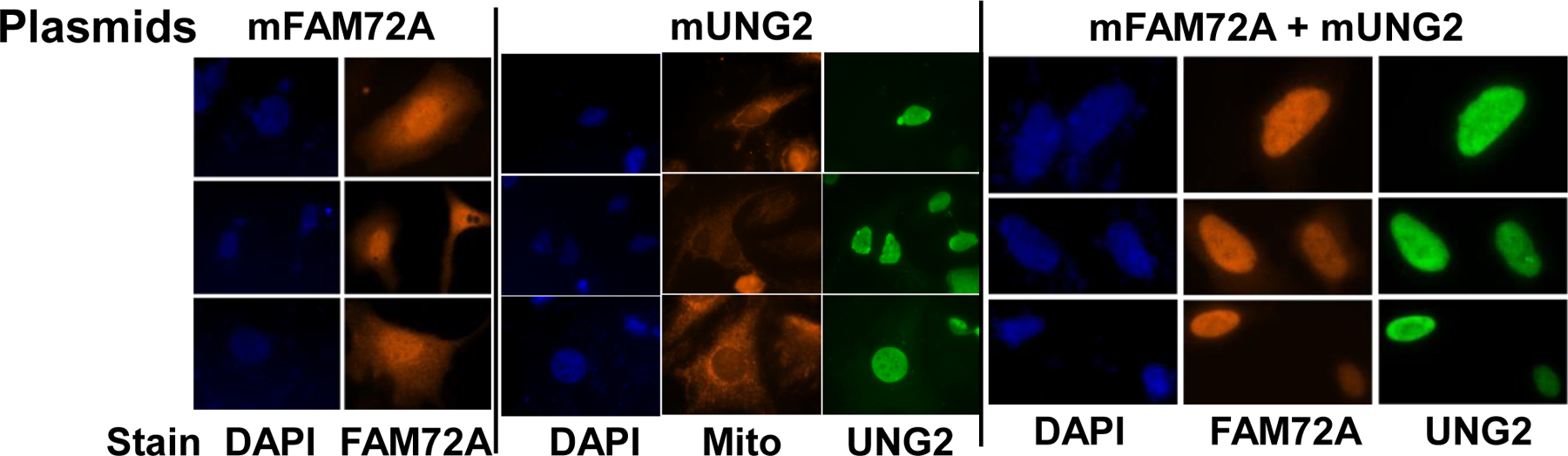
Subcellular localization of mUNG2 and mFAM72A in HeLa cells. Fluorescence images of cells stained with DAPI (nucleus), MitoTracker Red (mitochondria) or antibodies specific for UNG or FAM72A proteins are shown. The top line shows the plasmids that were transfected into cells, while the line at the bottom shows the stain or antibody used in each image.
In contrast to mFAM72A, mUNG2 was exclusively localized in the nucleus (Fig. 1, middle panel and Supplementary Fig. S3). Furthermore, when plasmids expressing mFAM72A and mUNG2 were co-transfected into HEK293T cells, both the proteins were found only in the nucleus (Fig. 1, right panel and Supplementary Fig. S4). These data suggest that binding of mUNG2 to mFAM72A helps transport the latter protein into nuclei or prevents nuclear export.
3.2. FAM72A binds tightly to N-terminal peptide from UNG2
Previous work showed that purified mUNG2 or hUNG2 binds mFAM72A in immunoprecipitation experiments, and expression of mFAM72A in the murine CH12F3 cell line or B lymphocytes promotes degradation of mUNG2, but not mUNG1 [7, 8]. To quantify the binding between mUNG2 and mFAM72A, we used biolayer interferometry (BLI) using proteins purified to near homogeneity (Supplementary Fig. S5). Poly-His tagged mFAM72A was immobilized on a BLI chip and the binding parameters for mUNG2 were determined. The association-dissociation kinetics of binding showed that mUNG2 binds to mFAM72A with a submicromolar dissociation constant (Fig. 2A and Table 1).
Figure 2.

Biolayer interferometry analysis of interaction between mUNG2, mFAM72A and Withaferin A. The molecular association-dissociation curves are shown. (A) Binding of mFAM72A (immobilized) to mUNG2. The concentrations of mUNG2 are shown. (B) Binding of mFAM72A (immobilized) to 25-mer polypeptide. The concentrations of the polypeptide are shown. (C) Binding of mFAM72A (immobilized) to Withaferin A. The concentrations of the Withaferin A are shown. (D) Binding of SUMO-mUNG2 (immobilized) to Withaferin A.
Table 1.
Binding studies using Biolayer Interferometry
| Bound protein | Ligand | ka (M−1 s−1) | kd (s−1) | KD |
|---|---|---|---|---|
| mFAM72A | mUNG2 | (4.3 ± 0.3)x104 | (2.3 ± 0.1)x10−2 | (0.6 ± 0.1) μM |
| mFAM72A | 25 aa peptide | (2.0 ± 0.0)x107 | (3.7 ± 0.8)x10−2 | (1.9 ± 0.3) nM |
| mFAM72A bound with Withaferin A | 25 aa peptide | 3.4×105 | 6.4×10−2 | 188 nM |
ka and kd are respectively rates of association and dissociation. KD is dissociation constant. When available, the numbers are reported from three independent BLI runs and show mean ± standard deviation.
Guo et al [4] showed that a polypeptide containing the first 25 amino acids of hUNG2 can compete with full-length hUNG2 for hFAM72A binding. These amino acids are strongly conserved in mUNG2 (Supplementary Fig. S6) and hence we studied the interaction of a synthetic polypeptide containing the first 25 aa of mUNG2 and mFAM72A using BLI. The rate of association of the 25 aa peptide was much faster than full-length mUNG2 and this resulted in a much lower KD value (Fig. 2B and Table 1). In contrast, BLI analysis of binding of mUNG2 catalytic domain [i.e. missing amino terminal 83 amino acids; Δ83mUNG2 [25]] binds mFAM72A very poorly (Supplementary Fig. S7). These results confirm and extend previous studies of interaction between the amino terminal amino acids of UNG2 with FAM72A and put them on a quantitative footing.
3.3. In silico models for interactions between mFAM72A and mUNG2
To obtain better insight into interaction of mFAM72A with mUNG2 we preformed homology modelling of mFAM72A using RaptorX [26, 27] and the models created by this software were in good agreement with the structure predicted by AlphaFold for this protein [21] (Supplementary Fig. S8). We then docked the AlphaFold-generated mFAM72A and mUNG2 structures using the pyDOCK Web server, without providing restraints. The best fit models suggested interaction of N-terminal 12 residues of mUNG2 with residues W125 and T131 of mFAM72A (Supplementary Fig. S9A and S9B).
Recently, another research group also docked a hFAM72A model with the first 45 amino acids of hUNG2 [18] and based on this analysis predicted that in addition to W125, the residues F61, F104 and I131 of hFAM72A should also interact with the N-terminus of hUNG2. These residues are conserved in the murine paralog, with the exception that it has a threonine at position 131 instead of isoleucine (Supplementary Figs. S9C and S10). The prediction that W125 of mFAM72A/hFAM72A may interact with mUNG2/hUNG2 is consistent with previous experimental results. mFAM72A, hFAM72A and hFAM72B have a Trp residue at position 125, while hFAM72C and hFAM72D have Arg instead (Supplementary Fig. S10). A Trp to Arg replacement was reported to abolish binding of hFAM72A to hUNG2 [4]. Moreover, the WT mFAM72A, but not its W125R mutant, causes degradation of mUNG2 upon a transduction into mFAM72A−/− B cells [7, 8]. We tested the effects of mutating these mFAM72A residues (Fig. 3A) on degradation of mUNG2 using a new assay.
Figure 3.

Degradation of mUNG2 by mFAM72A and its mutants. (A) Theoretical model of mFAM72A showing the residues that were mutated. (B) Upper panel- Western blot of mUNG1 and mUNG2 proteins in HEK293T cells transfected with different pairs of plasmids. The proteins expressed by the plasmids are indicated to the left and at top of the blot. The antibodies used to visualize the proteins are indicated on the right. Lower panel- Quantification of intensities of bands from three independent blots. Mean and standard deviations are shown. EV- empty vector. (C) Western blot of mUNG2 following co-transfection of cells with mUNG2 expression plasmid with mFAM72A mutant expression plasmids. The identity of the protein bands is indicated on the right of the blot. (D) Quantification of mUNG2 levels from independent blots such as shown in part C. Mean and standard deviations are shown.
3.4. Biochemical tests of models of mFAM72A-mUNG2 interaction
The previously used assay for the degradation of mUNG2 was cumbersome because it involved transduction of packaged lentiviral plasmids expressing mFAM72A or its mutants into murine CH12F3 cells [7, 8]. We replaced this assay with a simple transfection of plasmids expressing mFAM72A or its mutant into HEK293T cells along with an mUNG2 expression plasmid. Compared to transfection with empty vector (EV), the mFAM72A plasmid caused about 50% reduction in mUNG2 amount in cells, but the mUNG1 amount remained unchanged (Fig. 3B and Supplementary Fig. S11). These results suggest that the endogenous hUNG2 and hFAM72 proteins do not substantially affect degradation of either mUNG1 or mUNG2 in this hybrid mouse/human system.
The replacement of Trp125 with Arg eliminated the depletion of mUNG2, while its replacement with Ala reduced the depletion from 72% to 43% (Fig. 3C and 3D). However, confounding this conclusion we also found that the intracellular levels of W125R and W125A mutants were lower (Fig. 3C). When normalized for the amount of mFAM72A seen on the blot, the W125R mutant was defective in mUNG2 degradation, but the W125A mutant had little effect on mUNG2 levels (Supplementary Fig. S12). Therefore, W125 contributes both to stability or expression of mFAM72A as well its ability to promote mUNG2 depletion.
We also generated F61A, F104Y and T131A mutants of mFAM72A and tested them in the cellular transfection assay for degradation of mUNG2. F104Y mutation had a significant effect on cellular levels of mUNG2, while F61A and T131A mutations did not significantly change mUNG2 levels (Fig. 3C) However, the F104Y mutation also decreased cellular mFAM72A levels (Fig. 3C) again confounding the result. F61A and T131A changes had a smaller effect on mFAM72A protein levels (Fig. 3C) suggesting that these residues do not play a big role in protein expression or promotion of mUNG2 depletion.
Additionally, we tested a prediction regarding small molecule interactions with FAM72A. Renganathan et al performed in silico screening of small molecules that may bind to hFAM72A and predicted that withanolide analog Withferin B would bind hFAM72A and interfere with its binding with hUNG2 [18]. The same study also pointed out that a closely related natural products compound Withaferin A (Supplementary Fig. S13) with known anti-cancer properties [28, 29] may also disrupt hFAM72A-hUNG2 interaction [18]. We used the BLI assay to test this prediction using the murine homologs and found that Withaferin A binds mFAM72A with a micromolar dissociation constant while mUNG2 binds about 2000-fold worse (Table 2, Fig. 2C and 2D). However, when mFAM72A was pre-bound to Withaferin A and then challenged with the 25 aa peptide from mUNG2, the association kinetics was different than when Withaferin was absent (Supplementary Fig. S14). In the presence of Withaferin A, the ka for the association of 25-mer to mFAM72A was lower by almost two orders of magnitude while kd remained essentially unchanged (Table 1). This supports the suggestion [18] that Withaferin A (or Withaferin B) and mUNG2 interact with some of the same residues of mFAM72A and hence these withanolide derivatives may be used as lead molecules in the design of an inhibitor of FAM72A-UNG2 interaction.
Table 2.
Binding of Withaferin A to mFAM72A
| Bound protein | Ligand | ka (M−1 s−1) | kd (s−1) | KD |
|---|---|---|---|---|
| mFAM72A | Withaferin A | 8.9×103 | 2.1×10−1 | 24 μM |
| SUMO-mUNG2 | Withaferin A | 3.3×101 | 1.4×100 | 42 mM |
ka and kd are respectively rates of association and dissociation. KD is dissociation constant.
3.5. FAM72A has two potential metal coordination sites that regulate its activity
mFAM72A and the FAM72 human paralogs are only 149 aa in length but contain 14 cysteine residues (Supplementary Fig. S10). In our model of mFAM72A, we found two sites where four cysteine residues were positioned in a way that they could coordinate a metal ion or cofactor (Fig. 4A). Site A contains residues C65, C67, C89 and C92, and site B is made of residues C18, C21, C74 and C77 (Fig. 4A and Supplementary Fig. S10). We hypothesized that metal binding at one or both these sites may have a regulatory role in mFAM72A’s cellular function. Previously, Pramanik et al have suggested that site B may bind a Zn2+ ion [19]. To test the roles of these sites in mFAM72A activity, we replaced two cysteines in each of these potential metal-binding sites with alanines and tested the resulting mutants in the transfection assay.
Figure 4.

Role of potential metal-binding sites within FAM72A on its activity. (A) Center- The two sites, A and B, are shown within the predicted mFAM72A structure. Zoom in view of each site is shown on the side. (B and C) Western blots of cotransfection experiments using mUNG2 expression plasmid and metal-binding site mutants of mFAM72A are shown. Quantification of intensities of bands from three independent blots is presented below each blot as a bar graph. Mean and standard deviations are shown.
Substitutions at site A had a strong negative effect on depletion of mUNG2 by mFAM72A. The C65A mutant was ~50% less effective than WT and the C65/89A double mutant was completely ineffective at promoting UNG2 depletion (Fig. 4B, and S15A and S15B). However, like the other FAM72A mutants, the mutations at site A substantially decreased cellular abundance of mFAM72A (Fig. 4B). To compensate for this decrease, we normalized the mUNG2 depletion for protein abundance of mFAM72A and found that these mutations had little effect on the ability of mFAM72A to promote mUNG2 depletion (Supplementary Fig. S15E).
In sharp contrast with the site A mutants, mFAM72A mutants at site B had a strong positive effect on mFAM72A’s ability to deplete mUNG2 from cells (Fig. 4C, and S15C and S15D). This was true even though the site B mutations also decreased cellular mFAM72A levels (Fig. 4C). When mUNG2 levels were normalized for cellular levels of mFAM72A, the results were even more dramatic. While the C21A mutation caused mUNG2 levels to drop about 10-fold, the C18/21A mutant almost completely eliminated mUNG2 from cells (Supplementary Fig. S15F). These gain of function mutations show that the ion or co-factor at site B is plays a negative role in mFAM72A function. Using the metal ion-binding prediction and docking software, MIB (http://bioinfo.cmu.edu.tw/MIB/), we found that Zn2+ and Fe3+ had the best fit for the site B and this is consistent with the model of hFAM72A [19].
3.6. FAM72A has a redox sensitive Fe-S cluster that affects its activity
Based on the possibility that site B may contain an Fe3+, we hypothesized that it contains an Fe•S cluster. Although, we had previously expressed and purified mFAM72A using a pRSET plasmid, we re-cloned the gene into a pET vector and obtained a higher protein yield. Interestingly, cells containing induced mFAM72A protein appeared brown as a pellet (Fig. 5A), and the protein purified from these cells also maintained a yellowish color (Fig. 5B, sample 1). We had previously observed such a color in a known Fe-S-containing protein, UdgX [30, 31].
Figure 5. FAM72A has a redox sensitive iron-sulfur cluster.

The new pET28 mFam72a was used to purify the protein and the (A) cell pellet of mFam72a expressing E. coli was much darker in color than the mPCNA expressing cells. (B) Purified mFAM72A had a yellowish color (tube labelled 1) and when the iron-sulfur cluster was chemically reconstituted it had even darker color (tube labelled 2). (C) The UV-vis spectra the purified protein from pET28 expression system subjected to chemical reconstitution of the iron-sulfur cluster. For comparison, UV-VIS spectrum of older preparation of mFAM72A from pRSET expression system is also shown. (D) Effects of oxidation of the iron-sulfur cluster on UV- VIS absorption. The tube was opened for 10 minutes or 60 minutes prior to spectroscopy. (E) Effects of reduction of the iron-sulfur cluster on UV-VIS absorption. The protein was treated with sodium dithionite for 10 or 60 min prior to spectroscopy.
When the newly purified mFAM72A was subjected to UV-VIS spectroscopy, the protein displayed a absorption shoulder that starts at about 325 nm and a modest peak centered at 410 nm (Fig. 5C, green line). In contrast, the older preparation from the pRSET vector which had been stored in the freezer for over a year did not have a peak at 410 nm (Fig. 5C, blue line). We considered it likely that the protein preparations may lose the Fe•S cluster during purification and subsequent storage. To reverse this loss, we used ferrous ammonium sulfate and sodium sulfide in the presence of DTT to reconstitute the cluster in the protein obtained from the pRSET expression system. The chemical reconstitution resulted in a noticeably darker color for the protein (Fig. 5B, sample B) and an increase in in absorption across the visible spectrum (Fig. 5C, brown line). This translated into an increase in the molar extinction coefficient at 410 nm (ε410) from 6,850 to 15,800 M−1cm−1. Together these data strongly support the hypothesis that mFAM72A contains an Fe•S cluster that may be lost during purification or storage of the protein.
To determine whether the Fe-S cluster in the unreconstituted mFAM72A was redox sensitive, the protein was exposed to atmospheric oxygen and its UV-VIS spectrum was determined over time. There was a rapid increase in the absorption in the 400 to 500 nm range with a notable decrease in the peak definition (Fig. 5D). The ε410 increased 70% over the 60 min incubation (Fig. 5D). To reduce any Fe3+ within the protein to Fe2+, we treated the purified mFAM72A with sodium dithionite and observed a change in the opposite direction (Fig. 5E). Together these results demonstrate that mFAM72A’s Fe•S cluster is redox sensitive.
To determine whether these findings point to a role for oxidation/reduction of the Fe•S cluster on mFAM72A activity, we performed the mUNG2 depletion assay while cells were under oxidative stress. The HEK293T cells expressing mFAM72A and mUNG2 were treated with hydrogen peroxide to raise the level of cellular reactive oxygen species (ROS) and the amounts of cellular mUNG2 was quantified using a Western blot. The untransfected cells and cells transfected with mUNG2 plasmid were more sensitive to H2O2 treatment than those transfected with both mUNG2 and mFAM72A plasmids (Supplementary Fig. S16). H2O2 treatment decreased the viability of untransfected cells by about 30% at 250 μM and hence we did not test higher concentrations of the oxidizing agent.
When only mUNG2 expression plasmid was transfected into cells, H2O2 treatment resulted in only a modest change in mUNG2 levels with a slight decrease in protein level at the highest concentration of H2O2, 250 μM (Fig. 6A, top panel). When mFAM72A plasmid was co-transfected with mUNG2 plasmid, treatment of cells with 50 μM H2O2 resulted in less depletion of mUNG2 (Fig. 6A, bottom panel), but this change was not statistically significant (Fig. 6B). However, when the peroxide concentration was increased to 100 μM or higher, the depletion increased substantially (Fig. 6A, bottom panel). Treatment of cells with 100 μM H2O2 resulted in an increase in mUNG2 depletion that was statistically significant. It should be noted that the mFAM72A levels decreased following H2O2 treatment and normalization of the mUNG2 intensities to mFAM72A levels showed even more striking effect at 100 μM H2O2 (Supplementary Fig. S17). These results show that the ability of mFAM72A to promote mUNG2 degradation is modulated by oxidative stress within the cells.
Figure 6.

Effect of H2O2 treatment of cells on degradation mUNG2 promoted by mFAM72A. (A) Western blots of transfection experiments using mUNG2 expression plasmid with or without mFAM72A expression plasmid followed by treatment of cells with H2O2 at indicated concentrations. (B) Quantification of data from three independent experiments.
4. Discussion
Prior to this work, it was known that mFAM72A and hFAM72A respectively interact with the UNG2 in human and murine cells. It was also known that mFAM72A enhances both SHM and CSR by promoting degradation of mUNG2, but not mUNG1. The work presented here clarifies several aspects of interactions between mFAM72A and mUNG2, provides a quantitative assessment of this interaction and shows that mFAM72A is a redox-sensitive protein with an Fe•S cluster. It also provides a lead for the synthesis of small molecules that may disrupt hFAM72A-hUNG2 interaction with the aim of treating tumors with elevated FAM72A expression.
We found that mFAM72A binds mUNG2 at a submicromolar dissociation constant and binds the first 25 aa of mUNG2 at a nanomolar KD value (Table 1) confirming earlier suggestion that the mFAM72A interacts with mUNG2 principally through the N-terminus of the latter protein [4]. We were also able to confirm that Withaferin A binds mFAM72A and interacts very weakly with mUNG2 (Table 2; Figs. 2C and 2D) opening up the possibility of synthesizing inhibitors that disrupt the hFAM72A-hUNG2 interaction. However, our attempts to confirm predictions of molecular models of interaction between mUNG2 and mFAM72A through site-directed mutagenesis of the latter protein produced mixed results.
We mutated residues F61, F104, W125 and T131 residues of mFAM72A with the expectation that they may reduce interaction of the protein with mUNG2 and thereby reduce the depletion of mUNG2. While mutations at F104 and W125 decreased depletion of mUNG2, they also appeared to severely destabilize the protein (Fig. 3C and 3D). In contrast, mutations F61 and T131 did not affect depletion of mUNG2, and their amounts also remained unchanged in the Western blot (Fig. 3C and D). It is possible that F104 and W125 of mFAM72A are not only required for its interaction with mUNG2, but they are also required for protein stability or expression. Structural studies of mFAM72A are needed to clarify this issue.
The most intriguing finding of this study was that mFAM72A is an Fe-S containing protein and that this cluster may play a key role in the biochemical activity of this protein. We suspected that the protein had one or more metal cofactors based on the extremely high number of cysteines in the protein (Supplementary Fig. S10). On average, only 2.3% of residues in a human protein are cysteines, but this number is 9.4% for hFAM72A and mFAM72A. Additionally, there is on average one disulfide linkage per 100 aa residues in the human proteome [32] suggesting that 10 or more cysteines in mFAM72A may be free. In our modeling, we predicted two potential metal-binding sites (Fig. 4A). Heese and colleagues previously came to a somewhat different conclusion based on analysis of the protein using ligand-binding prediction algorithms [19]. They predicted a Zn2+ coordinated by C18, C21, C74, and C77, which we call site B (Fig. 4A) and they also predicted that residues Y83, V85, C96, and N97 of FAM72A would bind a small organic molecule [19]. Instead, our computations suggested that C65, C67, C89 and C92 may bind Zn2+ or Fe3+ (Site A; Fig. 4A).
Proteins containing [4Fe-4S] clusters typically display a broad absorption band between 400 and 420 nm, while proteins with [2Fe-2S] clusters show more complex spectra with peaks of absorption at lower and higher wavelengths [33, 34]. Newly purified mFAM72A showed a broad peak from 400–420 nm (Fig. 5C) which changed in magnitude- but not in shape- when the preparation was oxidized (Fig. 5D) or reduced (Fig. 5E). Furthermore, when mFAM72A was chemically reconstituted it had ε410 of 15,800 M−1cm−1, which is in the range expected for a [4Fe-4S] cluster (8,000–16,000 M−1cm−1) rather than a [2Fe-2S] cluster (6,000–11,000 M−1cm−1) [33, 35–37].
Activity of some proteins containing Fe-S clusters is redox sensitive, while others are insensitive to oxidation changes [38–40]. In the latter class of enzymes, the Fe-S cluster is thought to have only a structural role. We found that mFAM72A’s Fe-S was redox sensitive with dramatic changes in the UV-VIS spectra when purified protein was subjected to oxidative or reducing conditions. Based on these data, we tentatively conclude that mFAM72A has a [4Fe-4S] cluster that is redox sensitive. Further physical studies of this protein are required to confirm this finding.
Most redox sensitive proteins with Fe-S clusters are deactivated or downregulated in response to oxidative stress, but a few Fe-S proteins are activated in response to ROS [41, 42]. An example of this is the FBXL5, which binds and promotes proteasomal degradation of IRP2, an iron regulatory protein [42]. Oxidation of an [2Fe2S] cluster within FBXL5 to [2Fe-2S]2+ state is required for this process [42]. We speculated that the ability of mFAM72A to cause degradation of mUNG2 may also be regulated by the oxidation state of the Fe-S cluster within the former protein.
This was indeed the case. When the cells were exposed to ROS, mFAM72A showed a gain of function, i.e. an increase in ability to degrade mUNG2 which was dependent on mFAM72A. This makes sense for a protein that is expressed during B cell maturation because ROS plays a critical role in B cell development and immune cell function [43]. Consistent with these observations, we found that cells co-expressing mUNG2 and mFAM72a had no apparent change in cell viability or proliferation, even at detrimental amounts of ROS stress (Supplementary Fig. S16). These observations are also consistent with previous findings that hFAM72A lowered the cellular ROS in nasopharyngeal tumor cells [2]. Finally, disturbing the putative metal binding site A through site-directed mutagenesis of two cysteines within the site resulted in a gain of function similar to H2O2 treatment suggesting that the redox-sensitive Fe-S cluster lies in site A. Structural studies of the protein are needed to validate this conclusion.
The role of hFAM72A and its paralogs in tumorigenesis has largely been attributed to its effects on cellular proliferation [2, 3, 5]. However, it may also have other roles in cancer such as promoting error-prone processing of uracils created by AID/APOBEC enzymes [7, 8] which result in mutations that drive cancer development and tumor evolution [44–46]. Cancers cells are under oxidative stress and FAM72A may also lower cellular ROS to promote cell survival. Furthermore, it is likely that the FAM72 family of proteins may cause degradation of proteins other than UNG2. Using a yeast two-hybrid system, Heese showed that hFAM72A interacts with five additional human proteins [1]. A common feature of this group of proteins was the prevalence of their dysregulation in many different types of cancers [1]. We propose that FAM72 paralogs may bind these and other cellular proteins promoting their degradation, and this helps tumor proliferation. If this is found to be correct, small molecule inhibitors may be developed based on Withaferin A or Withaferin B to counteract tumor promoting activity of these proteins.
Supplementary Material
Funding
This work was supported by National Institutes of Health 1R21AI144708 and 1R21CA252858-01A1, and Bridge Funding grant from Wayne State University.
Footnotes
Conflicts of Interest
The authors declare that there are no conflicts of interest.
References
- [1].Heese K, The protein p17 signaling pathways in cancer, Tumour Biol, 34 (2013) 4081–4087. [DOI] [PubMed] [Google Scholar]
- [2].Wang LT, Lin CS, Chai CY, Liu KY, Chen JY, Hsu SH, Functional interaction of Ugene and EBV infection mediates tumorigenic effects, Oncogene, 30 (2011) 2921–2932. [DOI] [PubMed] [Google Scholar]
- [3].Rajan P, Stockley J, Sudbery IM, Fleming JT, Hedley A, Kalna G, Sims D, Ponting CP, Heger A, Robson CN, McMenemin RM, Pedley ID, Leung HY, Identification of a candidate prognostic gene signature by transcriptome analysis of matched pre- and post-treatment prostatic biopsies from patients with advanced prostate cancer, BMC Cancer, 14 (2014) 977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Guo C, Zhang X, Fink SP, Platzer P, Wilson K, Willson JK, Wang Z, Markowitz SD, Ugene, a newly identified protein that is commonly overexpressed in cancer and binds uracil DNA glycosylase, Cancer Res, 68 (2008) 6118–6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rahane CS, Kutzner A, Heese K, A cancer tissue-specific FAM72 expression profile defines a novel glioblastoma multiform (GBM) gene-mutation signature, J Neurooncol, 141 (2019) 57–70. [DOI] [PubMed] [Google Scholar]
- [6].Zhu D, Fang C, Li X, Geng Y, Li R, Wu C, Jiang J, Wu C, Predictive analysis of long non-coding RNA expression profiles in diffuse large B-cell lymphoma, Oncotarget, 8 (2017) 23228–23236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Feng Y, Li C, Stewart JA, Barbulescu P, Seija Desivo N, Alvarez-Quilon A, Pezo RC, Perera MLW, Chan K, Tong AHY, Mohamad-Ramshan R, Berru M, Nakib D, Li G, Kardar GA, Carlyle JR, Moffat J, Durocher D, Di Noia JM, Bhagwat AS, Martin A, FAM72A antagonizes UNG2 to promote mutagenic repair during antibody maturation, Nature, 600 (2021) 324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rogier M, Moritz J, Robert I, Lescale C, Heyer V, Abello A, Martin O, Capitani K, Thomas M, Thomas-Claudepierre AS, Laffleur B, Jouan F, Pinaud E, Tarte K, Cogne M, Conticello SG, Soutoglou E, Deriano L, Reina-San-Martin B, Fam72a enforces error-prone DNA repair during antibody diversification, Nature, 600 (2021) 329–333. [DOI] [PubMed] [Google Scholar]
- [9].Sohail A, Klapacz J, Samaranayake M, Ullah A, Bhagwat AS, Human activation-induced cytidine deaminase causes transcription-dependent, strand-biased C to U deaminations, Nucleic Acids Res, 31 (2003) 2990–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dickerson SK, Market E, Besmer E, Papavasiliou FN, AID mediates hypermutation by deaminating single stranded DNA, J Exp Med, 197 (2003) 1291–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW, Transcription-targeted DNA deamination by the AID antibody diversification enzyme, Nature, 422 (2003) 726–730. [DOI] [PubMed] [Google Scholar]
- [12].Bransteitter R, Pham P, Scharff MD, Goodman MF, Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase, Proc Natl Acad Sci U S A, 100 (2003) 4102–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pilzecker B, Jacobs H, Mutating for Good: DNA Damage Responses During Somatic Hypermutation, Front Immunol, 10 (2019) 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Methot SP, Di Noia JM, Molecular Mechanisms of Somatic Hypermutation and Class Switch Recombination, Adv Immunol, 133 (2017) 37–87. [DOI] [PubMed] [Google Scholar]
- [15].Siriwardena SU, Chen K, Bhagwat AS, Functions and Malfunctions of Mammalian DNA-Cytosine Deaminases, Chem Rev, 116 (2016) 12688–12710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjord JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jager N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, Lopez-Otin C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt AN, Valdes-Mas R, van Buuren MM, van ‘t Veer L, Vincent-Salomon A, Waddell N, Yates LR, Australian I Pancreatic Cancer Genome, I.B.C. Consortium, I.M.-S. Consortium, I. PedBrain, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR, Signatures of mutational processes in human cancer, Nature, 500 (2013) 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, Harris S, Shah RR, Resnick MA, Getz G, Gordenin DA, An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers, Nat Genet, 45 (2013) 970–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Renganathan S, Pramanik S, Ekambaram R, Kutzner A, Kim PS, Heese K, Identification of a Chemotherapeutic Lead Molecule for the Potential Disruption of the FAM72A-UNG2 Interaction to Interfere with Genome Stability, Centromere Formation, and Genome Editing, Cancers (Basel), 13 (2021) 5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pramanik S, Kutzner A, Heese K, Lead discovery and in silico 3D structure modeling of tumorigenic FAM72A (p17), Tumour Biol, 36 (2015) 239–249. [DOI] [PubMed] [Google Scholar]
- [20].Kallberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J, Template-based protein structure modeling using the RaptorX web server, Nat Protoc, 7 (2012) 1511–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D, Highly accurate protein structure prediction with AlphaFold, Nature, 596 (2021) 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A, Zidek A, Green T, Tunyasuvunakool K, Petersen S, Jumper J, Clancy E, Green R, Vora A, Lutfi M, Figurnov M, Cowie A, Hobbs N, Kohli P, Kleywegt G, Birney E, Hassabis D, Velankar S, AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models, Nucleic Acids Res, 50 (2022) D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cheng TM, Blundell TL, Fernandez-Recio J, pyDock: electrostatics and desolvation for effective scoring of rigid-body protein-protein docking, Proteins, 68 (2007) 503–515. [DOI] [PubMed] [Google Scholar]
- [24].Guo Z, Xu S, Chen X, Wang C, Yang P, Qin S, Zhao C, Fei F, Zhao X, Tan PH, Wang J, Xie C, Modulation of MagR magnetic properties via iron-sulfur cluster binding, Sci Rep, 11 (2021) 23941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nilsen H, Otterlei M, Haug T, Solum K, Nagelhus TA, Skorpen F, Krokan HE, Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene, Nucleic Acids Res, 25 (1997) 750–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang S, Li W, Liu S, Xu J, RaptorX-Property: a web server for protein structure property prediction, Nucleic Acids Res, 44 (2016) W430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Xu J, McPartlon M, Li J, Improved protein structure prediction by deep learning irrespective of co-evolution information, Nature machine intelligence, 3 (2021) 601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hahm ER, Kim SH, Singh KB, Singh K, Singh SV, A Comprehensive Review and Perspective on Anticancer Mechanisms of Withaferin A in Breast Cancer, Cancer Prev Res (Phila), 13 (2020) 721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hassannia B, Logie E, Vandenabeele P, Vanden Berghe T, Vanden Berghe W, Withaferin A: From ayurvedic folk medicine to preclinical anti-cancer drug, Biochem Pharmacol, 173 (2020) 113602. [DOI] [PubMed] [Google Scholar]
- [30].Sang PB, Srinath T, Patil AG, Woo EJ, Varshney U, A unique uracil-DNA binding protein of the uracil DNA glycosylase superfamily, Nucleic Acids Res, 43 (2015) 8452–8463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Stewart JA, Schauer G, Bhagwat AS, Visualization of uracils created by APOBEC3A using UdgX shows colocalization with RPA at stalled replication forks, Nucleic Acids Res, 48 (2020) e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wiedemann C, Kumar A, Lang A, Ohlenschlager O, Cysteines and Disulfide Bonds as Structure-Forming Units: Insights From Different Domains of Life and the Potential for Characterization by NMR, Frontiers in chemistry, 8 (2020) 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Freibert SA, Weiler BD, Bill E, Pierik AJ, Muhlenhoff U, Lill R, Biochemical Reconstitution and Spectroscopic Analysis of Iron-Sulfur Proteins, Methods Enzymol, 599 (2018) 197–226. [DOI] [PubMed] [Google Scholar]
- [34].Valer L, Rossetto D, Scintilla S, Hu YJ, Tomar A, Nader S, Betinol IO, Mansy SS, Methods to identify and characterize iron–sulfur oligopeptides in water, Can J Chem, in press (2022). [Google Scholar]
- [35].Dailey HA, Finnegan MG, Johnson MK, Human ferrochelatase is an iron-sulfur protein, Biochemistry, 33 (1994) 403–407. [DOI] [PubMed] [Google Scholar]
- [36].Emptage MH, Dreyers JL, Kennedy MC, Beinert H, Optical and EPR characterization of different species of active and inactive aconitase, J Biol Chem, 258 (1983) 11106–11111. [PubMed] [Google Scholar]
- [37].Stephens PJ, Thomson AJ, Dunn JB, Keiderling TA, Rawlings J, Rao KK, Hall DO, Circular dichroism and magnetic circular dichroism of iron-sulfur proteins, Biochemistry, 17 (1978) 4770–4778. [DOI] [PubMed] [Google Scholar]
- [38].Johnson DC, Dean DR, Smith AD, Johnson MK, STRUCTURE, FUNCTION, AND FORMATION OF BIOLOGICAL IRON-SULFUR CLUSTERS, Annual Review of Biochemistry, 74 (2005) 247–281. [DOI] [PubMed] [Google Scholar]
- [39].Bruska MK, Stiebritz MT, Reiher M, Analysis of differences in oxygen sensitivity of Fe–S clusters, Dalton Transactions, 42 (2013) 8729–8735. [DOI] [PubMed] [Google Scholar]
- [40].Fontecilla-Camps JC, Volbeda A, Cavazza C, Nicolet Y, Structure/Function Relationships of [NiFe]- and [FeFe]-Hydrogenases, Chemical Reviews, 107 (2007) 4273–4303. [DOI] [PubMed] [Google Scholar]
- [41].Mueller S, Iron regulatory protein 1 as a sensor of reactive oxygen species, BioFactors (Oxford, England: ), 24 (2005) 171–181. [DOI] [PubMed] [Google Scholar]
- [42].Wang H, Shi H, Rajan M, Canarie ER, Hong S, Simoneschi D, Pagano M, Bush MF, Stoll S, Leibold EA, Zheng N, FBXL5 Regulates IRP2 Stability in Iron Homeostasis via an Oxygen-Responsive [2Fe2S] Cluster, Mol Cell, 78 (2020) 31–41.e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhang H, Wang L, Chu Y, Reactive oxygen species: The signal regulator of B cell, Free radical biology & medicine, 142 (2019) 16–22. [DOI] [PubMed] [Google Scholar]
- [44].Revathidevi S, Murugan AK, Nakaoka H, Inoue I, Munirajan AK, APOBEC: A molecular driver in cervical cancer pathogenesis, Cancer Lett, 496 (2021) 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Robbiani DF, Nussenzweig MC, Chromosome translocation B cell lymphoma, and activation-induced cytidine deaminase, Annu Rev Pathol, 8 (2013) 79–103. [DOI] [PubMed] [Google Scholar]
- [46].Smith NJ, Fenton TR, The APOBEC3 genes and their role in cancer: insights from human papillomavirus, J Mol Endocrinol, 62 (2019) R269–r287. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.