

Abstract
Purpose
To explore the genetic background of choroidal and ciliary body melanoma among children and young adults, with special focus on BAP1 germline variants in this age group.
Methods
Patients under the age of 25 and with confirmed choroidal or ciliary body melanoma were included in this retrospective, multicenter observational study. Nuclear BAP1 immunopositivity was used to evaluate the presence of functional BAP1 in the tumor. Next-generation sequencing using Ion Torrent platform was used to determine pathogenic variants of BAP1, EIF1AX, SF3B1, GNAQ and GNA11 and chromosome 3 status in the tumor or in DNA extracted from blood or saliva. Survival was analyzed using Kaplan-Meier estimates.
Results
The mean age at diagnosis was 17 years (range 5.0–24.8). A germline BAP1 pathogenic variant was identified in an 18-year-old patient, and a somatic variant, based mainly on immunohistochemistry, in 13 (42%) of 31 available specimens. One tumor had a somatic SF3B1 pathogenic variant. Disomy 3 and the absence of a BAP1 pathogenic variant in the tumor predicted the longest metastasis-free survival. Males showed longer metastasis-free survival than females (P = 0.018).
Conclusions
We did not find a stronger-than-average BAP1 germline predisposition for choroidal and ciliary body melanoma among children and young adults compared to adults. Males had a more favorable survival and disomy 3, and the absence of a BAP1 mutation in the tumor tissue predicted the most favorable metastasis-free survival. A BAP1 germline pathogenic variant was identified in one patient (1%), and a somatic variant based mainly on immunohistochemistry in 13 (42%).
Keywords: children, adolescents, pediatric, uveal melanoma, BAP1 gene, germline mutation, monosomy 3
Uveal melanoma (UM) is the most common primary intraocular malignancy among adults1 with an estimated overall incidence of 6-7 per million in the Western world.2 The incidence varies from two to eight cases per million in whites, depending on latitude.3,4 Congenital UM is extremely rare, with only a few patients in the literature.5–9 Pediatric UM (PUM), which affects children and young adults (variably defined as <21 to 25 years) is also rare and comprises <1% to 2% of all UM.10–16
PUM and adult UM differ in some characteristics. Both are located primarily in the choroid, followed by iris and ciliary body,11–13 but the incidence of iris melanoma among young adults is higher.15 The mean tumor diameter is smaller in young adults than in patients >60 years.15 Higher percentage of females was evident in our large collaborative Pediatric Choroidal and Ciliary Body Melanoma Study of 299 PUM17 in line with a prior meta-analysis,10 whereas the prevalence of UM in adults is somewhat higher in males than in females.2
Most11,12,15,18 but not all14 series report that PUM has a lower metastatic rate and better prognosis than adult UM. In The Pediatric Choroidal and Ciliary Body Melanoma Study, 17 boys had a more favorable survival than girls, but this difference was not observed among young adults (18–24 years). Mortality increased with ciliary body involvement and a higher TNM (tumor, node, metastasis) stage.10,13,17 Moreover, extraocular extension and congenital oculo(dermal) melanocytosis were unfavorable prognostic factors.17
UM with monosomy 3 has a worse prognosis than that with disomy 3.19 Monosomy 3 is often accompanied by a gain of chromosome 8q,20,21 a combination associated with even worse prognosis.22,23 The tumor suppressor gene BRCA1-associated protein 1 (BAP1), located on chromosome 3, plays an important role in prognosis of UM.24 BAP1 is a deubiquitinating enzyme involved in the DNA repair mechanism.25 BAP1 additionally contributes to cell regulation, metabolism, and chromosome stability.26–28 Loss of BAP1 in UM is associated with higher transcriptome levels of CD38 (CD38 molecule), HLA-DRA (major histocompatibility complex, class II, DR alpha), IDO-1 (indoleamine 2,3-dioxygenase 1), and LAG-3 (lymphocyte activating 3), which are associated with immune suppressive pathways.29 Exactly how BAP1 contributes to the development of metastasis of UM is still unclear because of its many functions and interactions. Immunohistochemically demonstrable loss of nuclear BAP1 is a surrogate for a pathogenic variant30 and strongly associated with metastasis and survival.31,32 Grading systems for BAP1 IHC in lieu of a dichotomous score have been described, but none of the patients without metastatic disease had absence of BAP1.33
Not only somatic but also germline pathogenic variants in BAP1 are described in patients with UM. Patients carrying a BAP1 germline variant develop other cancers besides UM, especially mesothelioma, clear cell renal cell carcinoma, and cutaneous melanoma.34–36 Such a BAP1 tumor predisposition syndrome is present in about 2% of UM patients.34 No significant difference was found in the age of onset of UM in patients with a null and a missense BAP1 variant.35 However, the study did not include many patients with PUM, leaving the frequency of germline variants in this group unknown.
In The Pediatric Choroidal and Ciliary Body Melanoma Study,17 which did collect new genetic data, we reported that monosomy 3 was found in eight (54%) of 15 children 11 to 17 years old and in six (24%) of 25 young adults 18 to 24 years old. One of five patients in both groups had tested positive for somatic BAP1 pathogenic variants. The data suggested that the pathogenesis of PUM might differ by age group.
The present extended study aims to confirm, or exclude, that monosomy 3 predicts a higher risk for metastasis in PUM. We hypothesize that monosomy 3 is correlated with poor prognosis, as has been shown in adults as well.19,22 The other primary aim was to identify BAP1 somatic and germline variants in PUM. A secondary aim was to screen for other known UM driver mutations.
Material and Methods
Patient Selection
Eligibility for this retrospective cohort study included all patients in whom a choroidal and ciliary body melanoma was diagnosed at an age younger than 25 years and for whom a sample for genetic study and at least the following anonymous data were available: birth date, date of diagnosis, gender, treatment type, presence or absence of local or systemic tumor recurrence, last survival status, date of last known status, and cause of death (UM, second cancer, or nonmalignant cause) determined by reviewing the patient charts, registry data, histopathologic samples, and death certificates. Patients with iris melanomas were ineligible. All treatment methods were eligible. Informed consent from all patients was obtained before the samples were processed. This investigation was approved by the institutional review boards of the participating centers as required and it adhered to the tenets of the Declaration of Helsinki and was approved by the Rotterdam local ethics committee.
Data Collection
Anonymous data on consecutive eligible patients were collected from members of the European Ophthalmic Oncology Group. The data additionally acquired included presence of congenital oculo(dermal) melanocytosis or neurofibromatosis; visual acuity and intraocular pressure at diagnosis and at last visit; tumor thickness, largest basal diameter, ciliary body involvement, extraocular extension, distance from the center of the fovea and the margin of the optic disc, cell type, cytogenetic features; dates of any local tumor recurrence, secondary enucleation, and metastasis; and second primary malignancies. We staged the tumors according to the eighth edition of the TNM system of the American Joint Committee on Cancer.37,38 Participating ocular oncology services submitted their data through a secure survey website from patients diagnosed between 1968 and 2018.
Blood was withdrawn or saliva was collected from patients and parents whenever possible. Germline testing was performed on retinal tissue of the histopathologic specimen in cases where no blood or saliva was available. Tumor tissue from enucleated eyes (n = 50), resections (n = 6), or biopsy specimens (n = 2) were used for BAP1 immunohistochemistry (IHC) or DNA isolation when available.
DNA Extraction
Targeted next-generation sequencing (NGS) was performed on DNA extracted from blood, saliva, formalin-fixed paraffin-embedded tissue (FFPE) of the retina and the tumor. For DNA isolation of blood, the QIAmp DNA Blood kit was used (Qiagen, Hilden, Germany) according to the manufacturer's protocol. DNA isolation from saliva was performed with the Oragene DNA OG-500 kit for collection of human DNA (DNA Genotek, Ottawa, ON, Canada) following manufacturer's protocol. FFPE sections were used to isolate retinal and tumor tissue. Depending on the size of the tumor, four to nine sections 5 µm thick were deparaffinized and stained with hematoxylin before DNA isolation. DNA extraction was performed as described with 5% Chelex (Bio-Rad, Hercules, CA, USA) and Proteinase K (Qiagen).39
DNA was stored at −20C°, and concentrations were measured using the Quant-iT dsDNA Assay Kit, high sensitivity (Thermo Fisher Scientific, Waltham, MA, USA) as described by the manufacturer. Moreover, prior histopathological and genetic data were directly obtained of 28 PUM patients from the participating institutes.
Immunohistochemistry
BAP1 IHC was performed on FFPE tumor tissue as described previously.30 Sections 4 to 5 µm thick were used. All slides, except one, were evaluated by an ophthalmic pathologist (RMV) and one of the authors (NvP) for the presence of nuclear BAP1 expression. Agreement on all samples was reached. Lack of nuclear BAP1 immunoreactivity was taken to indicate a pathogenic variant in the BAP1 gene.30
Mutation Analysis
Germline analysis of BAP1 was performed on blood, or on retinal FFPE tissue from the histopathologic slides. NGS was performed using the Ion Torrent platform (Thermo Fisher Scientific). A panel covering exon 4 and 5 of GNAQ and GNA11, exon 1 and 2 of EIF1AX, exon 14 of SF3B1, and all exons of BAP1 was used as described before.39 The sequencing results were analyzed with Integrative Genomics Viewer (IGV; Broad Institute, Cambridge, MA, USA). A variant was considered when it occurred in at least 10% of the reads with a minimal read count of 50.
Copy Number Variation
Copy number variation analysis of chromosome 3 was performed with 21 amplicons covering highly polymorphic regions with a minor allele frequency of at least 45% as described previously.39 Scatter plots were used to display the frequency of variant coverage compared to total coverage. These data were extracted from the variant calling files.
Statistical Analysis
All analyses were performed with IBM SPPS Statistics Version 28.0.1.0 (IBM, Armonk, NY, USA). Kaplan-Meier plots and the log-rank test were used to compare survival between groups. P < 0.05 was considered significant.
Results
A total of 84 patients were enrolled, and specimens from 58 of them were collected from 12 ocular oncology centers to be analyzed at the Erasmus Medical Center (Rotterdam, the Netherlands), whereas samples from 26 French patients were analyzed at the Institute Curie (Paris, France). The samples that were sent to the Erasmus Medical Center were either blood, saliva, or FFPE tissue. Blood was collected from 12 patients, FFPE material was available for 36 patients, and saliva was used for analysis from nine patients. From one patient both blood and FFPE tissue were used. Thus 59 samples from 58 patients were analyzed at the Erasmus Medical Center. Fine-needle aspiration biopsy was performed on three patients (4%) before definitive treatment.
Clinical Characteristics
Our cohort consisted of 46 (55%) females and 37 (45%) males (data not available for one patient) with a mean age at diagnosis of 17 years (range 5.0–24.8; median = 18; Table 1, Fig. 1). The female/male ratio (1:0.8) was not different from 1 (P = 0.38, binomial test). The age at diagnosis was comparable between males and females (P = 0.53, independent samples t-test). The presence of extraocular extension and ciliary body involvement were comparable between genders (P = 0.95 and P = 0.71, respectively, Pearson's χ2 test).
Table 1.
Clinical and Histopathological Characteristics of Children and Young Adults With Primary Uveal Melanoma
| Characteristic | |
|---|---|
| Sex | |
| Male | 37 (45%) |
| Female | 46 (55%) |
| Mean age at diagnosis (y) | 17 (5.0–24.8) |
| Treatment* | |
| Surgery | 56 (67%) |
| Enucleation | 50 (60%) |
| Local resection | 6 (7%) |
| Endoresection† | 2 (2%) |
| Radiotherapy | 41 (49%) |
| Ruthenium | 12 (14%) |
| Iodine | 7 (8%) |
| Brachytherapy | 3 (4%) |
| Proton beam | 11 (13%) |
| External beam | 3 (4%) |
| Cyber knife | 3 (4%) |
| Gamma knife | 2 (2%) |
| Metastatic disease | |
| Yes | 12 (15%) |
| No | 68 (85%) |
| Mean metastasis free survival (y) | 9.0 (0.01–43.7) |
| Cell type | |
| Spindle cell | 17 (50%) |
| Epithelioid | 6 (18%) |
| Mixed | 11 (32%) |
| Ciliary body involvement | |
| Yes | 34 (41%) |
| No | 49 (59%) |
| Extraocular extension | |
| Yes | 7 (9%) |
| No | 75 (91%) |
| Mean largest basal tumor diameter (mm) | 12.7 (4.0–20.4) |
| Mean maximum tumor thickness (mm) | 8.1 (1.0–20.0) |
Combined treatment in some patients.
Enucleation as secondary treatment. The total amount of patients included in this study is not represented in all categories because of missing data.
Figure 1.

Histogram of age at diagnosis (left), and the treatment of children and young adults with primary uveal melanoma as a Venn diagram (right).
Histopathological Features
Most of the 34 UM with a known cell type were of spindle cell type (n = 17 [50%]). One-third consisted of mixed cell type (n = 11 [32%]), and six (18%) were epithelioid cell type. Ciliary body involvement was present histopathologically in 41% (34/83), and extraocular extension was described in seven (8.5%) of the 82 UM of which the data were available.
The median largest tumor basal diameter was 12.0 mm with a mean of 12.7 mm (range 4.0–20.4), and the median tumor thickness was 7.7 mm (mean = 8.1, range 1.0–20.0). The TNM anatomical category was described in 56 cases. The most common was T2a (n = 15) followed by T3a (n = 9) and T1a (n = 7). The highest TNM category was T4d, in one patient.
Treatment
Treatment data were available for 83 (99%) of patients and consisted of surgery, radiotherapy, or a combination of both (Table 2, Fig. 1). Surgery included enucleation in 50 patients (60%), of whom 10 received adjuvant treatment, endoresection in two patients followed by enucleation, local resection, and iridocyclectomy in one patient. Enucleation was also performed secondarily after primary treatment. Radiotherapy was performed in 41 patients (50%), of whom ruthenium brachytherapy was used most often, followed by proton beam therapy. Fourteen patients (17%) were treated with a combination of radiotherapy and surgery: ruthenium brachytherapy and resection (n = 4), external beam radiotherapy and enucleation (n = 3), proton beam therapy and enucleation (n = 2), proton beam therapy and endoresection (n = 1), cyber knife and enucleation (n = 1), ruthenium and enucleation (n = 1), and iodine and enucleation (n = 1). One patient was treated with iridocyclectomy, iodine radiotherapy, and enucleation. Extraocular extension was present in three patients who underwent combination therapy.
Table 2.
The Findings of GNAQ, GNA11, EIF1AX, SF3B1, BAP1 and Chromosome Status in Primary Uveal Melanoma Tumor Tissue and Germline of Children and Young Adults
| Tumor | Germline | |
|---|---|---|
| Gene | n = 12 | n = 26 |
| GNAQ | 626A>T, p.(Gln209Leu); 626A>C, p.(Gln209Pro); c.548G>A, p. (Arg183Gln) | None |
| GNA11 | c.626A>T, p.(Gln209Leu); c.626A>C, p.(Gln209Pro) | None |
| EIF1AX | None | None |
| SF3B1 | c.1874G>A, p.(Arg625His) | None |
| BAP1 | c.122+1G>A; c.38-1G>C; c.442G>T, p.(Glu148*); c.467del, p.(Gln156Argfs*31); .254del, p.(Gln85Argfs*2); c.2023_2037del, p.(Ile675_Ile679del) | c.1708C>G, p.(Leu570fs*40) |
| Chromosome 3 | n = 18 | n = 32 |
| Disomy | 9 (50.0%) | 32 (100%) |
| Loss of heterozygosity | 6 (33.3%) | 0 (0%) |
| Monosomy | 3 (16.7%) | 0 (0%) |
Metastases and Survival
Almost 80% of patients were alive at the time of data collection (n = 65 [77%]), and 15 patients had died (18%). The latest status of four patients (5%) was unknown. Of 12 patients who developed metastases, eight have died of them. One patient died of another cause, and the cause of death for six patients was reported as unknown and presumed to be UM. Two patients underwent liver surgery for metastatic disease. One patient developed another primary malignancy, an intestinal adenocarcinoma. We did not have enough data to confirm that females had metastases more frequently (22% vs. 6%; P = 0.058, Fisher's exact test).
Mean metastasis-free survival (MFS) was 9.0 years (range 0.01 to 43.7) and longer than the median MFS (5.5 years) because of a few very long survival times. Overall survival was comparable to MFS with a mean of 9.1 years and a median of 5.8 years (range 0.01–43.7). Males survived longer than females (P = 0.018, log-rank test, Fig. 2A). No overall difference in DFS among three age groups (0–17, 18–20, and 21–24 years; Fig. 2B) or when divided into younger and older than 18 (P = 0.56) was observed. No difference in DFS was confirmed by extraocular extension or ciliary body involvement (Figs. 2C, 2D).
Figure 2.

Kaplan-Meier estimates of disease-free survival in children and young adults with uveal melanoma. Metastasis-free survival in months according to (A) gender, (B) age at diagnosis, (C) ciliary body involvement, (D) extraocular extension, (E) BAP1 status of the tumor, and (F) chromosome 3 status of the tumor. LOH, loss of heterozygosity. P values from log-rank test. Ticks indicate censored observations.
When DFS was compared by gender among different age groups, a difference in the age group of 0–17 years was noted (Fig. 3). However, these subgroups are small and a lead time bias could not be excluded.
Figure 3.
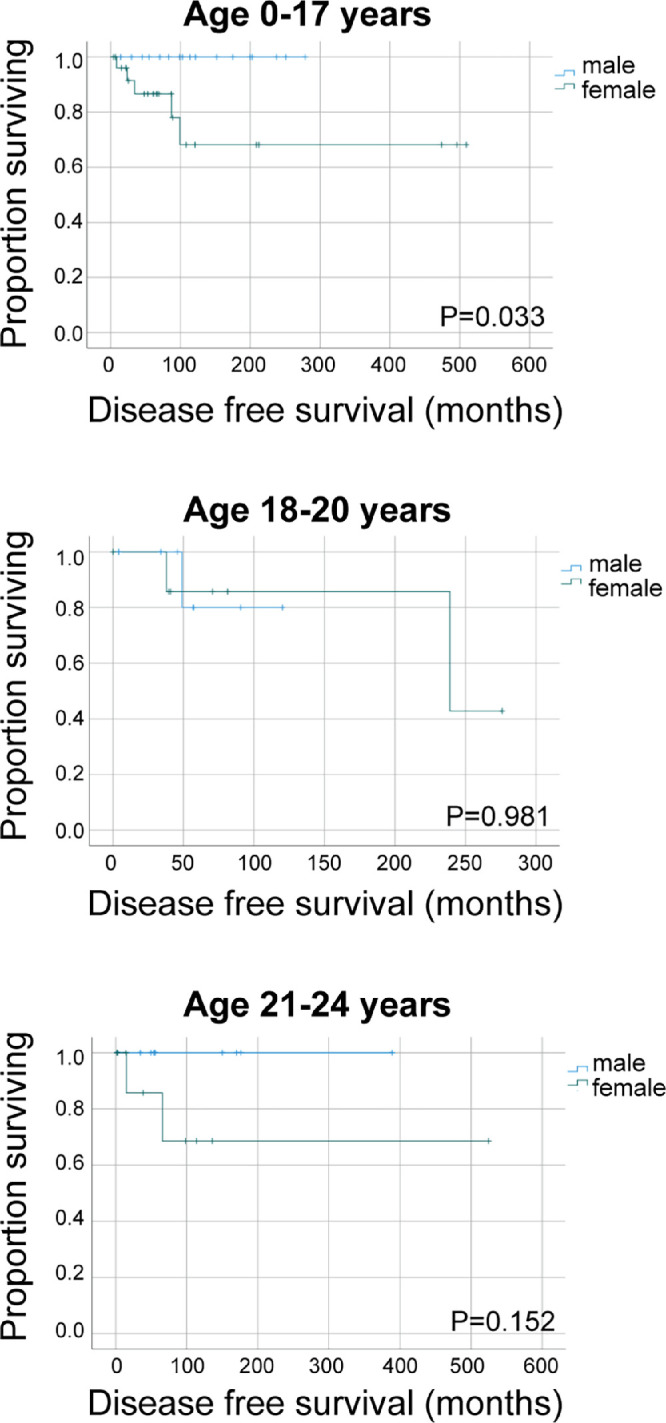
Kaplan-Meier estimates of disease-free survival in children and young adults with uveal melanoma. Survival is compared between males and females by age at diagnosis subgroups. P values from log-rank test are unadjusted. Ticks indicate censored observations.
BAP1 Immunohistochemistry
BAP1 IHC was performed on 31 sections (Fig. 4), none of them were obtained from fine-needle aspiration biopsy. No nuclear staining was observed in 13 (42%), interpreted as the presence of a BAP1 pathogenic variant. The other 18 UM showed preserved nuclear BAP1 immunoreaction (58%). These samples included six patients who underwent both surgical treatment, as well as radiotherapy, of which one sample (3%) did not show nuclear staining. The last-mentioned patient was treated with enucleation and external beam therapy because extraocular extension was present in this case. Metastatic disease developed in this patient.
Figure 4.

BAP1 immunohistochemistry of formalin-fixed paraffin-embedded tissue of uveal melanoma (magnification × 400). Lost (left) and preserved nuclear BAP1 immunoreactivity (right).
Genetic Analysis
NGS (Erasmus MC) was performed on DNA isolated from FFPE tissue of seven patients with PUM; none of them were irradiated. GNAQ/GNAQ11 somatic pathogenic variants were detected in four of them, BAP1 variants in three, and an SF3B1 variant was found in one tumor (Table 2). No EIF1AX pathogenic variants were identified. Genetic analysis performed at the Curie Institute revealed three BAP1 mutations (two wildtypes, of which one was irradiated before genomic testing), four GNAQ/GNA11 mutations and no SF3B1 nor EIF1AX mutations.
A reliable result of germline analysis of BAP1 was obtained from 12 blood samples, seven saliva samples, and eight FFPE samples. In one patient both blood and FFPE was tested, so 26 patients were tested for germline BAP1 analysis. The prevalence of the BAP1 germline pathogenic variants was 4% (1/26, 95% CI −0.04 to 0.13). The only germline c.1708C>G, p.(Leu570Val) pathogenic variant was detected in an 18-year-old female who underwent enucleation. This variant results in a frameshift with a stop codon after 40 amino acids. The BAP1 variant in this patient was described previously.40 No germline variants were detected in GNAQ, GNA11, SF3B1, or EIF1AX (Table 2).
The MFS of patients with a somatic BAP1 mutation (no BAP1 expression using IHC or mutation detected with sequencing) was shorter compared to the MFS of those with wild-type BAP1 (Fig. 2E).
Analysis of chromosome 3 was performed on 27 germline samples (blood, FFPE, or saliva), all with disomy 3. Disomy 3 in the tumor was shown in nine of 18 PUM (50%), three (17%) showed a loss of chromosome 3 (monosomy 3), and six (33%) showed a loss of heterozygosity (95% CI [0.37, 1.29]).
Tumors with disomy 3 developed metastasis less frequently than tumors with loss of heterozygosity (P = 0.027, not corrected for multiple pairwise comparisons). Log-rank testing across all groups did not show a difference in log rank test (Fig. 2F).
Discussion
In our collaborative study, were able to enroll 84 children and young adults with a PUM and genetic data from 13 centers. The number is reasonable given that this infrequent tumor is even more rare in children than in adults.15 In our series, 14% of patients developed metastatic disease by 15 years, which is roughly comparable to a previous study in which metastasis 15 years after diagnosis was described in 19% of patients 20 years old or younger at diagnosis of PUM.13 One patient developed metastatic disease even after 20 years in the aforementioned study. In another study, metastasis at 10 years occurred in 9% and at 20 years in 20% of children.11 The follow-up time in our cohort was relatively short with a median of 5.8 years.
In previous studies, females had PUM more frequently than males,10 although this was not significant in most studies,11,12,17 including the present one. We observed a longer DFS in young males compared to females (P = 0.018), indicating a more favorable prognosis in the former. This is in line with previous studies in which gender was an independent predictor of survival,10,17 of which one showed this to be the case, especially in children younger than 17 years.10
Of the genetic features analyzed, predictors for survival among children and young adults in our study was chromosome 3 status and presence of a BAP1 pathogenic variant in the tumor. The prognosis was better when both copies of chromosome 3 were present. This is in line with the previously reported series in which monosomy 3 was associated with worse survival.19,22 BAP1 pathogenic variants in the tumor were observed in 45% of the patients, and they had a shorter DFS (P = 0.004).
Not only do somatic BAP1 mutations occur in UM, but also germline pathogenic variants in BAP1 are described. Germline BAP1 variants are described in other studies to be present in about 2% of unselected patients, and in about 4% of patients who had a UM diagnosed before the age of 40 years.34,41 In another group of young patients with UM (age < 30 years) the incidence of BAP1 germline mutations was slightly higher (7%).42 We did not observe a higher percentage of germline BAP1 pathogenic variants in PUM, compared to the latter studies. This is in line with the observation that the age of diagnosis in adults is not lower for patients with an BAP1 germline pathogenic variant as compared to UM patients with a somatic BAP1 mutation.41 More research is needed to further evaluate the development of UM in children and young adults. This study did not shed light on the difference in biology because of no high prevalence of germline BAP1 mutations in this patient group.
In the BAP1 tumor predisposition syndrome, the age at diagnosis of cancer in general is associated with the type of BAP1 variant: it was lower in patients harboring a null variant compared to a missense variant. However, this difference was not observed specifically for UM and renal cell carcinoma.35 This suggests that the pathophysiology of BAP1 mutations in UM is different compared to some other BAP1-associated cancers. This could explain the not-higher-than average frequency of germline variants in BAP1 in PUM.
The main strength of our study is its collaborative nature with a relatively high number of patients with PUM. Its main but unavoidable weakness is its retrospective nature, resulting in variable practices as regards prognostic biopsy specimens and methods of prior genetic analysis across time and centers, for which reason full germline and somatic genetic data were not available from all patients. Moreover, the genetic analyses used in this study were performed in two centers, and only the tumors that had evidence of non-functional BAP1 were evaluated further for germline aberrations, which are caveats in our study. Tumors might have been incorrectly scored as BAP1 positive because of tumor heterogeneity which has been demonstrated for BAP1 in UM.43 This could be the case when an area with loss of nuclear BAP1 was not subject to evaluation of BAP1 IHC. This does not apply to the BAP1 negative cases; when there is nuclear BAP1 loss in an area, the tumor should be coded BAP1 negative.
It should be noted that the number of patients with ciliary body involvement is relatively high in this study. Moreover, present results do not apply to children younger than five years of age, and the pathogenesis of PUM remains incompletely known pending further research to elucidate all underlying genetic predisposing factors.
Acknowledgments
Supported by the KWF Dutch Cancer Society (Grant no. 6905).
Disclosure: N.M. van Poppelen, None; N. Cassoux, None; J.A. Turunen, None; N.C. Naus, None; R.M. Verdijk, None; J. Vaarwater, None; V. Cohen, None; V.P. Papastefanou, None; H. Kiratli, None; S.V. Saakyan, None; A.Y. Tsygankov, None; I. Rospond-Kubiak, None; H.S. Mudhar, None; S.M. Salvi, None; J.F. Kiilgaard, None; S. Heegaard, None; A.P. Moulin, None; M.A. Saornil, None; C. Garciá-Alvarez, None; M. Fili, None; N.A. Eide, None; P. Meyer, None; T.T. Kivelä, None; A. de Klein, None; E. Kilic, None; R.T. Al-Jamal, None
References
- 1. Spaeth EB. Ocular tumors; a study of incidence of the various types and their mortality rates. AMA Arch Ophthalmol. 1951; 46: 421–423. [PubMed] [Google Scholar]
- 2. Isager P, Osterlind A, Engholm G, et al.. Uveal and conjunctival malignant melanoma in Denmark, 1943-97: incidence and validation study. Ophthalmic Epidemiol. 2005; 12: 223–232. [DOI] [PubMed] [Google Scholar]
- 3. Singh AD, Bergman L, Seregard S.. Uveal melanoma: epidemiologic aspects. Ophthalmol Clin North Am. 2005; 18: 75–84, viii. [DOI] [PubMed] [Google Scholar]
- 4. Kivelä T. Incidence, prevalence and epidemiology of ocular melanoma. Ocular Melanoma: Advances in Diagnostic and Therapeutic Strategies. London: Future Medicine; 2014: 20–38. [Google Scholar]
- 5. Pukrushpan P, Tulvatana W, Pittayapongpat R.. Congenital uveal malignant melanoma. J AAPOS. 2014; 18: 199–201. [DOI] [PubMed] [Google Scholar]
- 6. Greer CH. Congenital melanoma of the anterior uvea. Arch Ophthalmol. 1966; 76: 77–78. [DOI] [PubMed] [Google Scholar]
- 7. Broadway D, Lang S, Harper J, et al.. Congenital malignant melanoma of the eye. Cancer. 1991; 67: 2642–2652. [DOI] [PubMed] [Google Scholar]
- 8. Posnick JC, Chen P, Zuker R, Greenberg ML, Becker LE, Phillips J.. Extensive malignant melanoma of the uvea in childhood: resection and immediate reconstruction with microsurgical and craniofacial techniques. Ann Plast Surg. 1993; 31: 265–270. [DOI] [PubMed] [Google Scholar]
- 9. Palazzi MA, Ober MD, Abreu HF, et al.. Congenital uveal malignant melanoma: a case report. Can J Ophthalmol. 2005; 40: 611–615. [DOI] [PubMed] [Google Scholar]
- 10. Al-Jamal RT, Kivela T. Uveal melanoma among Finnish children and young adults. J AAPOS. 2014; 18: 61–66. [DOI] [PubMed] [Google Scholar]
- 11. Shields CL, Kaliki S, Arepalli S, et al.. Uveal melanoma in children and teenagers. Saudi J Ophthalmol. 2013; 27: 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vavvas D, Kim I, Lane AM, Chaglassian A, Mukai S, Gragoudas E.. Posterior uveal melanoma in young patients treated with proton beam therapy. Retina. 2010; 30: 1267–1271. [DOI] [PubMed] [Google Scholar]
- 13. Petrovic A, Bergin C, Schalenbourg A, Goitein G, Zografos L.. Proton therapy for uveal melanoma in 43 juvenile patients: long-term results. Ophthalmology. 2014; 121: 898–904. [DOI] [PubMed] [Google Scholar]
- 14. Fry MV, Augsburger JJ, Correa ZM.. Clinical features, metastasis, and survival in patients younger than 21 years with posterior uveal melanoma. JAMA Ophthalmol. 2019; 137: 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shields CL, Kaliki S, Furuta M, Mashayekhi A, Shields JA.. Clinical spectrum and prognosis of uveal melanoma based on age at presentation in 8,033 cases. Retina. 2012; 32: 1363–1372. [DOI] [PubMed] [Google Scholar]
- 16. Liu YM, Li Y, Wei WB, Xu X, Jonas JB.. Clinical characteristics of 582 patients with uveal melanoma in China. PLoS One. 2015; 10(12): e0144562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Al-Jamal RT, Cassoux N, Desjardins L, et al.. The Pediatric Choroidal and Ciliary Body Melanoma Study: a survey by the European Ophthalmic Oncology Group. Ophthalmology. 2016; 123: 898–907. [DOI] [PubMed] [Google Scholar]
- 18. Kaliki S, Shields CL, Mashayekhi A, Ganesh A, Furuta M, Shields JA.. Influence of age on prognosis of young patients with uveal melanoma: a matched retrospective cohort study. Eur J Ophthalmol. 2013; 23: 208–216. [DOI] [PubMed] [Google Scholar]
- 19. Prescher G, Bornfeld N, Hirche H, Horsthemke B, Jockel KH, Becher R.. Prognostic implications of monosomy 3 in uveal melanoma. Lancet. 1996; 347(9010): 1222–1225. [DOI] [PubMed] [Google Scholar]
- 20. Prescher G, Bornfeld N, Becher R.. Nonrandom chromosomal abnormalities in primary uveal melanoma. J Natl Cancer Inst. 1990; 82: 1765–1769. [DOI] [PubMed] [Google Scholar]
- 21. Horsman DE, Sroka H, Rootman J, White VA.. Monosomy 3 and isochromosome 8q in a uveal melanoma. Cancer Genet Cytogenet. 1990; 45: 249–253. [DOI] [PubMed] [Google Scholar]
- 22. Sisley K, Rennie IG, Parsons MA, et al.. Abnormalities of chromosomes 3 and 8 in posterior uveal melanoma correlate with prognosis. Genes Chromosomes Cancer. 1997; 19: 22–28. [DOI] [PubMed] [Google Scholar]
- 23. Abdel-Rahman MH, Cebulla CM, Verma V, et al.. Monosomy 3 status of uveal melanoma metastases is associated with rapidly progressive tumors and short survival. Exp Eye Res. 2012; 100: 26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harbour JW, Onken MD, Roberson ED, et al.. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010; 330(6009): 1410–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yu H, Pak H, Hammond-Martel I, et al.. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc Natl Acad Sci USA. 2014; 111: 285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Daou S, Hammond-Martel I, Mashtalir N, et al.. The BAP1/ASXL2 histone H2A deubiquitinase complex regulates cell proliferation and is disrupted in Cancer. J Biol Chem. 2015; 290: 28643–28663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baughman JM, Rose CM, Kolumam G, et al.. NeuCode proteomics reveals Bap1 regulation of metabolism. Cell Rep. 2016; 16: 583–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zarrizi R, Menard JA, Belting M, Massoumi R.. Deubiquitination of gamma-tubulin by BAP1 prevents chromosome instability in breast cancer cells. Cancer Res. 2014; 74: 6499–6508. [DOI] [PubMed] [Google Scholar]
- 29. Figueiredo CR, Kalirai H, Sacco JJ, et al.. Loss of BAP1 expression is associated with an immunosuppressive microenvironment in uveal melanoma, with implications for immunotherapy development. J Pathol. 2020; 250: 420–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koopmans AE, Verdijk RM, Brouwer RW, et al.. Clinical significance of immunohistochemistry for detection of BAP1 mutations in uveal melanoma. Mod Pathol. 2014; 27: 1321–1330. [DOI] [PubMed] [Google Scholar]
- 31. Shah AA, Bourne TD, Murali R.. BAP1 protein loss by immunohistochemistry: a potentially useful tool for prognostic prediction in patients with uveal melanoma. Pathology. 2013; 45: 651–656. [DOI] [PubMed] [Google Scholar]
- 32. Kalirai H, Dodson A, Faqir S, Damato BE, Coupland SE.. Lack of BAP1 protein expression in uveal melanoma is associated with increased metastatic risk and has utility in routine prognostic testing. Br J Cancer. 2014; 111: 1373–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Szalai E, Wells JR, Ward L, Grossniklaus HE.. Uveal melanoma nuclear BRCA1-associated protein-1 immunoreactivity is an indicator of metastasis. Ophthalmology. 2018; 125: 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Repo P, Jarvinen RS, Jantti JE, et al.. Population-based analysis of BAP1 germline variations in patients with uveal melanoma. Hum Mol Genet. 2019; 28: 2415–2426. [DOI] [PubMed] [Google Scholar]
- 35. Walpole S, Pritchard AL, Cebulla CM, et al.. Comprehensive study of the clinical phenotype of germline BAP1 variant-carrying families worldwide. J Natl Cancer Inst. 2018; 110: 1328–13241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Abdel-Rahman MH, Pilarski R, Cebulla CM, et al.. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet. 2011; 48: 856–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Amin MB, Greene FL, Edge SB, et al.. The Eighth Edition AJCC Cancer Staging Manual: continuing to build a bridge from a population-based to a more "personalized" approach to cancer staging. CA Cancer J Clin. 2017; 67: 93–99. [DOI] [PubMed] [Google Scholar]
- 38. Kujala E, Damato B, Coupland SE, et al.. Staging of ciliary body and choroidal melanomas based on anatomic extent. J Clin Oncol. 2013; 31: 2825–2831. [DOI] [PubMed] [Google Scholar]
- 39. Smit KN, van Poppelen NM, Vaarwater J, et al.. Combined mutation and copy-number variation detection by targeted next-generation sequencing in uveal melanoma. Mod Pathol. 2018; 31: 763–771. [DOI] [PubMed] [Google Scholar]
- 40. Wadt K, Choi J, Chung JY, et al.. A cryptic BAP1 splice mutation in a family with uveal and cutaneous melanoma, and paraganglioma. Pigment Cell Melanoma Res. 2012; 25: 815–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ewens KG, Lalonde E, Richards-Yutz J, Shields CL, Ganguly A.. Comparison of germline versus somatic BAP1 mutations for risk of metastasis in uveal melanoma. BMC Cancer. 2018; 18: 1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cebulla CM, Binkley EM, Pilarski R, et al.. Analysis of BAP1 germline gene mutation in young uveal melanoma patients. Ophthalmic Genet. 2015; 36(2): 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stalhammar G, Grossniklaus HE.. intratumor heterogeneity in uveal melanoma BAP-1 expression. Cancers (Basel). 2021; 13: 1143. [DOI] [PMC free article] [PubMed] [Google Scholar]