

Abstract
Adeno-associated viral vectors (AAV) are frequently used by neuroscientists to deliver tools, such as biosensors and optogenetic and chemogenetic actuators, in vivo. Despite its widespread use, AAV vector characterization and quality control can vary between labs and viral vector cores leading to variable results and irreproducibility. This protocol describes some of the characterization and quality control assays necessary to confirm an AAV vector’s titer, genomic identity, serotype and purity.
Keywords: Adeno-associated viral vectors, quality control, rAAV, AAV, viral vectors
Introduction
Recombinant adeno-associated virus (rAAV) vectors are increasingly popular gene delivery tools for use in research and therapeutically. They are relatively easily produced in a well-equipped lab or production facility, but before in vivo use, rAAV vectors should be extensively characterized to ensure proper dosing, vector genome integrity and purity. The concentration of an rAAV vector is typically reported as a physical titer of the total viral particles present and does not take into account the presence of defective particles. Physical titer is frequently measured via quantitative polymerase chain reaction (qPCR) and reported as vector genomes per milliliter (VG/mL) or genome copies per milliliter (GC/mL) with values relative to a plasmid standard. Viral production facilities frequently design qPCR titration assays with primers and/or probes that target commonly occuring regions of the transfer plasmid such as promoters, polyadenylation signals, the Woodchuck Hepatitis Virus Posttranscriptional Regulatory Element (WPRE) or the inverted terminal repeats (ITRs) (1). By targeting common elements, once validated, the same assay can be used to titer multiple rAAV vectors in parallel. Production facilities using this method should validate the assay with an rAAV vector of known titer before use. Once validated, this rAAV reference virus should be included as a control in all titrations to confirm consistency across runs.
Recently, a droplet digital PCR-based titration (ddPCR) method was developed for rAAV vectors (2). Unlike qPCR, ddPCR does not rely on a standard curve and provides an absolute measurement. In contrast, qPCR results vary depending on the specific plasmid used and the overall quality of the standard preparation leading to variation across run, users and labs. In an analysis of replicate samples, ddPCR was determined to have a lower coefficient of variation and therefore a higher precision as compared to qPCR (3). ddPCR is also less prone to PCR inhibitions than qPCR. This resistance to PCR inhibition is thought to be due to the partitioning reaction that occurs in ddPCR (4). In ddPCR, a single droplet with minor to moderate PCR inhibition will still be read as positive whereas in a minor to moderately inhibited qPCR more cycles are required to reach a signal above the threshold causing underreported titers. Moreover, ddPCR is the superior titration method for self-complementary rAAV (scAAV) vector templates as it does not have the same self-annealing issues observed with qPCR. Previous studies have shown that the kinetics of ITR hairpin structure formation in the scAAV genome outcompetes those of primer and probe binding, impairing PCR efficiency and resulting in underreported titers (5)(6). Though ddPCR is gaining popularity, especially among viral vector cores, qPCR is still the most commonly used titration assay.
In addition to the physical titer, some users may wish to determine the specific infectivity of the vector. The specific infectivity is the ratio of physical-to-infectious particles and assesses the presence of defective viral particles. Physical titer is measured by qPCR or ddPCR as described above while a functional or infectious titer is determined using cell-based transduction assays. Stable cell lines are co-transduced with the rAAV vector and wild type adenovirus and transduced cells can be detected by a variety of methods such as flow cytometry or microscopy to detect fluorescent reporters, cellular staining to detect reporter enzyme expression such as β-galatcosidase, or PCR to detect of the gene of interest in target cells (7) (8). Of note, transduction efficiencies often vary between cell lines and tissues therefore the functional titer determined in a cell-based assay may not transfer precisely to an in vivo system. Specific infectivity is not routinely measured for rAAV vectors used in basic research but is critical in determining dosing regimens for clinical use in order to minimize adverse immune responses (8).
Production facilities or laboratories that produce a number of different rAAV vectors should confirm the identity and serotype of the vector. The identity of the vector can be easily confirmed via PCR of the genomic DNA with primers targeting critical regions such as promoter elements or the genes of interest. PCR products can be analyzed on a gel to confirm size or sent for Sanger sequencing for a more thorough analysis. For labs producing recombinase dependent vectors, it is recommended that they include additional reactions with primers inside and outside of the recombination sites in order to detect the presence of recombinase-independent recombination events. For a deeper analysis of vector genome integrity, labs may consider next generation sequencing (NGS). In addition to sequence confirmation of the vector’s genome, NGS has been used to detect contaminants, confirm serotype, and measure recombination rates in rAAV vectors (9) (10) (11) (12).
The serotype of an rAAV vector will determine its tropism, or tissue specificity. It is therefore critical that production facilities routinely working with a number of different capsids confirm that the correct capsid was used for each vector. Serotype confirmation can be done in a number of ways. Historically, western blotting and mass spectrophotometry were used, but these methods have some limitations. Western blotting relies on antibodies that tend to cross-react due to the high homology between some serotypes, while mass spectrophotometry is capsid specific but costly and labor intensive.
One of the simplest methods for serotype confirmation is AAV-ID, a thermostability-based approach that uses melting temperature to differentiate between serotypes (13). AAV-ID is fast and can be easily incorporated into most laboratories as it does not require specialized reagents or equipment. Of note, some serotypes naturally have similar melting temperatures and cannot be easily distinguished using this method. Moreover, the formulation buffer and purity of the vector will also affect the melting temperature. Recently, open-access Python software was developed to determine serotype from NGS data (9). The software takes advantage of the small amount of capsid plasmid DNA naturally packaged into the viral vector and probes NGS reads for serotype-specific, user-defined, seed sequence. This software is open-access and available at GitHub in Addgene’s Open Toolkit https://addgene.github.io/openbio/.
Much like specific infectivity, rAAV vector purity affects transduction efficiency, but in a serotype and tissue-specific manner (14). Common impurities include packaging cell proteins and residual plasmid or host cell DNA and can be routinely detected by most labs. Overall DNA impurities can be estimated using spectrophotometry by subtracting the known OD260 and OD280 contributed by the vector from the overall OD260 and OD280 values (14). To measure specific DNA contamination, qPCR with primers and probes targeting specific genes or genetic elements of interest from the plasmid or host cells can be used.
Protein impurities are easily observed following SDS-PAGE and protein staining of the rAAV vector. In a pure rAAV vector, only 3 bands corresponding to the VP1, VP2 and VP3 capsid proteins should be visible. Additional bands in the stain suggest packaging cell protein contamination during vector purification.
The presence of endotoxins in viral vector preparations can also impact downstream applications. Endotoxins arise from the lipid A portion of bacterial lipopolysaccharides and can be introduced to the viral vector through low quality plasmid DNA or bacterial contamination. The Limulus polyphemus amebocyte lysate turbidimetric assay detects endotoxin using a reagent derived from horseshoe crab blood that coagulates upon exposure to endotoxins. Even low activities of endotoxin can elicit an immune response underscoring the need for endotoxin screening before rAAV vector use in vivo (15).
rAAV vector contamination by microorganisms such as bacteria, yeast or fungi should also be ruled out. To confirm sterility, production facilities can inoculate microbial growth media such as Fluid Thioglycollate Medium or Soybean-Casein Digest Medium with the rAAV vector, and check for microbial growth after 2 weeks of cultivation. Production facilities may also consider checking vectors for mycoplasma contamination. Mycoplasma are common tissue culture contaminants and can be difficult to detect during routine cell culture work. There are several methods to detect mycoplasma including PCR based assays targeting the 16S rRNA, luciferase-based assays that detect mycoplasma-specific enzymes and mycoplasma-specific indicator cell lines (16) (17) (18). Mycoplasma contamination of producer cell lines can be difficult to treat, and it is recommended that viral production facilities routinely screen their cell lines before rAAV vector production.
The following protocol outlines some of the more widely used methods of characterizing rAAV vectors and assessing their purity and sterility (Figure 1). Many of the methods described can be modified for other types of vectors such as lentivirus, or adenovirus (19). The specific methods chosen do not require specialized lab equipment and can be easily applied to most laboratories and core facilities. They do not however encompass an exhaustive list and production facilities may prefer additional or alternative tests to meet their specific needs. It is worth noting that efficiency of rAAV mediated in vivo transduction depends on numerous factors, including animal species, strain, age, viral delivery route and targeting cell types. Differences in construct design, transgene of interest and vector serotype also make it difficult to directly predict an rAAV’s performance in vivo. In addition to the assays described in this paper, for a specific experimental paradigm, we recommend rAAV users design preliminary in vivo dosing experiments to determine the optimal dose before they begin any large-scale animal experiments.
Figure 1:
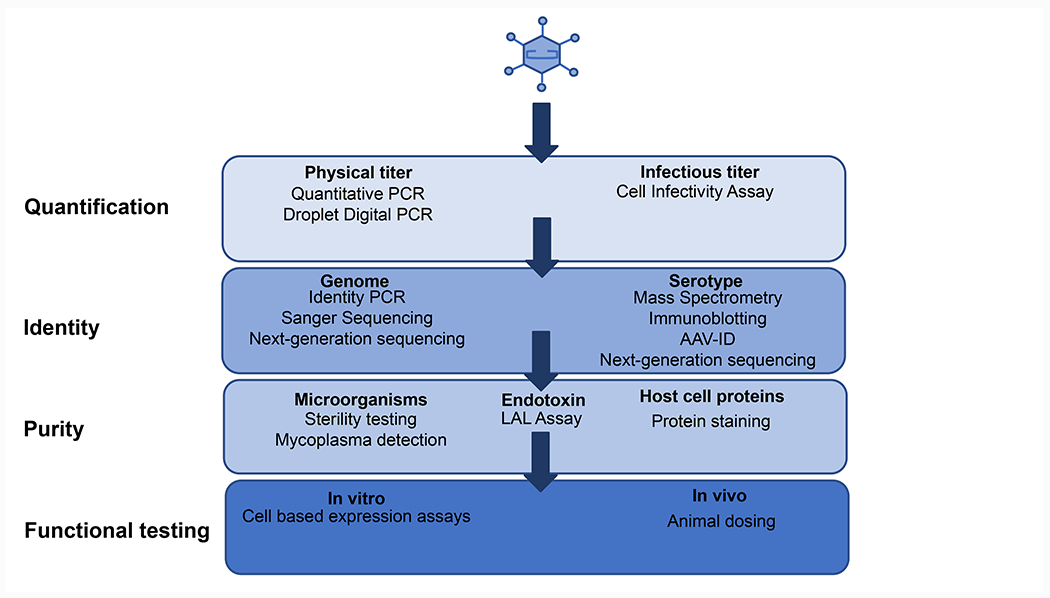
Overview of the rAAV quality control workflow and assays. rAAV quality control consists of 4 general categories, quantification, identity confirmation, purity and functional testing. A number of assays are possible for each category and choice of the assay will depend on the specific needs of the lab.
Materials
Titration by quantitative PCR
PowerUp SYBR Green Master Mix or alternative universal SYBR mastermix containing a high-quality DNA polymerase and a blend of dTTP/dUTP, protect from light (Thermo Fisher Scientific, A25776)
Plasmid for standard curve (containing ITRs or gene of interest)
Yeast tRNA (Thermo Fisher Scientific, AM7119)
100 μM forward and reverse primer (targeting ITRs or gene of interest)
AAV reference vector preparation, avoid multiple freeze-thaws, prepare individual aliquots for one time use
RNase-free DNase, keep on ice (Agilent Technologies, ST600031)
DNase buffer (10x) (Thermo Fisher Scientific, B43)
Nuclease-free water (Thermo Fisher Scientific, R0582)
Microcentrifuge tubes (Neptune, 3745.X)
96 well optical plate (Bio-Rad, HSP9601)
96 well plate for dilutions (Bio-Rad, MLL9601)
Plate sealing, adhesive film (Bio-Rad, MSB1001)
Reagent reservoirs (VWR, 89094-662)
Confirmation of packaged AAV genome by identity PCR
Platinum Hot Start PCR 2x Master Mix, thaw on ice (Thermo Fisher Scientific 13000013)
10 μM forward and reverse primers (IDT)
Nuclease-free water (Thermo Fisher Scientific, R0582)
PCR tubes (VWR, 89096-722)
Microcentrifuge tubes (Neptune, 3745.X)
Phosphate-Buffered Saline, 1X Without Calcium and Magnesium (PBS) (Corning, 21-040-CV)
Agarose (Bio-Rad, 1613101)
Gel loading dye (New England Biolabs, J62157)
Serotype determination via melting temperature (AAV-ID)
96 well-plate (Bio-Rad, HSP9601)
Microseal (Bio-Rad, MSB1001)
SYPRO Orange 5000x, protect from light (Thermo Fisher, S6651)
Phosphate-Buffered Saline, 1X with calcium and magnesium (Corning, 21-030-CV)
Lysozyme solution, 0.25 mg/mL (Sigma Aldrich, L6876)
Vector Purity by SDS-PAGE and Silver Staining
4-12% NuPage Novex bis-tris mini gel, 1mm thick, 10-well (Thermo Fisher Scientific, NP0321BOX)
20x MOPS SDS running buffer (Thermo Fisher Scientific, NP0001)
4x NuPage sample buffer (Thermo Fisher Scientific, NP0007)
10x NuPage sample reducing agent (Thermo Fisher Scientific, NP0009)
PageRuler Plus Prestained protein ladder, optional (Thermo Fisher Scientific, 26619)
Glacial acetic acid, corrosive to the skin and eyes, handle with extreme care (VWR, BDH3092-500mL)
Methanol, highly flammable and toxic, handle in a fume hood (VWR, BDH1135-1LP)
Microcentrifuge tubes (Neptune, 3745.X)
Gel loading tips (Neptune, 2016)
250 mL sterile bottles (Corning, 430281)
- SilverXpress Silver Staining Kit, some contents are corrosive, handle with extreme care (Thermo Fisher Scientific, LC6100)
- Solutions for Silver Staining (prepared freshly, according to kit instructions)
- Fixing solution: 90 mL deionized water + 100 mL methanol + 20 mL glacial acetic acid
- Sensitizing solution: 105 mL deionized water + 100 mL methanol + 5 mL sensitizer (component of SilverXPress kit)
- Staining solution: 5 mL stainer A (component of SilverXPress kit) + 5 mL stainer B (component of SilverXPress kit) + 90 mL deionized water
- Developing solution: 5 mL developer (component of SilverXPress kit) + 95 mL deionized water
- Stopper solution: 5 mL stopper solution (component of SilverXPress kit) directly added to staining solution when protein bands show sufficient intensity.
ImageJ or alternative imaging software
Limulus Amebocyte Lysate Chromogenic Endotoxin Test
Pierce Chromogenic Endotoxin Quant Kit (Thermo Fisher Scientific, A39552)
- Stop reagent
- Glacial acetic acid, corrosive to the skin and eyes, handle with extreme care (VWR, BDH3092-500mL): 25% v/v glacial acetic acid in water
Disposable sterile microplates (Corning, 9018)
Reagent reservoirs (VWR, 89094-662)
Disposable, endotoxin-free glass dilution tubes (Lonza, N207)
In vitro Sterility and Expression Assay
Media, e.g. DMEM high glucose (Corning, 10-013-CV)
HI-FBS (Seradigm, 89510-196)
GlutaGRO, Liquid 200 mM Solution (with 8.5 g/L Nacl) (Corning, 25-015-CI)
Flat-bottom 96-well tissue culture treated plate (Corning, 3596)
15 mL conical tubes (VWR, 21008-216)
Microcentrifuge tubes (Neptune, 3745.X)
0.4% Trypan blue Solution (Thermo Fisher Scientific, T10282)
Countess Cell Counting Chamber Slides (Thermo Fisher Scientific, C10228) (alternatively can use a hemocytometer)
Methods
Titration by quantitative PCR
A qPCR titration assay with primers and/or probes targeting regions of the transfer plasmid or the ITRs and SYBR green detection is commonly used to determine the physical titer of rAAV preparations. All values are calculated relative to a plasmid standard.
- Prepare plasmid standards for quantitative PCR titration.
- Use purified plasmid DNA that contains the ITR sequence or your sequence of interest that you can target.
- Linearize your selected plasmid by restriction digest. Verify your digest by running a sample on a 1% agarose gel.
- Calculate the plasmid concentration in molecules/μL. Concentration (molecules/μL) = (DNA conc (μg/μL) * 10E-06 gram / MW (g/mol)) * 6.023E+23 molecules/mole
- Prepare plasmid DNA standard by diluting your linearized plasmid to 2E+09 molecules/μL in nuclease-free water with 4μg/mL carrier RNA for stabilization. Store in small aliquots at −20°C.
- Use one aliquot to prepare 10-fold serial dilutions that range from 2E+02 to 2E+08 molecules/μL for your qPCR standard curve for each qPCR assay.
- Titration of viral vector genome titer by qPCR
- Prepare seven 10-fold serial dilutions of plasmid standard stock (see 1.d.)
- Thaw rAAV preparation of interest and rAAV reference vector preparation and keep on ice.
- Digest all purified rAAV samples (including the rAAV reference sample) with DNase to eliminate any contaminating plasmid DNA carried over from the production process. Set up digests by mixing 5 μL vector sample with 39 μL deionized water, 5 μL 10x DNase buffer and 1 μL DNase.
- Gently mix and incubate for 30 min at 37°C.
- Heat inactivate for 15 min at 75°C
- Dilute DNase treated rAAV sample and rAAV reference sample according to the dilution scheme in Table 1
- If your sample is expected to have a titer <1E+12 GC/mL, use dilutions 3-6 to load onto your qPCR plate
- If your sample is expected to have a titer >3E+13 GC/mL, use dilutions 4-7 to load onto your qPCR plate
- Prepare a master mix of 10 μL 2x Universal SYBR Master Mix, 0.15 μL 100uM forward primer, 0.15 μL 100uM reverse primer and 4.7 μL of nuclease-free water per reaction. Mix well.
- Set up and load the 96-well plate (Table 2)
- Load 5 μL of each standard in duplicate
- Load 5 μL of each sample in duplicate.
- Include a no template control (NTC = master mix + water).
- Add 15 μL of master mix per well and mix well by pipetting back and forth.
- Seal your 96-well plate with adhesive film.
- Centrifuge plate at 3,000 rpm for 2 min.
- Run your qPCR instrument with the following protocol using SYBR detection: 98°C - 3 min / 98°C - 15 sec / 58°C - 30 sec / read plate/ repeat 39x from step 3 / melt curve
- qPCR data analysis
- Perform data analysis using your instrument’s software. Determine the physical titer of your sample based on your standard curve and your sample dilutions.
- Set the program settings to an appropriate baseline to ensure that small amounts of background signal detected in initial PCR cycles will be removed.
- Make sure that a single peak is seen in your melt curve analysis (Figure 2C). The existence of a second peak could indicate the presence of primer dimers and impact the Ct values of your samples.
- Exclude any duplicates with a difference >0.5 in Ct value.
- Ensure that the Ct values for your NTCs are higher than any plasmid standard or sample Ct values on the plate.
- You should observe differences in Ct values that make sense for your dilutions (~3.3 difference Ct for a 10-fold dilution is appropriate).
Table 1:
Dilution series for AAV titration by qPCR.
| Dilution series | Sample volume (μl) | Volume of nuclease free water (μl) | Dilution factor | Total dilution |
|---|---|---|---|---|
| Dilution 1 | 5μl AAV stock | 45μl | 10x | 10x |
| Dilution 2 | 5μl dilution 1 | 95μl | 20x | 200x |
| Dilution 3 | 20μl dilution 2 | 80μl | 5x | 1000x |
|
| ||||
| Dilution 4 | 20μl dilution 3 | 80μl | 5x | 5000x |
| Dilution 5 | 20μl dilution 4 | 80μl | 5x | 25000x |
| Dilution 6 | 20μl dilution 5 | 80μl | 5x | 125000x |
| Dilution 7 | 20μl dilution 6 | 80μl | 5x | 625000x |
Table 2:
Example for qPCR plate set up. Plasmid standards are loaded in duplicate. The 10-fold diluted plasmid standard ranges from 2E+02 to 2E+08 molecules/μL. For each standard, 5μL are loaded for a range of 1E+03 to 1E+09 total molecules. For the AAV reference sample and all other AAV samples, 4 dilutions each are loaded in duplicate. A no template control (NTC) is included on the plate.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
|---|---|---|---|---|---|---|---|---|
| A | 1.00E+09 | 1.00E+08 | 1.00E+07 | 1.00E+06 | 1.00E+05 | 1.00E+04 | 1.00E+03 | NTC |
| B | ||||||||
| C | AAV reference | Sample 3 | ||||||
| D | ||||||||
| E | Sample 1 | Sample 4 | ||||||
| F | ||||||||
| G | Sample 2 | Sample 5 | ||||||
| H | ||||||||
Figure 2:
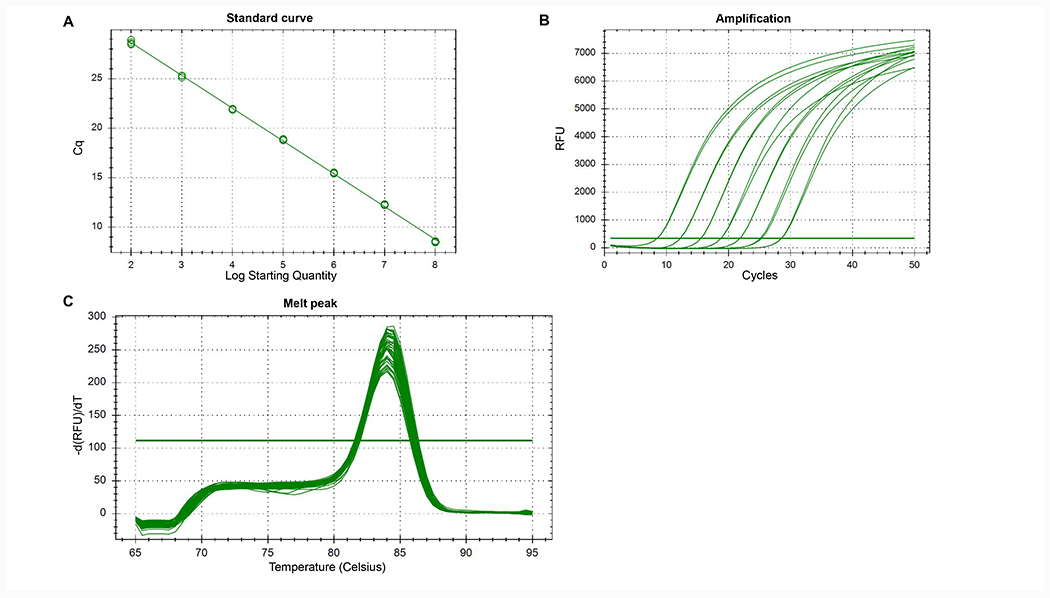
(A) Example of plasmid standard curve with R2=0.999, E=100.3%, slope=−3.316 and y-int=35.280. (B) Example of amplification plots obtained from a plasmid standard curve. Each curve represents a standard dilution. (C) Example of melt curve analysis with a single peak.
Confirmation of packaged AAV genome by identity PCR
Amplification of the packaged genomic DNA with primers targeting the promoter region or gene of interest can be used to confirm the identity of the vector.
- Start by selecting a suitable primer pair for your construct of interest.
- For non-FLEx/DIO constructs, target the area between the ITRs, including the fluorescent tag, with a forward and reverse primer pair of your choice (fwd1 + rev1) (Figure 3A). Select at least one primer pair for confirmation by PCR.
- For FLEx/DIO constructs, target the area between the loxP/lox2272 sites with one primer pair of your choice (fwd2 + rev2) (Figure 3B). In addition, select a primer pair that spans across the loxP/lox2272 sites, with forward and reverse primer pointing towards each other (fwd3 + rev2). As a control, select a third primer pair that again spans across the loxP/lox2272 sites, but with both primers pointing in the same direction (fwd3 + fwd2).
- Dilute a small sample of your rAAV preparation in PBS.
- For rAAV preparations with a titer between 1E+12 and 1E+13 GC/mL, prepare a 10-fold dilution by diluting 5 μL of your rAAV in 45 μL of PBS.
- For rAAV preparations with a titer higher than 1E+13 GC/mL, prepare a 25-fold dilution by diluting 4 μL of your rAAV in 96 μL of PBS.
Thaw PCR reagents on ice.
Prepare your master mix. Calculate the amount of reactions needed and prepare a master mix for each primer pair according to polymerase manufacturer instructions. Make sure to include one NTC for each primer pair.
- Set up PCR reactions
- Load 24 μL of master mix in appropriate wells.
- Load 1 μL of diluted rAAV sample in one of the reaction tubes with your master mix.
- For your NTC, load 1 μL of nuclease-free water in the other reaction tube with your master mix.
Run PCR protocol according to manufacturer instructions.
- To analyze the result of your PCR, mix 10 μL of each PCR reaction with loading dye and run on a 1.2% agarose gel with an appropriate ladder
- For non-FLEx/DIO constructs, ensure that your product runs at the expected size
- For FLEx/DIO constructs, ensure that your product from the primer pair targeting the region within the loxP/lox2272 sites and the primer pair spanning across the loxP/lox2272 sites, runs at the expected size. Confirm that the control reaction with the primers pointing in the same direction, does not show any bands on the gel. If unexpected bands show up in this reaction, you could be having issues with a PCR product due to recombination.
Figure 3:
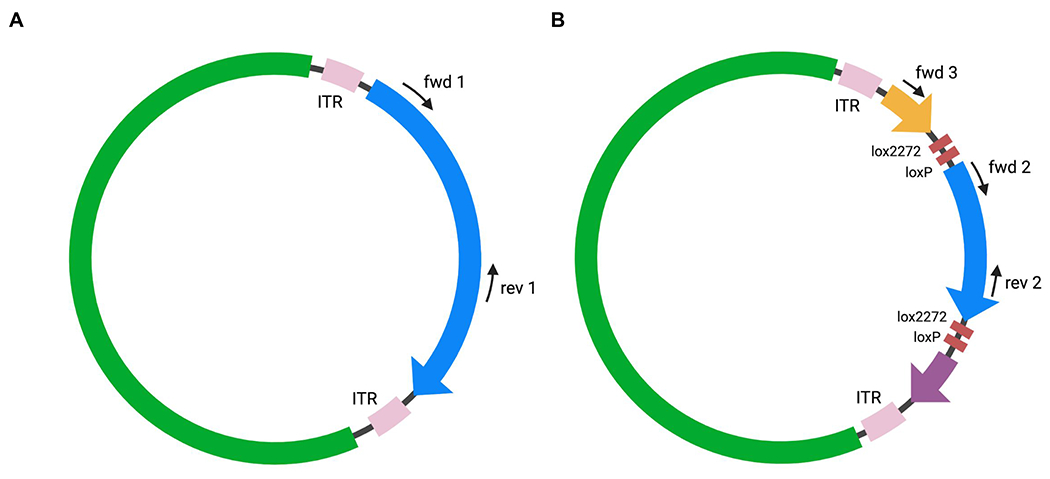
Plasmid with selected primer pairs. (A) For an identity PCR for a non-FLEx/DIO construct, choose forward (fwd) and reverse (rev) primers that target your insert between the ITRs. (B) For an identity PCR for a FLEx/DIO construct, choose one primer pair that targets your insert area (fwd2 + rev2), one primer pair with one primer being outside of and one in between the lox-sites (fwd3 + rev2) and one control primer pair (fwd3 + fwd 2).
Serotype determination via melting temperature (AAV-ID) - protocol adapted from Pacouret et al. 2017 (13)
AAV-ID is a thermostability-based approach that allows distinguishing between different AAV serotypes based on melting temperature.
- Prepare a 50x solution of SYPRO Orange solution as follows:
- Dilute 5 μL of SYPRO Orange 5000x into 495 μL Phosphate-Buffered Saline, 1X with calcium and magnesium.
Aliquot 45 μL of purified rAAV into a 96-well plate.
Aliquot 45 μL of 0.25 mg/mL Lysozyme solution into the 96-well plate as a positive control.
Add 5 μL of the 50x solution of SYPRO Orange to each well.
Pipet up and down 10-20x to mix.
Seal the plate with microseal film.
Centrifuge the plate for 2 minutes at 2000 rpm.
- Run the following PCR parameters:
- Ramp: 25°C to 99°C for 2 minutes
- Step and hold mode with 0.4°C increment (equivalent to ~0.2°C/min)
- Reporter: Rox
- Quencher: None
- Analyze the data as follows:
- Plot fluorescence as a function of temperature.
- If necessary, normalize the signal between 0 and 100% as follows:
- Step 1: S = 5 - min(S)
- Step 2: S = S/max(S)/100
- Calculate and plot the deterative fluorescence signal to get the melting temperature.
- dF/dT(T) = (F(T+ΔT) − F(T))/ΔT
Vector Purity by SDS-PAGE and Silver Staining
Potential residual protein impurities in rAAV preparations, e.g. from cellular proteins, can be detected by subjecting the sample to SDS-PAGE followed by a silver staining procedure. The presence of AAV capsid proteins VP1, VP2 and VP3 in correct stoichiometry and the absence of unwanted protein contaminants can be confirmed.
- rAAV sample preparation:
- Prepare a 10-fold dilution of your rAAV vectors if your titer is >5E+12 GC/mL. Dilute 2 μL of rAAV in 18 μL of PBS.
- Calculate the amount needed for 2E+10 vector particles and aliquot that amount into a fresh microcentrifuge tube. Add PBS to 13 μL.
- Add 5 μL of 4x sample buffer to each sample.
- Add 2 μL of 10x reducing agent to each sample.
- Spin the sample for 1 min at 10,000 x g in a microcentrifuge.
- Heat the sample for 5 min at 95°C in a heat block.
- Spin the sample for 1 min at 10,000 x g in a microcentrifuge and place on ice while preparing your gel.
- Preparation of SDS-PAGE and gel loading
- Prepare 1x MOPS buffer by diluting 25 mL of 20x MOPS buffer in 475 mL of deionized water. Gently invert to mix.
- Prepare the NuPage Novex bis-tris mini gel, place the gel in the electrophoresis chamber, and secure. Check the manufacturer’s instructions on the correct orientation of the gel.
- Fill the electrophoresis chamber with 1x MOPS running buffer.
- Rinse each well with 200 μL 1x MOPS running buffer.
- Optional: Load 5 μL of prestained protein ladder in the appropriate well.
- Load 20 μL of each prepared rAAV sample in the appropriate wells.
- Cover the electrophoresis chamber and attach to a power supply.
- Run the gel at constant voltage (~150V) until the dye from the loading buffer reaches the bottom of your gel (~1 hour).
- Turn off the power supply and remove the gel from the electrophoresis chamber.
- Optional: Use a razor blade and cut the wells on top of the gel and the bottom of the gel where dye is still visible.
- Silver staining procedure:
- Place the gel in a container and rinse for 5 minutes with deionized water while gently shaking.
- Follow the staining procedure according to manufacturer instructions.
- Fix gel in 200 mL fixing solution and shake gently for 10 min.
- Decant fixing solution and incubate gel in 100 mL sensitizing solution. Shake gently for 30 min.
- Decant solution and repeat sensitizing step with fresh sensitizing solution.
- Decant sensitizing solution and rinse gel with 200 mL deionized water while gently shaking for 10 min. Repeat wash step.
- Decant deionized water and stain gel with 100 mL staining solution for 15 min while gently shaking.
- Decant staining solution and rinse gel with 200 mL deionized water for 5 min while gently shaking. Repeat wash step.
- Develop gel with 100 mL developing solution while gently shaking. Observe the staining process and stop staining when reaching your desired band intensity for VP1, 2 and 3 by adding 5 mL stopper solution directly into the staining solution. Depending on purity of the vector, protein impurities may or may not be visible. Incubate for 10 minutes.
- Decant stopping solution and rinse the gel with 200 mL dH2O for 10 min while gently shaking. Repeat wash step two more times.
- Dry gel or image directly with a gel imager under white light (Figure 4).
- Use ImageJ or a similar photo software to determine the relative intensity of the capsid bands to the overall lane.
- Import the gel image into ImageJ.
- Select File.
- Select Open.
- Choose the location of the file to open.
- Change the image type to 8-bit.
- Select Image.
- Select Type.
- Choose 8-bit.
- Determine each lane of the gel as follows:
- Using the box tool, draw a box around the entire first gel lane.
- Select Analyze.
- Select Gels.
- Select First Lane.
- Drag the box to the next lane.
- Select Analyze.
- Select Gels.
- Select Next Lane.
- Repeat for all lanes.
- Plot the area under the curves for the protein bands as follows:
- Select Analyze.
- Select Gels.
- Select Plot Lanes.
- One graph per lane will appear with peaks representing each protein band.
- Use the line tool to connect the bottom of each peak.
- Select the wand tool.
- Fill in each peak by selecting the center of the peak.
- Export results as a csv file.
- In the results table select File and Save As.
- Determine the relative abundance of the VP proteins as compared to the entire lane using the integrated density (ID) as follows:
- ((ID VP1 + ID VP2 + ID VP3)/(Total ID of Lane))x100
- Bands for VP1:VP2:VP3 should be visible in a ratio of 1:1:10.
- VP protein bands should account for >90% of the protein abundance in the lane.
Figure 4:
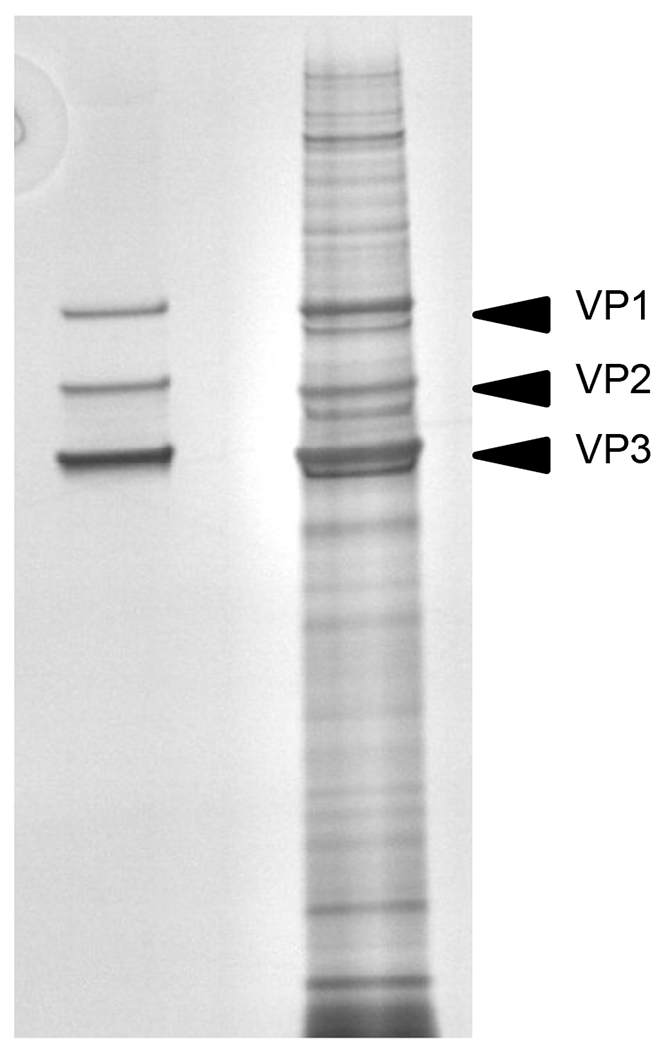
Silver staining of purified (left) and non-purified rAAV (right).
Limulus Amebocyte Lysate Chromogenic Endotoxin Test
An LAL chromogenic endotoxin test is used to detect bacterial endotoxins in rAAV preparations. Endotoxin is a lipopolysaccharide (LPS) molecule that can be found in the cell membrane of gram negative bacteria. Since the presence of endotoxin in rAAV preparations can cause an immune response when injected in an animal model, it is crucial to confirm no endotoxin contamination occurred during vector production.
- Follow all steps of the procedure according to manufacturer instructions.
- Allow all reagents to warm up to room temperature before use.
- Reconstitute endotoxin standard in endotoxin-free water at 10 EU/ml.
- Vortex the solution for at least 15 minutes.
- Prepare desired set of standards from reconstituted endotoxin stock solution by preparing an initial dilution and at least four two-fold serial dilutions in endotoxin-free glass vials, e.g. 0.01-0.1 EU/ml or 0.1-1 EU/ml.
- Vortex each dilution for 1-2 minutes before proceeding.
- Reconstitute lyophilized limulus amebocyte lysate (LAL) reagent in endotoxin-free water. Do not vortex. Avoid foaming.
- Reconstitute lyophilized chromogenic substrate in endotoxin-free water.
- Pre-warm a sufficient amount of substrate to 37°C for 5-10 minutes before use.
- Pre-equilibrate a microplate in a heat block at 37°C. Maintain the plate at 37°C throughout the procedure.
- In your assay, include the two-fold serial dilutions of endotoxin as a standard, dilutions of the sample of interest and endotoxin-free water as a negative control.
- Transfer 50 μL of each standard, rAAV sample or endotoxin-free water into the appropriate wells while carefully avoiding contaminations.
- Add 50 μL of LAL reagent solution to each well.
- Start your timer immediately once the LAL reagent solution is added to the first well. The appropriate time is indicated on the lysate vial.
- Take the microplate out of the heat block and gently tap the side of the plate to mix.
- Return the plate to the heat block and cover.
- After the appropriate time indicated on lysate vial, add 100 μL pre-warmed chromogenic substrate solution per well.
- Take the microplate out of the heat block and gently tap the side of the plate to mix.
- Return the plate to the heat block and cover.
- After 6 minutes, add 100 μL stop solution per well.
- Take the microplate out of the heat block adapter and gently tap the side of the plate to mix.
- Place the microplate in the plate reader and read the absorbance of each well at 405-410nm.
- Assay analysis:
- Analyze data graphically according to kit manufacturer instructions by plotting the mean absorbance and correlating endotoxin concentrations of your standards into a graph and drawing a line for your standard curve. Determine the endotoxin concentration for each rAAV sample based on their absorbance using linear regression.
- Alternatively, the data can be analyzed using a spreadsheet or calculator, by calculating the mean absorbance for each of the four standards and determining the corresponding endotoxin concentration for each sample by linear regression.
In vitro Sterility and Expression Assay
An in vitro assay in which small amounts of a rAAV preparation are used to transduce a target cell line. This assay can confirm in vitro expression of the expected fluorophore in fluorescently-labeled rAAV and allow detection of any potential bacterial, fungal or yeast contamination.
Determine the appropriate cell line for transgene expression based on the promoter in your construct (Table 3).
Warm up media.
Prepare a tissue culture treated 96-well plate in a biosafety cabinet.
- Determine the number of wells required for testing your rAAV sample.
- If the vector is not recombinase-dependent:
- Plan to transduce duplicate wells with your vector.
- Leave two wells untransduced as a negative control.
- If the vector is recombinase-dependent:
- Plan to transduce duplicate wells with the recombinase-dependent vector only.
- Plan to transduce duplicate wells with recombinase-expressing rAAV only.
- Plan to transduce duplicate wells with both the recombinase-dependent vector and the recombinase-expressing vector.
- Leave two wells untransduced as a negative control.
- If the vector is expressing a recombinase:
- Plan to transduce duplicate wells with the recombinase-expressing vector only.
- Plan to transduce duplicate wells with a recombinase-dependent rAAV only.
- Plan to transduce duplicate wells with both the recombinase-expressing vector and the recombinase-dependent vector.
- Leave two wells untransduced as a negative control.
- Prepare a cell stock solution to seed an appropriate amount of cells per well in 200 μL media each.
- For example, for HEK293T cells seed 5,000 cells per well in 200 μL DMEM. For N number of wells add (N+1) x 5,000 cells to (N+1) x 200 μL DMEM high glucose supplemented with 10% fetal bovine serum (D10) in a 15 mL conical tube. Mix gently.
Seed cell solution in appropriate wells on the 96-well plate.
Add 1 μL of rAAV sample to the appropriate wells.
Gently mix cells and rAAV by pipetting.
Return plate to incubator.
- Sterility and expression testing analysis:
- Look at each well under 20x magnification.
- After 24-48h, check for presence or absence of bacterial or fungal contamination.
- After 24-120h, depending on cell line and promoter, check the expression levels of your fluorescent tag.
- Check all fluorescent filters to ensure only the expected fluorophores are being expressed.
- For example, a GFP-expressing rAAV should not show any fluorescence in the RFP channel.
- Take images of each well with all filters, using the 20x objective.
- For example, brightfield, GFP, RFP, YFP
- Ensure that recombinase-dependent vectors are only expressing the transgene and fluorophore in the presence of a recombinase-expressing rAAV.
- Generate an overlay image.
- Carefully examine the images and look for the following (Figure 5):
- Untransduced sample: There should be no observable bacterial, fungal or yeast growth in the image. Bacterial growth is observed as dark specs in the background between cells, yeast are observed as round circles and fungi typically grows as strands. No fluorescence should be seen in this well.
- Recombinase only sample: If the recombinase is not fluorescently tagged, there should be no fluorescence visible in the well, as in Figure 5. If the recombinase has a fluorescent tag, such as RFP, you should observe RFP fluorescence but no fluorescence in the other filters. Ensure that your recombinase does not have the same fluorescent tag as your recombinase-dependent rAAV or you will not be able to interpret the results.
- Non-recombinase dependent fluorescently-tagged rAAV sample: You should observe fluorescence only with the appropriate filter. For example, if the rAAV has a GFP tag, fluorescence should be observed in the GFP filter only.
- Recombinase-dependent fluorescently-tagged rAAV only sample: Fluorescence should not be observed in any filter. If fluorescence is detected, it suggests that the rAAV has undergone recombinase-independent recombination. While low levels of recombinase-independent recombination are expected, levels should be significantly lower than for the dual recombinase-dependent fluorescently-tagged rAAV and recombinase sample.
- Recombinase-dependent fluorescently-tagged rAAV and recombinase sample: Fluorescence should be observed for this sample in the appropriate channels. For example, if the recombinase-dependent fluorescently-tagged rAAV has a GFPtag and the recombinase is untagged, then you should observe GFP fluorescence only. If the recombinase-dependent fluorescently-tagged rAAV has a GFP tag and the recombinase has a RFP tag, then you should observe overlapping GFP and RFP fluorescence.
Table 3:
Promoters along with their expression targets and recommended cell lines for testing.
| Promoter | Target | Cell Line |
|---|---|---|
| CAG/CBA | Ubiquitous | 293T |
| CMV | Ubiquitous | 293T |
| EF1alpha | Ubiquitous | 293T |
| PGK | Ubiquitous | 293T |
| GFAP104 | Ubiquitous/Astrocytes | 293T/C8-B4 |
| Synapsin | Neurons | N2A |
| CamKIIa | Neurons | N2A |
| GFAP | Astrocytes | N2A |
| Dlx enhancer | Interneurons | N2A |
| CD68 | Glia | C8-B4 |
| TBG | Liver | HepG2/Huh-7 |
Figure 5:
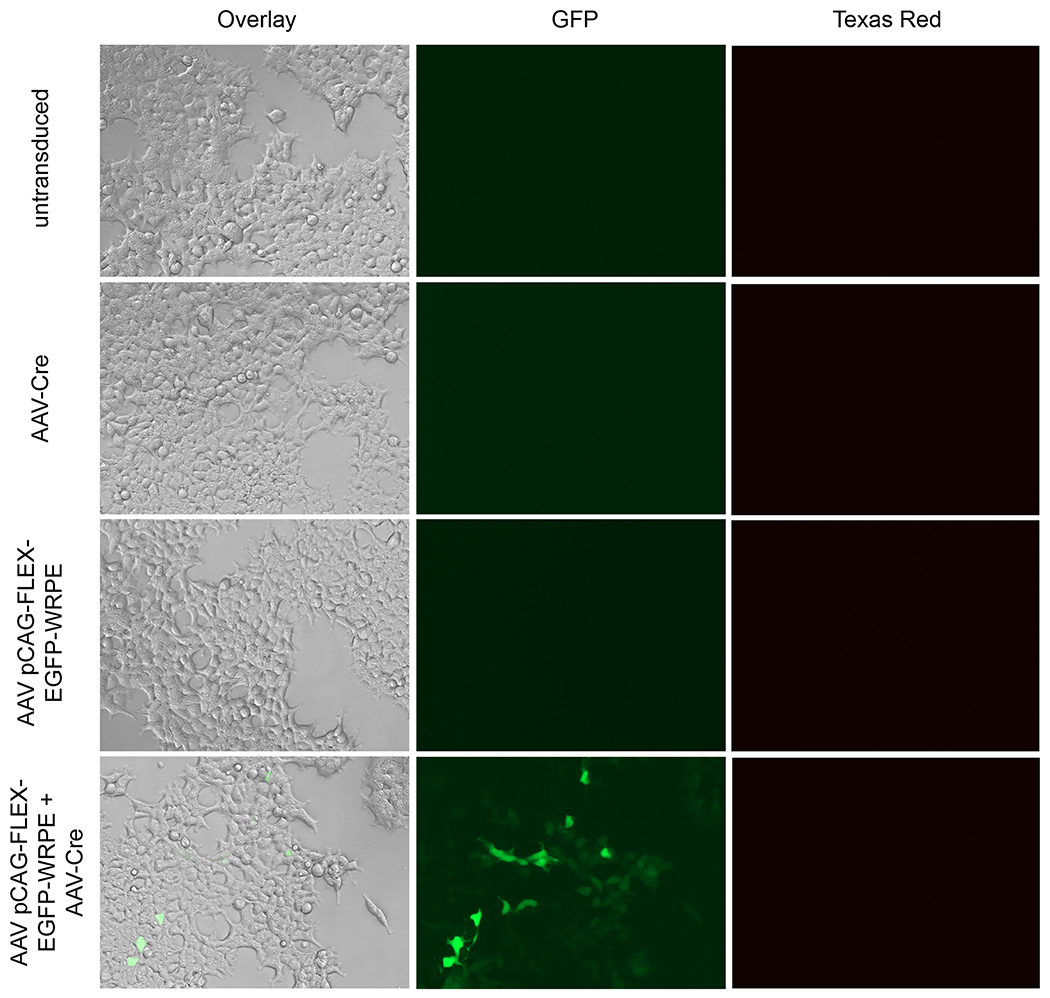
In vitro expression images for AAV pCAG-FLEX-EGFP-WPRE with and without AAV-Cre in AAVpro 293T cells.
Notes
Titration by quantitative PCR
Do not treat your plasmid standards with DNase.
The quality of the sample dilution series is critical. Make sure to pipet each dilution up and down at least 10 times, and use at least half of the final volume to mix.
Always include a no template control (NTC) in your run.
Run plasmid standard and samples at least in duplicate.
Plasmid standard: If only making 1 virus, use the transfer plasmid. If making many different ones, use a plasmid that contains a common element found in the different viruses. Store the standard in 4μg/mLyeast tRNA to increase stability.
Include a reference rAAV virus of a known titer in all assays to ensure run to run consistency. The reference virus should have a titer within 1 log of the expected titer of the sample.
- The following primer sequences can be used when detecting AAV2 ITRs (1):
- Forward ITR primer, 5’-GGAACCCCTAGTGATGGAGTT
- Reverse ITR primer, 5’-CGGCCTCAGTGAGCGA
Baseline removal: all samples will have some small amount of background signal that is most evident during initial PCR cycles. This background signal must be removed to accurately determine differences between samples.
Melt curve analysis: a single peak should be seen. The presence of a second peak at a temperature of −70-75°C usually indicates the presence of primer dimers which can increase background signal and alter the Ct values of your samples.
Pipetting less than 5 μL is not recommended. When preparing the master mix, prepare for enough reactions that the volume of each primer is >5 μL.
Confirmation of packaged AAV genome by identity PCR
To reduce the level of plasmid recombination, use a stable competent cell line for plasmid propagation and grow cultures at 30°C.
Vector Purity by SDS-PAGE and Silver Staining
Silver staining solution is considered hazardous and should not be poured down the sink.
The gel should be completely submerged in solution. A floating gel may not be stained properly.
The VP proteins should make up at least 90% of the band intensity of the lane. Depending on the downstream applications, some laboratories may require higher levels of purity.
Limulus Amebocyte Lysate Chromogenic Endotoxin Test
Reconstituted LAL reagent can be stored at −20°C for up to one week. Freeze and thaw only once. Thaw immediately before use.
Reconstituted endotoxin can be stored at 2-8°C for up to four weeks. Vortex vigorously for 15 minutes before every use.
Reconstituted chromogenic substrate can be stored at 2-8°C for up to four weeks.
Protect chromogenic substrate from exposure to light.
All materials used must be endotoxin-free.
In vitro Sterility and Expression Assay
Before setting up this assay, determine the optimal cell density for your cell line in a 96-well plate.
Recombinase activity can be driven by very low levels of protein. In some cases, when using a fluorescently-tagged recombinase and fluorescently-tagged recombinase-dependent rAAV you may observe expression of the recombinase-dependent rAAV in cells that do not express observable levels of the tagged-recombinase. This does not necessarily represent recombinase-independent expression as the recombinase may be present in the sample but at levels too low to observe. In this case, refer to the recombinase-dependent fluorescently-tagged rAAV only sample; if no fluorescence was detected in this sample then the fluorescence observed in the dual recombinase-dependent fluorescently-tagged rAAV and recombinase sample is likely being driven by low levels of the recombinase.
References
- 1.Aurnhammer C, Haase M, Muether N, et al. (2012) Universal Real-Time PCR for the Detection and Quantification of Adeno-Associated Virus Serotype 2-Derived Inverted Terminal Repeat Sequences. Human Gene Therapy Methods 23:18–28. [DOI] [PubMed] [Google Scholar]
- 2.Lock M, Alvira MR, Chen S-J, Wilson JM (2014) Absolute Determination of Single-Stranded and Self-Complementary Adeno-Associated Viral Vector Genome Titers by Droplet Digital PCR. Human Gene Therapy Methods 25:115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dobnik D, Kogovšek P, Jakomin T, et al. (2019) Accurate Quantification and Characterization of Adeno-Associated Viral Vectors. Front Microbiol 10:1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dingle T, Sedlak R, Cook L, et al. (2013) Tolerance of droplet-digital PCR versus real-time quantitative PCR to inhibitory substances. Clin Chem 59(11): 1670–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allay J, Sleep S, Long S, et al. (2011) Good manufacturing practice production of self-complementary serotype 8 adeno-associated viral vector for a hemophilia B clinical trial. Hum Gene Ther 22(5):595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fagone P, Wright J, Nathwani A, et al. (2012) Systemic errors in quantitative polymerase chain reaction titration of self-complementary adeno-associated viral vectors and improved alternative methods. Hum Gene Ther Methods 23(1):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhen Z, Espinoza Y, Bleu T, et al. (2004) Infectious Titer Assay for Adeno-Associated Virus Vectors with Sensitivity Sufficient to Detect Single Infectious Events. Human Gene Therapy 15:709–715. [DOI] [PubMed] [Google Scholar]
- 8.Zeltner N, Kohlbrenner E, Clément N, et al. (2010) Near-perfect infectivity of wild-type AAV as benchmark for infectivity of recombinant AAV vectors. Gene Ther 17:872–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guerin K, Rego M, Bourges D, et al. (2020) A Novel Next-Generation Sequencing and Analysis Platform to Assess the Identity of Recombinant Adeno-Associated Viral Preparations from Viral DNA Extracts. Human Gene Therapy 31:664–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maynard LH, Smith O, Tilmans NP, et al. (2019) Fast-Seq: A Simple Method for Rapid and Inexpensive Validation of Packaged Single-Stranded Adeno-Associated Viral Genomes in Academic Settings. Human Gene Therapy Methods 30:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tai PWL, Xie J, Fong K, et al. (2018) Adeno-associated Virus Genome Population Sequencing Achieves Full Vector Genome Resolution and Reveals Human-Vector Chimeras. Molecular Therapy - Methods & Clinical Development 9:130–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lecomte E, Toumaire B, Cogné B, et al. (2015) Advanced Characterization of DNA Molecules in rAAV Vector Preparations by Single-stranded Virus Next-generation Sequencing. Molecular Therapy - Nucleic Acids 4:e260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pacouret S, Bouzelha M, Shelke R, et al. (2017) AAV-ID: A Rapid and Robust Assay for Batch-to-Batch Consistency Evaluation of AAV Preparations. Molecular Therapy 25:1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ayuso E, Mingozzi F, Montane J, et al. (2009) High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther 17:503–510. [DOI] [PubMed] [Google Scholar]
- 15.Sampath V, (2018) Bacterial endotoxin-lipopolysaccharide; structure, function and its role in immunity in vertebrates and invertebrates. Agric Nat Resour 52:115–120. [Google Scholar]
- 16.Uphoff CC, Drexler HG (2011) Detecting Mycoplasma Contamination in Cell Cultures by Polymerase Chain Reaction. In: Methods in Molecular Biology. Humana Press, pp 93–103. [DOI] [PubMed] [Google Scholar]
- 17.Uphoff CC, Drexler HG (2012) Detection of Mycoplasma Contaminations. In: Basic Cell Culture Protocols. Humana Press, pp 1–13. [Google Scholar]
- 18.Spierenburg GT, Polak-Vogelzang AA, Bast BJEG (1988) Indicator cell lines for the detection of hidden mycoplasma contamination, using an adenosine phosphorylase screening test. Journal of Immunological Methods 114:115–119. [DOI] [PubMed] [Google Scholar]
- 19.King D, Hitchcock T, Krishnan R (2021) Evolution of Viral Vector Analytics for Gene Therapy Manufacturing. Insights on Successful Gene Therapy Manufacturing and Commercialization 34–3E [Google Scholar]
- 20.Hitoshi N, Ken-ichi Y, Jun-ichi M (1991) Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199. [DOI] [PubMed] [Google Scholar]
- 21.Jun-ichi M, Satoshi T, Kimi A, et al. (1989) Expression vector system based on the chicken β-actin promoter directs efficient production of interleukin-5. Gene 79:269–277. [DOI] [PubMed] [Google Scholar]
- 22.Nathanson JL, Yanagawa Y, Obata K, Callaway EM (2009) Preferential labeling of inhibitory and excitatory cortical neurons by endogenous tropism of adeno-associated vims and lentivirus vectors. Neuroscience 161:441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim DW, Uetsuki T, Kaziro Y, et al. (1990) Use of the human elongation factor 1α promoter as a versatile and efficient expression system. Gene 91:217–223. [DOI] [PubMed] [Google Scholar]
- 24.Adra CN, Boer PH, McBumey MW (1987) Cloning and expression of the mouse pgk-1 gene and the nucleotide sequence of its promoter. Gene 60:65–74. [DOI] [PubMed] [Google Scholar]
- 25.Hoesche C, Sauerwald A, Veh RW, et al. (1993) The 5’-flanking region of the rat synapsin I gene directs neuron-specific and developmentally regulated reporter gene expression in transgenic mice. J Biol Chem 268:26494–502 [PubMed] [Google Scholar]
- 26.Glover CPJ, Bienemann AS, Heywood DJ, et al. (2002) Adenoviral-Mediated, High-Level, Cell-Specific Transgene Expression: A SYN1-WPRE Cassette Mediates Increased Transgene Expression with No Loss of Neuron Specificity. Molecular Therapy 5:509–516. [DOI] [PubMed] [Google Scholar]
- 27.Kügler S, Kilic E, Bähr M (2003) Human synapsin 1 gene promoter confers highly neuron-specific long-term transgene expression from an adenoviral vector in the adult rat brain depending on the transduced area. Gene Ther 10:337–347. [DOI] [PubMed] [Google Scholar]
- 28.Dittgen T, Nimmerjahn A, Komai S, et al. (2004) Lentivirus-based genetic manipulations of cortical neurons and their optical and electrophysiological monitoring in vivo. Proceedings of the National Academy of Sciences 101:18206–18211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hioki H, Kameda H, Nakamura H, et al. (2007) Efficient gene transduction of neurons by lentivirus with enhanced neuron-specific promoters. Gene Ther 14:872–882. [DOI] [PubMed] [Google Scholar]
- 30.Dimidschstein J, Chen Q, Tremblay R, et al. (2016) A viral strategy for targeting and manipulating interneurons across vertebrate species. Nat Neurosci 19:1743–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brenner M, Kisseberth W, Su Y, et al. (1994) GFAP promoter directs astrocyte-specific expression in transgenic mice. J Neurosci 14:1030–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee Y, Messing A, Su M, Brenner M (2008) GFAPpromoter elements required for region-specific and astrocyte-specific expression. Glia 56:481–493. [DOI] [PubMed] [Google Scholar]
- 33.Greaves DR, Quinn CM, Seldin MF, Gordon S (1998) Functional Comparison of the Murine Macrosialin and Human CD68 Promoters in Macrophage and Nonmacrophage Cell Lines. Genomics 54:165–168. [DOI] [PubMed] [Google Scholar]
- 34.Yan Z, Yan H, Ou H (2012) Human thyroxine binding globulin (TBG) promoter directs efficient and sustaining transgene expression in liver-specific pattern. Gene 506:289–294. [DOI] [PubMed] [Google Scholar]