

Abstract
Lactam rings are found in many biologically active natural products and pharmaceuticals, including important classes of antibiotics. Methods for the asymmetric synthesis of these molecules are therefore highly desirable, particularly through the selective functionalization of unreactive aliphatic C–H bonds. Here we show the development of a strategy for the asymmetric synthesis of β-, γ-, and δ-lactams via hemoprotein-catalysed intramolecular C-H amidation reaction with readily available dioxazolone reagents. Engineered myoglobin variants serve as excellent biocatalysts for this transformation yielding the desired lactam products in high yields, high enantioselectivity, and on preparative scale. Mechanistic and computational studies elucidate the nature of the C–H amination and enantiodetermining steps and provide insights into protein-mediated control of regioselectivity and stereoselectivity. Additionally, an alkaloid natural product and a drug molecule were synthesized chemoenzymatically in much fewer steps (7–8 vs. 11–12) than previously reported, further demonstrating the power of biosynthetic strategy for the preparation of complex bioactive molecules.
Introduction
The selective functionalization of ubiquitous yet unreactive carbon-hydrogen (C–H) bonds via both chemical and enzymatic methods constitutes a powerful strategy for the diversification of organic molecules and enabling the devise of new disconnections and routes for the construction of complex molecules and natural products.1–7 Due to the large prevalence of amine-based functionalities in bioactive molecules and pharmaceuticals, a highly desirable and sought-after transformation in organic and medicinal chemistry is the selective amination of aliphatic C–H bonds.8–10 Notable advances in this field have led to the development of organometallic catalysts for catalyzing the insertion of nitrene species into C−H bonds, resulting in the formation of new carbon-nitrogen bonds (Figure 1b).8–10 These transformations are mediated by reactive metal-nitrenoid species generated upon reaction of the transition metal catalyst with nitrene precursor reagents such as iminoiodinane, azides, and hydroxylamine derivatives.8–10 Using this strategy, a variety of cyclic amines, including oxazolidinones, sulfamates, sultams, and pyrrolidines have been accessible. Despite this progress, extension of this C–H amination strategy to the synthesis of cyclic amides (lactams), which are key structural motifs in many pharmaceuticals, agrochemicals, and other fine chemicals (Figure 1a),11,12 has represented a major challenge.13 The difficulty of this transformation can be attributed to the instability of the acyl nitrene intermediate that undergoes facile decomposition to isocyanates through a Curtius-type rearrangement, thereby outcompeting the desired C–H nitrene insertion process.13 Recently, Chang and coworkers has reported a breakthrough in this area through the development of an iridium-based system for enabling this transformation.13 This progress notwithstanding, asymmetric versions of this methodology are restricted to 5-membered rings (γ-lactams) and require the use of rare and toxic metals (i.e., Ir, Ru).14,15
Figure 1. Asymmetric lactam synthesis via enzyme catalyzed C(sp3)-H amidation.

a) Bioactive molecules containing β- and γ-lactam rings. b) General catalytic cycle for C-N bond formation via nitrene transfer c) Enzyme-catalyzed intramolecular C–H amination reactions. d) Biocatalytic construction β-, γ-, and δ-lactam rings via myoglobin-catalyzed intramolecular C–H amidation of dioxazolones (this work).
Inspired by the chemistry of metalloporphyrins,16 our group and the Arnold group have recently demonstrated the ability of engineered hemoproteins to serve as biocatalysts for intramolecular17–22 and intermolecular23–25 C-H aminations via nitrene transfer (Figure 1c). Specifically, engineered cytochrome P450 enzymes have been shown catalyze the cyclization of sulfonyl azides, carbonazidates, and sulfonazidates26 to produce sultams, cyclic carbamates, and cyclic sulfamides, respectively.17–22 Furthermore, iridium-substituted P450s27 and non-heme Fe-dependent enzymes28,29 were also found to catalyze similar intramolecular C-H amination reactions. Despite this progress, the synthesis of lactam rings using biocatalytic nitrene transfer approaches has remained elusive, largely due to the aforementioned difficulty in controlling the reactivity of acyl nitrene intermediates while disfavoring other competing, unproductive reactions (e.g., nitrene reduction) known to affect these abiological enzyme-catalyzed reactions.21
Here, we report the development of a general biocatalytic methodology for the asymmetric synthesis of enantioenriched lactams via intramolecular C-H amidation of dioxazolones (Figure 1d), a safe and readily accessible class of nitrene donor reagents.30 This strategy provides an efficient and scalable approach to the selective construction of γ-lactam molecules with high enantioselectivity. Furthermore, the scope of this biocatalytic system could be extended to the construction of optically active β- and δ-lactam motifs with high enantiocontrol. The power of this methodology is further showcased through the implementation of concise chemoenzymatic routes for the total synthesis of an alkaloid natural product and a drug molecule. Comprehensive mechanistic studies elucidate the nature of the C–H amination step as well as the role of the protein scaffold in controlling the regio- and enantioselectivity of the reaction. Leveraging a direct C–H amination strategy, this methodology is mechanistically distinct yet complementary to recently reported biocatalytic approaches for γ-lactam synthesis that rely on radical-mediated cyclizations of alkene-containing substrates31–33 or carbene C–H insertion with artificial enzymes.34
Results
Biocatalyst discovery.
Inspired by prior work of Chang and coworkers,13,35 we envisioned the possibility to execute an enzyme-catalyzed γ-C–H amidation reaction via nitrene transfer with dioxazolone reagent 1a (Figure 2a–b). Particularly attractive features of dioxazolones as nitrene precursors include their facile synthesis from commodity carboxylic acids and their stability and safety compared to azide-based reagents previously used in biocatalytic nitrene transfer reactions.17–22 To identify an initial biocatalyst for this reaction, we tested various heme-containing enzymes and proteins, including wild-type myoglobin (Mb), cytochromes P450s, peroxidases, and cytochromes c, under anaerobic conditions (Supplementary Table 1). While many of these reactions produced the acyclic amide 2b as byproduct, none of these biocatalysts displayed any activity toward formation of the desired γ-lactam product (Supplementary Table 1). Previous studies from our group demonstrated that mutations of the distal His64 residue in Mb (Figure 2c) can enhance its activity toward non-native carbene and nitrene transfer reactions.36,37 Promisingly, Mb(H64V) was found to react with 1a to yield minute yet detectable amounts of the desired lactam 2a (2% GC yield) with good levels of enantioselectivity (96% ee; Figure 2d).
Figure 2. Biocatalytic intramolecular C(sp3)‒H amidation with dioxazolones.

(a) Reaction with 3-phenylpropyl-dioxazolone 1a. Box: Structures of alternative nitrene precursors and corresponding yields for 2a using Mb* under standard reaction conditions. (b) Envisioned hemoprotein-catalyzed nitrene transfer process for γ-lactam ring formation. (c) Crystal structure of wild-type Mb (pdb 1JW8) with residues near the Fe center highlighted in yellow. (d) Activity, chemoselectivity, and enantioselectivity (for 2a) of engineered Mb variants in the reaction with 1a. Reaction conditions: 20 μM protein, 10 mM 1a, 10 mM Na2S2O4 in potassium phosphate buffer (50 mM, pH 7), 3 hours, room temperature, under anaerobic conditions. Yields and product distribution as determined by GC using calibration curves with isolated product. * With acetonitrile as co-solvent. ** Using sodium borate buffer (pH 9) with 5% (v/v) acetonitrile (= standard reaction conditions or s.r.c).
Encouraged by these results, we extended the screening to a broader panel of engineered Mb variants (Supplementary Table 2) containing mutations at the level of the distal His64 residue (Ala, Val, Gly) along with additional mutations within the active site of the hemoprotein (Figure 2c). From this screening, multiple Mb variants were found to exhibit increase C–H amidation activity compared to Mb(H64V) (10–50% yield; Supplementary Table 2). Among them, Mb(H64V,V68A) (called Mb*), which was previously developed for stereoselective cyclopropanation,36 emerged as the best biocatalyst for this reaction, producing 2a in 50% yield with excellent enantioselectivity (>99% ee) (Figure 2d). The configuration of the γ-lactam product was determined to be S by crystallography (Figures 2a and Supplementary Figure 11). In addition to the C–H amidation product, the Mb*-catalyzed reaction also produced a significant amount (40%) of the amide byproduct 2b (Figure 2a), which likely arises from reduction and protonation of the nitrene intermediate (SupplementaryFigure 15) as observed previously for C–H amination reactions with azide-based precursors.18,22 Unexpectedly, a minor product (10%) corresponding to the γ-lactone 2c was also formed in the reaction (Figure 2a).
Method optimization.
Compared to Mb*, Mb variants containing Ala and/or Gly mutations at the 64 or 68 positions showed significant decreases in activity and/or selectivity (Entries 3–6 vs. 7; Supplementary Table 2) and a similar effect was observed upon introduction of additional mutations to Mb*, suggesting that the reaction is sensitive to subtle changes in the shape of the enzyme’s active site. Based on prior mechanistic studies on P450-mediated C–H amination21 and given the robustness of Mb biocatalysts to organic solvents,38 we hypothesize that the addition of an organic co-solvent could favor the desired C–H amidation reaction by disfavoring formation of the amide byproduct 2b. Accordingly, screening of various organic co-solvents showed that acetonitrile (ACN) is beneficial toward increasing the yield of the C–H amidation product (50→65% yield), while reducing the undesired reduction reaction (30%) and without affecting enantioselectivity (Figures 2d and Supplementary Figure 1). Further optimization of the reaction conditions revealed that slightly alkaline conditions (pH 9) further increase the enzyme’s C–H amidation activity (75% yield of 2a) at the expenses of byproduct 2b (10%), while retaining excellent enantioselectivity (Figure 2d). These trends are consistent with our hypothesis that formation of the amide byproduct 2b involves protonation of the nitrene intermediate, which should be disfavored under more alkaline conditions and in the presence of organic solvent. Under catalyst limiting conditions (0.07 mol%), Mb* was determined to catalyze the C–H amidation of 1a in 58% yield and 2,175 TON with excellent enantioselectivity. In addition to dioxazolone 1a, we also evaluated other nitrene precursors such as dioxazole 1aa, acyl-protected hydroxylamine 1ab, and dioxathiazole 1ac (Figure 2a). Whereas 1ac and 1ab were inactive and less effective than 1a, respectively, for the cyclization reaction (Figure 2c), dioxazole 1aa produced the desired γ-lactam product in good yield (75% 2a), thereby constituting another viable nitrene precursor for this biocatalytic C-H amidation strategy.
Substrate scope.
To assess the substrate scope of this methodology, Mb* was tested against an array of substituted dioxazolone substrates (1d-p). As summarized in Figure 3a, these experiments revealed a large tolerance of the enzyme toward substitutions at the para position of the aryl ring affording the desired γ-lactam products (2d-i) with high to excellent enantioselectivity (73–99% ee). While both electron-withdrawing and donating substitutions are accepted by the enzyme, increased activity was observed for substrates containing electron-donating substituents (1h-i). Substitutions at the ortho and meta positions were also well tolerated by the enzyme, producing the corresponding γ-lactam products (2j-l) in good yields (68–88%) and high enantiopurity (99% ee, Figure 3a). In addition to substituted phenyl groups, the enzyme is able to catalyze the cyclization of substrates containing heteroaryl groups such as thiophenyl group (2m-n) with good to high levels of activity (75–90% yields) and enantioselectivity (66–99% ee). On the other hand, enzymatic synthesis of 2p in 28% yield and 83:17 diastereomeric ratio showed the tolerance of the enzyme also to substitutions in α to the dioxazolone core. Allylic C–H amidation was also possible as demonstrated by the successful preparation of 2o in 38% yield and 99% ee from 1o (7:3 E/Z mixture), with the other products corresponding only to the reduction (amide) byproducts. Notably, no reactivity toward the double bond in 1o was observed in this reaction, which highlights the chemoselectivity of the iron-based biocatalyst toward C–H nitrene insertion over aziridination, typically favored by Rh-based catalysts in nitrene transfer reactions.39 Lastly, isoindolinone 9 and 11 could be successfully obtained from cyclization of phenyl-dioxazolones 10 and 12, respectively (Supplementary Figure 14), albeit in high yield but no enantioselectivity (10) or with high enantioselectivity but low yield (12). Conversely, dioxazolone substrates lacking an aromatic group (e.g., 16-17; Supplementary Figure 14) or containing bulky aryl substituents proximal to the γ-C–H bond (e.g. 15, 18; Supplementary Figure 14) eluded Mb*-catalyzed C–H amidation, indicating that further optimization of the biocatalyst for these types of substrates would be required in the future.
Figure 3. Substrate scope for Mb*-catalysed C-H amidation reaction for the synthesis of lactams with various ring sizes.

(a) Scope of γ-lactams, (b) Scope of β-lactams, (c) Scope of δ-lactams via. * Using Mb(L29T,H64T,V68L) as the catalyst. n.a. = not active.
Enantiodivergent biocatalyst.
Enantiodivergent biocatalysts are highly desirable yet often hard to develop.40 Notably, screening of the initial Mb active-site mutant library revealed a variant, Mb(L29T,H64V,V68L), that catalyzes the cyclization of 1a with inverted enantioselectivity compared to Mb*, producing the R-configured γ-lactam product ent-2a in 65% ee, albeit in modest yield (15%; Supplementary Table 2). To improve the performance of this biocatalyst, Mb(L29T,H64V,V68L) was subjected to active-site mutagenesis, ultimately leading to Mb(L29T,H64T,V68L), which produces ent-2a with both an improved enantioselectivity of 91% ee and two-fold higher activity compared to the parent enzyme. To explore the substrate promiscuity of this enantiocomplementary biocatalyst, Mb(L29T,H64T,V68L) was tested against the panel of dioxazolones 1d-p. Albeit in more moderate yields compared to the Mb* reactions, the majority of these substrates (10/14) could be converted to the R-configured γ-lactam products with good to high enantioselectivity (57–99% ee; Figure 3a). Of note, ent-2k, ent-2l, and the thiophenyl containing substrate ent-2m were all obtained in 99% enantiomeric excess. Altogether, these results highlighted the broad substrate scope and predictable enantiocomplementarity of the two Mb-based biocatalysts for γ-lactam ring formation.
Synthesis of β-lactams and δ-lactams.
Next, we targeted the synthesis of β-lactams which are highly desirable building blocks for medicinal chemistry as well as key pharmacophores in β-lactam antibiotics.12 Notably, β-lactam formation via intramolecular nitrene transfer has not been reported to date. Upon challenging Mb* with substrate 3a, the desired β-lactam 4a was obtained in high yield (85%) and excellent enantioselectivity (99% ee) (Figure 3b). Mirroring the S-enantioselectivity in γ-lactam formation, the enzyme maintains S-enantiopreference for the formation of 4j, as determined by X-ray crystallography (Figure 3b). These findings prompted us to further explore the substrate scope of this reaction (3b-j; Figure 3b). Remarkably, variously substituted substrates could be converted into the desired β-lactam products with excellent enantioselectivity (99% ee; Figure 3b) and up to 93% yield. Unlike the γ-lactams, substrates bearing electron-withdrawing groups on the aryl ring were cyclized more efficiently than those containing electron-donating groups (e.g., 32% yield for 4d vs. 75% for 4c) and para substitutions were better tolerated than meta substitutions (e.g., 75% yield for 4g vs 32% for 4d). These differences likely arise from the differential role of electronic and steric constraints in the 4- vs. 5-membered ring formation. Furanyl- and thiophenyl-containing substrates 3i and 3j, respectively, were also cyclized very efficiently (75% and 93% yields, respectively) and with high enantiocontrol (99% ee).
To explore the reactivity of the Mb biocatalyst toward synthesis of δ-lactams, a reaction was carried out using substrate 5a, which resulted in a mixture of δ-lactam (6aa) and γ-lactam (6ab) in a 1:3.7 ratio, in addition to the amide 6b as the major product (7:26:67 ratio for 6aa:6ab:6b; Figure 3c). These results revealed the enzyme’s preference for amidation of the homobenzylic γ-C–H bond (to give 6ab) over the benzylic δ-C–H bond (to give 6aa) despite the higher bond dissociation energy (BDE) of the latter (~95 vs. 90 kcal/mol) (Figure 3c). While the observed formation of both the γ- and δ-lactam product (3.7:1 ratio) suggests that the enzyme’s regioselectivity could be tuned via protein engineering, these findings inspired us to steer the enzyme’s regioselectivity via substrate engineering, i.e., by substituting the γ-C–H bond with an O atom, as a means to favor δ-lactam formation (Figure 3c). Gratifyingly, the Mb* reaction with the corresponding substrate 5c produced the desired δ-lactam 6c with significantly improved efficiency (78% yield) as well as excellent enantioselectivity (99% ee; Figure 3c). This biocatalytic reaction was also found to be tolerant toward substitution on the aryl ring, as demonstrated by the synthesis of δ-lactams 6d-f in 45–93% yields and high enantiomeric excess (90–99% ee). In addition, benzooxazinone 14 could be also obtained in high yield from the Mb*-catalyzed cyclization of 3-phenyl-dioxazolone based substrate 13, albeit only in racemic form (Supplementary Figure 14).
Taken together, these results reveal a remarkable generality of the Mb* catalyst toward enabling the stereoselective synthesis of lactams of varying sizes and with different substitutions. Noteworthy is also the consistent and predictable S stereoselectivity of the Mb*-catalyzed C–H amidation reaction not only across the different substrates but also across the β-, γ-, and δ-lactam rings, which adds to the synthetic utility of this biocatalytic system.
Mechanistic Studies.
Studies were then performed to gain insights into the mechanism of this enzyme-catalyzed reaction. To probe the nature of the C–H amidation step, the Mb*-catalyzed reaction was carried out in the presence of the Z-configured dioxazolone 1q, which resulted in the formation of the lactam product 2q in the E configuration (Figure 4a). This result rules out a concerted C–H nitrene insertion process and is consistent with a stepwise hydrogen atom abstraction (HAA)/radical rebound mechanism proceeding via an allylic radical that undergoes Z→E isomerization to yield trans-2q prior to radical recombination. Of note, complete isomerization of the double bond in the cyclization product (no cis-2q was observed) shows that the radical intermediate is relatively long-lived.
Figure 4. Mechanistic studies.
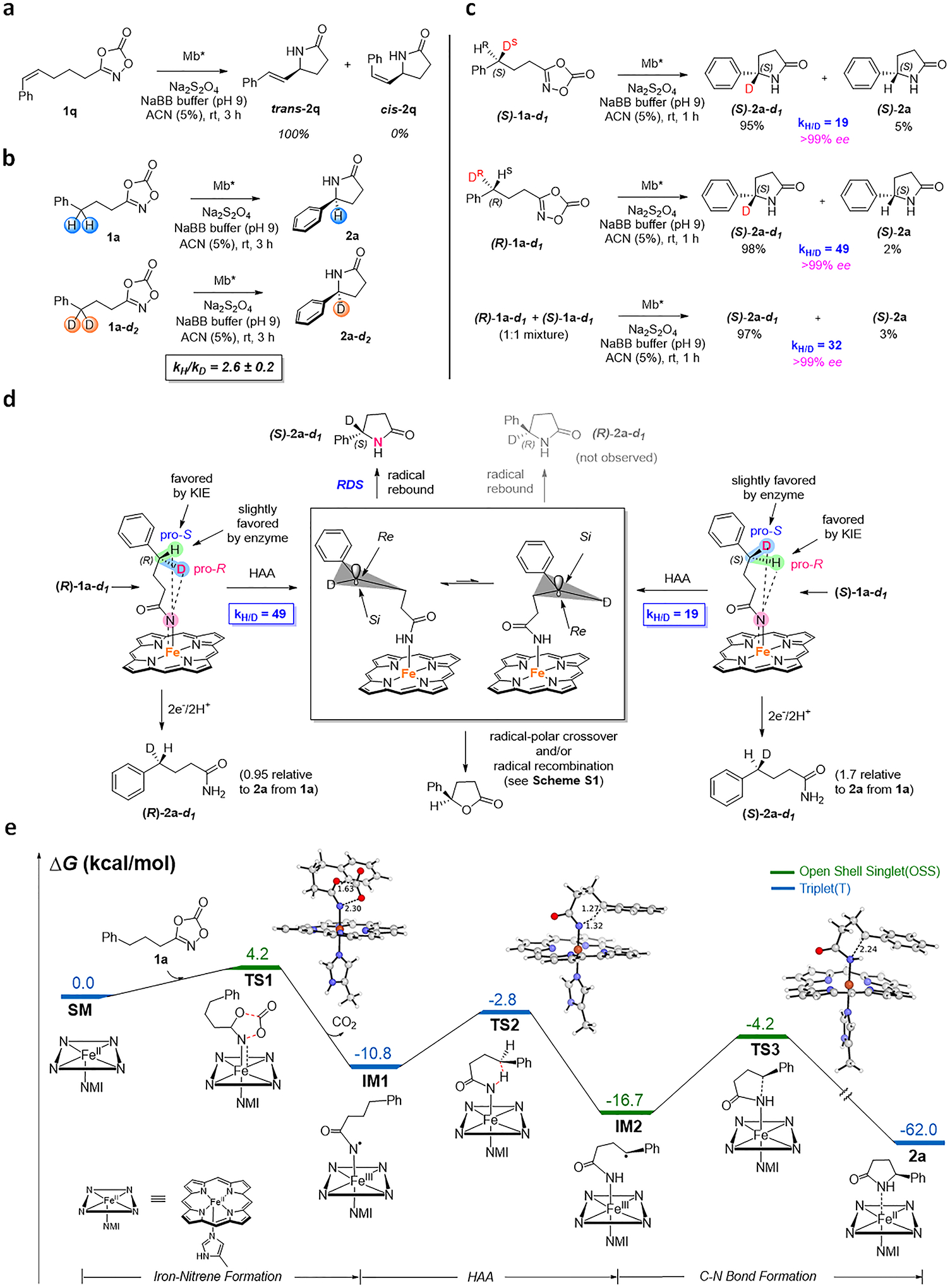
a) Mb-catalyzed cyclization of substrate (Z)-1q shows complete isomerization to the E isomer product trans-2p (see Supplementary Figure 2 for details). b) Non-competitive intermolecular KIE experiments as determined via comparison of product formation rates from parallel reactions with protiated and deuterated substrate. See Supplementary Figure 3 for details. c) H/D competition experiments with enantiopure isotopomeric dioxazolidinones. KH/D values were determined by NMR (see Supplementary Figure 4 for details). d) Mechanistic model for Mb*-controlled asymmetric induction. HAA = H atom abstraction. e) Gibbs free energy diagram for the Mb-catalyzed γ-lactam formation. ΔG values were calculated at the uB3LYP-D3BJ/def2tzvp(SMD, solvent=water)//uB3LYP-D3BJ/6–31G(d)+SDD(Fe) level using a truncated computational model of the enzyme (see Supplementary Figure 5). See Supplementary Figure 5 for additional spin states. NMI=5-methyl-imidazole. Distances in Å.
To further investigate the kinetic role of the C–H cleavage step, non-competitive intermolecular H/D competition experiments were carried out using substrate 1a and 1a-d1 in parallel reactions (Figure 4b). These experiments yielded a kinetic isotope effect (KIE) value of 2.6 ± 0.2 (Figure 4b and Supplementary Figure 3), which is lower than that determined for C−H amination reactions with azide-based substrates catalyzed by engineered P450s (kH/kD: 3.4–5.3)20,41, but higher than that determined for P450-catalyzed cyclization of sulfonylazides (kH/kD: 0.9), where the azide activation was established to be rate-determining.21 Overall, these results indicated that the C–H cleavage step in the present system is only partially rate determining, with other steps contributing to control the overall rate of the reaction.
To gain insight into the enantioselectivity determining step(s) in this reaction, isotopomeric dioxazolones (S)-1a-d1 and (R)-1a-d1 were synthesized in enantiopure form (Supplementary Figure 4) and subjected to Mb*-catalyzed C–H amidation. Interestingly, both reactions led to the accumulation of deuterated lactam (S)-2a-d1 as the largely predominant product over the protiated counterpart (S)-2a (95:5 and 98:2 ratio from (S)-1a-d1 and (R)-1a-d1, respectively) as determined by NMR (Figure 4c and Supplementary Figure 4). In addition, both reactions proceed with high enantioselectivity (99% ee). From these results, two conclusions can be derived, first, H abstraction is strongly favored over D abstraction regardless of the configuration of the C–H amination site and secondly, protein-mediated enantioinduction in the C–N bond forming process must occur at the level of the radical rebound step (Figure 4d). Indeed, formation of (S)-2a-d1 as the major product from either (S)-1a-d1 or (R)-1a-d1, along with the preserved high % ee in both cases, imply that, after HAA, the pro-S and pro-R conformations of the radical intermediate can undergo rapid interconversion, with the enzyme active site enforcing radical rebound through the pro-S intermediate (Si face attack) (Figure 4c).
The kH/D values derived from the reactions with the isotopomeric substrates (Figure 4c) result from a combination of the KIE (kH/kD) and enzyme’s enantiopreference in the HAA step (i.e., kS/kR). The larger kH/D measured for (R)-1a-d1 vs. (S)-1a-d1 (49 vs. 19) indicates a higher chirality-match for H abstraction in the former substrate vs. the latter, i.e., abstraction of Hpro-S in (R)-1a-d1 is favored by both KIE and the enzyme, whereas abstraction of Hpro-R in (S)-1a-d1 is favored by KIE but disfavored by the enzyme (Figure 4d). Consistent with this chirality match effect, the relative amount of reduction byproduct (i.e., (R)-2a-d1 or (S)-2a-d1), when compared to that of 2a from the reaction with 1a, is slightly reduced (−5%) in the case of (R)-1a-d1 and markedly increased (+70%) in the case (S)-1a-d1 (Figure 4d). However, the mere 2.5-fold difference between the kH/D values for (R)-1a-d1 vs. (S)-1a-d1 (in contrast to the 50- to 220-fold difference determined with other systems41,42) suggests that the HAA step is barely stereoselective and thus that asymmetric induction is largely controlled by the enzyme during the radical recombination step. As such, this system shows a distinct enantioinduction mechanism compared to that described for asymmetric C–H aminations of sulfonyl azide substrates catalyzed by Co-porphyrins42 and P411 catalysts22,41.
Computational analysis (DFT) of the reaction mechanism.
Density functional theory (DFT) calculations were performed to explore the formation of the γ-lactam product 2a from dioxazolone 1a. The quintet (high spin), triplet (intermediate spin), closed-shell singlet (low spin) and open-shell singlet (low spin) spin states of each intermediate and transition state structure were considered in the calculations. Figure 4e reports energies of the lowest energy spin-state for different intermediates and transition-states, with energies of other spin states (Supplementary Figure 5).DFT calculations describe the iron-nitrenoid active species in this system as a triplet ground state, in analogy with what found for other iron-nitrenoid species.22,25 Calculations indicate that formation of iron-nitrenoid complex IM1 through activation of dioxazolone substrate, and concomitant loss of CO2, has an energy barrier of only 4.2 kcal/mol and involves an open-shell singlet (OSS) spin state. After spin crossing, the hydrogen atom abstraction (HAA) step is favored in the triplet state, giving a carbon radical intermediate IM2, with an 8.0 kcal/mol energy barrier. The following step, involving a radical rebound to give the final γ-lactam product, was found to be most favorable in the open-shell singlet state with an energy barrier of 12.5 kcal/mol. The IM2 has a heavy spin contamination (S2=1.10), indicating that the OSS is not a pure singlet spin state, but 50:50 singlet:triplet, often observed in calculations on diradicals. Based on the energy profile, the C-N bond forming step is predicted to be rate determining, exhibiting a 4.5 kcal/mol higher energy barrier than the C–H bond cleavage event. These findings are in excellent agreement with the results from the mechanistic experiments (Figure 4) and explain the long-lived nature of the radical intermediate, as suggested by the complete double bond isomerization observed with trans-2q (Figure 4a).
This mechanistic model provides a framework also for defining a plausible mechanism for the formation of the unexpected γ-lactone product 2c from 1 (Figure 2a).. After the HAA step, single electron transfer from the C-centered radical to the heme (or protein matrix) can generate a benzylic carbocation which can then react with the amide group (via the carbonyl group) to form a dihydrofuranimine ring. Hydrolysis of the latter produces 3c (Supplementary Figure 15). In addition to this radical-polar crossover mechanism, a radical pathway can be envisioned that proceeds via the same dihydrofuranimine intermediate produced via reaction of the benzylic radical with the amidyl group (Supplementary Figure 15). In either case, a slow C-N bond forming radical rebound step as revealed by our mechanistic and DFT studies is expected to enable this competing side reaction to occur under suboptimal reaction conditions.
Enzyme-controlled regio- and enantioselectivity.
To investigate the role of the enzyme in controlling the enantioselectivity of the reaction, we explored the heme-bound iron-nitrenoid intermediate IM1 docked in the active site of Mb* using the available crystal structure of this protein.43 As shown in Figure 5a, the N atom of IM1 can abstract either the H1 or H2, leading to the S and R lactam product, respectively, under fast rebound conditions. We measured the evolution of distances between the N atom and the H1 and H2 atoms during 1000 ns MD simulations (Figure 5b). These studies show that the average distance from N atom to H1 and H2 are 3.96–4.01 Å and 4.06–4.10 Å, respectively, suggesting little to no preference for abstraction of H1 vs. H2 (leading to the S vs. R product) by the nitrene intermediate.
Figure 5. Computational analysis for enantio- and regiocontrol.

a. H atom abstraction step in the C–H amidation of 1a to give the corresponding γ-lactam product. b. Representative snapshot of IM1 intermediate in Mb* active site and plot of the nitrene N∙∙∙H1 and nitrene N∙∙∙H2 distances over the entire 1000 ns MD simulations. See Supplementary Figure 6for additional replicas. c. Most populated conformation of the benzylic radical intermediate IM2 in Mb* heme pocket over the entire 1000 ns simulation. Key residues implicated in stabilizing IM2 conformation leading to the S-configured product 2a after radical rebound are highlighted. d. DFT calculated energy barrier difference for γ-H vs. δ-H atom abstraction by nitrene intermediate IM1 derived from substrate 5a. Structures of the transition states are shown with key distances given in Å. e. Representative snapshots of 5a-derived IM1 intermediate in Mb* active site and distance plots and Boltzmann distribution of the nitrene N∙∙∙γ-H atoms and nitrene N∙∙∙δ-H atoms distances over the entire 1000 ns MD simulation. See Supplementary Figure 9 for additional replicas.
Next, the carbon radical intermediate IM2 was docked in Mb* active site in both a conformation that could lead to the S lactam product (IM2pro(S)) and a conformation that could lead to the R lactam product (IM2pro(R)), as shown in Supplementary Figure 7. After 1000 ns MD simulation, both complexes converged in a conformation of the intermediate that favors radical rebound through the Si face of the benzylic radical species (Figure 5c) leading to the (S)-γ-lactam 2a. Inspection of this structure indicates that the active site residues Ile107, Val64 and Ala68 contribute to orient the heme-bound intermediate in the pro-(S) conformation. Furthermore, DFT calculations show that TS3 for formation of the R-configured product is disfavored by 3.1 kcal/mol versus that leading to the S product (Supplementary Figure 8). This ΔΔG‡ is consistent with the high S-enantioselectivity induced by the enzyme (>99:1 e.r.) in the intramolecular C–H amidation reaction. The models for both the HAA and radical rebound step show the aryl ring projecting into the core of the protein between Ile107 and Ala68 (Figure 5b–c). While this arrangement can accommodate substitution at different positions of the aryl ring, it also shows potential steric constraints as the size of the para substituent increases, providing a plausible rationale for the structure-activity trends observed experimentally (i.e., –F (2d) > -Cl/Br (2e-f) > -I (2g); Figure 4a).
To understand the regioselectivity of the enzyme, we studied the nitrene intermediate IM1 generated from substrate 5a (Figure 5d). In this case, the nitrene intermediate can abstract either the γ- or δ-H atom, leading to the 5- and 6-membered lactam product, respectively. DFT calculations show that the energy barrier for hydrogen abstraction leading to the δ-lactam is 2.4 kcal/mol higher than for the formation of γ-lactam (Figure 5d). We also studied the proximity of the γ and δ H atoms to the nitrene N atom via 1000 ns MD simulation. The γ-H…N average distance is 2.8–3.9 Å, whereas for δ-H…N distance is significantly larger, namely 4.3–5.2 Å (Figures 5e and Supplementary Figure 10). Thus, both the lower energy barrier for Hγ abstraction and a closer nitrene N…Hγ atom distance contribute to favor formation of the γ-lactam product, which can explain the regioselectivity of the Mb*-catalyzed C–H amidation of 5a observed experimentally (Figure 4c).
Chemoenzymatic total synthesis of bioactive molecules.
The present strategy was then applied for the chemoenzymatic syntheses of bioactive alkaloid (S,S)-(−)-Homaline (7) and FDA-approved drug (S)-Dapoxetine (8) (Figure 6). Specifically, we envisioned that the key β-lactam intermediate 4a, previously accessible only in low yields and after lengthy routes (7–8 steps, 7% overall yield44,45; Figure 6b), could be produced in a more efficient and step-economical manner by enzymatic means using the present method. Accordingly, enantiopure β-lactam 4a (>99% ee) was produced from 3a and isolated on a preparative scale (0.5 g) from a scaled up reaction with Mb* (Figure 6a). From 4a, (S,S)-(−)-Homaline (7) and (S)-Dapoxetine (8) could be synthesized in only four and five steps, respectively, using known routes (Figure 6b; see Supplementary Figure 16for further details). Overall, asymmetric Mb-catalyzed C–H amidation enabled the chemoenzymatic synthesis of the alkaloid natural product and drug molecule in a total of 7 and 8 steps, respectively, compared to 11 and 12 steps required in previous total synthesis strategies. In addition, with the present approach, the key enantiopure β-lactam intermediate 4a was readily obtained from the achiral, commodity chemical hydrocinnamic acid as opposed to more expensive optically active precursors required in the previously reported routes (Figure 6b). These results further showcase the synthetic utility and scalability of the present methodology for the synthesis of biologically active molecules.
Figure 6. Applications of Mb*-catalysed C-H amidation reaction.

(a) Gram-scale synthesis of β-lactam (b) Chemoenzymatic total synthesis of the alkaloid natural product (S,S)-(−)-Homaline (7) and synthetic drug (S)-Dapoxetine (8).
Discussion.
In summary, we have developed a biocatalytic strategy for the asymmetric construction of lactam molecules via nitrene transfer. Starting from readily accessible dioxazolones, this strategy could be leveraged to afford a broad range of β−, γ-, and δ-lactam scaffolds in good yields and high enantiomeric excess using a single Mb-based biocatalyst. In addition, we demonstrated the possibility to obtain enantiopodes of the γ-lactam products using an alternate engineered Mb variant with enantiodivergent selectivity. Our mechanistic investigations revealed that these reactions proceed via a HAA/radical rebound pathway, with the enzyme binding site controlling the stereo- and regioselectivity (β-, γ-, δ-C-H amidation) of the process. Furthermore, while the HAA step is generally assumed to be enantiodetermining in enzymatic C–H aminations46,47, our studies show that protein-induced enantioselectivity in the present system is largely controlled at the level of the radical rebound step. The power of the present methodology was further showcased by the concise chemoenzymatic total synthesis of an alkaloid natural product and a drug molecule in about half of the steps required previously, while offering higher overall yields and starting from a commodity chemical instead of optically active precursors. This work expands the available biocatalytic toolbox for the asymmetric synthesis of amine-containing molecules and paves the way to the development of other asymmetric enzyme-catalyzed nitrene transfer reactions involving dioxazolones as nitrene precursors.
Methods
Molecular Cloning.
pET22b(+) (Novagen) was used as the recipient plasmid vector for expression of all of the myoglobin variants. In this construct, the Mb gene is C-terminally fused to a polyhistidine tag and it is under the control of an IPTG-inducible T7 promoter. Cloning of the Mb variants tested in this study was described previously.48–50
Protein Expression and Purification.
Engineered Mb variants were expressed in E. coli C41(DE3) or BL21(DE3) cells as described previously.48 Briefly, cells were grown in terrific broth (TB) medium (ampicillin, 100 mg L−1) at 37 °C (150 rpm) until OD600 reached 0.9–1.2. Cells were then induced with 0.25 mM β-d-1-thiogalactopyranoside (IPTG) and 0.3 mM δ-aminolevulinic acid (ALA). After induction, cultures were shaken at 180 rpm and 27 °C and harvested after 18–20 h by centrifugation at 4000 rpm at 4°C. After cell lysis by sonication, the proteins were purified by Ni-affinity chromatography. The lysate was transferred to a Ni-NTA column equilibrated with Ni-NTA Lysis Buffer. The resin was washed with 50 mL of Ni-NTA Lysis Buffer and then 50 mL of Ni-NTA Wash Buffer (50 mM KPi, 250 mM, NaCl, 20 mM imidazole, pH 8.0). Proteins were eluted with Ni-NTA Elution Buffer (50 mM KPi, 250 mM, NaCl, 250 mM histidine, pH 7.0). After elution, the proteins were buffer exchanged against 50 mM KPi buffer (pH 7.0 or 8.0) using 10 KDa Centricon filters. Myoglobin concentration was determined using an extinction coefficient (Fe(III)) ε410 = 157 mM−1 cm−1.
Purified Protein Reactions.
Analytical reactions were carried out at a 400 μL scale using 20 μM myoglobin, 10 mM dioxazolone compound, and 10 mM sodium dithionite under anaerobic conditions unless otherwise noted. In a typical procedure, 24-well plates or crimp vials containing a concentrated amount of Mb were introduced to an anaerobic chamber. Then, a corresponding amount of degassed potassium phosphate buffer (KPi, 50 mM, pH 7.0) or sodium borate buffer (NaBB 50 mM, pH 9) was added to each well/vial followed by the addition of 40 μL of sodium dithionite solution (100 mM stock solution) in KPi or NaBB, producing a 20 μM myoglobin solution. The reactions were initiated by addition of 10 μL of the dioxazolone compound (400 mM stock solution in organic solvent). The plates were covered with aluminum foil (vials were capped) and left shaking at 120 rpm (or under magnetic agitation for vials) for 3–16 hours at room temperature. The reactions were then analyzed outside of the chamber following the Product Analysis protocol shown below. Reactions with hemin or Fe(TPP)(Cl) were carried out using an identical procedure with the exception that the purified Mb was replaced by hemin (20 μM in DMF) or Fe(TPP)(Cl) (20 μM in DCM).
Product Analysis.
Enzymatic reactions were analyzed by adding 20 μL of internal standard (50 mM benzodioxole in EtOH) to the reaction mixture, following extraction 400 μL of CH2Cl2. The organic phase was analyzed by GC-FID using a Shimadzu GC-2010 gas chromatograph equipped with an FID detector, and a chiral Cyclosil-B column (30 m × 0.25 mm × 0.25 μm film) and the following method: 1 μL injection, injector temperature: 250 °C, detector temperature: 300 °C. Gradient: column temperature set at 180 °C for 3 min, then to 185 °C for 1.0 °C/min then 190 for 2.0 °C/min then to 245 °C at 80 °C/min with a 0 min hold. Total run time: 16.19 min. Stereoselectivity determination was performed via chiral GC-FID of HPLC as in the Supporting Information. Assay yields were determined using calibration curves constructed with authentic standards prepared via preparative-scale reactions with purified Mb(H64V,V68A) as described in Synthetic Procedures. All measurements were performed at least in duplicate.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the U.S. National Institute of Health grant GM098628 (R.F.). D.V. acknowledges support from the National Science Foundation Graduate Fellowship Program. K. N.H. acknowledges support from the US National Science Foundation (CHE-1764328) and Natural Science Foundation of China (22103060) provided computational resources used in QM analyses. S.R. and R.F. acknowledge support from the National Science Foundation, Center for Chemical Innovation (CCI) grant (CHE 2221346). The authors are grateful to Dr. William Brennessel (University of Rochester) for assistance with crystallographic analyses. MS and X-ray instrumentation at the University of Rochester are supported by U.S. National Science Foundation grants CHE-0946653 and CHE-1725028 and the U.S. National Institute of Health grant S10OD030302.
Footnotes
COMPETING INTERESTS
The authors have no competing interests.
DATA AVAILABILITY
Crystallographic data for small molecules have been deposited in the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers CCDC 1893087 (2a), 2157011 (2m), 2157007 (4j), 2157010 (6c). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All other data supporting the findings of this study are available within the article, its Supplementary Information or from the authors upon reasonable request.
References
- 1.Guillemard L, Kaplaneris N, Ackermann L & Johansson MJ Late-stage C-H functionalization offers new opportunities in drug discovery. Nat. Rev. Chem 5, 522–545, (2021). [DOI] [PubMed] [Google Scholar]
- 2.Hong BK, Luo TP & Lei XG Late-Stage Diversification of Natural Products. ACS Cent. Sci 6, 622–635, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gutekunst WR & Baran PS C-H functionalization logic in total synthesis. Chem. Soc. Rev 40, 1976–1991, (2011). [DOI] [PubMed] [Google Scholar]
- 4.Wencel-Delord J & Glorius F C-H bond activation enables the rapid construction and late-stage diversification of functional molecules. Nat. Chem 5, 369–375, (2013). [DOI] [PubMed] [Google Scholar]
- 5.Upp DM & Lewis JC Selective C-H bond functionalization using repurposed or artificial metalloenzymes. Curr. Opin. Chem. Biol 37, 48–55, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ren XK & Fasan R Engineered and artificial metalloenzymes for selective C-H functionalization. Curr. Opin. Green Sust 31, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chakrabarty S, Wang Y, Perkins JC & Narayan ARH Scalable biocatalytic C-H oxyfunctionalization reactions. Chem. Soc. Rev 49, 8137–8155, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roizen JL, Harvey ME & Du Bois J Metal-Catalyzed Nitrogen-Atom Transfer Methods for the Oxidation of Aliphatic C—H Bonds. Acc. Chem. Res 45, 911–922, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dequirez G, Pons V & Dauban P Nitrene Chemistry in Organic Synthesis: Still in Its Infancy? Angew. Chem. Int. Ed 51, 7384–7395, (2012). [DOI] [PubMed] [Google Scholar]
- 10.Park Y, Kim Y & Chang S Transition Metal-Catalyzed C-H Amination: Scope, Mechanism, and Applications. Chemical Reviews 117, 9247–9301, (2017). [DOI] [PubMed] [Google Scholar]
- 11.Royer J Chiral Amine Synthesis. Methods, Developments and Applications, (WILEY-VCH Verlag GmbH & Co., 2010). [Google Scholar]
- 12.Tahlan K & Jensen SE Origins of the beta-lactam rings in natural products. J. Antibiot 66, 401–410, (2013). [DOI] [PubMed] [Google Scholar]
- 13.Hong SY, Park Y, Hwang Y, Kim YB, Baik MH & Chang S Selective formation of gamma-lactams via C-H amidation enabled by tailored iridium catalysts. Science 359, 1016–1021, (2018). [DOI] [PubMed] [Google Scholar]
- 14.Park Y & Chang S Asymmetric formation of gamma-lactams via C-H amidation enabled by chiral hydrogen-bond-donor catalysts. Nat. Catal 2, 219–227, (2019). [Google Scholar]
- 15.Zhou ZJ, Chen SM, Hong YB, Winterling E, Tan YQ, Hemming M, Harms K, Houk KN & Meggers E Non-C-2-Symmetric Chiral-at-Ruthenium Catalyst for Highly Efficient Enantioselective Intramolecular C(sp(3))-H Amidation. J. Am. Chem. Soc 141, 19048–19057, (2019). [DOI] [PubMed] [Google Scholar]
- 16.Breslow R & Gellman SH Tosylamidation of Cyclohexane by a Cytochrome-P-450 Model. J. Chem. Soc. Chem. Comm, 1400–1401, (1982). [Google Scholar]
- 17.McIntosh JA, Coelho PS, Farwell CC, Wang ZJ, Lewis JC, Brown TR & Arnold FH Enantioselective Intramolecular C-H Amination Catalyzed by Engineered Cytochrome P450 Enzymes In Vitro and In Vivo. Angew. Chem. Int. Ed 52, 9309–9312, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh R, Bordeaux M & Fasan R P450-Catalyzed Intramolecular sp3 C—H Amination with Arylsulfonyl Azide Substrates. ACS Catal 4, 546–552, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hyster TK, Farwell CC, Buller AR, McIntosh JA & Arnold FH Enzyme-Controlled Nitrogen-Atom Transfer Enables Regiodivergent C—H Amination. J. Am. Chem. Soc 136, 15505–15508, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh R, Kolev JN, Sutera PA & Fasan R Enzymatic C(sp(3))—H Amination: P450-Catalyzed Conversion of Carbonazidates into Oxazolidinones. ACS Catal 5, 1685–1691, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steck V, Kolev JN, Ren XK & Fasan R Mechanism-Guided Design and Discovery of Efficient Cytochrome P450-Derived C-H Amination Biocatalysts. J. Am. Chem. Soc 142, 10343–10357, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Y, Cho I, Qi XT, Liu P & Arnold FH An Enzymatic Platform for the Asymmetric Amination of Primary, Secondary and Tertiary C(sp(3))—H Bonds. Nat. Chem 11, 987–993, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prier CK, Zhang RJK, Buller AR, Brinkmann-Chen S & Arnold FH Enantioselective, Intermolecular Benzylic C—H Amination Catalysed by an Engineered Iron-Haem Enzyme. Nat. Chem 9, 629–634, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Athavale SV, Gao SL, Liu Z, Mallojjala SC, Hirschi JS & Arnold FH Biocatalytic, Intermolecular C-H Bond Functionalization for the Synthesis of Enantioenriched Amides. Angew. Chem. Int. Ed 60, 24864–24869, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Z, Qin ZY, Zhu LD, Athavale SV, Sengupta A, Jia ZJ, Garcia-Borras M, Houk KN & Arnold FH An Enzymatic Platform for Primary Amination of 1-Aryl-2-alkyl Alkynes. J. Am. Chem. Soc 144, 80–85, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Y, Cho I, Qi X, Liu P & Arnold FH An enzymatic platform for the asymmetric amination of primary, secondary and tertiary C(sp3)–H bonds. Nature Chemistry 11, 987–993, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dydio P, Key HM, Hayashi H, Clark DS & Hartwig JF Chemoselective, Enzymatic C—H Bond Amination Catalyzed by a Cytochrome P450 Containing an Ir(Me)-PIX Cofactor. J. Am. Chem. Soc 139, 1750–1753, (2017). [DOI] [PubMed] [Google Scholar]
- 28.Goldberg NW, Knight AM, Zhang RJK & Arnold FH Nitrene Transfer Catalyzed by a Non-Heme Iron Enzyme and Enhanced by Non-Native Small-Molecule Ligands. J. Am. Chem. Soc 141, 19585–19588, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vila MA, Steck V, Giordano SR, Carrera I & Fasan R C-H Amination via Nitrene Transfer Catalyzed by Mononuclear Non-Heme Iron-Dependent Enzymes. Chembiochem 21, 1981–1987, (2020). [DOI] [PubMed] [Google Scholar]
- 30.van Vliet KM & de Bruin B Dioxazolones: Stable Substrates for the Catalytic Transfer of Acyl Nitrenes. ACS Catal 10, 4751–4769, (2020). [Google Scholar]
- 31.Biegasiewicz KF, Cooper SJ, Gao X, Oblinsky DG, Kim JB, Garfinkle SE, Joyce LA, Sandoval BA, Scholes GD & Hyster TK Photoexcitation of flavoenzymes enables a stereoselective radical cyclization. Science 364, 1166–+, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Black MJ, Biegasiewicz KF, Meichan AJ, Oblinsky DG, Kudisch B, Scholes GD & Hyster TK Asymmetric redox-neutral radical cyclization catalysed by flavin-dependent ‘ene’-reductases. Nat. Chem 12, 71–75, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou Q, Chin M, Fu Y, Liu P & Yang Y Stereodivergent atom-transfer radical cyclization by engineered cytochromes P450. Science 374, 1612–+, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rumo C, Stein A, Klehr J, Tachibana R, Prescimone A, Haussinger D & Ward TR An Artificial Metalloenzyme Based on a Copper Heteroscorpionate Enables sp(3) C-H Functionalization via Intramolecular Carbene Insertion. J. Am. Chem. Soc 144, 11676–11684, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kweon J & Chang S Highly Robust Iron Catalyst System for Intramolecular C(sp(3))-H Amidation Leading to gamma-Lactams. Angew. Chem. Int. Ed 60, 2909–2914, (2021). [DOI] [PubMed] [Google Scholar]
- 36.Bordeaux M, Tyagi V & Fasan R Highly Diastereoselective and Enantioselective Olefin Cyclopropanation Using Engineered Myoglobin-Based Catalysts. Angew. Chem. Int. Ed 54, 1744–1748, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bordeaux M, Singh R & Fasan R Intramolecular C(sp(3))—H Amination of Arylsulfonyl Azides with Engineered and Artificial Myoglobin-Based Catalysts. Bioorg. Med. Chem 22, 5697–5704, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pineda-Knauseder AJ, Vargas DA & Fasan R Organic solvent stability and long-term storage of myoglobin-based carbene transfer biocatalysts. Biotechnol. Appl. Bioc 67, 516–526, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deng T, Mazumdar W, Yoshinaga Y, Patel PB, Malo D, Malo T, Wink DJ & Driver TG Rh(2)(II)-Catalyzed Intermolecular N-Aryl Aziridination of Olefins Using Nonactivated N Atom Precursors. J. Am. Chem. Soc 143, 19149–19159, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sangster JJ, Marshall JR, Turner NJ & Mangas-Sanchez J New Trends and Future Opportunities in the Enzymatic Formation of C-C, C-N, and C-O bonds. Chembiochem 23, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mai BK, Neris NM, Yang Y & Liu P C-N Bond Forming Radical Rebound Is the Enantioselectivity- Determining Step in P411-Catalyzed Enantioselective C(sp(3))-H Amination: A Combined Computational and Experimental Investigation. J. Am. Chem. Soc 144, 11215–11225, (2022). [DOI] [PubMed] [Google Scholar]
- 42.Lang K, Hu Y, Lee W-CL & Zhang XP Combined radical and ionic approach for the enantioselective synthesis of beta-functionalized amines from alcohols. Nat. Synth 1, 548–557, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tinoco A, Wei Y, Bacik J-P, Carminati DM, Moore EJ, Ando N, Zhang Y & Fasan R Origin of High Stereocontrol in Olefin Cyclopropanation Catalyzed by an Engineered Carbene Transferase. ACS Catal 9 1514–1524 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crombie L, Haigh D, Jones RCF & Matzin AR Synthesis of the Alkaloid Homaline in (+/−) and Natural (S,S)-(−) Forms, Using Amination and Transamidative Ring Expansion in Liquid-Ammonia. J. Chem. Soc. Perk, 2047–2054, (1993). [Google Scholar]
- 45.Chincholkar PM, Kale AS, Gumaste VK & Deshmukh ARAS An efficient formal synthesis of (S)-dapoxetine from enantiopure 3-hydroxy azetidin-2-one. Tetrahedron 65, 2605–2609, (2009). [Google Scholar]
- 46.Wang JP, Gao H, Yang LJ & Gao YQ Role of Engineered Iron-haem Enzyme in Reactivity and Stereoselectivity of Intermolecular Benzylic C-H Bond Amination. ACS Catal 10, 5318–5327, (2020). [Google Scholar]
- 47.Kalita S, Shaik S & Dubey KD MD simulations and QM/MM calculations reveal the key mechanistic elements which are responsible for the efficient C-H amination reaction performed by a bioengineered P450 enzyme. Chemical Science 12, 14507–14518, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bajaj P, Sreenilayam G, Tyagi V & Fasan R Gram-Scale Synthesis of Chiral Cyclopropane-Containing Drugs and Drug Precursors with Engineered Myoglobin Catalysts Featuring Complementary Stereoselectivity. Angew. Chem. Int. Ed 55, 16110–16114, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ren XK, Chandgude AL & Fasan R Highly Stereoselective Synthesis of Fused Cyclopropane-gamma-Lactams via Biocatalytic Iron-Catalyzed Intramolecular Cyclopropanation. ACS Catal 10, 2308–2313, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chandgude AL, Ren X & Fasan R Stereodivergent Intramolecular Cyclopropanation Enabled by Engineered Carbene Transferases. J. Am. Chem. Soc 141, 9145–9150, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Crystallographic data for small molecules have been deposited in the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers CCDC 1893087 (2a), 2157011 (2m), 2157007 (4j), 2157010 (6c). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All other data supporting the findings of this study are available within the article, its Supplementary Information or from the authors upon reasonable request.