

Abstract
Idiopathic multicentric Castleman disease (iMCD) is a rare hematologic disorder characterized by generalized lymphadenopathy with atypical histopathological features and systemic inflammation caused by a cytokine storm involving interleukin-6 (IL-6). Three clinical subtypes are recognized: thrombocytopenia, anasarca, fever, renal dysfunction, organomegaly (iMCD-TAFRO); idiopathic plasmacytic lymphadenopathy (iMCD-IPL), involving thrombocytosis and hypergammaglobulinemia; and iMCD-not otherwise specified (iMCD-NOS), which includes patients who do not meet criteria for the other subtypes. Disease pathogenesis is poorly understood, with potential involvement of infectious, clonal, and/or autoimmune mechanisms. To better characterize iMCD clinicopathology and gain mechanistic insights into iMCD, we analyzed complete blood counts, other clinical laboratory values, and blood smear morphology among 63 iMCD patients grouped by clinical subtype. Patients with iMCD-TAFRO had large platelets, clinical severity associated with lower platelet counts, and transfusion-resistant thrombocytopenia, similar to what is observed with immune-mediated destruction of platelets in immune thrombocytopenic purpura. Conversely, elevated platelet counts in iMCD-IPL were associated with elevated IL-6 and declined following anti-IL-6 therapy. Our data suggest that autoimmune mechanisms contribute to the thrombocytopenia in at least a portion of iMCD-TAFRO patients wheareas IL-6 drives thrombocytosis in iMCD-IPL, and these mechanisms likely contribute to disease pathogenesis.
Keywords: Idiopathic multicentric Castleman disease, platelets, cytokines, peripheral blood smear
Introduction
Idiopathic multicentric Castleman disease (iMCD) is a hematological disorder characterized by generalized lymphadenopathy, polyclonal lymphoproliferation, and inflammation driven by excessive cytokines that can lead to multi-organ dysfunction and death (1). Though the proinflammatory cytokine, interleukin-6 (IL-6) is an established pathogenic driver in a proportion of patients, there is limited understanding of the etiology and pathophysiology underlying iMCD. Infectious, neoplastic, and autoimmune mechanisms have all been hypothesized to play a role in iMCD. The poor understanding of underlying mechanisms has limited the development of specific diagnostic tests and effective therapies beyond IL-6 inhibitors (2). Among patients who do not respond to first-line therapy with IL-6 blockers, few effective treatment options exist (1). Thus, a better understanding of the mechanisms involved in iMCD is needed to help identify new diagnostic tests and additional therapies.
Consensus diagnostic criteria for iMCD were developed in 2017 and require two major criteria which include characteristic lymph node histopathology and enlarged lymph nodes at multiple stations. In addition, at least 2 of 11 minor criteria must be met including at least one laboratory abnormality (3). Given the poor specificity of the diagnostic criteria, providers must also exclude infectious, malignant, and autoimmune disorders to diagnose iMCD (1). Diagnostic lymph node histopathologic features include regressed or hyperplastic germinal centers, hypervascularization, follicular dendritic cell prominence, and plasmacytosis. Lymph nodes are classified as hypervascular (regressed germinal centers and prominent vascularization), plasmacytic (hyperplastic germinal centers with prominent plasmacytosis), or mixed (overlapping features of both histopathologies) (1). However, there is frequent discordance among pathologists about the presence or absence of these subjective histopathological features, highlighting the importance of identifying new diagnostic biomarkers.
Despite iMCD patients sharing a spectrum of histopathological features, iMCD describes a heterogeneous group of inflammatory disorders (2, 4, 5) that can be further divided into three clinical subtypes (Table 1). The most aggressive and clinically severe subtype involves a constellation of abnormal laboratory and clinical features including thrombocytopenia (T), anasarca (A), fever/elevated C-reactive protein (F), reticulin myelofibrosis/renal failure (R), and organomegaly (O) also known as iMCD-TAFRO (6–9). Without prompt therapy, disease progression in iMCD-TAFRO patients can rapidly escalate to multi-organ dysfunction and death (10). Other subtypes include idiopathic plasmacytic lymphadenopathy (iMCD-IPL), which involves hypergammaglobulinemia and thrombocytosis (11, 12), and iMCD-not otherwise specified (iMCD-NOS), which does not meet the criteria for either iMCD-TAFRO or iMCD-IPL (13). Rather than exhibiting acute, life-threatening symptoms, iMCD-IPL and iMCD-NOS patients tend to experience moderate, persistent symptoms (14).
Table 1:
Criteria used to classify patients according to clinical subtypes of iMCD
| iMCD-TAFRO | iMCD-IPL | iMCD-NOS |
|---|---|---|
|
Thrombocytopenia (<100 k/uL) Anasarca Fever/ elevated CRP (>20 mg/L) Renal dysfunction (Creatinine >1.1 (F) >1.3 (M) mg/dL) / Reticulin fibrosis Organomegaly (includes lymphadenopathy) |
Idiopathic Plasmacytic Lymphadenopathy Hypergammaglobulinemia (γ globulin > 1.7 g/dL; IgG > 1700 mg/dL) Thrombocytosis (>400 k/uL) |
Not Otherwise Specified |
| Definition: (T+A+F+O) + (R|R) | Definition: Hypergammaglobulinemia + thrombocytosis | Definition: Does not achieve criteria for TAFRO or IPL |
The heterogeneous clinical features across the three clinical subtypes of iMCD suggest that divergent pathological mechanisms may lead to the defining lymph node enlargement, lymph node histology, and systemic inflammation. To identify mechanisms leading to these clinically distinct phenotypes, we analyzed quantitative clinical laboratory values and qualitative blood smear features from iMCD patients. Low platelet counts in iMCD-TAFRO were associated with the presence of large platelets with elevated mean platelet volume (MPV) and in at least a portion of patients, a lack of improvement in platelet count after platelet transfusion. Conversely, high platelet counts in iMCD-IPL were associated with elevated levels of IL-6. The data presented here uncover new insights into iMCD clinical subtypes and suggest varying mechanisms that underly iMCD pathogenesis.
Materials and Methods
Clinical and Laboratory Data
Clinical and laboratory data were obtained for 63 iMCD patients enrolled in the ACCELERATE natural history registry (NCT02817997) who met clinical and laboratory criteria for iMCD (1). Based on clinical and laboratory data using current diagnostic guidelines (1, 8), patients were categorized into iMCD-TAFRO (n=38), iMCD-IPL (n=11), and iMCD-NOS (n=14) (Table 1). Complete blood counts (CBC), included total white blood cells, monocytes, lymphocytes, neutrophils, basophils, eosinophils, platelet counts, and MPV measurements. Clinical data evaluated included hemoglobin, C-reactive protein (CRP), albumin, creatinine, immunoglobulin G (IgG) levels. CBC counts, clinical values, and peripheral blood smears evaluated for this study were all performed as part of routine clinical care. Peripheral blood count data were obtained from CBC tests performed within 90 days of disease diagnosis. Date of disease diagnosis was defined as date of lymph node resection and histopathologic confirmation of iMCD. For patients with multiple CBC tests available within 90 days of disease diagnosis, CBC data closest to date of disease diagnosis were selected for analysis. All available data on peripheral blood smear morphologic findings were included in the analysis based on the report provided by the local pathologist (Supplementary Table 1). For patients with multiple blood smears available, all abnormalities were reported once, even if not present on all smears or present on multiple smears. Multiple smears were available for 45/63 patients. Platelet transfusion data were available for 11 iMCD-TAFRO patients. Patients were considered to be refractory if the platelet count increased by less than 10 × 109/L following transfusion (15). Anti-platelet antibody tests were available for 5 iMCD-TAFRO patients.
Serum Proteomics Data
Serum samples were obtained from an independent cohort of 88 iMCD patients, with 73 pre-treatment disease flare samples collected as part of the siltuximab phase 2 study (NCT01024036) and 15 disease flare samples collected in real-world practice from 6 sites (16, 17). Samples collected in real-world practice were included to better represent the full spectrum of iMCD. Due to a lack of available clinical data, patients in the serum proteomics study were categorized into clinical subtypes by platelet counts and IgG levels only: patients with low platelets (<150 k/uL) and normal IgG (TAFRO), those with high platelets (>400 k/uL) and/or high IgG (>1700 mg/dL) (IPL), and those that didn’t fall into either category (NOS). Quantification of 1,305 protein analytes was performed using the SomaLogic SOMAscan assay, a DNA-based aptamer technology per manufacturer’s protocol (18). Values were standardized and log2 transformed. Values below the limit of detection were capped with the least detectable dose. IL-6 and TPO levels were included in the current report.
Statistical Analysis
A one-way ANOVA was used to compare laboratory values between subgroups and p-values were reported. For platelet counts and IgG, a Bonferonni’s correction was applied to account for multiple comparisons and determine significance between individual groups. For patient demographics, clinical characteristics, and peripheral blood morphologic findings, a Fisher’s exact test was used to compare features between the three clinical subtypes. To compare platelet counts pre- and post-treatment with siltuximab or placebo, paired two-sided t-tests were used. Spearman’s correlation coefficient was used to determine the relationship between log2-transformed IL-6 values and platelets or TPO levels. A one-way ANOVA was used to compare IL-6 levels between the clinical subgroups. A Bonferroni correction was applied to account for multiple comparisons. All analyses were performed using GraphPad Prism Version 9.3.1.
Results
Patient Characteristics
To identify differences among iMCD clinical subtypes, we first compared patient characteristics and clinical laboratory values. Of the 63 iMCD patients included in this study, 34 were male and 29 were female with an average age at diagnosis of 35.6 years (range: 1.8–55.8 years) (Table 2). Histopathological analysis showed that lymph nodes from iMCD-TAFRO patients were more frequently hypervascular in appearance compared to iMCD-IPL patients (p=0.0003), whereas plasmacytic lymph node features were more common in iMCD-IPL patients (p=0.0007). The analysis also identified differences in the proportions of individuals from various races across the three clinical subgroups (Table 2).
Table 2:
Patient Demographics and Clinical Characteristics
| iMCD Total n=63 |
iMCD-TAFRO n=38 |
iMCD-IPL n=11 |
iMCD-NOS n=14 |
p-value‡ | |
|---|---|---|---|---|---|
| Gender, n (%) | |||||
| Male | 34 (54) | 22 (58) | 4 (36) | 8 (57) | 0.492 |
| Female | 29 (46) | 16 (42) | 7 (64) | 6 (43) | |
| Age at diagnosis | |||||
| Mean (SD) | 35.6 (16) | 33.6 (18) | 42.5 (10) | 35.5 (14) | 0.294 |
| Self-reported race, n (%) | |||||
| Black | 7 (11) | 3 (8) | 0 | 4 (29) | 0.0183 |
| White | 39 (62) | 27 (71) | 4 (36) | 8 (57) | |
| Asian | 9 (14) | 3 (8) | 5 (46) | 1 (7) | |
| Other | 8 (13) | 5 (13) | 2 (18) | 1 (7) | |
| Histopathological subtype, n (%) | |||||
| Hypervascular | 39 (62) | 29 (76) | 1 (9) | 9 (64) | <0.0001 |
| Mixed | 16 (25) | 9 (24) | 3 (27) | 4 (29) | |
| Plasmacytic | 5 (8) | 0 | 5 (46) | 0 | |
| Unknown | 3 (5) | 0 | 2 (18) | 1 (7) | |
| Constitutional symptoms, n (%) | |||||
| Yes | 60 (95) | 37 (97) | 10 (91) | 13 (93) | 0.345 |
| No | 3 (5) | 1 (3) | 1 (9) | 1 (7) | |
| Not assessed | 0 | 0 | 0 | 0 | |
| Organomegaly, n (%) | |||||
| Yes | 49 (79) | 35 (92) | 4 (36) | 10 (77) | 0.0005 |
| No | 13 (21) | 3 (8) | 7 (64) | 3 (23) | |
| Not assessed | 1 | 0 | 0 | 1 | |
| Fluid accumulation, n (%) | |||||
| Yes | 51 (86) | 38 (100) | 5 (56) | 8 (67) | 0.0001 |
| No | 8 (14) | 0 | 4 (44) | 4 (33) | |
| Not assessed | 4 | 0 | 2 | 2 |
p-values were generated from Fisher’s exact analysis of iMCD-TAFRO, iMCD-IPL, and iMCD-NOS.
As expected because specific clinical criteria were used to group patients, iMCD-TAFRO patients had greater proportions of organomegaly and fluid accumulation as well as lower platelet counts compared to the other two clinical subtypes (Table 2 and Table 3). Accordingly, iMCD-IPL patients had the highest platelets and highest IgG levels (Table 3 and Supplemental Figure 1). iMCD-TAFRO patients had the lowest platelet counts among the three groups and had a significantly higher MPV compared to iMCD-IPL patients (Figure 1A and Supplemental Figure 1). Interestingly, the platelet count was negatively associated with MPV (Figure 1B). The mean platelet count of 122.6 × 109/L for iMCD-TAFRO patients was higher than expected. One possible explanation for this is that the platelet counts used were observed closest to the date of iMCD diagnosis, which does not necessarily capture the patients’ lowest platelet value. We repeated our analysis using the lowest platelet levels ever recorded per patient among the subgroups (Supplemental Figure 2). In this analysis, the median platelet count for iMCD-TAFRO patients was 22.5 × 109/L with 9/38 (23.4%) exhibiting platelet counts less than 10 × 109/L. Additionally, the differences in platelet count between iMCD-TAFRO and the other two subtypes were more pronounced. The platelet counts in iMCD-TAFRO were, on average, more than 7-fold and 4.5-fold lower compared to iMCD-IPL or iMCD-NOS patients, respectively. These data suggest that the differences reported among the subgroups may be even more significant when patients experience the worst of their disease.
Table 3:
Laboratory values across iMCD subtypes
| Normal Range | iMCD Total | iMCD-TAFRO | iMCD-IPL | iMCD-NOS | p-value‡ | |
|---|---|---|---|---|---|---|
|
White blood cells (109/L) Mean (SD), n |
4.5 – 11.0 | 10.9 (5.8), 63 | 12.3 (6.4), 38 | 7.83 (3.3), 11 | 9.50 (4.7), 14 | 0.0460 |
|
Absolute neutrophils (109/L) Mean (SD), n |
1.8 – 7.7 | 6.9 (3.9), 60 | 7.43 (3.9), 35 | 5.52 (3.5), 11 | 6.41 (4.1), 14 | 0.331 |
|
Absolute lymphocytes (109/L) Mean (SD), n |
1.0 – 4.8 | 1.80 (1.1), 58 | 1.85 (1.2), 35 | 1.31 (0.4), 10 | 2.23 (1.0), 13 | 0.131 |
|
Absolute monocytes (109/L) Mean (SD), n |
0.2 – 1.2 | 0.73 (0.6), 59 | 0.79 (0.6), 35 | 0.63 (0.4), 10 | 0.63 (0.5), 14 | 0.551 |
|
Absolute eosinophils (109/L) Mean (SD), n |
0.0 – 0.9 | 0.10 (0.1), 57 | 0.08 (0.1), 33 | 0.11 (0.1), 10 | 0.15 (0.1), 14 | 0.0965 |
|
Absolute basophils (109/L) Mean (SD), n |
0.0 – 0.3 | 0.03 (0.1), 56 | 0.04 (0.1), 34 | 0.04 (0.1), 9 | 0.02 (0.02), 13 | 0.776 |
|
Platelets (109/L) Mean (SD), n |
150 – 400 | 212 (173), 61 | 123 (100), 36 | 440 (200), 11 | 271 (107), 14 | <0.0001 |
|
MPV (fL) Mean (SD), n |
6.0 – 13.2 | 9.64 (2.1), 60 | 10.3 (1.8), 38 | 7.65 (1.5), 10 | 9.09 (2.2), 12 | 0.0019 |
|
Hemoglobin (g/dL) Mean (SD), n |
13.5 – 17.5 | 10.2 (2), 63 | 9.7 (2), 38 | 10.3 (3), 11 | 11.5 (2), 14 | 0.0385 |
|
CRP (mg/L) Mean (SD), n |
> 10 | 99.3 (93), 48 | 111 (101), 33 | 112 (68), 8 | 31.0 (39), 7 | 0.107 |
|
Albumin (g/dL) Mean (SD), n |
3.5 – 5.5 | 2.7 (1), 62 | 2.4 (1), 38 | 2.9 (1), 11 | 3.6 (1), 13 | <0.0001 |
|
Creatinine (mg/dL) Mean (SD), n |
0.6 – 1.3 | 1.2 (1), 62 | 1.3 (1), 38 | 0.8 (0.3), 11 | 1.2 (1), 13 | 0.129 |
|
IgG (mg/dL) Mean (SD), n |
600 – 1700 | 1,989 (1,758), 51 | 1,254 (927), 32 | 4,773 (1,728), 10 | 1,504 (716), 9 | <0.0001 |
p-values were generated from one-way ANOVA analysis of iMCD-TAFRO, iMCD-IPL, and iMCD-NOS.
Figure 1. Higher MPV values detected in iMCD patients with thrombocytopenia.
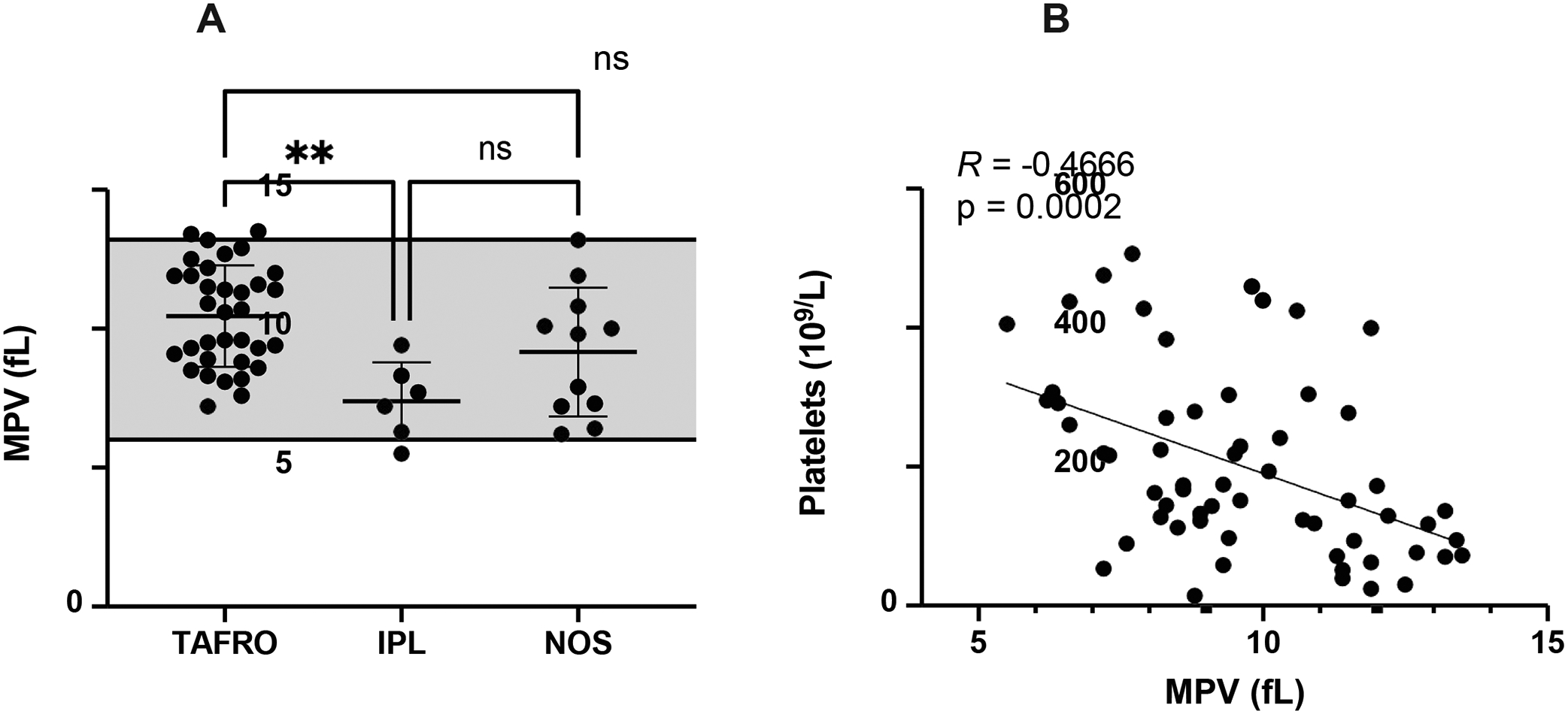
(A) MPV values in iMCD subtypes. The shaded region represents the normal range. (B) The relationship between platelet count and MPV in iMCD as determined by Spearman correlation (R = −0.4666; p=0.0002). TAFRO, n=38; IPL, n=6; NOS, n=11. **p<0.01.
iMCD-TAFRO patients also had the lowest hemoglobin and albumin levels, which is consistent with the published literature (6, 10). Though many blood analytes, such as C-reactive protein, hemoglobin, albumin, and creatinine were abnormal or borderline abnormal across iMCD patients, white blood cell count was the only cellular measurement different between subgroups, which was slightly above the normal range in iMCD-TAFRO patients (Table 3).
Large platelets in iMCD-TAFRO are among the most common peripheral blood morphologic abnormalities
Inflammation and autoimmunity can cause changes in blood cell size, shape, and color (19). To identify potential blood morphological features, we quantified abnormalities described by local pathologists on blood smears and compared their frequencies between subtypes. While iMCD-TAFRO patients demonstrated nominally higher frequencies of the most common morphological irregularities, a range of abnormalities in platelets, red blood cells (RBCs), and white blood cells was noted in all three clinical subtypes (Table 4). The phenotypes captured would not be expected in healthy individuals but can be found in a variety of other diseases (19).
Table 4.
Peripheral blood smear morphologic findings in iMCD
| iMCD Total n = 63 |
iMCD-TAFRO n = 38 |
iMCD-IPL n = 11 |
iMCD-NOS n = 14 |
p-value‡ | |
|---|---|---|---|---|---|
| Platelets | |||||
| Large platelets, n (%) | 30 (47.6) | 23 (60.5) | 2 (18.2) | 5 (35.7) | 0.0260 |
| RBC shape | |||||
| Poiklocytosis, n (%) | 27 (42.9) | 21 (55.2) | 4 (36.4) | 2 (14.3) | 0.0253 |
| Schistocytes, n (%) | 21 (33.3) | 18 (47.4) | 1 (9.09) | 2 (14.3) | 0.0175 |
| Teardrops, n (%) | 20 (31.7) | 19 (50) | 0 (0) | 1 (7.1) | <0.001 |
| Ovalocytes, n (%) | 27 (47.6) | 20 (52.6) | 4 (36.4) | 3 (21.4) | 0.118 |
| RBC size | |||||
| Anisocytosis, n (%) | 37 (58.7) | 27 (71.1) | 3 (27.3) | 7 (50) | 0.0240 |
| Microcytosis, n (%) | 32 (50.8) | 20 (52.6) | 6 (54.5) | 6 (42.9) | 0.827 |
| Macrocytosis, n (%) | 20 (31.7) | 15 (39.5) | 2 (18.1) | 3 (21.4) | 0.348 |
| RBC color | |||||
| Hypochromia, n (%) | 36 (57.1) | 27 (71.1) | 4 (36.4) | 5 (35.7) | 0.0218 |
| Polychromasia, n (%) | 37 (58.7) | 26 (68.4) | 6 (54.5) | 5 (35.7) | 0.0988 |
| WBC features | |||||
| Atypical lymphocytes, n (%) | 8 (12.7) | 5 (13.2) | 1 (9.1) | 2 (14.3) | 1 |
| Dohle Bodies, n (%) | 4 (6.3) | 4 (10.5) | 0 | 0 | 0.464 |
| Hypersegmented polymorphonuclear leukocytes, n (%) | 5 (7.9) | 5 (13.2) | 0 | 0 | 0.266 |
| Left Shift, n (%) | 3 (4.8) | 3 (7.9) | 0 | 0 | 0.752 |
| Pseudo-Pelger-Huët, n (%) | 1 (1.6) | 1 (2.6) | 0 | 0 | 1 |
| Smudge cells, n (%) | 5 (7.9) | 4 (10.5) | 0 | 1 (7.1) | 0.815 |
| Toxic granulation, n (%) | 11 (17.5) | 11 (28.9) | 0 | 0 | 0.0073 |
| Toxic vacuolization, n (%) | 8 (12.7) | 8 (21.1) | 0 | 0 | 0.0715 |
p-values were generated from Fisher’s exact analysis of iMCD TAFRO, iMCD-IPL, and iMCD-NOS.
Red blood cell morphologic findings across iMCD patients included abnormalities in size, such as anisocytosis (n = 37/63, 59%), microcytosis (n = 32/63, 51%), macrocytosis (n = 20/63, 32%), and abnormalities in color, such as hypochromia (n = 36/63, 57%), and polychromasia (n = 37/63, 59%). Abnormalities in shape, such as poikilocytosis (n = 27/63, 43%), schistocytosis, teardrops and ovalocytes, were also reported frequently. White blood cell morphologic abnormalities, such as toxic granulation and toxic vacuolization, were more prominent in iMCD-TAFRO cases but were noted less frequently overall.
Between clinical subtypes, we discovered a range of differences in blood smear features. In particular, large platelets were observed to be approximately 3-times and 2-times more frequent in iMCD-TAFRO than iMCD-IPL and iMCD-NOS, respectively. The higher frequency of large platelets reported in iMCD-TAFRO patients mirrored the higher MPV values measured in iMCD-TAFRO patients (Table 3 and Figure 1). Together, these data suggest that newly formed platelets, which tend to be larger, are associated with the characteristic thrombocytopenia in iMCD-TAFRO; however, the cause of these platelet abnormalities remains unknown.
iMCD-TAFRO patients can exhibit refractoriness to platelet transfusion
Megakaryocytic hyperplasia in the bone marrow is a common feature of iMCD-TAFRO suggesting that platelet production is likely increased to compensate for the loss of platelets in circulation (20). Potential causes of thrombocytopenia in iMCD-TAFRO other than a decrease in platelet production include increased platelet turnover or activation, peripheral destruction, or sequestration. Given the large platelets with elevated MPV and single-digit platelet counts observed in iMCD-TAFRO, which are also characteristics of immune thrombocytopenic purpura (ITP) (21–23), we hypothesized that one mechanism contributing to the severe thrombocytopenia in iMCD-TAFRO could be immune-mediated platelet destruction that occurs in ITP. We evaluated this by assessing changes in platelet counts following platelet transfusions, as counts do not significantly increase following transfusions in ITP due to immune-mediated destruction (24). Among the eleven iMCD-TAFRO patients where platelet counts were measured within 24 hours after receiving platelet transfusions, only one (9.1%) achieved a clinically significant increase (>30 × 109/L) following transfusion (Figure 2). Three iMCD-TAFRO patients had platelet increments, the change in platelet counts before and after transfusion, under 5 × 109/L within 4 hours after transfusion and were considered refractory. An additional 7 iMCD-TAFRO patients had a change in platelet count less than 30 × 109/L. Anti-platelet antibodies were measured in only five iMCD-TAFRO patients and three of these patients had positive tests. One of the three patients was tested prior to platelet transfusion and can be confirmed to have platelet autoantibodies while the others were tested after at least one platelet transfusion so they may have developed anti-platelet alloantibodies against the transfusion. More data are needed to determine the frequency of iMCD-TAFRO patients with self-reactive anti-platelet antibodies. These data suggest immune-mediated destruction of platelets, which occurs in ITP, may contribute to thrombocytopenia in at least a proportion of iMCD-TAFRO patients. In addition to anti-platelet antibodies, patients with ITP also typically have antibodies against RBCs, suggesting that the immune-mediated destruction is non-specific. Interestingly, 8/14 (57%) of iMCD-TAFRO patients tested for the Coomb’s test had positive results, indicating the presence of antibodies against RBCs. The presence of antibodies against RBCs may support the possibility of immune-mediated mechanisms underlying the hematological abnormalities in at least a portion of iMCD-TAFRO patients. However, further work is needed to investigate this possibility.
Figure 2. Minimal change in platelet count following platelet transfusion in iMCD-TAFRO patients.
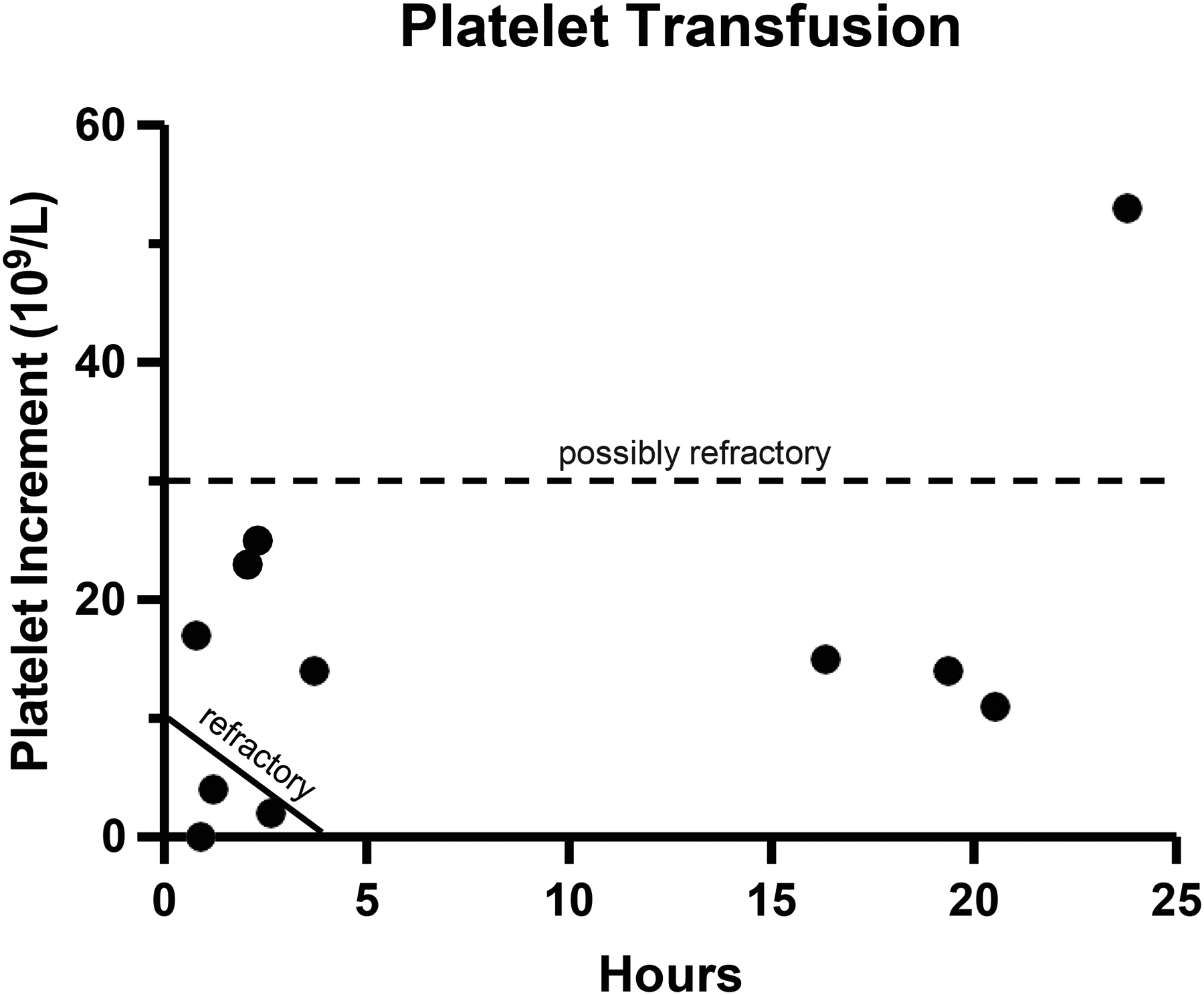
(A) Platelet increment, the change in platelet count pre- and post-transfusion, is provided versus time in hours after platelet transfusion. Each dot indicates one patient. n=11. The platelet count taken closest to the time of transfusion was used if multiple platelet counts were measured.
Increased IL-6 levels are associated with elevated platelets in iMCD-IPL
Unlike iMCD-TAFRO patients who exhibit severe thrombocytopenia and large platelets that may, in part, be driven by immune-mediated destruction, iMCD-IPL patients have thrombocytosis that occurs by an unknown mechanism. Given that IL-6 is known to promote megakaryopoiesis and platelet production in other contexts (25–28), we investigated whether elevated IL-6 levels could account for thrombocytosis in iMCD-IPL. To determine if IL-6 also promotes platelet production in iMCD, we utilized data from a previously published study where serum samples were analyzed before and after IL-6 blockade with siltuximab. Before siltuximab treatment, we discovered a significant association between serum IL-6 and platelet counts across iMCD clinical subtypes (Figure 3A). Interestingly, IL-6 was also positively correlated with the megakaryopoiesis-driver thrombopoietin (TPO) (Figure 3B). Although these patients could not be appropriately subtyped due to limited clinical information at the time of diagnosis, we used available clinical laboratory data to group iMCD patients into likely clinical subtypes and compared their serum IL-6 levels. Patients with high platelets and/or high IgG, considered iMCD-IPL, had significantly higher levels of IL-6 in circulation compared to patients with low (iMCD-TAFRO) or normal platelets (iMCD-NOS) (Figure 3C). After treatment with siltuximab, both platelet and TPO levels fell indicating that IL-6 drives platelet production by promoting megakaryopoiesis in iMCD (Figure 3D–E). Together, these data suggest that elevated serum IL-6 mediates thrombocytosis in iMCD-IPL.
Figure 3. IL-6 levels associated with increased platelet count in iMCD.
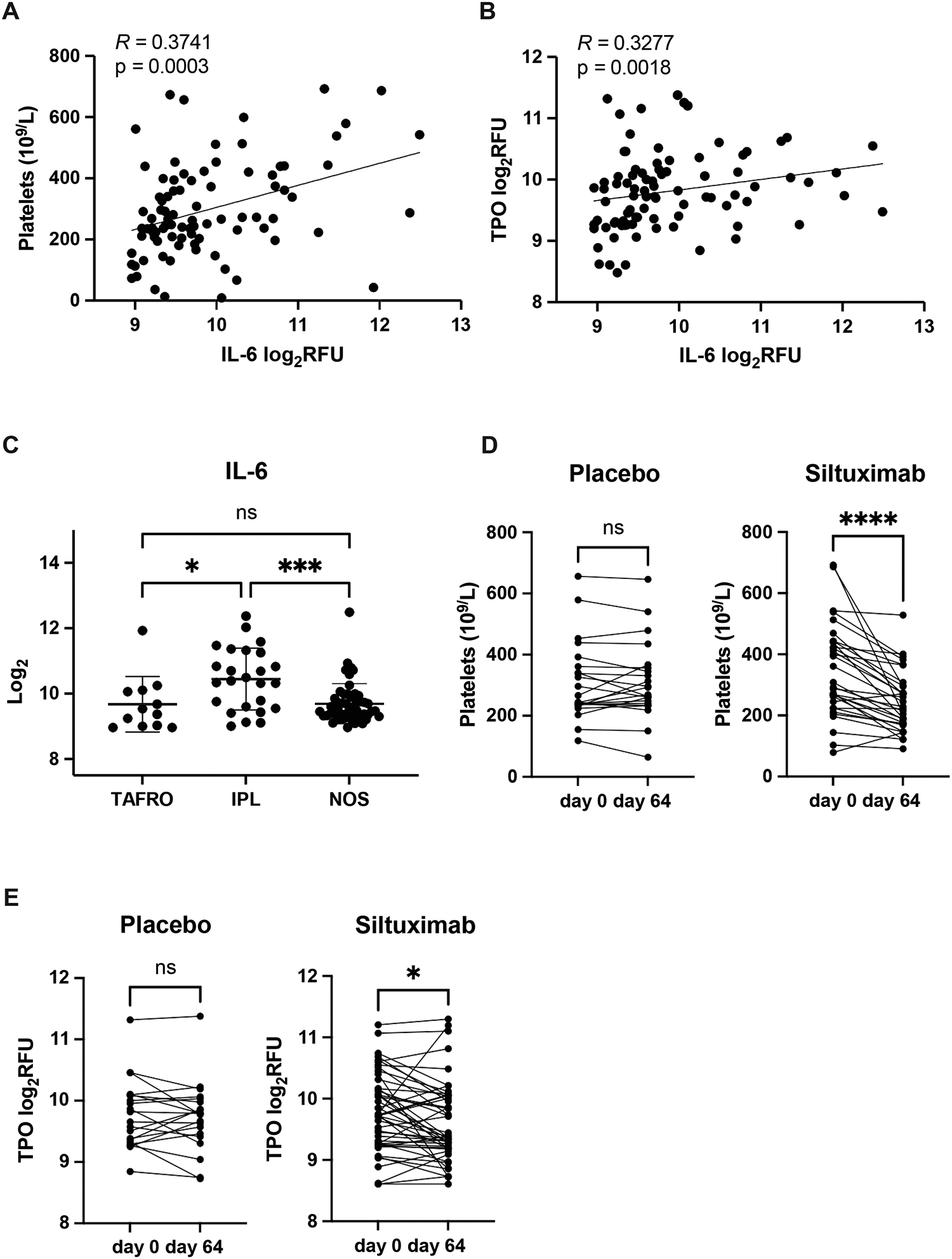
Spearman correlation between log2-transformed IL-6 values and (A) platelet counts (R = 0.3741; p=0.0003) and (B) thrombopoietin (TPO) (R = 0.3277; p=0.0018) in n=88 iMCD patients. (C) Log2-transformed IL-6 values across TAFRO-(n = 12), IPL- (n = 25), and NOS- (n = 51) iMCD patients. Data are mean ± SD. The change in (D) platelet count and (E) TPO in iMCD patients following administration of anti-IL-6 blockade with siltuximab (platelets, n=50; TPO, n=48) or placebo (platelets, n=22; TPO, n=20). **p < 0.01, ***p < 0.001, ****p < 0.0001.
Discussion
In this study, we report laboratory abnormalities and morphological findings in a large cohort of iMCD patients and suggest mechanisms underlying abnormalities in platelet counts often present in iMCD-TAFRO and iMCD-IPL clinical subtypes. This study is the largest and most comprehensive analysis of peripheral blood morphologic findings in iMCD to date.
Interestingly, we observed large platelets with elevated MPV in a majority of iMCD-TAFRO patients, which has not previously been reported. Large platelets can be found in other disorders including ITP, myeloproliferative disorders, pseudothrombocytopenia, and inherited platelet disorders (22, 29–33) and suggests accelerated platelet production and early release into circulation (33). Megakaryocytic hyperplasia in the bone marrow, which was previously reported in iMCD-TAFRO (20), would be expected in patients with large platelets with elevated MPV and likely represents a compensatory response to the severe thrombocytopenia.
The minimal response to platelet transfusion (platelet increments of less than 5 ×109/L) in 3/11 iMCD-TAFRO patients suggests immune-mediated platelet destruction as one mechanism promoting thrombocytopenia in a portion of iMCD-TAFRO patients. Although platelet refractoriness calculations typically take into account body surface area and the number of platelets transfused, an increase of less than 10 × 109/L is considered refractory (15). As has been described previously (34), we reasoned that the very small increase (< 5 × 109/L) in platelet count following transfusion observed in a portion of iMCD-TAFRO patients was likely immune-mediated, which can be caused by anti-platelet antibodies. The remaining 7 patients (63.6%) that had platelet increments under 30 × 109/L likely exhibited platelet refractoriness, but the mechanism underlying the platelet refractoriness is currently unknown. We found anti-platelet antibodies in three of the five iMCD-TAFRO patients for which we had data and at least one definitively had anti-platelet autoantibodies since the screening was done prior to at least one platelet transfusion. It is possible that other immune-mediated mechanisms, such as inflammation-driven platelet activation or T-cell mediated platelet destruction that has been implicated in rare cases of ITP (35), or non-immune-mediated mechanisms such as peripheral platelet sequestration may account for severe thrombocytopenia in iMCD-TAFRO. However, the mechanism underlying the thrombocytopenia across all iMCD-TAFRO patients is still not known.
Patients with the iMCD-IPL subtype demonstrate thrombocytosis and hypergammaglobulinemia from an unknown cause. IL-6, which is a known disease driver in a portion of iMCD patients, has been shown in other settings to stimulate thrombopoiesis. Accordingly, administration of IL-6 is associated with an increase in circulating platelet counts (36–42). Likewise, we found in an independent cohort that IL-6 promoted platelet production in iMCD and was significantly higher in patients with high platelets and that platelet counts reduced significantly upon IL-6 inhibition. Interestingly, we found that TPO, which drives megakaryopoiesis, was also positively associated with IL-6. These data suggest that IL-6-driven thrombopoiesis is one mechanism that could explain the thrombocytosis observed in iMCD-IPL. Among patients with iMCD-NOS, who do not have thrombocytopenia or thrombocytosis, it is interesting to consider the possibility that there is simultaneous immune-mediated platelet destruction and IL-6-driven thrombopoeisis.
There were also a number of morphological changes beyond large platelets worth noting. The most common findings in this cohort included abnormalities in RBC shape, size, and color, such as poikilocytosis, schistocytes, teardrops, and ovalocytes, which can be seen with immune-mediated RBC destruction and myelofibrosis, as well as anisocytosis, microcytosis, macrocytosis, hypochromia, and polychromia. These abnormalities can be found in a number of conditions, including myelofibrosis, autoimmune diseases, and other inflammatory conditions (19). In iMCD, they may be related to cytokine-driven anemia of chronic inflammation. While the WBC morphological abnormalities were less common, they almost all occurred in iMCD-TAFRO. The high proportion of toxic granulation and vacuolization in neutrophils, which is a non-specific indication of excessive inflammation, is striking and warrants future studies.
Our study has a number of limitations. There was no concomitant control group of healthy individuals against which to compare blood smear findings in iMCD. Nonetheless, it is well-established that the peripheral blood smear abnormalities frequently observed in this cohort are rarely observed in healthy individuals (19). An additional limitation is that the blood smears were analyzed by multiple hematopathologists, potentially leading to variability in results. However, given that guidelines for blood smear analysis are widely standardized this effect is unlikely to alter the results.
In summary, we observed abnormal blood cell morphologies and other laboratory tests in iMCD with notable differences between subtypes. A portion of iMCD-TAFRO patients demonstrated thrombocytopenia that was resistant to platelet transfusion as well as large platelets with elevated MPV. In contrast, elevated platelet counts in iMCD-IPL patients were associated with high levels of IL-6 and declined following anti-IL-6 treatment with siltuximab. Our study revealed fundamental differences in laboratory findings in a large cohort of iMCD patients and suggests that distinct disease mechanisms underlie the platelet abnormalities in iMCD subtypes and may play further mechanistic roles in disease pathogenesis. Future studies are needed to determine the therapeutic implications of these findings.
Supplementary Material
Acknowledgments
The authors would like to thank all the patients and their families for their participation in the registry. This research was supported by the National Heart, Lung, & Blood Institute (R01HL141408) and the Castleman Disease Collaborative Network. The ACCELERATE natural history registry has received funding from Janssen Pharmaceuticals (2016 - 2018), EUSA Pharma, LLC (US), which has merged with Recordati Rare Diseases Inc. (2018 - 2022), and is now supported by the U.S. Food & Drug Administration (R01FD007632) (2022 - Present).
Funding Statement
The funders of this study had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest Disclosure
D.C.F. has received research funding for the ACCELERATE registry and consulting fees from EUSA Pharma and has two provisional patents pending related to the diagnosis and treatment of iMCD.
Footnotes
Ethics Approval Statement
The study protocol was approved by the University of Pennsylvania IRB. All procedures performed in this study involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Patient Consent Statement
Informed consent was obtained from all individual participants included in the study.
Permission to Reproduce Material from Other Sources
Permission to reproduce copyrighted material from other sources has been obtained, and proper acknowledgments are provided in the manuscript.
Clinical Trial Registration
This study is part of the ACCELERATE natural history registry (NCT02817997). The trial was registered on June 29, 2016 at ClinicalTrials.gov.
Data Availability
All source data reported in this study are available by contacting the ACCELERATE team at accelerate@uphs.upenn.edu.
References
- 1.Fajgenbaum DC, Uldrick TS, Bagg A, Frank D, Wu D, Srkalovic G, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood. 2017;129(12):1646–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fajgenbaum DC. Novel insights and therapeutic approaches in idiopathic multicentric Castleman disease. Blood. 2018;132(22):2323–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dispenzieri A, Fajgenbaum DC. Overview of Castleman disease. Blood. 2020;135(16):1353–64. [DOI] [PubMed] [Google Scholar]
- 4.Dispenzieri A, Armitage JO, Loe MJ, Geyer SM, Allred J, Camoriano JK, et al. The clinical spectrum of Castleman’s disease. Am J Hematol. 2012;87(11):997–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oksenhendler E, Carcelain G, Aoki Y, Boulanger E, Maillard A, Clauvel JP, et al. High levels of human herpesvirus 8 viral load, human interleukin-6, interleukin-10, and C reactive protein correlate with exacerbation of multicentric castleman disease in HIV-infected patients. Blood. 2000;96(6):2069–73. [PubMed] [Google Scholar]
- 6.Iwaki N, Fajgenbaum DC, Nabel CS, Gion Y, Kondo E, Kawano M, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J Hematol. 2016;91(2):220–6. [DOI] [PubMed] [Google Scholar]
- 7.Iwaki N, Sato Y, Takata K, Kondo E, Ohno K, Takeuchi M, et al. Atypical hyaline vascular-type castleman’s disease with thrombocytopenia, anasarca, fever, and systemic lymphadenopathy. J Clin Exp Hematop. 2013;53(1):87–93. [DOI] [PubMed] [Google Scholar]
- 8.Masaki Y, Kawabata H, Takai K, Tsukamoto N, Fujimoto S, Ishigaki Y, et al. Proposed diagnostic criteria, disease severity classification, and treatment strategy for a novel disorder; TAFRO syndrome. Rinsho Ketsueki. 2016;57(10):2029–37. [DOI] [PubMed] [Google Scholar]
- 9.Takai K, Nikkuni K, Shibuya H, Hashidate H. [Thrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegaly]. Rinsho Ketsueki. 2010;51(5):320–5. [PubMed] [Google Scholar]
- 10.Nishimura Y, Fajgenbaum DC, Pierson SK, Iwaki N, Nishikori A, Kawano M, et al. Validated international definition of the thrombocytopenia, anasarca, fever, reticulin fibrosis, renal insufficiency, and organomegaly clinical subtype (TAFRO) of idiopathic multicentric Castleman disease. Am J Hematol. 2021;96(10):1241–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawabata H, Takai K, Kojima M, Nakamura N, Aoki S, Nakamura S, et al. Castleman-Kojima disease (TAFRO syndrome) : a novel systemic inflammatory disease characterized by a constellation of symptoms, namely, thrombocytopenia, ascites (anasarca), microcytic anemia, myelofibrosis, renal dysfunction, and organomegaly : a status report and summary of Fukushima (6 June, 2012) and Nagoya meetings (22 September, 2012). J Clin Exp Hematop. 2013;53(1):57–61. [DOI] [PubMed] [Google Scholar]
- 12.Nishikori A, Nishimura MF, Nishimura Y, Otsuka F, Maehama K, Ohsawa K, et al. Idiopathic Plasmacytic Lymphadenopathy Forms an Independent Subtype of Idiopathic Multicentric Castleman Disease. Int J Mol Sci. 2022;23(18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pierson SK, Stonestrom AJ, Shilling D, Ruth J, Nabel CS, Singh A, et al. Plasma proteomics identifies a ‘chemokine storm’ in idiopathic multicentric Castleman disease. Am J Hematol. 2018;93(7):902–12. [DOI] [PubMed] [Google Scholar]
- 14.Bustamante S, Pierson SK, Ren Y Bagg A, Brandstadter JD, Srkalovic G, Mango N, Alapat d., Lechowicz MJ, Lee H, van Rhee F, Lim MS, Fajgenbaum DC Longitudinal, natural history study reveals the disease burden of idiopathic multicentric Castleman disease. Haematologica. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Slichter SJ, Davis K, Enright H, Braine H, Gernsheimer T, Kao KJ, et al. Factors affecting posttransfusion platelet increments, platelet refractoriness, and platelet transfusion intervals in thrombocytopenic patients. Blood. 2005;105(10):4106–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pierson SK, Shenoy S, Oromendia AB, Gorzewski AM, Langan Pai RA, Nabel CS, et al. Discovery and validation of a novel subgroup and therapeutic target in idiopathic multicentric Castleman disease. Blood Adv. 2021;5(17):3445–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Rhee F, Wong RS, Munshi N, Rossi JF, Ke XY, Fossa A, et al. Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2014;15(9):966–74. [DOI] [PubMed] [Google Scholar]
- 18.Gold L, Ayers D, Bertino J, Bock C, Bock A, Brody EN, et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One. 2010;5(12):e15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lynch EC. Peripheral Blood Smear. In: Walker HK, Hall WD, Hurst JW, editors. Clinical Methods: The History, Physical, and Laboratory Examinations. 3rd ed. Boston: 1990. [PubMed] [Google Scholar]
- 20.Belyaeva E, Rubenstein A, Pierson SK, Dalldorf D, Frank D, Lim MS, et al. Bone marrow findings of idiopathic Multicentric Castleman disease: A histopathologic analysis and systematic literature review. Hematol Oncol. 2022;40(2):191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med. 2002;346(13):995–1008. [DOI] [PubMed] [Google Scholar]
- 22.Illes I, Pfueller SL, Hussein S, Chesterman CN, Martin JF. Platelets in idiopathic thrombocytopenic purpura are increased in size but are of normal density. Br J Haematol. 1987;67(2):173–6. [DOI] [PubMed] [Google Scholar]
- 23.McMillan R Antiplatelet antibodies in chronic adult immune thrombocytopenic purpura: assays and epitopes. J Pediatr Hematol Oncol. 2003;25 Suppl 1:S57–61. [DOI] [PubMed] [Google Scholar]
- 24.Neunert C, Lim W, Crowther M, Cohen A, Solberg L Jr, Crowther MA, et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011;117(16):4190–207. [DOI] [PubMed] [Google Scholar]
- 25.Ishibashi T, Kimura H, Uchida T, Kariyone S, Friese P, Burstein SA. Human interleukin 6 is a direct promoter of maturation of megakaryocytes in vitro. Proc Natl Acad Sci U S A. 1989;86(15):5953–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lotem J, Shabo Y, Sachs L. Regulation of megakaryocyte development by interleukin-6. Blood. 1989;74(5):1545–51. [PubMed] [Google Scholar]
- 27.Stahl CP, Zucker-Franklin D, Evatt BL, Winton EF. Effects of human interleukin-6 on megakaryocyte development and thrombocytopoiesis in primates. Blood. 1991;78(6):1467–75. [PubMed] [Google Scholar]
- 28.Warren MK, Conroy LB, Rose JS. The role of interleukin 6 and interleukin 1 in megakaryocyte development. Exp Hematol. 1989;17(11):1095–9. [PubMed] [Google Scholar]
- 29.Bolton-Maggs PH, Chalmers EA, Collins PW, Harrison P, Kitchen S, Liesner RJ, et al. A review of inherited platelet disorders with guidelines for their management on behalf of the UKHCDO. Br J Haematol. 2006;135(5):603–33. [DOI] [PubMed] [Google Scholar]
- 30.Cortelazzo S, Barbui T, Bassan R, Dini E. Abnormal aggregation and increased size of platelets in myeloproliferative disorders. Thromb Haemost. 1980;43(2):127–30. [PubMed] [Google Scholar]
- 31.Lardinois B, Favresse J, Chatelain B, Lippi G, Mullier F. Pseudothrombocytopenia-A Review on Causes, Occurrence and Clinical Implications. J Clin Med. 2021;10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noris P, Klersy C, Gresele P, Giona F, Giordano P, Minuz P, et al. Platelet size for distinguishing between inherited thrombocytopenias and immune thrombocytopenia: a multicentric, real life study. Br J Haematol. 2013;162(1):112–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robier C Platelet Morphology. Journal of Laboratory Medicine. 2020;44(5):231–9. [Google Scholar]
- 34.Pai RL, Japp AS, Gonzalez M, Rasheed RF, Okumura M, Arenas D, et al. Type I IFN response associated with mTOR activation in the TAFRO subtype of idiopathic multicentric Castleman disease. JCI Insight. 2020;5(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vrbensky JR, Nazy I, Clare R, Larche M, Arnold DM. T cell-mediated autoimmunity in immune thrombocytopenia. Eur J Haematol. 2022;108(1):18–27. [DOI] [PubMed] [Google Scholar]
- 36.D’Hondt V, Humblet Y, Guillaume T, Baatout S, Chatelain C, Berliere M, et al. Thrombopoietic effects and toxicity of interleukin-6 in patients with ovarian cancer before and after chemotherapy: a multicentric placebo-controlled, randomized phase Ib study. Blood. 1995;85(9):2347–53. [PubMed] [Google Scholar]
- 37.Gordon MS, Nemunaitis J, Hoffman R, Paquette RL, Rosenfeld C, Manfreda S, et al. A phase I trial of recombinant human interleukin-6 in patients with myelodysplastic syndromes and thrombocytopenia. Blood. 1995;85(11):3066–76. [PubMed] [Google Scholar]
- 38.Lazarus HM, Winton EF, Williams SF, Grinblatt D, Campion M, Cooper BW, et al. Phase I multicenter trial of interleukin 6 therapy after autologous bone marrow transplantation in advanced breast cancer. Bone Marrow Transplant. 1995;15(6):935–42. [PubMed] [Google Scholar]
- 39.Schrezenmeier H, Marsh JC, Stromeyer P, Muller H, Heimpel H, Gordon-Smith EC, et al. A phase I/II trial of recombinant human interleukin-6 in patients with aplastic anaemia. Br J Haematol. 1995;90(2):283–92. [DOI] [PubMed] [Google Scholar]
- 40.van Gameren MM, Willemse PH, Mulder NH, Limburg PC, Groen HJ, Vellenga E, et al. Effects of recombinant human interleukin-6 in cancer patients: a phase I-II study. Blood. 1994;84(5):1434–41. [PubMed] [Google Scholar]
- 41.Veldhuis GJ, Willemse PH, Sleijfer DT, van der Graaf WT, Groen HJ, Limburg PC, et al. Toxicity and efficacy of escalating dosages of recombinant human interleukin-6 after chemotherapy in patients with breast cancer or non-small-cell lung cancer. J Clin Oncol. 1995;13(10):2585–93. [DOI] [PubMed] [Google Scholar]
- 42.Weber J, Yang JC, Topalian SL, Parkinson DR, Schwartzentruber DS, Ettinghausen SE, et al. Phase I trial of subcutaneous interleukin-6 in patients with advanced malignancies. J Clin Oncol. 1993;11(3):499–506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All source data reported in this study are available by contacting the ACCELERATE team at accelerate@uphs.upenn.edu.